

Abstract
Background
Plant height is one of the most important agronomic traits in many crops due to its influence on lodging resistance and yield performance. Although progress has been made in the use of dwarfing genes in crop improvement, identification of new dwarf germplasm is still of significant interest for breeding varieties with increased yield.
Results
Here we describe a dominant, dwarf mutant G7 of Brassica napus with down-curved leaves derived from tissue culture. To explore the genetic variation responsible for the dwarf phenotype, the mutant was crossed to a conventional line to develop a segregating F2 population. Bulks were formed from plants with either dwarf or conventional plant height and subjected to high throughput sequencing analysis via mutation mapping (MutMap). The dwarf mutation was mapped to a 0.6 Mb interval of B. napus chromosome C05. Candidate gene analysis revealed that one SNP causing an amino acid change in the domain II of Bna.IAA7.C05 may contribute to the dwarf phenotype. This is consistent with the phenotype of a gain-of-function indole-3-acetic acid (iaa) mutant in Bna.IAA7.C05 reported recently. GO and KEGG analysis of RNA-seq data revealed the down-regulation of auxin related genes, including many other IAA and small up regulated response (SAUR) genes, in the dwarf mutant.
Conclusion
Our studies characterize a new allele of Bna.IAA7.C05 responsible for the dwarf mutant generated from tissue culture. This may provide a valuable genetic resource for breeding for lodging resistance and compact plant stature in B. napus.
Keywords: Brassica napus, Dwarf, IAA7, MutMap, Auxin, Tissue culture
Background
Plant height is a key trait related to lodging resistance, harvest index and fertilizer response [1]. Within important crops, such as wheat and rice, the green revolution brought about significant increases in yield by combining the breeding of high yielding dwarf varieties with agricultural mechanisation and fertilizer application [2]. Extreme dwarf varieties are usually correlated with poor agronomic performance and traits such as smaller grains, excessive tillering, or narrow leaves [3, 4], therefore semi-dwarf varieties are used to enhance yield and lodging resistance [5, 6].
Specific genes contributing to plant height are widely used in crop improvement. In rice, the semi-dwarf gene sd1, which regulates a key step in gibberellic acid (GA) biosynthesis, has been used worldwide in rice production [5]. The use of Rht (Reduced height) genes, involved in GA signaling transduction, was instrumental in bringing about the “green revolution” in wheat as well as other crops [7]. Many phytohormones, including GA, brassinosteriod (BR), strigolactone (SL), auxin, abscisic acid (ABA) and ethelyne (ETH), have been reported to influence crop height [3, 8–12]. Additional dwarfing genes involved in other pathways have also been shown to determine plant height [13]. The mechanisms underlying this complex trait are still largely not understood.
B. napus (oilseed rape) is one of the most important oilseed crops in China and the second most important oilseed worldwide [14]. Plant height is a key agronomic trait for rapeseed production as its heavy canopy makes it prone to lodging. Dwarfing can increase both lodging resistance and yield performance [15]. Within B. napus several dwarf mutants have been identified and the causal genes have been cloned. Dwarf gene BREIZH, derived from oilseed rape through chemical mutagenesis, was mapped by RAPD and RFLP markers [16, 17]. Dwarf gene Brrga1-d, encoding a DELLA protein, was first identified in the diploid, Brassica rapa, before being transferred to B. napus [18, 19]. Genetic analysis showed that mutation of Brrga1-d alters GA signaling pathway thus reducing plant height [20]. Semi-dwarf gene DS-1, mapped to chromosome A06, also encodes a DELLA protein. The single amino acid substitution of proline to leucine in the VHYNP motif causes a gain-of-function mutation in GA signaling [21]. Another semi-dwarfing gene, ds-3, encoding a mutant DELLA protein, also has the substitution of proline to leucine in the conserved VHYNP motif [6]. The recessive dwarfing gene, BnaC.dwf, was demonstrated to be insensitive to exogenous GA3 [22]. Dwarf mutant “NDF-1” in B. napus was found to be controlled by one major gene with three base pair mutations in the pyrimidine box of GID1 promoter [23]. Another dwarf mutant with down-curved leaf (Bndwf/dcl1) was mapped to a 175 kb region on B. napus chromosome C05 [15].
Auxin regulates many aspects of plant development [24]. Auxin signaling controlled by ARFs and Aux/IAA has been well studied in Arabidopsis [25]. Under low auxin concentration, Aux/IAA proteins interact and inhibit the activity of AUXIN RESPONSE FACTOR (ARFs), thereby repressing the auxin response gene expression [26]. At high intracellular auxin concentrations, auxin is perceived by TIR1/AFB1–3 receptors and the Aux/IAA proteins are then degraded by the ubiquitin-proteasome pathway. This releases the repression of ARFs and auxin response genes are activated [25]. Loss-of-function mutants of any of the twelve Aux/IAA genes in Arabidopsis do not show an obvious phenotype [26]. However, amino acid mutation in the conserved motif of domain II in Aux/IAA proteins causes dramatic gain-of-function phenotypes [27]. Gain-of-function mutants of 10 out of 29 Arabidopsis Aux/IAA, including iaa1, 3, 6, 7, 12, 14, 17, 18, 19 and 28, have been reported [28–39]. All mutants were caused by one amino acid substitution in the conserved GWPPV motif of domain II. This leads to reduced TIR binding, disrupting the degradation of Aux/IAA and therefore increasing the suppression of ARFs [40, 41].
In the present study, one dominant dwarf mutant was identified during the genetic transformation in B. napus. Transgenic element and expression results showed that this effect was not due to T-DNA insertion and therefore was most likely caused by somatic mutation generated during tissue culture. Following the construction of a segregating population, we performed MutMap to map the candidate region to one 0.6 Mb interval on chromosome BnaC05. Further, phenotype and correlation analysis of this region was consistent with the region for a B. napus dwarfism mutant reported recently [42, 43]. Eighteen candidate genes in this region were found to contain non-synonymous SNPs within coding regions. A single nucleotide substitution (G to A) in the conserved domain II of candidate gene Bna.IAA7.C05 resulted in changing the GWPPV motif to EWPPV. A C to T substitution in the conserved domain II of BnaIAA7 changing the GWPPV motif to GWLPV has been shown to cause a dwarfism phenotype recently [42, 43]. Thus, we speculated that another allelic mutation in the conserved domain II of Bna.IAA7.C05 leads to the dwarfism phenotype of G7. Exploitation of this dwarf mutant, with a compact plant stature and reduced plant height phenotype, will be valuable to assist breeding lodging resistant varieties.
Results
Generation and phenotype of the G7 dwarf mutant
To determine the function of miR169d in B. napus, we previously performed overexpression of miR169d in oilseed rape using the vector illustrated in Fig. 1a. We obtained 14 transgenic plants after genetic transformation, only one of which exhibited a dwarf phenotype with down-curved leaves. Detection for NPT and NOS presence within the vector (Fig. 1a) showed that the dwarf mutant contained no transgenic element (Fig. 1b), indicating this phenotype was not caused by overexpression of miR169d. Stem-loop RT-qPCR to check expression level of miR169d showed no significant increase in expression (Fig. 1c). Therefore, we hypothesized that this mutant was generated during the tissue culture process.
Fig. 1.
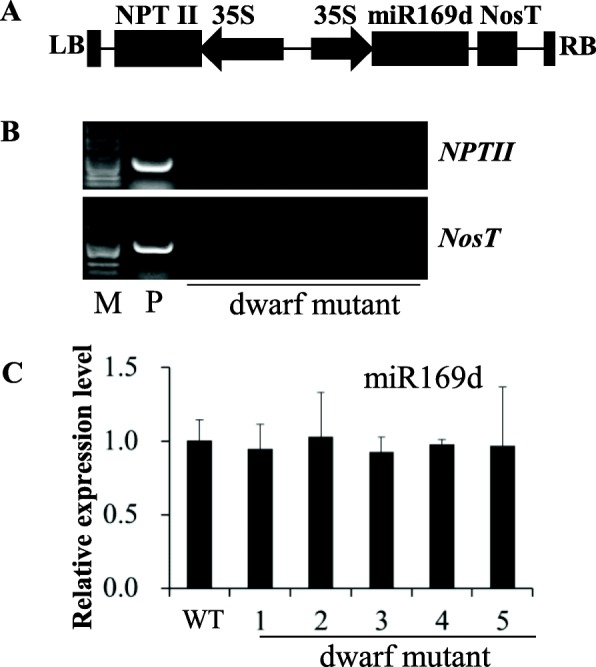
a. Schematic representations of the key components of the transformation vector. b. Transgenic elements kanamycin (NPTII) and NosT could not be detected in dwarf mutants. P. positive transgenic plant. M. marker. c. Expression level detection of miR169d by RT-qPCR showed no difference between G7 dwarf mutants and WT
This dwarf mutant displayed down-curved leaves and reduced height compared to WT at seedling stage (Fig. 2a). Following the floral transition, mutant plants exhibited a compact and significantly dwarf plant stature (Fig. 2b). Plant height of the dwarf mutant was significantly shorter (~ 30 cm) than WT (~ 150 cm) due to reduction of internode length. The leaves of this dwarf mutant became slightly crinkled and down-curved (Fig. 2c). Microscopy revealed that the cell size in dwarf plant leaves (Fig. 2e) was significantly decreased compared to WT (Fig. 2d).
Fig. 2.

Wild-type and mutant plant phenotype in a. the field after one months growth and b. the growth chamber. Left = WT, right = G7 dwarf mutant. c. The leaves of the G7 dwarf mutant displayed a down curved phenotype. Anatomy of leaves in WT (d) and mutant plants (e). Bar = 0.1 mm showed significant differences in cell size
Inheritance of the dwarf phenotype
The dwarf mutant, henceforth known as “G7”, and the WT variety “48,557” were used to construct a segregating population. The resulting F1 plants (48,557 × G7) exhibited down-curved leaves and crinkled phenotype, indicating the G7 mutation was controlled by a dominant gene. Mutant G7 displayed curled and wavy leaves and the yield-related traits were decreased compared with normal variety 48,557. The F1 plants showed intermediate plant height between two parents. Though the branch number and total silique number was decreased, higher density of pod layer were observed in the F1 plants. Plants from the F2 population could be divided into two distinct groups: WT (tall plants with normal leaves) and those displaying the G7 phenotype (dwarf plants with down-curved leaves). At seedling stage, 255 plants from the F2 population contained 190 G7 plants to 65 WT plants. A Chi-squared test revealed that this segregation pattern agreed with the 3:1 Mendelian segregation ratio (P = 0.857 > 0.05, χ2 0.05 = 0.0327 < χ2 0.05 = 3.842). Twenty-four G7 mutant plants from the F2 population died before flowering. Plant height of the remaining F2 individuals displayed a bimodal distribution (Fig. 3) again demonstrating a 3:1 Mendelian segregation ratio (P = 0.2443 > 0.05, χ2 0.05 = 1.356 < χ2 0.05 = 3.842). Therefore, we considered that the G7 dwarfism phenotype is likely controlled by one single dominant gene.
Fig. 3.
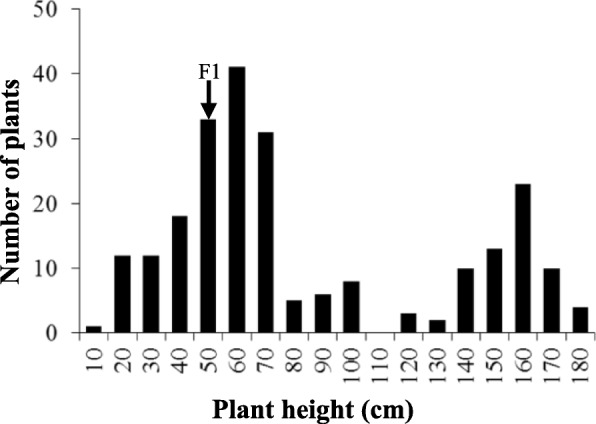
Distribution of plant height in the F2 population derived from the cross of G7 and 48,557 showing bimodal distribution. Arrow indicated the plant height of F1 from dwarf mutant G7 and normal plant 48,557 which was about 50 to 60 cm
Identification of candidate genomic region by MutMap
Genomic DNA of the two parents (G7 and 48,557) and the two pools (dwarf-pool and WT-pool) was sequenced, resulting in 510,946,104, 523,601,254, 438,302,148 and 411,082,480 clean reads, respectively. After aligning clean reads with the reference genome sequence, we acquired 232,279,673 and 215,670,613 unique mapped reads from dwarf-pool and WT-pool respectively, corresponding to 53 and 52.46% coverage of the genome. Ultimately, 5,388,850 and 5,370,965 SNPs were identified between the two DNA pools and the reference genome. After calculating the SNP-index from the dwarf-pool and WT-pool, the Δ SNP-index was plotted against the A and C sub-genome positions (Fig. 4a). At 99% significance level, one significant locus for dwarf phenotype on chromosome C05 (from 28.0–28.6 Mb) was identified with a peak Δ SNP-index value (Fig. 4b).
Fig. 4.
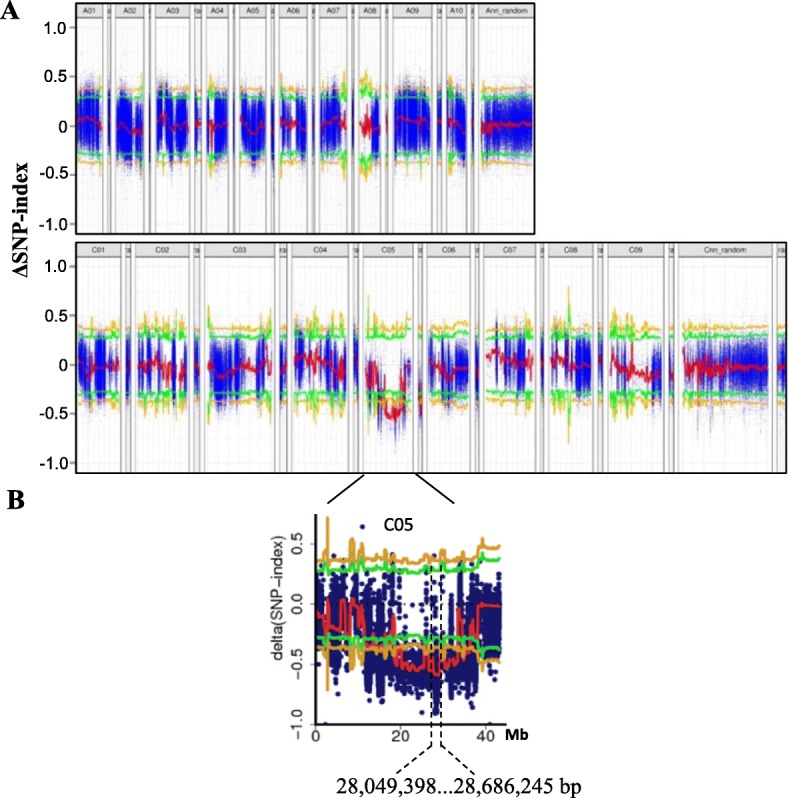
a. Plots showing the ΔSNP-index value (blue) and the average value of the ΔSNP-index for all SNP within a 4 Mb sliding window (red) for all chromosome in the B. napus A sub-genome (upper panel) and C sub-genome (middle panel). Green and yellow lines indicates the 95% (P < 0.05) and 99% confidence level (P < 0.01). b. The candidate region on chromosome C05 showing a ΔSNP-index peak between 28,049,398 to 28,686,245 bp
Candidate gene identification by MutMap
Within the candidate region on BnaC05, SNPs resulting in amino acid variation were identified within 18 candidate genes (Additional file 1: Table S1). Putative gene function prediction revealed none of the candidate genes had been implicated in the control of plant architecture except BnaC05g29300D, a homolog of AtIAA7 in Arabidopsis. The SNP in BnaC05g29300D results in a glycine to glutamic acid (G to E) amino acid substitution in Bna.IAA7.C05 (Fig. 5a) within conserved domain II resulting in the core sequence change from GWPPV to EWPPV. Many auxin gain-of-function mutant alleles of aux/iaa which exhibit morphological abnormalities, including decreased apical dominance, reduced plant height and severe stunting, have been reported in Arabidopsis. Thus, this gene is the most likely candidate gene for the G7 dwarf mutation. Expression analysis showed that Bna.IAA7.C05 gene was constitutively expressed in leaf, flower, stem and pod (Fig. 5b). No obvious difference of expression level was detected between the mutant and wild type (Fig. 5b).
Fig. 5.
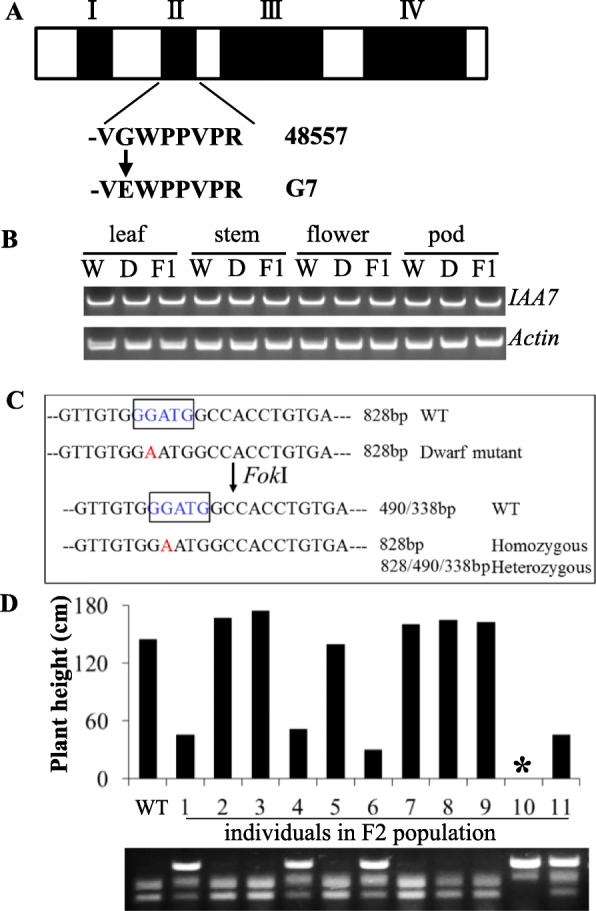
a. IAA7 gene structure is divided into four domains. A SNP identified between the G7 dwarf mutant and WT caused a G (WT) to E (G7 mutant) amino acid substitution within domain II of Bna.IAA7.C05. b. Gene expression of Bna.IAA7.C05 between WT (W), dwarf mutant (D) and F1 lines across multiple tissues was detected by RT-PCR. BnaActin was used as control. c. Variation within the recognition site of restriction enzyme FokI (blue boxed sequence) was used to develop a dCAPs marker for the SNP within Bna.IAA7.C05. d. Plant height of F2 individuals co-segregated with the dCAPs marker (* plant died at pod maturation)
Dwarf phenotype co-segregates with the dCAPs marker for SNP variation in Bna.IAA7.C05
The Bna.IAA7.C05 gene has been cloned from two dwarf EMS mutants and the function confirmed by genetic transformation [42, 43]. Sequence analysis showed that the dwarf phenotype of both these mutants were due to amino acid mutation in the GWPPV motif (GWPPV to GWLPV) of Bna.IAA7, thus leading to the same dwarfism phenotype. Based on the SNP within Bna.IAA7.C05, we developed a dCAPs marker to classify the individual type from F2 population (Fig. 5c). This marker co-segregated with plant height (Fig. 5d) and therefore is a suitable early diagnostic tool for the G7 dwarf mutation.
RNA-seq analysis revealed enrichment of the auxin signaling pathway in the G7 dwarf mutant
To further understand the regulatory mechanism, we performed transcriptome analysis of the G7 dwarf and WT plants. A total of 9516 differentially expressed genes (DEGs) were found between G7 and 48,557 (Fig. 6a). Within the biological process classification, auxin activated signaling pathway was detected to be overrepresented (Fig. 6b). To investigate the potential role of DEGs, we performed KEGG (Kyoto Encyclopedia of Genes and Genomes) analysis. Five processes including cellular, metabolism, genetic environment and organismal were found to be enriched for DEGs (Fig. 6c). We also found that the signaling transduction pathway category was overrepresented in environment processes (Fig. 6c).
Fig. 6.

Transcriptome analysis of dwarf mutant and wild type.a. Differentially expressed genes detected between G7 and westar. b. GO analysis of DEG showing enrichment of the auxin-activated signaling pathway. c. KEGG analysis of DEGs showing enrichment of signal transduction within the environment process category
Auxin plays a major role in regulating numerous processes for plant growth and development. Mutation of domain II of IAA proteins significantly suppressed auxin induced expression of other IAA genes in Arabidopsis [41]. In the present study, we found that 12 IAA genes were downregulated in the G7 dwarf mutant compared with WT (Fig. 7a). The expression of small auxin up RNAs (SAUR) was triggered by auxin. Overexpression of various Arabidopsis SAURs leads to induction of cell elongation and growth [44]. The expression level of many SAURs genes was also suppressed in the dwarf mutant (Fig. 7b).
Fig. 7.

The heatmap of genes that are involved in auxin signaling transduction pathway showing up-regulation (purple) and down regulation (yellow) in dwarf mutants (D1, D2, D3) and WT (W1, W2, W3). Color key represents log2 transformed FPKM (fragments per kilo base of exon per million fragments mapped) values, from low (yellow) to high (purple). SAUR (small auxin up RNAs)
Discussion
Many studies have been performed for plant height, but the application of dwarfing genes is still rare. Newly identified genes for dwarfism offered important sources of variation for breeding crops with lodging resistance and compact plant architecture [45]. Dwarfism and compact mutants have also been shown to possess improved response to fertilizer applications [1]. In the present study, one dwarf mutant was casually obtained in tissue culture when we conducted miR169d overexpression transformation. Genetic variation can be induced by chemical mutagens and physical treatment or tissue culture [46], therefore tissue culture alone or combined with chemical and biological agents can be utilized to increase genetic variability and provide resources for new commercial cultivar production [47, 48]. Within tissue culture itself, mutations can arise from calli, organ cultures, protoplasts and somatic embryogenesis [48]. Mutants induced from somatic variation have been successfully utilized to create potential new varieties in potato, millet and other crops [49–51]. The dwarf mutant generated from tissue culture in our study has the potential for breeding varieties with increased lodging resistance and improved fertilizer response.
MutMap can be used to easily screen and map genes to precise location by sequencing of DNA pools and has been successfully used in many species, such as Arabidopsis, rice, cucumber and oilseed rape [45, 52, 53]. In the present study, one significant locus with a peak Δ SNP-index value for the G7 dwarf phenotype on chromosome C05 (28.0–28.6 Mb) was identified by MutMap (Fig. 4b). A new dwarf mutant Bndwf/dcl1 from EMS-mutagenesis has been recently reported. Mutant Bndwf/dcl1 displayed a sharply down-curved and crinkled phenotype with short petioles at the seedling stage [15]. In the present study, we also observed the same phenotype in the G7 mutant. Bndwf/dcl1 has been mapped to a 6.58-cM interval on BnaC05. Further mapping narrowed the interval of Bndwf/dcl1 to 175 kb (C05: 29.76–29.94 Mb) in length. This region is adjacent to the peak of the ΔSNP-index value in our study. Putative gene function prediction revealed that one of candidate genes, BnaC05g29300D encoding IAA7, has been reported to cause dwarfing and other plant architecture variation in Arabidopsis. Meanwhile, fine mapping of two EMS dwarf mutant also identified causal variation within Bna.IAA7.C05 [42, 43]. Sequence analysis revealed that these two mutants both have a SNP variation causing an amino acid change in the conserved GWPPV motif (GWPPV to GWLPV) of domain II in Bna.IAA7.C05. In present study, the nucleotide variation in conserved motif leads to change of GWPPV to EWPPV. One another EMS mutant of IAA7 gene in B. napus A3 genome also has one SNP in the conserved GWPPV domain (GWPPV to EWLPV) [54]. Thus, this is an allelic mutation in the conserved GWPPV motif of IAA7 genes in B. napus that caused dwarfism phenotype.
In Arabidopsis, the AUX/IAA family contains four conserved domains and exhibit strong gene redundancy, with even triple loss-of-function mutants showing no defects in plant phenotype [26]. Conversely, AUX/IAA gain-of-functional mutations reveal a dramatic change to development as compared to WT. Many gain-of-function mutants of Aux/IAA have been reported in Arabidopsis [39], including those caused by single amino acid substitutions in the conserved GWPPV motif of domain II [55–57]. Gain-of-function mutation of IAA mutants in Arabidopsis exhibit decreased apical dominance with shorter stems. The dCAPs marker developed from the sequence variation in the GWPPV motif co-segregated with the dwarf phenotype of G7. Thus, we hypothesize that the dwarf phenotype with down-curved leaf of G7 is most likely to be caused by the G to E amino acid substitution in the GWPPV motif of Bna.IAA7.C05.
In wild type plants, auxin will combine with TIR (auxin-transport inhibitor response) when auxin concentration is increased, causing Aux/IAA ubiquitination and degradation. This releases ARFs and eventually their expression is promoted [24]. However, the amino acid change in the GWPPV motif reduces the TIR binding activity and the degradation of Aux/IAA is disrupted, resulting in Aux/IAA protein accumulation and increased repression of ARFs [40]. Small auxin up RNAs (SAUR), are the largest gene family triggered by auxin [58]. SAUR proteins are involved in cell expansion, growth and development of plants [59–61]. In present study, auxin-induced response genes, including many SAURs, were suppressed in the dwarf mutant (Fig. 7b). The low expression level of ARFs and other auxin response genes eventually leads to auxin overproduction and developmental defects.
Conclusions
In summary, a gain-of-function mutant with dwarfism and down-curved leaf was isolated from tissue culture processes. The candidate region for dwarfism was mapped to a 0.6 Mb region of B. napus chromosome C05 through the MutMap method. These results are consistent with previous dwarf mutant mapping results reported in B. napus. Further candidate genes analysis revealed that one amino acid substitution from G to E in in the conserved motif GWPPV of Bna.IAA7.C05 might also lead to dwarfism phenotype. This mutation of Bna.IAA7.C05 resulted in decreased expression of other IAA and auxin response genes expression. Our findings identified one new allele of Bna.IAA7.C05 responsible for plant dwarf phenotype and provide insights for understanding a dominant dwarfism mutant in B. napus.
Methods
Plant materials and phenotyping for plant height
The B. napus dwarf mutant was originally isolated from tissue culture (Gansu Academy of Agricultural Sciences). A cross was made between 48,557 (wild type, female parent) and the G7 dwarf (pollen donor) to create F1 plants which were subsequently selfed to create a segregating F2 population. All plant materials were grown at the field in Oil Crops Research Institute of the Chinese Academy of Agricultural Sciences (OCRI-CAAS), Wuhan, China. Plant height was measured when plants attained maximum height at the final flowering stage. DNA samples from 50 of 277 F2 plants with the dwarf phenotype were mixed to form the dwarf bulk, and from 50 tall plants to form the tall bulk for MutMap analysis as described by [52].
Microscopy analysis
Stem segment of Brassica napus at the early flowering time stage was fixed by 50% FAA (Formalin–acetic acid–alcohol) solution. Samples were then embedded in Paraffin Plus after dehydration and infiltration steps. Tissues were sliced into about 8 to 10 μm (Leica RM2265) and stained by 0.05% toluidine blue. Image was observed under microscope (Nikon).
Generation and analysis of NGS data of MutMap analysis
DNA was extracted by DNA sample preparation Kits (Tiangen, Beijing, China) according to the manufacturer’s instructions. The quantification and quality verification was detected by Nanodrop one (Massachusetts, America). About 2 μg genomic DNA from two DNA bulks or two parents were prepared for sequencing library construction. Sequence data were generated by Illumina HiSeq X ten (San Diego, California, USA) with paired-end (PE150) and 350 bp of reads length. Sequence quality and adaptor trimming was conducted by SOAPnuke 1.4 (BGI, Shenzhen, China) and analyzed by Benagen company (Wuhan, China) for both DNA bulks and parental lines. Raw reads including adapter sequences and low-quality reads were removed after processed by Trimmomatic software [45]. Then clean reads with high quality were then aligned to the B. napus reference genome by the BWA (Burrows-Wheeler Aligner) software [62–64]. BAM files were created from alignment file by using SAMtools software [65].
MutMap analysis to determine candidate gene variation
SNP calling and SNP index was performed as previous study reported [52]. After aligned GFF3 files to the B. napus genome with ANNOVAR, homozygous SNPs between the dwarf mutant and the normal line were realized from VCF files generated by Variant Filtration-GATK software using defaulting settings [45, 66]. SNP index is the ratio of reads with SNP (harboring nucleotide different to reference genome) to the total reads contained the SNP [52]. The Δ SNP-index was obtained by subtracting the SNP-index between dwarf and normal DNA pools. Sliding window method was conducted to detect the SNP-index across the B. napus genome [45]. Δ SNP-index across the chromosome of B. napus genome was performed by sliding-window analysis with 1 Mb window size and 10 kb step size [45]. Statistical confidence intervals of the ΔSNP index were defined at 95 and 99% following the description reported before [53, 67].
Expression analysis of dwarfing candidate genes by semi-quantitative RT-PCR and RT-qPCR
To detect expression pattern of candidate gene, different samples including stem, leaf and floral bud, were taken from five individuals from dwarf mutant G7 and Westar. RNA was extracted by RNAprep Pure kit (Tiangen, China). After quality detection, reverse transcription was conducted by using FastQuant RT kit according to the instruction (Tiangen, China). Semi-quantitative RT-PCR for gene Bna.IAA7.C05 (BnaC05g29300D) was performed for 32 cycles by using the primers (AHC5F and AHC5R) listed in Additional file 2: Table S2. BnaActin gene (BnaC02g00690D) from B.napus was used as the control for the RNA sample. The reaction of semi-quantitative RT-PCR was performed for 32 cycles, with 30 s at 95 °C, 45 s at 57 °C and 50 s at 72 °C. Stem-loop RT-qPCR was used to examine miR169d expression level according to previous method [68]. Stem-loop qRT-PCR was performed in CFX96 Real Time System (Bio-Rad, Hercules, California, USA) using SYBR Green mix (Transgen Biotech, Beijing). The reactions were performed as following program: 30 s at 95 °C, 40 cycles of 5 s at 95 °C, and 30 s at 60 °C. Primers used for stem-loop RT-qPCR were listed in Additional file 2: Table S2.
RNA-seq of differential gene expression
Seedlings of G7 dwarf and WT (Westar) were selected to extract the RNA. Samples were collected and frozen in liquid nitrogen and stored at − 70 °C for RNA preparation. Total RNA from bulked samples was extracted in accordance with the manufacturer protocol (RNA-kit Tiangen, China). The integrity of the total RNA was detected by 1% agarose gel electrophoresis. All RNA samples were detected by Nanodrop 2000 to analyze A260/A280 value for protein contamination and A230/A280 value for reagent contamination. The concentration was detected by Nano-Drop (Thermo Scientific, La Jolla, CA, USA) and purity of RNA was also checked by Agilent 2100 Bio-analyzer (Agilent-USA). 10 μl RNA sample was used to construct the sequencing library according to manual instruction. Sequence data was generated by Illumina HiSeq X ten (San Diego, California, USA) with paired-end (PE150) and 350 bp of reads length. Sequence quality and adaptor trimming was conducted by SOAPnuke 1.4 (BGI, Shenzhen, China). Clean reads were mapped to the reference genome of B. napus by BWA software [62–64].
Transcriptome analysis was mostly conducted as the description in the previous study [68]. False discovery rate (FDR) cutoff of less than 0.01 was used to determine the DEGs. The absolute value of log2 Ratio ≥ 2 between different samples with FDR ≤ 0.001 were determined as DEGs by using DEseq2 software [67]. All annotated genes were mapped to the GO and KEGG database. DEGs in the KEGG pathway were enriched by KOBAS software [69]. GO annotation was performed by Blast2GO software [70]. For each sample, three biological replicates were conducted. The value of log2 transformed FPKM value was used to create heat maps. Color key from yellow to purple represented the expression level with log2 transformed FPKM values. Negative and positive values represented the low and high level, respectively. For each gene, the log2 transformed FPKM value was normalized in each row.
Statistical analysis
The analysis of RT-qPCR results was carried out by Student’s t-test (P < 0.05) or (P < 0.01). The RT-qPCR was performed for three biological repeats.
Supplementary information
Additional file 1: Table S1. Genes on the candidate region of chromosome C05 of Brassica napus.
Additional file 2: Table S2. Primers used in this study.
Acknowledgments
Not applicable.
Abbreviations
- DEGs
differentially expressed genes
- iaa
indole-3-acetic-acid
- Mutmap
mutation mapping
- SAUR
small up regulated response
Authors’ contributions
HTC, YD and QH designed and carried out the research. BLD, YD and HTC investigated the plant height and extracted the DNA. MYH and FWJ performed the expression pattern analysis. YH and YW analyzed the data. HTC wrote the manuscript. QZ, RW and QH revised the manuscript. All authors read and approved.
Funding
This work was supported by the Agricultural Science and Technology Innovation Project of Gansu Academy of Agricultural Sciences (2018GAAS04), the Key program of Technological Innovation in Hubei Province (No. 2016ABA084), the Earmarked Fund for Gansu Agriculture Research System, Open Research Fund of Key Laboratory for Biological Sciences and Genetic Improvement of Oil Crops (Ministry of Agriculture) (KF2017010), Fundamental Research Funds for Central Non-profit Scientific Institution (1610172017005). The funding bodies were involved in the material creation, designing the study, data collection and writing the manuscript.
Availability of data and materials
All data in this study are included in this published article and its supplementary information files Additional file 1: Table S1 and Additional file 2: Table S2. Sequences data of RNA-seq were deposited to the NCBI Sequence Read Archive (SRA) repository under accession number (SRR10090703, SRR10090704, SRR10090705 for Westar sample; SRR10090706, SRR10090707, SRR10090708 for G7 mutant sample). Genome re-sequencing data could be achieved under accession number (SRR10189121, SRR10189122 for 48557 sample; SRR10189123, SRR10189124 for G7 sample).
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Hongtao Cheng and Fenwei Jin contributed equally to this work.
Contributor Information
Hongtao Cheng, Email: chenghongtao@caas.cn.
Fenwei Jin, Email: jinfwei@gsagr.ac.cn.
Qamar U. Zaman, Email: qamaruzamanch@gmail.com
Bingli Ding, Email: dingbl91@163.com.
Mengyu Hao, Email: haomengyu@caas.cn.
Yi Wang, Email: wangyi@gsagr.ac.cn.
Yi Huang, Email: huangyi@caas.cn.
Rachel Wells, Email: rachel.wells@jic.ac.uk.
Yun Dong, Email: dongyungs@163.com.
Qiong Hu, Phone: +86-27-8671-7152, Email: huqiong01@caas.cn.
Supplementary information
Supplementary information accompanies this paper at 10.1186/s12870-019-2094-2.
References
- 1.Khush GS. Green revolution: the way forward. Nat Rev Genet. 2001;2:815–822. doi: 10.1038/35093585. [DOI] [PubMed] [Google Scholar]
- 2.Hargrove TR, Cabanilla VL. The impact of Semidwarf varieties on Asian Rice-breeding programs. Bioscience. 1979;29:731–735. doi: 10.2307/1307667. [DOI] [Google Scholar]
- 3.Tanabe S, Ashikari M, Fujioka S, Takatsuto S, Yoshida S, Yano M, Yoshimura A, Kitano H, Matsuoka M, Fujisawa Y, Kato H, Iwasaki Y. A novel cytochrome P450 is implicated in brassinosteroid biosynthesis via the characterization of a rice dwarf mutant, dwarf11, with reduced seed length. Plant Cell. 2005;17:776–790. doi: 10.1105/tpc.104.024950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ueguchi-Tanaka M, Ashikari M, Nakajima M, Itoh H, Katoh E, Kobayashi M, Chow TY, Hsing YI, Kitano H, Yamaguchi I, Matsuoka M. GIBBERELLIN INSENSITIVE DWARF1 encodes a soluble receptor for gibberellin. Nature. 2005;437:693–698. doi: 10.1038/nature04028. [DOI] [PubMed] [Google Scholar]
- 5.Hedden P. The genes of the green revolution. Trends Genet. 2003;19:5–9. doi: 10.1016/S0168-9525(02)00009-4. [DOI] [PubMed] [Google Scholar]
- 6.Zhao B, Li H, Li J, Wang B, Dai C, Wang J, Liu K. Brassica napus DS-3, encoding a DELLA protein, negatively regulates stem elongation through gibberellin signaling pathway. Theor Appl Genet. 2017;130(4):727–741. doi: 10.1007/s00122-016-2846-4. [DOI] [PubMed] [Google Scholar]
- 7.Salamini F. Plant biology. Hormones and the green revolution. Science. 2003;302:71–72. doi: 10.1126/science.1090811. [DOI] [PubMed] [Google Scholar]
- 8.Ashikari M, Sasaki A, Ueguchi-Tanaka M, Itoh H, Nishimura A, Datta S, Ishiyama K, Saito T, Kobayashi M, Khush GS. Loss-of-function of a rice gibberellin biosynthetic gene, GA20 oxidase (GA20ox-2), led to the rice ‘green revolution’. Breed Sci. 2002;52:143–150. doi: 10.1270/jsbbs.52.143. [DOI] [Google Scholar]
- 9.Ueguchi-Tanaka M, Fujisawa Y, Kobayashi M, Ashikari M, Iwasaki Y, Kitano H, Matsuoka M. Rice dwarf mutant d1, which is defective in the alpha subunit of the heterotrimeric G protein, affects gibberellin signal transduction. Proc Natl Acad Sci U S A. 2000;97:11638–11643. doi: 10.1073/pnas.97.21.11638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hong Z, Ueguchi-Tanaka M, Umemura K, Uozu S, Fujioka S, Takatsuto S, Yoshida S, Ashikari M, Kitano H, Matsuoka M. A rice brassinosteroid-deficient mutant, ebisu dwarf (d2), is caused by a loss of function of a new member of cytochrome P450. Plant Cell. 2003;15:2900–2910. doi: 10.1105/tpc.014712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Itoh H, Tatsumi T, Sakamoto T, Otomo K, Toyomasu T, Kitano H, Ashikari M, Ichihara S, Matsuoka M. A rice semi-dwarf gene, Tan-Ginbozu (D35), encodes the gibberellin biosynthesis enzyme, ent-kaurene oxidase. Plant Mol Biol. 2004;54:533–547. doi: 10.1023/B:PLAN.0000038261.21060.47. [DOI] [PubMed] [Google Scholar]
- 12.Lin H, Wang R, Qian Q, Yan M, Meng X, Fu Z, Yan C, Jiang B, Su Z, Li J, Wang Y. DWARF27, an iron-containing protein required for the biosynthesis of strigolactones, regulates rice tiller bud outgrowth. Plant Cell. 2009;21:1512–1525. doi: 10.1105/tpc.109.065987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu F, Wang P, Zhang X, Li X, Yan X, Fu D, Wu G. The genetic and molecular basis of crop height based on a rice model. Planta. 2018;247(1):1–26. doi: 10.1007/s00425-017-2798-1. [DOI] [PubMed] [Google Scholar]
- 14.Hu Q, Hua W, Yin Y, Zhang X, Liu L, Shi J, Zhao Y, Qin L, Chen C, Wang HZ. Rapeseed research and production in China. The Crop Journal. 2017;5:127–135. doi: 10.1016/j.cj.2016.06.005. [DOI] [Google Scholar]
- 15.Wang Y, Chen W, Chu P, Wan S, Yang M, Wang M, Guan R. Mapping a major QTL responsible for dwarf architecture in Brassica napus using a single-nucleotide polymorphism marker approach. BMC Plant Biol. 2016;16(1):178. doi: 10.1186/s12870-016-0865-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Foisset N, Delourme R, Barret P, Renard M. Molecular tagging of the dwarf BREIZH (Bzh) gene in Brassica napus. Theor Appl Genet. 1995;91(5):756–761. doi: 10.1007/BF00220955. [DOI] [PubMed] [Google Scholar]
- 17.Barret P, Delourme R, Foisset N, Renard M. Development of a SCAR (sequence characterised amplified region) marker for molecular tagging of the dwarf BREIZH (Bzh) gene in Brassica napus L. Theor Appl Genet. 1998;97:828–833. doi: 10.1007/s001220050962. [DOI] [PubMed] [Google Scholar]
- 18.Muangprom A, Mauriera I, Osborn TC. Transfer of a dwarf gene from Brassica rapa to oilseed B. napus, effects on agronomic traits, and development of a ‘perfect’ marker for selection. Mol Breed. 2006;17(2):101–110. doi: 10.1007/s11032-005-3734-9. [DOI] [Google Scholar]
- 19.Muangprom A, Osborn TC. Characterization of a dwarf gene in Brassica rapa, including the identification of a candidate gene. Theor Appl Genet. 2004;108(7):1378–1384. doi: 10.1007/s00122-003-1551-2. [DOI] [PubMed] [Google Scholar]
- 20.Muangprom A, Thomas SG, Sun TP, Osborn TC. A novel dwarfing mutation in a green revolution gene from Brassica rapa. Plant Physiol. 2005;137(3):931–938. doi: 10.1104/pp.104.057646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu C, Wang J, Huang T, Wang F, Yuan F, Cheng X, Zhang Y, Shi S, Wu J, Liu K. A missense mutation in the VHYNP motif of a DELLA protein causes a semi-dwarf mutant phenotype in Brassica napus. Theor Appl Genet. 2010;121(2):249–258. doi: 10.1007/s00122-010-1306-9. [DOI] [PubMed] [Google Scholar]
- 22.Zeng X, Zhu L, Chen Y, Qi L, Pu Y, Wen J, Yi B, Shen J, Ma C, Tu J, et al. Identification, fine mapping and characterisation of a dwarf mutant (bnaC.dwf) in Brassica napus. Theor Appl Genet. 2011;122(2):421–428. doi: 10.1007/s00122-010-1457-8. [DOI] [PubMed] [Google Scholar]
- 23.Li H, Wang Y, Li X, Gao Y, Wang Z, Zhao Y, Wang M. A GA-insensitive dwarf mutant of Brassica napus L. correlated with mutation in pyrimidine box in the promoter of GID1. Mol Biol Rep. 2011;38(1):191–197. doi: 10.1007/s11033-010-0094-2. [DOI] [PubMed] [Google Scholar]
- 24.Strader LC, Zhao Y. Auxin perception and downstream events. Curr Opin Plant Biol. 2016;33:8–14. doi: 10.1016/j.pbi.2016.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Weijers D, Wagner D. Transcriptional responses to the Auxin hormone. Annu Rev Plant Biol. 2016;67:539–574. doi: 10.1146/annurev-arplant-043015-112122. [DOI] [PubMed] [Google Scholar]
- 26.Lau S, Jurgens G, De Smet I. The evolving complexity of the auxin pathway. Plant Cell. 2008;20:1738–1746. doi: 10.1105/tpc.108.060418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mockaitis K, Estelle M. Auxin receptors and plant development: a new signaling paradigm. Annu Rev Cell Dev Biol. 2008;24:55–80. doi: 10.1146/annurev.cellbio.23.090506.123214. [DOI] [PubMed] [Google Scholar]
- 28.Uehara T, Okushima Y, Mimura T, Tasaka M, Fukaki H. Domain II mutations in CRANE/IAA18 suppress lateral root formation and affect shoot development in Arabidopsis thaliana. Plant Cell Physiol. 2008;49:1025–1038. doi: 10.1093/pcp/pcn079. [DOI] [PubMed] [Google Scholar]
- 29.Tian Q, Reed JW. Control of auxin-regulated root development by the Arabidopsis thaliana SHY2/IAA3 gene. Development. 1999;126:711–721. doi: 10.1242/dev.126.4.711. [DOI] [PubMed] [Google Scholar]
- 30.Kim BC, Soh MS, Kang BJ, Furuya M, Nam HG. Two dominant photomorphogenic mutations of Arabidopsis thaliana identified as suppressor mutations of hy2. Plant J. 1996;9:441–456. doi: 10.1046/j.1365-313X.1996.09040441.x. [DOI] [PubMed] [Google Scholar]
- 31.Liscum E, Reed JW. Genetics of aux/IAA and ARF action in plant growth and development. Plant Mol Biol. 2002;49:387–400. doi: 10.1023/A:1015255030047. [DOI] [PubMed] [Google Scholar]
- 32.Wilson AK, Pickett FB, Turner JC, Estelle M. A dominant mutation in Arabidopsis confers resistance to auxin, ethylene, and abscisic acid. Mol Gen Genet. 1990;222:377–383. doi: 10.1007/BF00633843. [DOI] [PubMed] [Google Scholar]
- 33.Nagpal P, Walker LM, Young JC, Sonawala A, Timpte C, Estelle M, Reed JW. AXR2 encodes a member of the aux/IAA protein family. Plant Physiol. 2000;123:563–573. doi: 10.1104/pp.123.2.563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hamann T, Mayer U, Jürgens G. The auxin-insensitive bodenlos mutation affects primary root formation and apical-basal patterning in the Arabidopsis embryo. Development. 1999;126:1387–1395. doi: 10.1242/dev.126.7.1387. [DOI] [PubMed] [Google Scholar]
- 35.Hamann T, Benkova E, Bäurle I, Kientz M, Jürgens G. The Arabidopsis BODENLOS gene encodes an auxin response protein inhibiting MONOPTEROS-mediated embryo patterning. Genes Dev. 2002;16:1610–1615. doi: 10.1101/gad.229402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fukaki H, Tameda S, Masuda H, Tasaka M. Lateral root formation is blocked by a gain-of-function mutation in the SOLITARY-ROOT/IAA14 gene of Arabidopsis. Plant J. 2002;29:153–168. doi: 10.1046/j.0960-7412.2001.01201.x. [DOI] [PubMed] [Google Scholar]
- 37.Leyser HMO, Pickett FB, Dharmasiri S, Estelle M. Mutations in the AXR3 gene of Arabidopsis result in altered auxin response including ectopic expression from the SAUR-AC1 promoter. Plant J. 1996;10:403–413. doi: 10.1046/j.1365-313x.1996.10030403.x. [DOI] [PubMed] [Google Scholar]
- 38.Rouse D, Mackay P, Stirnberg P, Estelle M. Leyser O. Changes in auxin response from mutations in an AUX/IAA gene: Science; 1998. [DOI] [PubMed] [Google Scholar]
- 39.Yang X, Lee S, So JH, Dharmasiri S, Dharmasiri N, Ge L, Jensen C, Hangarter R, Hobbie L, Estelle M. The IAA1 protein is encoded by AXR5 and is a substrate of SCFTIR1. Plant J. 2004;40:772–782. doi: 10.1111/j.1365-313X.2004.02254.x. [DOI] [PubMed] [Google Scholar]
- 40.Calderon-Villalobos LI, Tan X, Zheng N, Estelle M. Auxin perception--structural insights. Cold Spring Harb Perspect Biol. 2010;2:a005546. doi: 10.1101/cshperspect.a005546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rinaldi MA, Liu J, Enders TA, Bartel B, Strader LC. A gain-of-function mutation in IAA16 confers reduced responses to auxin and abscisic acid and impedes plant growth and fertility. Plant Mol Biol. 2012;79:359–373. doi: 10.1007/s11103-012-9917-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zheng M, Hu M, Yang H, Tang M, Zhang L, Liu H, Li X, Liu J, Sun X, Fan S, Zhang J, Terzaghi W, Pu H, Hua W. Three BnaIAA7 homologs are involved in auxin/brassinosteroid-mediated plant morphogenesis in rapeseed (Brassica napus L.). Plant Cell Rep. 2019. [DOI] [PMC free article] [PubMed]
- 43.Zhao B, Wang B, Li Z, Guo T, Zhao J, Guan Z, Liu K. Identification and characterization of a new dwarf locus DS-4 encoding an aux/IAA7 protein in Brassica napus. Theor Appl Genet. 2019;132(5):1435–1449. doi: 10.1007/s00122-019-03290-8. [DOI] [PubMed] [Google Scholar]
- 44.Stortenbeker N, Bemer M. The SAUR gene family: the plant's toolbox for adaptation of growth and development. J Exp Bot. 2019;70:17–27. doi: 10.1093/jxb/ery332. [DOI] [PubMed] [Google Scholar]
- 45.Wang H, Cheng H, Wang W, Liu J, Hao M, Mei D, Zhou R, Fu L, Hu Q. Identification of BnaYUCCA6 as a candidate gene for branch angle in Brassica napus by QTL-seq. Sci Rep. 2016;6:38493. doi: 10.1038/srep38493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bairu MW, Aremu AO, Van Staden J. Somaclonal variation in plants: causes and detection methods. Plant Growth Regul. 2011;63:147–173. doi: 10.1007/s10725-010-9554-x. [DOI] [Google Scholar]
- 47.Rasheed S, Tahira F, Khurram B, Tayyab H, Shiekh R. Agronomical and physiochemical characterization of somaclonal variants in Indica basmati rice. Pak J Biol Sci. 2003;6:844–848. doi: 10.3923/pjbs.2003.844.848. [DOI] [Google Scholar]
- 48.Orbović V, Ćalović M, Viloria Z, Nielsen B, Gmitter F, Castle W, Grosser J. Analysis of genetic variability in various tissue culture-derived lemon plant populations using RAPD and flow cytometry. Euphytica. 2008;161:329–335. doi: 10.1007/s10681-007-9559-3. [DOI] [Google Scholar]
- 49.Thieme R, Griess H. Somaclonal variation in tuber traits of potato. Potato Res. 2005;48:153–165. doi: 10.1007/BF02742373. [DOI] [Google Scholar]
- 50.Wang Y, Wang F, Zhai H, Liu Q. Production of a useful mutant by chronic irradiation in sweetpotato. Sci Hortic. 2007;111:173–178. doi: 10.1016/j.scienta.2006.10.007. [DOI] [Google Scholar]
- 51.Baer G, Yemets A, Stadnichuk N, Rakhmetov D, Blume Y. Somaclonal variability as a source for creation of new varieties of finger millet (Eleusine coracana (L.) Gaertn.) Cytol Genet. 2007;41:204–208. doi: 10.3103/S0095452707040020. [DOI] [PubMed] [Google Scholar]
- 52.Abe A, Kosugi S, Yoshida K, Natsume S, Takagi H, Kanzaki H, Matsumura H, Yoshida K, Mitsuoka C, Tamiru M, Innan H, Cano L, Kamoun S, Terauchi R. Genome sequencing reveals agronomically important loci in rice using MutMap. Nat Biotechnol. 2012;30:174–178. doi: 10.1038/nbt.2095. [DOI] [PubMed] [Google Scholar]
- 53.Takagi H, Tamiru M, Abe A, Yoshida K, Uemura A, Yaegashi H, Obara T, Oikawa K, Utsushi H, Kanzaki E, Mitsuoka C, Natsume S, Kosugi S, Kanzaki H, Matsumura H, Urasaki N, Kamoun S, Terauchi R. MutMap accelerates breeding of a salt-tolerant rice cultivar. Nat Biotechnol. 2015;33:445. doi: 10.1038/nbt.3188. [DOI] [PubMed] [Google Scholar]
- 54.Li H, Li J, Song J, Zhao B, Guo C, Wang B, Zhang Q, Wang J, King GJ, Liu K. An auxin signaling gene BnaA3.IAA7 contributes to improved plant architecture and yield heterosis in rapeseed. New Phytol. 2019;222:837–851. doi: 10.1111/nph.15632. [DOI] [PubMed] [Google Scholar]
- 55.Kepinski S, Leyser O. Auxin-induced SCFTIR1-aux/IAA interaction involves stable modification of the SCFTIR1 complex. Proc Natl Acad Sci U S A. 2004;101:12381–12386. doi: 10.1073/pnas.0402868101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kepinski S, Leyser O. Plant development: auxin in loops. Curr Biol. 2005;15:208–210. doi: 10.1016/j.cub.2005.03.012. [DOI] [PubMed] [Google Scholar]
- 57.Dharmasiri N, Dharmasiri S, Estelle M. The F-box protein TIR1 is an auxin receptor. Nature. 2005;435:441–445. doi: 10.1038/nature03543. [DOI] [PubMed] [Google Scholar]
- 58.Ren H, Gray WM. SAUR proteins as effectors of hormonal and environmental signals in plant growth. Mol Plant. 2015;8:1153–1164. doi: 10.1016/j.molp.2015.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chae K, Isaacs CG, Reeves PH, Maloney GS, Muday GK, Nagpal P, Reed JW. Arabidopsis SMALL AUXIN UP RNA63 promotes hypocotyl and stamen filament elongation. Plant J. 2012;71:684–697. doi: 10.1111/j.1365-313X.2012.05024.x. [DOI] [PubMed] [Google Scholar]
- 60.Spartz AK, Lee SH, Wenger JP, Gonzalez N, Itoh H, Inzé D, Peer WA, Murphy AS, Overvoorde PJ, Gray WM. The SAUR19 subfamily of SMALL AUXIN UP RNA genes promote cell expansion. Plant J. 2012;70:978–990. doi: 10.1111/j.1365-313X.2012.04946.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kong Y, Zhu Y, Gao C, She W, Lin W, Chen Y, Han N, Bian H, Zhu M, Wang J. Tissue-specific expression of SMALL AUXIN UP RNA41 differentially regulates cell expansion and root meristem patterning in Arabidopsis. Plant & Cell Physiology. 2013;54:609–621. doi: 10.1093/pcp/pct028. [DOI] [PubMed] [Google Scholar]
- 62.Li H, Durbin R. Fast and accurate short read alignment with burrows-wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chalhoub B, Denoeud F, Liu S, Parkin IA, Tang H, et al. Plant genetics. Early allopolyploid evolution in the post-Neolithic Brassica napus oilseed genome. Science. 2014;345:950–953. doi: 10.1126/science.1253435. [DOI] [PubMed] [Google Scholar]
- 64.Sun F, Fan G, Hu Q, Zhou Y, Guan M, Tong C, Li J, Du D, Qi C, Jiang L, Liu W, Huang S, Chen W, Yu J, Mei D, Meng J, Zeng P, Shi J, Liu K, Wang X, Wang X, Long Y, Liang X, Hu Z, Huang G, Dong C, Zhang H, Li J, Zhang Y, Li L, Shi C, Wang J, Lee SM, Guan C, Xu X, Liu S, Liu X, Chalhoub B, Hua W, Wang H. The high-quality genome of Brassica napus cultivar 'ZS11' reveals the introgression history in semi-winter morphotype. Plant J. 2017;92:452–468. doi: 10.1111/tpj.13669. [DOI] [PubMed] [Google Scholar]
- 65.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G. Durbin R; 1000 genome project data processing subgroup. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Takagi H, Uemura A, Yaegashi H, Tamiru M, Abe A, Mitsuoka C, Utsushi H, Natsume S, Kanzaki H, Matsumura H, Saitoh H, Yoshida K, Cano LM, Kamoun S, Terauchi R. MutMap-gap: whole-genome resequencing of mutant F2 progeny bulk combined with de novo assembly of gap regions identifies the rice blast resistance gene Pii. New Phytol. 2013;200:276–283. doi: 10.1111/nph.12369. [DOI] [PubMed] [Google Scholar]
- 68.Cheng Hongtao, Hao Mengyu, Wang Wenxiang, Mei Desheng, Wells Rachel, Liu Jia, Wang Hui, Sang Shifei, Tang Min, Zhou Rijin, Chu Wen, Fu Li, Hu Qiong. Integrative RNA- and miRNA-Profile Analysis Reveals a Likely Role of BR and Auxin Signaling in Branch Angle Regulation of B. napus. International Journal of Molecular Sciences. 2017;18(5):887. doi: 10.3390/ijms18050887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Mao XZ, Cai T, Olyarchuk JG, Wei LP. Automated genome annotation and pathway identification using the KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics. 2005;21(19):3787–3793. doi: 10.1093/bioinformatics/bti430. [DOI] [PubMed] [Google Scholar]
- 70.Conesa A, Götz S, García-Gómez JM, Terol J, Talón M, Robles M. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics. 2005;21:3674–3676. doi: 10.1093/bioinformatics/bti610. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: Table S1. Genes on the candidate region of chromosome C05 of Brassica napus.
Additional file 2: Table S2. Primers used in this study.
Data Availability Statement
All data in this study are included in this published article and its supplementary information files Additional file 1: Table S1 and Additional file 2: Table S2. Sequences data of RNA-seq were deposited to the NCBI Sequence Read Archive (SRA) repository under accession number (SRR10090703, SRR10090704, SRR10090705 for Westar sample; SRR10090706, SRR10090707, SRR10090708 for G7 mutant sample). Genome re-sequencing data could be achieved under accession number (SRR10189121, SRR10189122 for 48557 sample; SRR10189123, SRR10189124 for G7 sample).