

Abstract
Objective
The Phenotype Risk Score (PheRS) is a method to detect Mendelian disease patterns using phenotypes from the electronic health record (EHR). We compared the performance of different approaches mapping EHR phenotypes to Mendelian disease features.
Materials and Methods
PheRS utilizes Mendelian diseases descriptions annotated with Human Phenotype Ontology (HPO) terms. In previous work, we presented a map linking phecodes (based on International Classification of Diseases [ICD]-Ninth Revision) to HPO terms. For this study, we integrated ICD-Tenth Revision codes and lab data. We also created a new map between HPO terms using customized groupings of ICD codes. We compared the performance with cases and controls for 16 Mendelian diseases using 2.5 million de-identified medical records.
Results
PheRS effectively distinguished cases from controls for all 15 positive controls and all approaches tested (P < 4 × 1016). Adding lab data led to a statistically significant improvement for 4 of 14 diseases. The custom ICD groupings improved specificity, leading to an average 8% increase for precision at 100 (-2% to 22%). Eight of 10 adults with cystic fibrosis tested had PheRS in the 95th percentile prio to diagnosis.
Discussion
Both phecodes and custom ICD groupings were able to detect differences between affected cases and controls at the population level. The ICD map showed better precision for the highest scoring individuals. Adding lab data improved performance at detecting population-level differences.
Conclusions
PheRS is a scalable method to study Mendelian disease at the population level using electronic health record data and can potentially be used to find patients with undiagnosed Mendelian disease.
Keywords: Electronic health record, Data mining, Mendelian genetics, Diagnosis
INTRODUCTION
Recognizing clinical patterns is a cornerstone of medical diagnosis.1 Physicians are trained to look at patient symptoms, signs, laboratory values, and diseases in relation to each other in order to identify underlying causes that can explain seemingly divergent conditions. Patterns of these phenotypes can be particularly useful for recognizing Mendelian conditions, as genetic mutations often manifest as a constellation of phenotypes.2,3 The knowledge of these patterns, which formed the basis for medical genetics, has been recorded in resources like the Online Mendelian Inheritance in Man (OMIM).4 In this article, we evaluate modifications to the phenotype risk score (PheRS), a high-throughput, automated approach to study Mendelian disease by applying the phenotypic patterns described in OMIM to electronic health record (EHR) data.
EHRs are an important resource for genetic research and have been used to discover genetic associations for a wide variety of phenotypes.5,6 EHRs have also been used to provide insight into the pathogenicity of rare genetic variants.7 We recently described a method called the Phenotype Risk Score (PheRS) as a means to assess the phenotypic overlap between a patient and the clinical profile of a Mendelian disease.8 For example, the PheRS calculation for cystic fibrosis (CF) is based on the diverse set of features that are commonly observed in patients with CF, such as bronchiectasis, asthma, infertility, and pneumonia. PheRS enables the recognition of patients who are similar to Mendelian disease profiles, without relying on the diagnosis label itself.
The original PheRS method was based on phenotypes represented as phecodes, which are aggregated International Classification of Diseases-Ninth Revision (ICD-9) codes designed for performing phenome-wide association studies (PheWAS).6,9 We found that the PheRS was significantly elevated among patients with Mendelian disease vs controls. Applying PheRS to an EHR-linked cohort of genotyped patients, we showed that we could generate evidence of pathogenicity for variants of unknown significance in a high-throughput manner.
Our initial work on PheRS demonstrated the problem of underdiagnosis of Mendelian disease, a finding that has also been seen in other studies.10,11 We found that the majority of patients with rare genetic variants did not receive genetic testing, despite having significant morbidity. Patients with atypical or mild presentations are often not tested for genetic disease due to lack of clinical suspicion.12 As we discover more pathogenic variants in Mendelian disease–causing genes, the number of patients who can be diagnosed will increase.13 Because of the rapid pace of discovery, clinical practice has not fully responded to the growing diagnostic power of genetic testing. PheRS may be used as a tool to help clinicians find undiagnosed patients.
In this article, we tested 3 specific potential advances to PheRS. First, we integrated ICD-Tenth Revision (ICD-10) codes using the recently released ICD-10 to phecode map.14 Second, we created a new map that directly relates custom groupings of ICD codes to Human Phenotype Ontology (HPO) terms, eliminating the use of intermediary phecodes. Third, we integrated laboratory measurements. Using a gold standard set of clinically diagnosed patients, we evaluated these modifications using a series of tests intended to cover 2 use cases. First, we tested the ability of PheRS discriminate between cases and controls at a population level, a task analogous to testing rare genetic variants for pathogenicity. Second, we tested the ability of PheRS to identify cohorts that are highly enriched for individuals affected by Mendelian disease, a capability that is necessary if PheRS is to be used in the clinical setting to help identify undiagnosed patients. Finally, we demonstrate the utility of this approach by testing PheRS on diagnosed patients who do not have a relevant ICD code in their record and on patient data ascertained before diagnosis.
MATERIALS AND METHODS
Setting
We performed this analysis in the Vanderbilt University Medical Center (VUMC) Synthetic Derivative (SD), a de-identified version of the EHR containing about 2.5 million patients.15,16 The SD contains essentially all EHR data, including clinical notes. The data have been stripped of patient identifiers.
Gold standard curation
We defined a gold standard set of cases for 16 genetic diseases (see Table 1). These diseases were chosen based on the following criteria:
Table 1.
Mendelian diseases tested in this study
| Gold standard counts |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| OMIM ID | Disease | Abbrev | Gene | HPO terms | Mapped HPO terms | Cases | Exclude | Mean age at last visit (y) | Mean unique years | Female (%) |
| 100800 | Achondroplasia | ACH | FGFR3 | 26 | 16 | 107 | 610 | 19 | 7.3 | 55 |
| 613490 | Alpha-1 antitrypsin deficiency | A1A | SERPINA1 | 6 | 6 | 250 | 10 013 | 46 | 5.8 | 43 |
| 219700 | Cystic fibrosis | CF | CFTR | 15 | 14 | 766 | 1648 | 23 | 8.9 | 50 |
| 188400 | DiGeorge syndrome | DGS | 22q11.2 | 52 | 48 | 284 | 349 | 9 | 6.2 | 53 |
| 190685 | Down syndrome | DS | Chr 21 | 30 | 17 | 2301 | 1429 | 16 | 6.4 | 44 |
| 310200 | Duchenne muscular dystrophy | DMD | DMD | 20 | 19 | 199 | 1316 | 17 | 7.5 | 3 |
| 300624 | Fragile X syndrome | FXS | FMR1 | 21 | 14 | 106 | 1667 | 19 | 6.2 | 23 |
| 235200 | Hereditary hemochromatosis | HH | HFE | 26 | 26 | 401 | 4454 | 55 | 6.6 | 37 |
| 187300 | Hereditary hemorrhagic telangiectasia | HHT | ACVRL1 & ENG | 37 | 30 | 159 | 205 | 45 | 5.8 | 64 |
| 154700 | Marfan syndrome | MS | FBN1 | 53 | 43 | 449 | 996 | 33 | 6.1 | 48 |
| 162200 | Neurofibromatosis, type 1 | NF1 | NF1 | 30 | 29 | 722 | 841 | 21 | 6.6 | 51 |
| 101000 | Neurofibromatosis, type 2 | NF2 | NF2 | 19 | 19 | 104 | 373 | 44 | 6.5 | 47 |
| 261600 | Phenylketonuria | PKU | PAH | 26 | 17 | 218 | 160 | 20 | 7.6 | 59 |
| 263300 | Polycythemia vera | PV | JAK2 | 14 | 11 | 219 | 4047 | 65 | 7.0 | 51 |
| 603903 | Sickle cell anemia | SCA | HBB | 17 | 16 | 491 | 1229 | 21 | 8.3 | 51 |
| 191100 | Tuberous sclerosis | TS | TSC1 & 2 | 32 | 25 | 207 | 204 | 22 | 7.9 | 53 |
| — | ALL SD | — | — | — | — | 2 493 408 | — | 36 | 3.2 | 53 |
We identified diagnosed cases among the 2.5 million patients at Vanderbilt University Medical Center for 16 diseases. This table includes basic demographics for this cohort, as well as information about the gold standard diseases. The number of HPO terms indicates the number of HPO terms used to describe the clinical features of each disease. The number of mapped HPO terms indicates the number of HPO terms we were able to capture using International Classification of Diseases codes.
HPO: Human Phenotype Ontology; OMIM: Online Mendelian Inheritance in Man; SD: Synthetic Derivative.
Profile for disease present in OMIM
Feature set from HPO mapped to at least 3 unique EHR-derived phenotypes
At least 100 clinically diagnosed cases in the SD
We included the 5 diseases from the original PheRS analysis that met the above criteria (achondroplasia, CF, hereditary hemochromatosis [HH], Marfan syndrome [MS], phenylketonuria [PKU]); Li–Fraumeni syndrome was excluded due to lack of cases. To find the additional 10 diseases, we used prevalence estimates from the 2019 Orphanet Report Series.17 Starting with the most prevalent disease, we worked our way down the list to identify diseases that met our criteria. In this way, we added Down syndrome (DS), DiGeorge syndrome, fragile X syndrome (FX), polycythemia vera (PV), sickle cell anemia, alpha-1 antitrypsin deficiency (A1A), hereditary hemorrhagic telangiectasia (HHT), Duchenne muscular dystrophy (DMD), and tuberous sclerosis.
We identified the ICD-9 and ICD-10 codes that are used for each diagnosis (Supplementary Table S1). The charts of individuals who had a mention of the disease in their problem list, ≥4 ICD codes, or a diagnostic genetic report were reviewed. A positive assertion of the diagnosis was required for each case. We retrieved all available genetic testing in pathology results for HH, A1A, PV, FX, and CF. For HH, A1A, CF, and PV, we required cases to have a positive genetic test (defined as homozygosity or compound heterozygosity for pathogenic variants in the HFE gene for HH or CFTR for CF; the ZZ genotype for A1A; positive JAK2 for PV). We did not require a positive genetic test for FX due to the limited number of cases. Those who were clinically diagnosed with HH or A1A but had negative genetics were excluded (neither a case nor a control). We required that sickle cell cases have a specific mention of hemoglobin SS disease.
For controls, we used the nearly 2.5 million individuals in the SD, excluding those who had a negative genetic test for the disorder, 1 or more ICD diagnosis codes, or mention of the disease in the problem list, and lacked a clear diagnosis in their notes.
Identifying relevant HPO terms
We used the HPO annotation table to find the HPO terms used to annotate our 16 gold standard diseases, using the OMIM mapping (Supplementary Table S2). Collectively, 359 unique HPO terms were used to describe these 16 diseases. We excluded terms that did not have any correlated ICD codes or labs, such as “long face” and “reduced phenylalanine hydroxylase activity.” The following work is focused on the 276 remaining HPO terms.
Updating the HPO-phecode map to include ICD-10 codes
Phecodes were generated from ICD-9 codes using the 1.2 version of the phecode map (available at http://phewascatalog.org). ICD-10 codes (which were not included in our prior PheRS study) were incorporated using the ICD-10 to phecode map.14 We refer to this original map as the HPO-phecode map (see Supplementary Table S3 for HPO-phecode map used in this study).
Creating HPO-ICD maps
We developed a new map that directly relates ICDs to HPO terms (referred to as the HPO-ICD map). This map consists of 2 submaps: one for ICD-9 to HPO and the other for ICD-10 to HPO. A list of candidate HPO-ICD pairs was created by combining pairs found in the following sources:
All ICDs that were related to HPO terms via phecodes in the original map
Cases in which the HPO term name and the ICD string name were an exact match or one was a substring for the other.
ICD-HPO terms that were annotated with the same CUI in UMLS (version 2018AB) OR related via the MRREL with the rel specified as CHD, RQ, SY, or RO
ICDs found using WikiMedMap,18 a tool that finds ICD codes based on free text strings
ICDs related via a child HPO term (eg, ICDs mapped to the HPO term “tachycardia” were also added to the HPO term for the parent concept of arrhythmia)
A team of specialists (3 physicians and a bioinformatician) reviewed these candidate pairs, keeping ICD codes that would be used if a patient had a particular disease or symptom. During review, ICD codes that were not relevant to the HPO term were removed. For example, the HPO term for dolichocephaly was mapped to the phecode 749—“Congenital anomalies of face and neck”—which comprises 66 ICD-9 and 83 ICD-10 codes, including codes for “webbing of neck” and “microtia” that had no relation to the HPO term. The new map includes ICD-9 754.0 (Congenital musculoskeletal deformities of skull, face, and jaw [of which dolichocephaly is a synonym, according to the ICD9-CM index] andICD-10 Q67.2 (Dolichocephaly). ICDs not present in the map were added based on text searches of the ICD-9 and ICD-10 billing codes.
In the original HPO-to-phecode map, only a single phecode—the one that best represented the HPO term—could map to a single HPO term. While this restriction made the process of creating and using the map, it also led to situations in which relevant phecodes were left out of the map. For example, ICD-9 codes often encode a distinction between acquired vs congenital forms of disease, and these ICD codes were often grouped into different phecodes. For example, pes planus has an ICD-9 referring to congenital pes planus (754.61 [Pes planus, congenital]) and acquired (ICD-9 734.0 [Flat foot]), which map to phecodes 755.1 and 735.1, respectively (see Supplementary Tables 4 and 5 for HPO-ICD maps). We published the HPO-ICD maps, version 1.0-beta, on phewascatalog.org. Upon completing the new HPO-ICD map, we calculated the overlap between the HPO-ICD map and the HPO-phecode map.
Creating HPO-lab map
A total of 23 of the HPO terms used to describe our gold standard diseases were present in the HPO-to-LOINC (Logical Observation Identifiers Names and Codes) map produced by Zhang et al.19 From this list, we excluded hypertension (mapped to blood pressure) and arrythmia (mapped to heart rate). These values are frequently measured in a variety of contexts in which they may be temporarily abnormal (eg, due to pain) without reflecting underlying physiology. “Increased red blood cell mass” was not used because our EHR did not have any labs relevant to this feature. We also added 4 HPO terms that were not in the HPO-LOINC map. Because data in the SD is not consistently mapped to LOINC terms, we used lab identifiers specific to VUMC; we include the long-form descriptions of these identifiers as well as the HPO-lab itself in Supplementary Table S6, and includes 24 lab-related features. This map also specifies whether the HPO term pertains to an abnormally low or abnormally high lab.
We mapped each abnormal lab value to the corresponding HPO term, taking into consideration if the lab was low or high (using reference ranges included in lab data). For example, a high iron lab result was mapped to the HPO term for “Increased serum iron,” while low or normal labs were not mapped to this term. Thus, individuals were annotated with the HPO term for “Increased serum iron” if they had 1 or more elevated iron measurements. Individuals with normal or low iron measurements, as well as those without relevant lab data, were not labeled with the HPO term.
Height was the only measured value that did not have a reported standard reference range. To ascertain concepts pertaining to short or tall stature, we created a table with the most recent height for all individuals in the SD. We split adult (>21 years of age) and pediatric populations, excluding all individuals younger than 2 years of age. We ran a regression model to predict height using age (splined with 8 knots) and gender. We calculated the residual for each individual in the set. Individuals whose most recently measured height was greater than 2 standard deviations from the expected value were annotated with the HPO term for tall stature and those with a height >2 standard deviations from the expected value were annotated with the HPO term for short stature.
Calculating PheRS
We calculated the PheRS of each individual for the 16 gold standard diseases using 4 different maps: HPO-phecode, HPO-ICD, HPO-phecode+lab and HPO-ICD+lab. Using each of these maps, we generated tables translating the source data (ie, ICDs, phecodes, or labs) into HPO terms (Figure 1). We then calculated the prevalence of each of these in the population by dividing the number of unique individuals with the HPO term by number of individuals in our cohort. We calculated the -1og10 of the prevalence to use as the weight for the feature. The PheRS for a particular disease was calculated by summing up the weights of each feature that is present for an individual.
Figure 1.
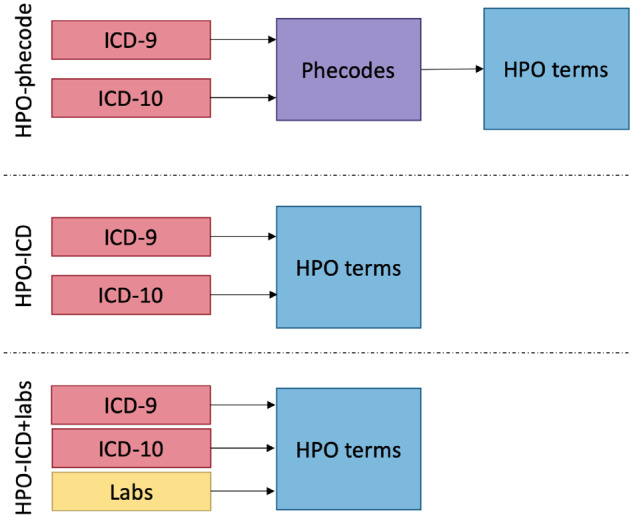
Diagram of Human Phenotype Ontology (HPO) maps tested in this article. Each map was used to translate phenotypic information contained in International Classification of Diseases (ICD) codes and labs into HPO terms. In the original HPO-phecode map, ICD codes were first translated the phecodes and then HPO terms. The new HPO-ICD map translates custom groupings of ICD codes to HPO without the intermediary phecodes. New information can be integrated into the Phenotype Risk Score, creating a map between the data elements and HPO terms, as we have done for labs. The HPO-phecode+labs table is not shown in this diagram. ICD-9: International Classification of Diseases-Ninth Revision; ICD-10: International Classification of Diseases-Tenth Revision.
To calculate the ICD+labs PheRS, we merged the HPO terms derived from ICD codes with those derived from ICDs. When an HPO term was linked to both an HPO term and a lab (as was the case for hematuria and others), we included individuals who had either an ICD or lab value.
Normalizing PheRS
We produced a residualized PheRS (rPheRS) using a linear regression model adjusted for age, sex, presence of ICD-9 codes, presence of ICD-10 codes, and the number of unique years for which they had billing data in the EHR (ie, PheRS ∼ Age + Sex + Race + has_icd9 + has_icd10 + uniq_encounter_years). We used a cubic spline with 3 knots for age. The rPheRS is defined as the studentized residual of the PheRS (expected vs actual) from this model.
Testing ability of PheRS to distinguish cases from controls at the population level
We used a 1-sided Wilcoxon rank sum test to test the hypothesis that the rPheRS of cases was greater than controls for the 4 maps: HPO-phecode, HPO-ICD, HPO-phecode+labs, and HPO-ICD+labs. We created a receiver-operating characteristic (ROC) curves and calculated the area under the curve (AUC-ROC) and the AUC for precision recall.
Testing sensitivity of PheRS
We calculated the precision at K for the 10, 100, 1000, and 10 000 top-scoring individuals by dividing the number of true positives by the number of individuals. We calculated the % of cases with normal PheRS scores, defined as rPheRS <1.
Comparing performance of different maps
We tested for a shift in the distribution of rPheRS among cases between the HPO-phecode map vs the HPO-ICD map and the HPO-ICD map vs the HPO-ICD+lab map. We used Wilcox rank sum test for this comparison.
Testing PheRS on underdocumented and prediagnosis cases
We identified cases in our gold standard that did not have any relevant ICD codes indicating their Mendelian disease diagnosis. We calculated the average rPheRS for these individuals, using the HPO-ICD+lab map (we used the HPO-ICD for FX and NF2, which have no relevant lab features). We counted the percent of under-documented cases with highly elevated PheRS scores (95th percentile and 99th percentile).
We identified CF patients who were diagnosed at VUMC and had at least 1 month and at least 2 clinic visits before diagnosis. For that subset of patients, we determined the date of their diagnosis by chart review. We calculated rPheRS for these patients at each clinic visit, using the HPO-ICD map.
Statistical tools
All statistically analyses were performed using R (version 3.4.1; R Foundation for Statistical Computing, Vienna, Austria). Plots were generated using ggplot2. Given that some of our P values were reported as 0 in R, we obtained the smallest representable number in R using the command Machine$double.xmin. Wilcoxon rank sum tests were performed using the wilcox.test function. ROC curves and AUCs were generated using the precrec package. Other figures were generated using ggplot2.
RESULTS
Composition HPO-ICD and HPO-phecode maps
We found the original HPO-phecode map and the new HPO-ICD map were quite divergent in terms of the ICDs related to a particular HPO term (Supplementary Figure S1). 51% of the ICD codes included in the HPO-phecode map were excluded from the new HPO-ICD map. ICDs pertaining to congenital conditions were most likely to be excluded, with 93% of the ICDs in the HPO-phecode map excluded from the HPO-ICD map. This is not surprising, given that many specific (and individually rare) congenital ICD codes are condensed into a single phecode. Many codes were also added to the HPO-ICD map; 56% of the ICDs included in the HPO-ICD map are not included in the HPO-phecode map. The majority of these codes were added due to a small number of very broad HPO terms, such as “Recurrent infections” (2610 ICD codes) and “Arthropathy” (1026 codes). In other cases, ICDs were added to the new map because the relevant codes were split across phecodes. For example, the HPO term “Bronchiectasis” has corresponding ICD codes in chapters relating to respiratory system, infectious disease, and conditions originating in the perinatal period, and no single phecode contains all of these phenotypes (Table 2).
Table 2.
Select examples of differences between the HPO-phecode map and HPO-ICD map
| HPO term ID | HPO term name | Phecode | Included in both | HPO-phecode only | HPO-ICD only |
|---|---|---|---|---|---|
| 1508 | Failure to thrive | (264.2) Failure to thrive (childhood) | (783.41/R62.51) Failure to thrive (child) | None | (779.34/P92.6) Failure to thrive in newborn |
| (783.7/R62.7) Adult failure to thrive | |||||
| 2099 | Asthma | (495) Asthma | (493*/J45*) Asthma | None | (E945.7) Adverse effects of antiasthmatics in therapeutic use |
| (T48.6X6*) Underdosing of antiasthmatics | |||||
| 2110 | Bronchiectasis | (496.3) Bronchiectasis | (494*/J47*) Bronchiectasis | None | (011.5*) Tuberculous bronchiectasis |
| (748.61/Q33.4) Congenital bronchiectasis | |||||
| 1738 | Exocrine pancreatic insufficiency | (577) Diseases of pancreas | (577.8) Other specified diseases of pancreas | (577.2/K86.2) Cyst of pancreas | None |
| (K86.81) Exocrine pancreatic insufficiency | + 5 ICD-9 codes & 34 ICD-10 codes | ||||
| 4401 | Meconium ileus | (656.6) Perinatal disorders of digestive system | (777.1) Meconium obstruction in fetus or newborn | None | (777.5*/P77*) Necrotizing enterocolitis in newborn |
| (P76.0) Meconium plug syndrome | (777.6/P78.0) Perinatal intestinal perforation |
This table contains 5 of the HPO terms used to describe cystic fibrosis. Each HPO term has a corresponding phecode from the HPO-phecode map. Each phecode contains 1 or more ICD code. The ICD codes that were included in both the HPO-phecode map and HPO-ICD map are listed, as well as the ICD codes that are exclusive to a single map.
HPO: Human Phenotype Ontology; ICD: International Classification of Diseases.
The candidate HPO-ICD pairs came from a number of sources, including the Unified Medical Language System (UMLS) and WikiMedMap. A total of 39% of the HPO-ICD9 pairs in the final map were present in the UMLS, while 807 HPO-ICD9 pairs were excluded from the map; 21% of the HPO-ICD9 pairs were found using WikiMedMap (214 were not present in the UMLS), and 151 of the pairs found in WikiMedMap were excluded from the map. The remaining HPO-ICD9 pairs identified via the HPO-phecode map, using substrings and manual entry from curators. Proportions for HPO-ICD10 were similar.
Using PheRS to distinguish cases vs controls: Both the HPO-ICD map and the HPO-phecode map generated PheRSs distinguished cases from controls for 15 of the 16 diseases (all P < 4 × 10−16) (Table 3). The only exception was PKU, in which the PheRS from both maps were not elevated among diagnosed cases. This was an expected result. A similar analysis of PKU was included in the original PheRS study, and cases were no different than controls in that case as well. We recognized that the patients with PKU were asymptomatic due to early detection with newborn screening and effective control of the disease through dietary means. PheRS from HPO-ICD+lab and HPO-phecode+lab also effective in distinguishing all 14 diseases tested (2 diseases had no relevant lab data), including PKU (all P < 1 × 10−56) (Figure 2; Supplementary Figure S2A-N).
Table 3.
Results from case/control and method comparison
| Performance of maps, cases vs controls |
Comparison between maps |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| HPO-phecode |
HPO-ICD |
HPO-ICD+lab |
HPO-ICD vs HPO-phe |
HPO-ICD+lab vs HPO-ICD |
||||||
| P | loc diff | P | loc diff | P | loc diff | P | loc diff | P | loc diff | |
| Achondroplasia | 2.4 × 10−31 | 3.21 | 4.6 × 10−34 | 4.59 | 1.4 × 10−59 | 5.53 | .309 | .54 | .063 | 1.32 |
| Alpha-1-antitrypsin deficiency | 1.5 × 10−68 | 3.21 | 9.3 × 10−86 | 2.77 | 2.2 × 10−120 | 3.26 | .401 | −.14 | .175 | .31 |
| Cystic fibrosis | <5 × 10−324 | 7.06 | <5 × 10−324 | 7.33 | <5 × 10−324 | 7.92 | .607 | .10 | 2.1 × 10−33 | 2.72 |
| Di George's syndrome | 2.7 × 10−160 | 6.30 | 3.8 × 10−155 | 6.88 | 1.7 × 10−158 | 6.29 | .151 | .57 | .160 | −.58 |
| Down Syndrome | <5 × 10−324 | 3.52 | <5 × 10−324 | 3.91 | <5 × 10−324 | 4.36 | .121 | .11 | 1.1 × 10−11 | .72 |
| Duchenne muscular dystrophy | 4.0 × 10−82 | 4.77 | 2.4 × 10−110 | 5.22 | 6.7 × 10−111 | 5.22 | .826 | −.07 | .860 | .05 |
| Fragile X syndrome | 4.1 × 10−16 | 2.65 | 5.0 × 10−19 | 2.94 | — | — | .584 | .12 | — | — |
| Hereditary hemochromatosis | 3.0 × 10−27 | .71 | 4.6 × 10−40 | .96 | 3.8 × 10−133 | 2.14 | .308 | .14 | 3.9 × 10−13 | 1.09 |
| Hereditary hemorrhagic telangiectasia | 4.6 × 10−59 | 3.19 | 1.7 × 10−59 | 3.03 | 6.0 × 10−59 | 2.97 | .813 | .09 | .888 | −.05 |
| Marfan syndrome | 1.5 × 10−177 | 3.98 | 3.0 × 10−203 | 4.81 | 1.1 × 10−211 | 5.23 | 3.2 × 10−5 | .95 | .251 | .29 |
| Neurofibromatosis, type 1 | 3.0 × 10−137 | 2.17 | 6.6 × 10−148 | 2.53 | 6.0 × 10−152 | 2.55 | .050 | .23 | .944 | .01 |
| Neurofibromatosis, type 2 | 5.8 × 10−49 | 1.02 | 9.2 × 10−53 | 8.51 | — | — | .274 | .97 | — | — |
| Phenylketonuria | 1 | −.38 | 1 | −.37 | 2.5 × 10−105 | 2.71 | .982 | .00 | 8.4 × 10−48 | 3.07 |
| Polycythemia vera | 2.3 × 10−58 | 2.19 | 1.8 × 10−38 | 2.55 | 4.3 × 10−86 | 2.96 | .993 | .00 | .361 | .26 |
| Sickle cell anemia | 1.5 × 10−191 | 4.64 | 2.4 × 10−237 | 4.66 | 1.1 × 10−261 | 4.57 | .416 | .22 | .353 | −.20 |
| Tuberous sclerosis | 2.8 × 10−86 | 4.64 | 1.1 × 10−89 | 5.23 | 2.2 × 10−89 | 5.16 | .277 | .43 | .865 | −.04 |
We used the Wilcoxon rank sum test to test the ability of PheRS to differentiate between cases and controls using 3 different maps (HPO-phecode, HPO-ICD, and HPO-ICD+lab). We compared the performance of HPO-ICD vs HPO-phecode and HPO-ICD+lab vs HPO-ICD by testing the differences between the PheRS of cases. Location shifts and P values were generated using a 2-sided Wilcoxon rank sum test. P values equal to <5 × 10−324 are listed as such because the exact P value was lower than the smallest representable double in R.
HPO: Human Phenotype Ontology; ICD: International Classification of Diseases; PheRS: Phenotype Risk Score.
Figure 2.

Boxplots of Phenotype Risk Score cases for 2 diseases: These boxplots compare the residualized Phenotype Risk Score (rPheRS) of cases generated form the different maps vs controls (scored with Human Phenotype Ontology [HPO]-phecode map). For hereditary hemochromatosis (HH), the addition of labs improved performance. However, phecodes produces the highest percentage of outliers. For Marfan syndrome (MS), HPO-International Classification of Diseases (ICD) resulted in a higher median than HPO-phecode.
Comparison of HPO-ICD vs HPO-phecode maps: The comparison PheRS for HPO-ICD vs HPO-phecode demonstrated a positive location shift for all diseases tested except A1A and DMD. However, this difference was only significant for MS. Adding labs resulted in a positive location shift for all labs except DMD, HHT, sickle cell anemia, and tuberous sclerosis. Four positive shifts were significant (CF, DS, HHT, and PKU) (Table 3).
Compared with the HPO-phecode map, using the HPO-ICD map led to increased precision among top scorers. The precision at K (K = 10, 100, 1000, and 10 000) was consistently higher for 11 diseases using the HPO-ICD map vs the HPO-phecode map; the reverse was true for only 2 diseases (A1A and FX). The HPO-ICD+labs map yielded the highest results across all K values in 5 cases (Figure 3).
Figure 3.

Precision @ K. Graphs of the precision for each disease tested at K = 10, 100, 1000 and 10000. The table includes the combined percentages for each map.
The HPO-ICD produced higher ROC-AUCs than HPO-phecode in for 12 of the 16 diseases, though the difference was often very small. The PRC-AUCs improved for 14 of 16 diseases, and this improvement was substantial for diseases like CF (PRC-AUC 0.13 vs 0.29). The HPO-ICD+lab map produced the overall best ROC-AUC for 6 diseases and the best PRC-AUC for 6 diseases (Figure 4; Table 4).
Figure 4.

The receiver-operating characteristic curves for each disease testing the ability of Phenotype Risk Score (PheRS) to classify cases vs controls. The red line indicates the PheRS generated by the Human Phenotype Ontology [HPO]-phecode map; the blue line indicates the PheRS generated by the HPO-International Classification of Diseases (ICD) map; green is HPO-ICD+phecode; purple is HPO-ICD+lab. ACH: achondroplasia; A1A: alpha-1 antitrypsin deficiency; CF: cystic fibrosis; DGS: DiGeorge syndrome; DS: Down syndrome; DMD: Duchenne muscular dystrophy; FXS: fragile X syndrome; HH: hereditary hemochromatosis; HHT: hereditary hemorrhagic telangiectasia; MS: Marfan syndrome; NF1: neurofibromatosis, type 1; NF2: neurofibromatosis, type 2; PKU: phenylketonuria; PV: polycythemia vera; SCA: Sickle cell anemia; TS: tuberous sclerosis.
Table 4.
ROC-AUC and PRC-AUC results
| ROC-AUC |
PRC-AUC |
|||||||
|---|---|---|---|---|---|---|---|---|
| HPO-phe | HPO-ICD | HPO-phe+lab | HPO-ICD+lab | HPO-phe | HPO-ICD | HPO-phe+lab | HPO-ICD+lab | |
| Achondroplasia | 0.82 | 0.84 | 0.94 | 0.95a | 0.007 | 0.029 | 0.009 | 0.031a |
| Alpha-1-antitrypsin deficiency | 0.82 | 0.86 | 0.91 | 0.93a | 0.005a | 0.004 | 0.004 | 0.003 |
| Cystic fibrosis | 0.95 | 0.95 | 0.96 | 0.96 | 0.127 | 0.294 | 0.210 | 0.403a |
| Di George's syndrome | 0.96a | 0.95 | 0.96 | 0.96 | 0.035 | 0.104a | 0.027 | 0.075 |
| Down syndrome | 0.88 | 0.85 | 0.91a | 0.90 | 0.056 | 0.080 | 0.083 | 0.121a |
| Duchenne muscular dystrophy | 0.89 | 0.96 | 0.91 | 0.96a | 0.009 | 0.014 | 0.011 | 0.016a |
| Fragile X syndrome | 0.73 | 0.75a | — | — | 0.001 | 0.002a | — | — |
| Hereditary hemochromatosis | 0.65 | 0.69 | 0.84 | 0.85a | 0.002 | 0.001 | 0.002a | 0.002 |
| Hereditary hemorrhagic telangiectasia | 0.87 | 0.87 | 0.87a | 0.87 | 0.012 | 0.015a | 0.010 | 0.013 |
| Marfan syndrome | 0.89 | 0.91 | 0.90 | 0.92a | 0.017 | 0.038 | 0.018 | 0.048a |
| Neurofibromatosis, type 1 | 0.77 | 0.78 | 0.77 | 0.78a | 0.022 | 0.036a | 0.021 | 0.035 |
| Neurofibromatosis, type 2 | 0.91 | 0.93a | — | — | 0.018 | 0.050a | — | — |
| Phenylketonuria | 0.32 | 0.34 | 0.93a | 0.93 | 0.000 | 0.000 | 0.001a | 0.001 |
| Polycythemia vera | 0.81 | 0.75 | 0.90a | 0.88 | 0.002 | 0.007 | 0.002a | 0.003 |
| Sickle cell anemia | 0.88 | 0.93 | 0.93 | 0.95a | 0.050 | 0.057 | 0.029 | 0.038a |
| Tuberous sclerosis | 0.89 | 0.90a | 0.89 | 0.90 | 0.036 | 0.060a | 0.034 | 0.058 |
| Best | 1 | 3 | 5 | 7 | 1 | 6 | 3 | 6 |
AUC: area under the curve; PRC: precision-recall curve; ROC: receiver-operating characteristic.
aHighest value among the 4 methods tested.
Underdocumented and prediagnosed case analysis
A total of 535 patients in our gold standard set (8%) lacked any ICD codes indicating diagnosis. A1A had the highest proportion of cases without an ICD (25%), followed by FX (20%). The mean rPheRS was elevated for underdocumented cases for all diseases, and 33% of the 535 cases had rPheRS >95th percentile (Supplementary Table S7).
We found 32 CF patients who had EHR data before diagnosis. The average age of diagnosis for these patients was 21 (range, 1-80) years of age. They had an average of 24 (range, 3-167) clinic visits before their diagnosis. Sixteen of the 32 patients had PheRS greater than the 95th percentile before diagnosis at an average 886 days before their diagnosis (range, 3-3689 days). Among the 10 patients diagnosed as adults (≥21 years of age), 80% had a PheRS in the 95th percentile before their diagnosis (mean = 1222 days; range, 51-3389 days). Three of these adults had a PheRS in the 99th percentile for over 2 years before diagnosis (Figure 5, Supplementary Figure S3A, B; Supplementary Table S8).
Figure 5.

Phenotype Risk Score of adults with cystic fibrosis over time. Each row represents a patient. The dots indicate a clinic visit. The line is pink during the period before diagnosis and blue after diagnosis. Residualized Phenotype Risk Score (rPheRS) was calculated at each new clinical encounter using the Human Phenotype Ontology-International Classification of Diseases map.
DISCUSSION
PheRS is a method that measures the similarity between a patient and an idealized description of Mendelian disease. In this article, we sought to improve the method in 3 ways: integrating ICD-10 codes, integrating lab data, and mapping ICDs directly into HPO terms instead of using intermediary phecodes. We tested the performance of PheRS using several metrics to simulate different applications of the method. All maps tested were found to have significant differences between groups of cases and controls. The maps we tested demonstrated different strengths depending on the performance metric.
If PheRS is used to assess pathogenicity of genetic variants in a population, then its performance needs to be tuned to finding differences between cases and controls in aggregate. All of the maps that we tested performed well in this regard, as demonstrated by the strong and statistically significant differences found between cases and controls. While we observed some improvement using the HPO-ICD map vs the HPO-phecode map, the differences between the 2 were small and only statistically significant for MS. Adding lab data, on the other hand, led to robust improvements across several diseases. This was particularly evident for PKU; while ICD features were entirely unsuccessful at differentiating PKU cases and controls, high serum phenylalanine was highly effective. Other diseases with multiple lab abnormalities such as A1A and HH were also improved with lab features.
To identify subsets enriched for patients with Mendelian disease—a “phenotype-first approach”—PheRS must have high specificity so that affected patients have high ranking scores. For this application, the HPO-ICD map had better performance compared with the HPO-phecode map, leading to an average 8% increase in the number of cases identified in the 100 top-scoring patients.
Diagnoses are not always well documented in the EHR, which is a challenge for EHR-based research. PheRS may help find these patients. We found that 8% of the patients in our gold standard set had no ICD codes relevant to their Mendelian disease diagnoses. However, these patients did have elevated PheRS compared with controls.
Mendelian diseases can be difficult to diagnose, particularly in patients with symptoms that are milder or develop in adulthood.12 We found that 36% of children and 80% of adults diagnosed with CF at VUMC had PheRS in the 95th percentile before their diagnosis. Three of the adult cases had scores in the 99th percentile for over 2 years before diagnosis.
The HPO-ICD map used in this study is presented as a starting point. It will evolve as we learn more about the way patient features are reflected in their claims data. In future work, we will attempt to refine the map with an empirical approach using EHR data. Further improvement to the method may also be realized by integrating additional sources of phenotypic information. Many of the features of the Mendelian diseases studied in this article are not readily captured by ICD and lab data. Examples include epicanthus for DS, short femoral neck for achondroplasia, and increased axial length of the globe for MS. Some of these features may be captured from clinical narratives or reports from radiology or pathology.20,21 Our work integrating lab data demonstrates a method by which this can be accomplished. If phenotypes from new sources are mapped to HPO terms, the scoring algorithm will not require modification.
While expanding the sources of phenotypic information will likely further improve the performance characteristics of PheRS, the fact that the algorithm works well with ICDs alone is an important finding. ICD billing codes are ubiquitous, easy to manipulate, and are more easily shared across institutions than clinical narratives or laboratory data.22 They do note require sophisticated and often site-specific natural language processing techniques to extract and can be de-identified in a relatively simple manner. Advances in and increased adoption of common data models may ameliorate current challenges in utilizing clinical notes.23,24 In researching rare genetic diseases and variants, large cohorts are an absolute requirement. Many of these large cohorts, such as the UK Biobank or China Kadoorie Biobank, do not have narrative or EHR laboratory data.25,26 Thus, using an algorithm that relies on readily available data such as ICD codes will likely have utility over large datasets.
The goal of precision medicine is to tailor treatment for an individual patient. Genetic sequencing and molecular diagnostics are increasingly used in pursuit of personalized treatment. However, even in the context of these powerful technologies, the phenotype is, and will likely always be, essential to the practice of medicine. Although computational phenotypes such as PheRS will never replace the careful observation of a good clinician, they may be useful in increasing clinical suspicion for patients who are difficult to diagnose.
Much of what we know about Mendelian disease has been discovered through studying individual patients and their families.27 While their methods were distinctly “low-tech,” the pioneers of genetic medicine assembled an impressive body of knowledge by viewing nuanced phenotypes through the lens of Mendelian inheritance. With the massive amount of medical data that is being aggregated today in EHRs and research cohorts like the All of Us Research Program28 and the UK Biobank, we now have an opportunity to study phenotypic patterns at the population level. From this new perspective, we can enhance our understanding of Mendelian disease so that we can better detect and diagnose individual patients.
FUNDING
This work was supported by grants R01-LM010685 from the National Library of Medicine (LB, JJH, LAT, JCD), P50-GM115305 from the National Institute for General Medical Sciences (DMR), T32 CA160056 Vanderbilt Training in the Molecular and Genetic Epidemiology of Cancer (CZ), and U01 grants supporting Vanderbilt's participation in the eMERGE (Electronic Medical Records and Genomics) network (HG004603, HG006378, and HG008672). BioVU received and continues to receive support through the National Center for Research Resources (UL1-RR024975), which is now the National Center for Advancing Translational Sciences (UL1-TR000445).
AUTHOR CONTRIBUTIONS
LB, DMR, and JCD contributed to the conceptual design of the work and methods. LB and JH developed the statistical tests and figures. LB, JB, SD, and NCZ created the HPO-ICD map. LB and JG developed the HPO-lab map. NCZ and LAT assisted with implementation and interpretation of the results. All authors contributed to the drafting and editing of the manuscript.
SUPPLEMENTARY MATERIAL
Supplementary material is available at Journal of the American Medical Informatics Association online.
COMPETING INTEREST STATEMENT
None declared.
Supplementary Material
References
- 1. Ledley RS, Lusted LB.. Reasoning foundations of medical diagnosis. Science 1959; 1303366: 9–21. [DOI] [PubMed] [Google Scholar]
- 2. McKusick VA. On lumpers and splitters, or the nosology of genetic disease. Perspect Biol Med 1969; 122: 298–312. [DOI] [PubMed] [Google Scholar]
- 3.OMIM clinical synopsis—#219700—CYSTIC FIBROSIS; CF. https://www.omim.org/clinicalSynopsis/219700Accessed May 21, 2019.
- 4.OMIM - Online Mendelian Inheritance in Man. http://omim.org/ Accessed May 20, 2014.
- 5. Crawford DC, Crosslin DR, Tromp G, et al. eMERGEing progress in genomics—the first seven years. Front Genet 2014; 5; 184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wei W-Q, Bastarache LA, Carroll RJ, et al. Evaluating phecodes, clinical classification software, and ICD-9-CM codes for phenome-wide association studies in the electronic health record. PLoS One 2017; 127: e0175508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Van Driest SL, Wells QS, Stallings S, et al. Association of arrhythmia-related genetic variants with phenotypes documented in electronic medical records. JAMA 2016; 3151: 47–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bastarache L, Hughey JJ, Hebbring S, et al. Phenotype risk scores identify patients with unrecognized Mendelian disease patterns. Science 2018; 3596381: 1233–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Denny JC, Bastarache L, Ritchie MD, et al. Systematic comparison of phenome-wide association study of electronic medical record data and genome-wide association study data. Nat Biotechnol 2013; 3112: 1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Groopman EE, Marasa M, Cameron-Christie S, et al. Diagnostic utility of exome sequencing for kidney disease. N Engl J Med 2019; 3802: 142–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Splinter K, Adams DR, Bacino CA, et al. Effect of genetic diagnosis on patients with previously undiagnosed disease. N Engl J Med 2018; 37922: 2131–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bastarache L, Bastarache JA, Denny JC.. Case 40-2018: a woman with recurrent sinusitis, cough, and bronchiectasis. N Engl J Med 2019; 38014: 1382–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wenger AM, Guturu H, Bernstein JA, et al. Systematic reanalysis of clinical exome data yields additional diagnoses: implications for providers. Genet Med 2017; 192: 209–14. [DOI] [PubMed] [Google Scholar]
- 14. Wu P, Gifford A, Meng X, et al. Developing and evaluating mappings of ICD-10 and ICD-10-CM codes to phecodes. bioRxiv 2018 Nov 12 [E-pub ahead of print].
- 15. Danciu I, Cowan JD, Basford M, et al. Secondary use of clinical data: the Vanderbilt approach. J Biomed Inform 2014; 52: 28–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Roden DM, Pulley JM, Basford MA, et al. Development of a large-scale de-identified DNA biobank to enable personalized medicine. Clin Pharmacol Ther 2008; 843: 362–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Orphanet: an online database of rare diseases and orphan drugs. http://www.orpha.net Accessed April 9, 2019.
- 18. Sulieman L, Wu P, Denny J, Bastarache L. WikiMedMap: expanding the phenotyping mapping toolbox using Wikipedia. bioRxiv 2019 Aug 6 [E-pub ahead of print].
- 19. Zhang XA, Yates A, Vasilevsky N, et al. Semantic integration of clinical laboratory tests from electronic health records for deep phenotyping and biomarker discovery. npj Digit Med 2019; 2: 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Teixeira PL, Wei W-Q, Cronin RM, et al. Evaluating electronic health record data sources and algorithmic approaches to identify hypertensive individuals. J Am Med Inform Assoc 2017; 241: 162–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Song W, Huang H, Zhang C-Z, et al. Using whole genome scores to compare three clinical phenotyping methods in complex diseases. Sci Rep 2018; 81: 11360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hripcsak G, Albers DJ.. Next-generation phenotyping of electronic health records. J Am Med Inform Assoc 2013; 201: 117–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Deisseroth CA, Birgmeier J, Bodle EE, et al. ClinPhen extracts and prioritizes patient phenotypes directly from medical records to expedite genetic disease diagnosis. Genet Med 2019; 21: 1585–1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Rosenbloom ST, Carroll RJ, Warner JL, et al. Representing knowledge consistently across health systems. Yearb Med Inform 2017; 26: 139–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sudlow C, Gallacher J, Allen N, et al. UK biobank: an open access resource for identifying the causes of a wide range of complex diseases of middle and old age. PLoS Med 2015; 123: e1001779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chen Z, Chen J, Collins R, et al. China Kadoorie Biobank of 0.5 million people: survey methods, baseline characteristics and long-term follow-up. Int J Epidemiol 2011; 406: 1652–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Amberger J, Bocchini CA, Scott AF, et al. McKusick’s online Mendelian inheritance in man (OMIM®). Nucleic Acids Res 2009; 37 (Database issue): D793–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sankar PL, Parker LS.. The Precision Medicine Initiative’s All of Us Research Program: an agenda for research on its ethical, legal, and social issues. Genet Med 2017; 197: 743–50. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.