

Abstract
Traumatic brain injury (TBI) induces cerebrovascular oxidative stress, which is associated with neurovascular uncoupling, autoregulatory dysfunction, and persisting cognitive decline in both pre-clinical models and patients. However, single mild TBI (mTBI), the most frequent form of brain trauma, increases cerebral generation of reactive oxygen species (ROS) only transiently. We hypothesized that comorbid conditions might exacerbate long-term ROS generation in cerebral arteries after mTBI. Because hypertension is the most important cerebrovascular risk factor in populations prone to mild brain trauma, we induced mTBI in normotensive and spontaneously hypertensive rats (SHR) and assessed changes in cytoplasmic and mitochondrial superoxide (O2-) production by confocal microscopy in isolated middle cerebral arteries (MCA) 2 weeks after mTBI using dihydroethidine (DHE) and the mitochondria-targeted redox-sensitive fluorescent indicator dye MitoSox. We found that mTBI induced a significant increase in long-term cytoplasmic and mitochondrial O2- production in MCAs of SHRs and increased expression of the nicotinamide adenine dinucleotide phosphate (NADPH) oxidase subunit Nox4, which were reversed to the normal level by treating the animals with the cell-permeable, mitochondria-targeted antioxidant peptide SS-31 (5.7 mg kg−1 day−1, i.p.). Persistent mTBI-induced oxidative stress in MCAs of SHRs was significantly decreased by inhibiting vascular NADPH oxidase (apocyinin). We propose that hypertension- and mTBI-induced cerebrovascular oxidative stress likely lead to persistent dysregulation of cerebral blood flow (CBF) and cognitive dysfunction, which might be reversed by SS-31 treatment.
Keywords: brain trauma, free radicals, hypertension, MCA, mitochondrion
Introduction
Traumatic brain injury (TBI) is a significant public health issue that often leads to lifelong deficits in brain function and disability. It has been suggested that pre-existing comorbid medical conditions exacerbate the deleterious effects of TBI, resulting in longer intensive care unit (ICU) stays and increased risk for complications and re-hospitalization.1 The most frequently identified comorbid condition in TBI is hypertension (in 55 and older individuals its prevalence is ∼38.8%).2,3 In experimental animals, it has been shown that pre-existing hypertension increases mortality post-TBI.4 These observations expand the conclusions in recent studies that comorbid hypertension exacerbates neuronal damage and worsens outcomes in ischemic stroke.5 Despite these advances, the cellular mechanisms by which hypertension exacerbates TBI-induced brain dysfunction remain elusive.
Cerebrovascular dysregulation plays a critical role in secondary damage of brain tissue induced by TBI.6 The mechanism by which TBI promotes cerebrovascular dysfunction includes an increased oxidative stress.7 In particular, recent studies demonstrate that in the walls of cerebral arteries, mitochondria are an important source for TBI-induced overproduction of reactive oxygen species (ROS).8 Another possible source of ROS, which might be linked to mitochondrial ROS generation,9 is nicotinamide adenine dinucleotide phosphate (NADPH) oxidase. TBI enhances the expression of Nox4,10 and TBI-related N-methyl-d-aspartate (NMDA) receptor activation can trigger NADPH-dependent ROS production.11 Cerebrovascular oxidative stress importantly contributes to a decline in cerebral blood flow (CBF), neurovascular uncoupling, and autoregulatory dysfunction associated with TBI, all of which likely play a role in induction of neuronal damage.12 Importantly, hypertension has been shown to synergize with other pathological conditions to exacerbate oxidative stress in the cerebral vasculature.13 For example, hypertension also exacerbates aging-induced mitochondrial oxidative stress in mouse cerebral arteries.14 Yet, the synergistic roles of pre-existing hypertension and TBI induction of cerebrovascular oxidative stress have not been explored. This especially applies to mild TBI (mTBI), the most frequent form of brain trauma, which has shown only a transient increase in cerebral oxidative stress.15
The present study was designed to test the hypothesis that pre-existing arterial hypertension exacerbates mTBI-induced cerebrovascular oxidative stress by upregulating TBI-induced mitochondrial ROS (mtROS) production in vascular cells, and to explore the role of NADPH oxidase. To achieve this goal, mild TBI was induced in normotensive Wistar and spontaneously hypertensive rats (SHR). Cellular oxidative stress was compared in isolated segments of the middle cerebral arteries (MCAs) using the fluorescent indicator dye dihydroethidium (DHE). mtROS production was assessed using the mitochondria-targeted redox-sensitive fluorescent indicator dye MitoSox. The protective effects of in vivo treatment with the potent mitochondrial-targeted antioxidative Szeto-Schiller (SS) peptide SS–31 was assessed.16
Methods
mTBI in normotensive and hypertensive rats and SS-31 treatment
All procedures were approved by the Institutional Animal Use and Care Committee of the University of Pecs Medical School and the National Scientific Ethical Committee on Animal Experimentation, Hungary (BAI/35/51-107/2016) and in accordance with the Animal Research: Reporting of in Vivo Experiments (ARRIVE) guidelines. Male SHR (300–350 g, n = 25) and age-matched normotensive male Wistar rats (300–350 g, n = 15) were purchased from Janvier Labs (Le Genest-Saint-Isle, France) and Toxi-Coop (Budapest, Hungary). Mild impact acceleration diffuse brain injury was induced by Marmarou's weight drop model. In brief, with the rats under isoflurane (2%) anesthesia, the skull was exposed by a midline incision between lambda and bregma, and a steel disc was fixed with cement on the skull. A 450 g cylindrical weight from 200 mm was dropped onto the disc causing mild diffuse TBI to the animals. All animals survived the procedure. SHRs with mTBI had been treated with either vehicle (physiological saline, n = 5) or the antioxidative tetrapeptide SS-31 (elamipretide; 5.7 mg/kg/day, i.p., dissolved in physiological saline, n = 5) for 14 days after mTBI.16 The SS compound SS-31 is a tetrapeptide with an alternating aromatic-cationic amino acid motif (H-D-Arg-Dmt-Lys-Phe-NH2), which concentrates in the inner mitochondrial membrane >1000-fold compared with the cytosolic concentration.17 Treatment of laboratory rodents with SS-31 was shown to effectively decrease mitochondrial oxidative stress in the cerebral microvasculature.16 Blood pressure was measured at the end of the treatment period before further experiments in all groups of animals using the tail cuff method.
Measurement of ROS and mtROS production in isolated MCAs
Two weeks after mTBI,18 rats were decapitated, the brains were removed, and segments of the MCAs were isolated under an operating microscope using microsurgical instruments for imaging studies, as reported.8 To characterize cytosolic ROS production in cells within the walls of cerebral arteries, we used the DHE fluorescence method. To study mitochondrial oxidative stress, we applied MitoSOX Red, a mitochondrion-specific hydroethidine-derivative fluorescent redox indicator dye, as previously reported.19 In brief, artery segments were immediately placed, upon dissection, into physiological Kreb's solutions containing fresh, non-oxidized DHE (10 μM/L; Sigma Aldrich Hungary, Budapest, Hungary) or MitoSOX (10 μM/L, Thermo Fisher Scientific, Life Technologies, Budapest, Hungary). The vessel segments were incubated for 30 min (at 37°C, protected from light). In a different series of experiments, arterial segments isolated from SHRs with and without mTBI (n = 5 in each group) were pre-treated with the NADPH-oxidase inhibitor apocynin (10 mM/L for 90 min; Sigma Aldrich Hungary, Budapest, Hungary, n = 6 in each group). After incubation, the vessels were washed in Krebs' buffer three times for 15 min. Then, MCAs were placed on slides, cover-slipped, and imaged with a laser scanning confocal microscope (Olympus Fluoview FV1000, excitation 488 nm, emission between 540 and 640 nm) using a × 40 objective. Average fluorescence intensities of the arteries measured in the optical sections of the endothelial and smooth muscle layers were quantified with ImageJ software, and expressed as a fold change compared with vessels from Wistar rats without mTBI (controls). The experimenter was blinded to the treatment groups.
Quantitative reverse transcription polymerase chain reaction (RT-PCR)
Total RNA from isolated MCAs was isolated using NucleoSpin RNA (Macherey-Nagel GmbH) and purity and concentration were analyzed by NanoDrop. RNA was reverse transcribed into cDNA with the High Capacity cDNA RT Kit (Life Technologies). RT-PCR was run on an ABI-PRISM 7500 device in duplicates using SensiFast SYBR Green (BioLine). The following primers were used: Nox4, forward: CTT TCC GTC CCA AGC ACC G, reverse: GGG ACA GCC AAA CAA GCA GA; β-actin, forward: GTA ACC CGT TGA ACC CCA TT, and reverse: CCA TCC AAT CGG TAG TAG CG. Results are shown as a percentage of the housekeeping gene. Quantification was performed using the ΔΔCT method.
Statistical analysis
Data were analyzed by analysis of variance (ANOVA). A p value <0.05 was considered statistically significant. Data are expressed as mean ± standard error of the mean (SEM).
Results
mTBI induces chronic mitochondrial oxidative stress in cerebral arteries of hypertensive rats
SHRs had significantly higher blood pressure than normotensive rats (Wistar: 109 ± 3 mm Hg, SHR:183 ± 3 mm Hg, p < 0.001). Blood pressure was not influenced by mTBI in either normotensive or hypertensive rats (Wistar + mTBI: 109 ± 5 mm Hg, SHR + mTBI: 174 ± 5 mm Hg, p < 0.001). Treatment with SS-31 also did not elicit significant changes in systolic blood pressure (SHR + mTBI + SS-31: 181 ± 5 mm Hg).
We found that in MCAs of normotensive rats 2 weeks after mTBI, both in the smooth muscle cells (Fig. 1A)and the endothelial layer (Fig. 1B) DHE fluorescence (indicating cytoplasmic superoxide production) had already returned to control levels. In contrast, 2 weeks after mTBI smooth muscle (Fig. 1A) and endothelial cells (Fig. 1B) in MCAs of SHRs exhibited significantly increased oxidative stress as compared with vessels isolated from non-TBI-challenged animals. In SHRs, SS-31 treatment initiated after TBI significantly reduced cerebrovascular oxidative stress both in the endothelial (Fig. 1B) and smooth muscle cells (Fig. 1A).
FIG. 1.

Mild traumatic brain injury (mTBI) induces persistent oxidative stress in the cerebral arteries of hypertensive rats, which is reversed by treatment with SS-31.(A) Bar graphs depict summary data of dihydroethidine (DHE) fluorescence indicating production of reactive oxygen species (ROS) in the smooth muscle layer of middle cerebral arteries (MCA) isolated from Wistar rats, Wistar rats 2 weeks after mTBI, spontaneously hypertensive rats (SHR), and SHRs after 2 weeks of mTBI treated with vehicle or the mitochondria-targeted antioxidant peptide SS-31. The treatment started after TBI was applied and lasted for 2 weeks (5.7 mg/kg−1day−1 i.p.). Data are means ± standard error of the mean (SEM) (n = 5 in each group). *p < 0.05 versus Wistar, #p < 0.05 versus Wistar + mTBI, &p < 0.05 versus SHR, $p < 0.05 versus SHR + mTBI. Lower panel shows representative confocal images of red DHE fluorescence in the smooth muscle layer (identified in the outer layer of arteries by the spindle-shaped nuclei that are perpendicular to the long axis of the artery) of MCAs in each group. (B) Summary data of endothelial DHE fluorescence in MCAs from each group after the same treatment protocols. Data are means ± SEM (n = 5 in each group). *p < 0.05 versus Wistar, #p < 0.05 versus Wistar + mTBI, &p < 0.05 versus SHR, $p < 0.05 versus SHR + mTBI. Lower panel demonstrates confocal images of red DHE fluorescence indicating ROS production in the endothelium (identified as the inner layer of arteries by elongated nuclei that are mostly parallel to the long axis of the artery) of MCAs in each group. Scale bar is 50 μm. Color image is available online.
To substantiate these findings, we assessed mitochondrial ROS production by using the mitochondria-targeted redox-sensitive dye MitoSox. We found that MitoSox fluorescence was also significantly increased in both the endothelial (Fig. 2B)and smooth muscle cells (Fig. 2A) in MCAs derived from SHRs 2 weeks after mTBI, whereas it did not change significantly in MCAs of normotensive rats (Fig. 2A,B). These findings suggest that pre-existing hypertension exacerbates TBI-induced mitochondrial oxidative stress in cerebral arteries. In SHRs, SS-31 treatment initiated after TBI significantly decreased MitoSox fluorescence in both the endothelial (Fig. 1B) and smooth muscle cells (Fig. 1A).
FIG. 2.

Mild traumatic brain injury (mTBI) induces persistent mitochondrial oxidative stress in cerebral arteries of hypertensive rats, which is reversed by SS-31. (A) Bar graphs show summary data of MitoSox staining (representing mitochondrial production of reactive oxygen species [ROS]) in the smooth muscle layer of middle cerebral arteries (MCA) isolated from Wistar rats, Wistar rats 2 weeks after mTBI, spontaneously hypertensive rats (SHR), and SHRs after 2 weeks of mTBI treated with vehicle or the mitochondria-targeted antioxidant peptide SS-31. The treatment started after TBI was applied and lasted for 2 weeks (5.7 mg/kg−1day−1 i.p.). Data are means ± standard error of the mean (SEM) (n = 5 in each group). *p < 0.05 versus Wistar, &p < 0.05 versus SHR, $p < 0.05 versus SHR + mTBI. Lower panel shows representative confocal images of red MitoSox fluorescence in the smooth muscle layer (identified in the outer layer of arteries by the spindle-shaped nuclei that are perpendicular to the long axis of the artery) of MCAs in each group. (B) Summary data of endothelial MitoSox fluorescence in MCAs from each group after the same treatment protocols. Data are means ± SEM (n = 5 in each group). *p < 0.05 versus Wistar, &p < 0.05 versus SHR, $p < 0.05 versus SHR + mTBI. Lower panel demonstrates confocal images of red MitoSox indicating mitochondrial ROS production in the endothelium (identified as the inner layer of arteries by elongated nuclei that are mostly parallel to the long axis of the artery) of MCAs in each group. Scale bar is 50 μm. Color image is available online.
Role of NADPH oxidase in mTBI-induced persistent mitochondrial oxidative stress in cerebral arteries of hypertensive rats
We found that mTBI-induced cerebrovascular oxidative stress indicated by DHE and MitoSox fluorescence in the smooth muscle and endothelial layers of isolated MCAs of SHRs was significantly reduced in the presence of the NADPH oxidase inhibitor apocynin (Fig. 3). The role of NADPH oxidase in mTBI-induced persistent mitochondrial oxidative stress in cerebral arteries of hypertensive rats was further substantiated by our findings that expression of the NADPH oxidase subunit Nox4 was significantly enhanced in MCAs of SHRs after mTBI, which was reversed by the mitochondria-targeted antioxidant peptide SS-31(Fig. 3).
FIG. 3.
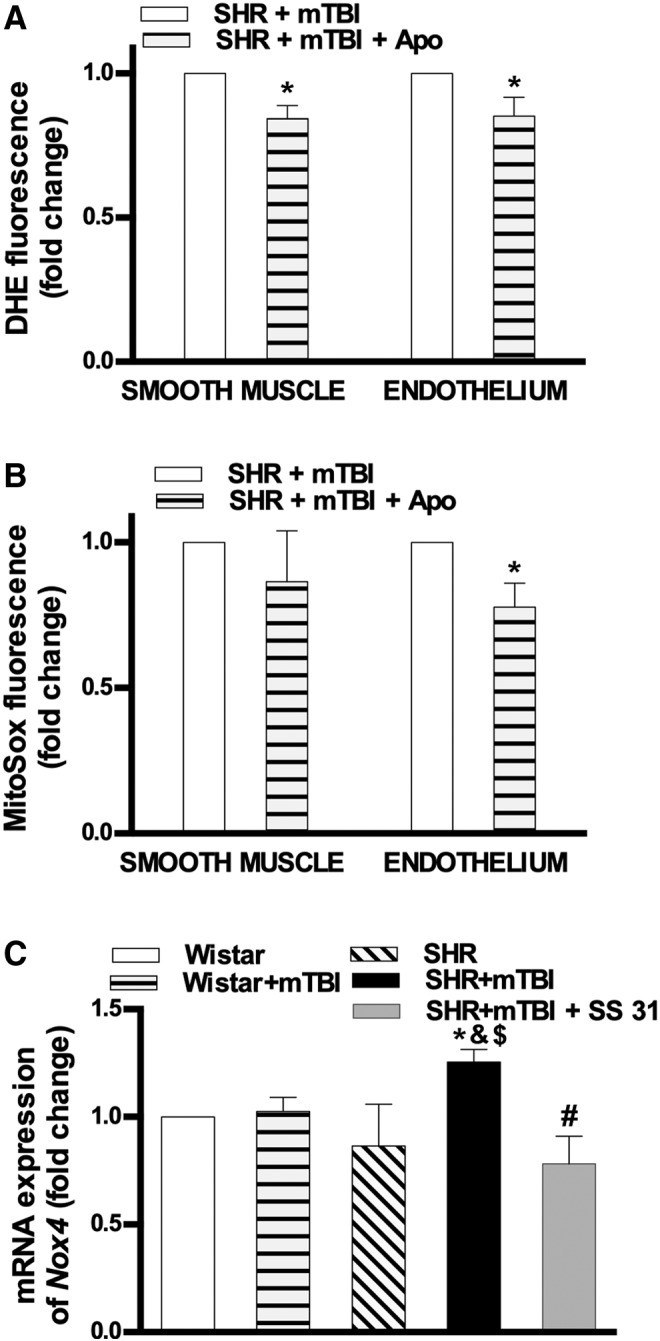
Role of nicotinamide adenine dinucleotide phosphate (NADPH) oxidase in mild traumatic brain injury (mTBI)-induced persistent mitochondrial oxidative stress in cerebral arteries of hypertensive rats. (A) Bar graphs depict summary data of dihydroethidine (DHE) and (B) MitoSox fluorescence in the smooth muscle and endothelial layers of middle cerebral arteries (MCA) isolated from spontaneously hypertensive rats (SHR) 2 weeks after mTBI in the absence and presence of the NADPH oxidase inhibitor apocynin. Data are means ± standard error of the mean (SEM) (n = 5 in each group). *p < 0.05 versus SHR + mTBI. (C) mRNA expression of the NADPH oxidase subunit Nox4 in cerebral arteries isolated from Wistar rats, Wistar rats 2 weeks after mTBI, SHR, and SHR after 2 weeks of mTBI treated with vehicle or the mitochondria-targeted antioxidant peptide SS-31. The treatment started after mTBI was applied and lasted for 2 weeks (5.7 mg/kg−1day−1 i.p.). Data are means ± SEM (n = 5 in each group). *p < 0.05 versus Wistar, &p < 0.05 versus Wistar + mTBI, $p < 0.05 versus SHR, #p < 0.05 versus SHR + mTBI.
Discussion
In the absence of comorbidities, single mild brain trauma, unlike severe or repetitive mTBI, has been shown to induce only transient increases in ROS production in the brain, which result in transient dysfunction of cerebrovascular regulation and transient deficit in cognitive function.20,21 The salient finding of the present study is that in the presence of pre-existing comorbid hypertension, a single episode of mTBI is sufficient to induce persisting oxidative stress in cerebral arteries (Figs 1 and 2). Our findings have important translational relevance. Increased vascular oxidative stress has been linked to exacerbation of neuronal damage and poor outcome in TBI,22 suggesting that mTBI likely has more deleterious consequences on brain function in hypertensive subjects.
The functional consequences of mTBI-induced persisting cerebrovascular mitochondrial oxidative stress are likely multifaceted. Increased ROS production has been shown to impair regulation of CBF. By altering nitric oxide (NO) and prostanoid production and/or bioavailability, it impairs endothelial function,23 decreases basal CBF,24 promotes neurovascular uncoupling,25 and contributes to autoregulatory dysfunction8; all of which are associated with cognitive decline. Our findings warrant further investigations to determine how comorbid hypertension and mTBI interact in patients to promote dysregulation of CBF, contributing to cognitive decline, and to elucidate the protective effects of treatments targeting ROS-dependent pathways, especially in the elderly.
The cellular and molecular mechanisms responsible for exacerbation of TBI-induced mitochondrial oxidative stress in hypertensive cerebral arteries are likely multifaceted. We show here that activation of NADPH oxidase is involved in the persistent cerebrovascular mitochondrial oxidative stress induced by single mTBI in hypertensive rats (Fig. 3). Supporting our results, recent studies suggest that a bidirectional link may exist between ROS generation by NADPH oxidases (NOX) and mtROS production.26 Nox4 is localized to the mitochondrial membrane, can be activated by mechanical forces,27 and its activation is associated with mitochondrial oxidative stress.28 In addition to the direct production of ROS by Nox4, activation of Nox4 may also promote ROS production by the mitochondrial electron transports chain. Nox4 expression is also increased after TBI,10 TBI-related NMDA-receptor activation can trigger NADPH-dependent ROS production,11 and there are studies showing that deletion of Nox429 and administration the NADPH oxidase inhibitor apocyinin30,31 attenuates the severity of secondary injury after TBI. The role of Nox4 is underlined by our results that cerebrovascular expression of Nox4 is persistently increased in SHRs after mTBI, which is reversed by targeted attenuation of mitochondrial oxidative stress with SS-31. Accordingly, there is growing evidence that mitochondria-derived ROS regulate the expression and activity of plasma membrane-associated NADPH oxidases in vascular cells,32 and that mitochondria-targeted antioxidants, by disrupting this regulatory cycle, can reduce cellular NADPH oxidase activity.33
The interaction of mTBI and hypertension in promoting cerebrovascular mitochondrial oxidative stress may include mechanosensitive pathways34 (e.g., integrin-mediated signal transduction, Ca2+ signaling, activation of protein kinase [PK]C, Rho kinases, and mitogen-activated protein [MAP] kinases). The sites of mechanosensitive mtROS production in the mitochondria that are sensitive to both hypertension and TBI should be explored in future studies. Potential sites include complex I, the entry point for electrons from NADH into the respiratory chain, and complex III, which funnels electrons from the coenzyme Q (CoQ) pool to cytochrome c. Mitochondrial O2- may also be dismutated to H2O2,8 which penetrates the mitochondrial membrane, and may promote further ROS production in the cytoplasm, for example by uncoupling endothelial nitric oxide synthase (eNOS).35 These possibilities should be studied in the future.
We provide proof of concept, that increased mTBI-induced cerebrovascular oxidative stress can be potentially attenuated by targeted pharmacological interventions in a high-risk population, and have identified a novel treatment regimen (SS-31) that specifically targets mtROS production,16 which effectively attenuates TBI-induced cerebrovascular oxidative stress in hypertensive rats. Our previous results, that increased mtROS is causally linked to impairment of endothelium-dependent vasodilation of cerebral arterioles during functional hyperemia16 and cognitive dysfunction, taken together with our findings that hypertension is associated with increased pressure-induced vasoconstriction of cerebral arteries,36 led to the formulation of the hypothesis that persisting exacerbation of mitochondrial oxidative stress in the cerebral vasculature leads to neurovascular dysfunction and dysregulation of local CBF and thereby cognitive decline after mTBI in hypertension. This should be tested by future studies. In addition, SS-31 also readily penetrates the blood–brain barrier, and there are important studies to suggest that in addition to its significant vasoprotective effects, SS-31 treatment exerts important direct neuroprotective effects as well.
Our results provide the basis for further investigations studying the interaction between hypertension and mTBI and characterizing the efficacy of SS-31 treatment in various pre-clinical models of TBI to prevent neurovascular dysregulation, attenuate secondary neuronal damage, and facilitate post-TBI recovery of higher cortical function.
Acknowledgments
This work was supported by grants from the National Research, Development, and Innovation Office to P.T. (NKFI-FK123798) and A.K. (K108444); the Hungarian Academy of Sciences Bolyai Research Scholarship BO/00634/15 to P.T.; the PTE AOK-KA 3/2016 04.01/F to P.T.; the ÚNKP-18-4-PTE-6 New National Excellence Program of the Ministry of Human Capacities, Higher Education Institutional Excellence Programme – Grant No. 20765-3/2018/FEKUTSTRAT, FIKP II/S, EFOP-3.6.2.-16-2017-00008, GINOP-2.3.2-15-2016-00048, and GINOP-2.3.3-15-2016-00032 to P.T. and A.B.; the Hungarian National Brain Research Program Grant No. 2017-1.2.1-NKP-2017-00002 to A.B.; the Marie Curie Actions SMARTER 7th Framework Program of the European Union 606998 to N.S. and A.K.; the National Institute on Aging (R01-AG055395, R01-AG047879); and the National Institute of Neurological Disorders and Stroke (NINDS) (R01-NS056218, R01-NS100782).
Author Disclosure Statement
No competing financial interests exist.
References
- 1. Kumar R.G., Juengst S.B., Wang Z., Dams-O'Connor K., Dikmen S.S., O'Neil-Pirozzi T.M., Dahdah M.N., Hammond F.M., Felix E.R., Arenth P.M., and Wagner A.K. (2018). Epidemiology of comorbid conditions among adults 50 years and older with traumatic brain injury. J. Head Trauma Rehabil. 33, 15–24 [DOI] [PubMed] [Google Scholar]
- 2. Centers for Disease Control and Prevention (2015). Report to Congress on Traumatic Brain Injury in the United States: Epidemiology and Rehabilitation. National Center for Injury Prevention and Control; Division of Unintentional Injury Prevention: Atlanta [Google Scholar]
- 3. Thompson H.J., Dikmen S., and Temkin N. (2012). Prevalence of comorbidity and its association with traumatic brain injury and outcomes in older adults. Res. Gerontol. Nurs. 5, 17–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lu Y.C., Liu S., Gong Q.Z., Hamm R.J., and Lyeth B.G. (1997). Inhibition of nitric oxide synthase potentiates hypertension and increases mortality in traumatically brain-injured rats. Mol. Chem. Neuropathol. 30, 125–137 [DOI] [PubMed] [Google Scholar]
- 5. Ergul A., Hafez S., Fouda A., and Fagan S.C. (2016). Impact of comorbidities on acute injury and recovery in preclinical stroke research: focus on hypertension and diabetes. Transl. Stroke Res. 7, 248–260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Overgaard J., and Tweed W.A. (1974). Cerebral circulation after head injury. 1. Cerebral blood flow and its regulation after closed head injury with emphasis on clinical correlations. J. Neurosurg. 41, 531–541 [DOI] [PubMed] [Google Scholar]
- 7. Marcano D.C., Bitner B.R., Berlin J.M., Jarjour J., Lee J.M., Jacob A., Fabian R.H., Kent T.A., and Tour J.M. (2013). Design of poly(ethylene glycol)-functionalized hydrophilic carbon clusters for targeted therapy of cerebrovascular dysfunction in mild traumatic brain injury. J. Neurotrauma 30, 789–796 [DOI] [PubMed] [Google Scholar]
- 8. Szarka N., Pabbidi M.R., Amrein K., Czeiter E., Berta G., Pohoczky K., Helyes Z., Ungvari Z., Koller A., Buki A., and Toth P. (2018). Traumatic brain injury impairs myogenic constriction of cerebral arteries: role of mitochondria-derived H2O2 and TRPV4-dependent activation of BKca channels. J. Neurotrauma [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Briones A.M., Tabet F., Callera G.E., Montezano A.C., Yogi A., He Y., Quinn M.T., Salaices M., and Touyz R.M. (2011). Differential regulation of Nox1, Nox2 and Nox4 in vascular smooth muscle cells from WKY and SHR. J. Am. Soc. Hypertens. 5, 137–153 [DOI] [PubMed] [Google Scholar]
- 10. Cooney S.J., Bermudez-Sabogal S.L., and Byrnes K.R. (2013). Cellular and temporal expression of NADPH oxidase (NOX) isotypes after brain injury. J. Neuroinflammation 10, 155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Brennan A.M., Suh S.W., Won S.J., Narasimhan P., Kauppinen T.M., Lee H., Edling Y., Chan P.H., and Swanson R.A. (2009). NADPH oxidase is the primary source of superoxide induced by NMDA receptor activation. Nat. Neurosci. 12, 857–863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Toth P., Szarka N., Farkas E., Ezer E., Czeiter E., Amrein K., Ungvari Z., Hartings J.A., Buki A., and Koller A. (2016). Traumatic brain injury-induced autoregulatory dysfunction and spreading depression-related neurovascular uncoupling: Pathomechanisms, perspectives, and therapeutic implications. Am. J. Physiol. Heart Circ. Physiol. 311, H1118–H1131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Pinto C.C., Silva K.C., Biswas S.K., Martins N., De Faria J.B., and De Faria J.M. (2007). Arterial hypertension exacerbates oxidative stress in early diabetic retinopathy. Free Radic. Res. 41, 1151–1158 [DOI] [PubMed] [Google Scholar]
- 14. Springo Z., Tarantini S., Toth P., Tucsek Z., Koller A., Sonntag W.E., Csiszar A., and Ungvari Z. (2015). Aging exacerbates pressure-induced mitochondrial oxidative stress in mouse cerebral arteries. J. Gerontol. A. Biol. Sci. Med. Sci., 70, 1355–1359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Fehily B., and Fitzgerald M. (2017). Repeated mild traumatic brain injury: potential mechanisms of damage. Cell Transplant. 26, 1131–1155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Tarantini S., Valcarcel-Ares N.M., Yabluchanskiy A., Fulop G.A., Hertelendy P., Gautam T., Farkas E., Perz A., Rabinovitch P.S., Sonntag W.E., Csiszar A., and Ungvari Z. (2018). Treatment with the mitochondrial-targeted antioxidant peptide SS-31 rescues neurovascular coupling responses and cerebrovascular endothelial function and improves cognition in aged mice. Aging Cell 17 [Epub ahead of print; DOI: 10.1111/acel.12731.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zhao K., Zhao G.M., Wu D., Soong Y., Birk A.V., Schiller P.W., and Szeto H.H. (2004). Cell-permeable peptide antioxidants targeted to inner mitochondrial membrane inhibit mitochondrial swelling, oxidative cell death, and reperfusion injury. J. Biol. Chem. 279, 34,682–34,690 [DOI] [PubMed] [Google Scholar]
- 18. Adams C., Bazzigaluppi P., Beckett T.L., Bishay J., Weisspapir I., Dorr A., Mester J.R., Steinman J., Hirschler L., Warnking J.M., Barbier E.L., McLaurin J., Sled J.G., and Stefanovic B. (2018). Neurogliovascular dysfunction in a model of repeated traumatic brain injury. Theranostics 8, 4824–4836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Csiszar A., Labinskyy N., Perez V., Recchia F.A., Podlutsky A., Mukhopadhyay P., Losonczy G., Pacher P., Austad S.N., Bartke A., and Ungvari Z. (2008). Endothelial function and vascular oxidative stress in long-lived GH/IGF-deficient Ames dwarf mice. Am. J. Physiol. Heart Circ. Physiol. 295, H1882–1894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Uryu K., Laurer H., McIntosh T., Pratico D., Martinez D., Leight S., Lee V.M., and Trojanowski J.Q. (2002). Repetitive mild brain trauma accelerates Abeta deposition, lipid peroxidation, and cognitive impairment in a transgenic mouse model of Alzheimer amyloidosis. J. Neurosci. 22, 446–454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Conte V., Uryu K., Fujimoto S., Yao Y., Rokach J., Longhi L., Trojanowski J.Q., Lee V.M., McIntosh T.K., and Pratico D. (2004). Vitamin E reduces amyloidosis and improves cognitive function in Tg2576 mice following repetitive concussive brain injury. J. Neurochem. 90, 758–764 [DOI] [PubMed] [Google Scholar]
- 22. Abdul-Muneer P.M., Schuetz H., Wang F., Skotak M., Jones J., Gorantla S., Zimmerman M.C., Chandra N., and Haorah J. (2013). Induction of oxidative and nitrosative damage leads to cerebrovascular inflammation in an animal model of mild traumatic brain injury induced by primary blast. Free Radic. Biol. Med. 60, 282–291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Toth P., Tarantini S., Tucsek Z., Ashpole N.M., Sosnowska D., Gautam T., Ballabh P., Koller A., Sonntag W.E., Csiszar A., and Ungvari Z. (2014). Resveratrol treatment rescues neurovascular coupling in aged mice: role of improved cerebromicrovascular endothelial function and downregulation of NADPH oxidase. Am. J. Physiol. Heart Circ. Physiol. 306, H299–308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hortobagyi L., Kis B., Hrabak A., Horvath B., Huszty G., Schweer H., Benyo B., Sandor P., Busija D.W., and Benyo Z. (2007). Adaptation of the hypothalamic blood flow to chronic nitric oxide deficiency is independent of vasodilator prostanoids. Brain Res. 1131, 129–137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Park L., Anrather J., Zhou P., Frys K., Pitstick R., Younkin S., Carlson G.A. and Iadecola C. (2005). NADPH-oxidase-derived reactive oxygen species mediate the cerebrovascular dysfunction induced by the amyloid beta peptide. J Neurosci 25, 1769–1777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Dikalov S.I., and Ungvari Z. (2013). Role of mitochondrial oxidative stress in hypertension. Am. J. Physiol. Heart Circ. Physiol. 305, H1417–1427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Touyz R.M., and Montezano A.C. (2012). Vascular Nox4: a multifarious NADPH oxidase. Circ. Res. 110, 1159–1161 [DOI] [PubMed] [Google Scholar]
- 28. Dai D.F., Chen T., Szeto H., Nieves-Cintron M., Kutyavin V., Santana L.F., and Rabinovitch P.S. (2011). Mitochondrial targeted antioxidant Peptide ameliorates hypertensive cardiomyopathy. J. Am. Coll. Cardiol. 58, 73–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ma M.W., Wang J., Dhandapani K.M., and Brann D.W. (2018). Deletion of NADPH oxidase 4 reduces severity of traumatic brain injury. Free Radic. Biol. Med. 117, 66–75 [DOI] [PubMed] [Google Scholar]
- 30. Ma M.W., Wang J., Dhandapani K.M., Wang R., and Brann D.W. (2018). NADPH oxidases in traumatic brain injury – Promising therapeutic targets? Redox Biol. 16, 285–293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Chandran R., Kim T., Mehta S.L., Udho E., Chanana V., Cengiz P., Kim H., Kim C., and Vemuganti R. (2018). A combination antioxidant therapy to inhibit NOX2 and activate Nrf2 decreases secondary brain damage and improves functional recovery after traumatic brain injury. J. Cereb. Blood Flow Metab. 38, 1818–1827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Dai D.F., Rabinovitch P.S., and Ungvari Z. (2012). Mitochondria and cardiovascular aging. Circ. Res. 110, 1109–1124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Dikalov S. (2011). Cross talk between mitochondria and NADPH oxidases. Free Radic. Biol. Med. 51, 1289–1301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Springo Z., Tarantini S., Toth P., Tucsek Z., Koller A., Sonntag W.E., Csiszar A., and Ungvari Z. (2015). Aging exacerbates pressure-induced mitochondrial oxidative stress in mouse cerebral arteries. J. Gerontol. A Biol. Sci. Med. Sci. 70, 1355–1359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Chalupsky K., and Cai H. (2005). Endothelial dihydrofolate reductase: critical for nitric oxide bioavailability and role in angiotensin II uncoupling of endothelial nitric oxide synthase. Proc. Natl. Acad. Sci. U. S. A. 102, 9056–9061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Szarka N., Amrein K., Horvath P., Ivic I., Czeiter E., Buki A., Koller A., and Toth P. (2017). Hypertension-induced enhanced myogenic constriction of cerebral arteries is preserved after traumatic brain injury. J. Neurotrauma [DOI] [PubMed] [Google Scholar]