

Graphical abstract

Keywords: Malaria, Plasmodium falciparum, cGMP, Protein kinase G, Imidazopyridine, SAR
Abstract
Focussed studies on imidazopyridine inhibitors of Plasmodium falciparum cyclic GMP-dependent protein kinase (PfPKG) have significantly advanced the series towards desirable in vitro property space. LLE-based approaches towards combining improvements in cell potency, key physicochemical parameters and structural novelty are described, and a structure-based design hypothesis relating to substituent regiochemistry has directed efforts towards key examples with well-balanced potency, ADME and kinase selectivity profiles.
Malaria is one of the most prevalent infectious diseases of the developing world in humans, whose causative agent is the protozoan parasite Plasmodium, with most deaths caused by P. falciparum. Despite being largely preventable and treatable, it was responsible for 435,000 deaths in 2017; young children and pregnant women in sub-Saharan Africa are particularly at risk.1 In addition to continuing challenges in the contexts of policy development and socio-economic impact,2 the observation of increasing resistance to current standard-of-care treatments is significant. This is driving research and development efforts to uncover new mechanisms by which the disease can be controlled and prevented.3
Studies on the malarial kinome continue to provide well characterised and credible new targets for antimalarial small molecule drug discovery.4, 5 The cGMP-dependent kinase PfPKG is one kinase which meets many of the criteria for such a target. Pharmacological characterisation using early chemical inhibitors in combination with reverse genetics has demonstrated the important role of this enzyme in numerous critical processes in the malaria life cycle.6, 7, 8, 9, 10, 11 Following previous experience with progressing chemical inhibitors of other important malarial kinases,12, 13, 14, 15 we have recently begun to disclose our efforts to develop a series of PfPKG inhibitors based upon both bicyclic16 and monocyclic scaffolds.17 In the bicyclic series, a number of advanced analogues were shown to possess promising in vitro activity, a well-defined mechanism of action and property profiles which translated to target-driven efficacy in vivo.16 An ongoing objective is to develop this chemical series with a view to improving key physiochemical parameters and compound novelty whilst retaining cell potency and lipophilic ligand efficiency (LLE).19
A recent report from us described initial efforts towards these goals by evaluating the aminopyrimidine hinge binding motif, bicyclic core structure and basic substituent positioning.20 Investigation of each of those structural features was found to be both necessary and productive, and the resulting compound profiles pointed strongly to retaining these motifs in their original forms. As a result, the profiles of analogues such as 1 (Figure 1) challenged us to consider additional strategies for re-positioning the series in suitable ADME property space whilst maintaining suitable levels of in vitro activity and improving compound novelty. A first approach was to reduce the size of the 4-fluorophenyl motif to lower lipophilicity and hence increase lipophilic ligand efficiency (LLE)19 (Figure 1 – A). Previous SAR20 suggested potency could be regained by enlarging the pyrimidine substituent, if needed (Figure 1 – B). In a second set of analogues, it was anticipated that re-design of the benzylic dimethylaminomethyl side chain in the prototypical inhibitor compound 2 would enable lowering of logD (for example by increasing chain length and basicity) and could also address one likely point of metabolic liability (for example by replacing the benzylic carbon atom with a heteroatom) (Figure 1 – C). Here we discuss the results of these investigations and show their significant beneficial impact against the above criteria.
Figure 1.

In vitro profiles of imidazopyridine 1 and 2, and design modifications to be applied to 2: A – truncate the aryl group; B – enlarge the pyrimidine substituent if required; C – re-design the basic substituent. ADME data: mLogD = measured logD; MLM = % remaining after 30 min incubation with mouse liver microsomes.
We first examined the possibility of improving the lipophilic efficiency by focusing on the large 4-fluorophenyl motif, and initially retained the original basic substituent at the 7-position of the bicyclic core in doing so. The main design emphasis was to attempt to balance the size of the pyrimidine substituent with a smaller lipophilically efficient replacement for the 4-fluorophenyl group. Among a small set of initial replacements, prepared by the general route shown in Scheme 1, the cyclopropyl analogue 9 was of lower potency in a biochemical assay21 as compared to 2, but significantly also showed a lower mLogD value of 1.7 of 1.7 (Table 1).22 Given that potency was lower than desirable, further analogues incorporating the cyclopropyl group were designed to combine a lower mlogD with improvements in potency and LLE. Hence a set of compounds with larger groups appended to the aminopyrimidine nitrogen was prepared using variations of the same chemical approach. Small alkyl groups such as that in 10 did not provide any further boost in activity or LLE but, in line with previous SAR, arylaminopyrimidines such as 11 and 12 were more biochemically active and possessed the anticipated trend towards lower mLogD. The most balanced profile was achieved in 13,23 which showed similar levels of both biochemical potency and anti-malarial activity in a blood stage hypoxanthine incorporation (HXI) cell assay21 compared to 2, coupled with improvements in mLogD and LLE.
Scheme 1.

Reagents and conditions: (i) LiHMDS, R1CO2Et, THF, −78 °C – rt, 3 h, 27–76%; (ii) Bu4NBr3 or NBS, CH2Cl2, rt, 2 h; (iii) 2-aminopyridine-4-methanol, EtOH, 4 Å sieves, 100 °C, 18 h, 12–44% for two steps; (iv) MsCl, Et3N, THF, 0 °C, 1 h or SOCl2, CH2Cl2, 50 °C, 1 h; (v) Me2NH, THF, 0 °C – rt, 33–65% for two steps; (vi) H2O2, Na2WO4·2H2O, AcOH, MeOH, 0 °C – rt, 3 h; (vii) for 7–9: NH4OAc, melt, 130 °C, 3 h, 5–27% for two steps; for 10: iPrNH2 (neat), 60 °C 3 h, 23% for two steps; for 11: 2-aminopyridine (excess), NMP, microwave, 150 °C, 3 h, 8% for two steps; for 12: 4-(4-methylpiperazino)aniline, neat, microwave, 170 °C, 15 min, 5% for two steps; for 13: 4-(4-aminophenyl)piperazine-1-carboxylic acid tert-butyl ester, TFA, sBuOH, 110 °C, 6 h, then TFA, CH2Cl2, rt, 2 h, 10% for three steps.
Table 1.
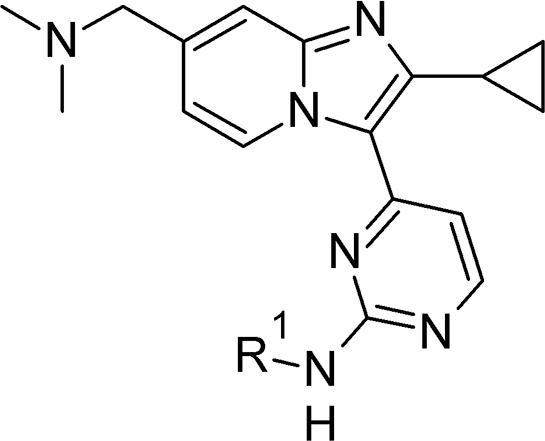
| Compound | R1 | PfPKG pIC50 | Pf HXI pEC50a | LLE | mLogD |
|---|---|---|---|---|---|
| 2 | – | 8.70 | 6.44 | 6.3 | 2.4 |
| 9 | H | 7.45 | nt | 5.8 | 1.7 |
| 10 | 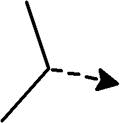 |
7.36 | nt | 4.4 | 3.0 |
| 11 | 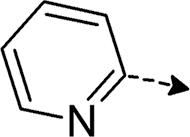 |
8.06 | 5.76 | 5.8 | 2.5 |
| 12 | 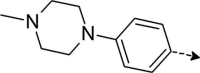 |
8.14 | 6.95 | 6.0 | 2.1 |
| 13 |  |
8.35 | 6.70 | 6.8 | 1.5 |
nt = not tested.
Turning next to the basic substituent on the bicyclic core, a small number of molecules were initially designed to identify the optimum position at which to locate this motif. We decided to employ the benzylic dimethylaminomethyl group present in 2 for this analysis. Docking of 2 and its 5-, 6- and 8- regioisomers into an apo-structure of PfPKG (PDB:5DYK24) suggested that the best site for that substituent was at the 7-position (Figure 2). The location of the positively charged basic center between two acidic protein residues (E625 and D682) was judged to be optimal for that particular group. Whilst relocating to the 6- or 8- positions appeared to be spatially tolerable, sub-optimal interaction with the acids and a subsequent loss in affinity was predicted. Appending several possible groups at the 5-position appeared to result in a significant steric clash with the pyrimidine hinge binding motif (data not shown); this was predicted to cause a significant loss of activity and hence was not pursued.
Figure 2.

Docking of 2 (left), the 8-regioisomer 15 (centre) and the 6-regioisomer 16 (right) into an apo-PfPKG crystal structure (PDB:5DYK24), with protein surface coloured by electrostatic potential. H-bonds and charge interactions are shown as dashed lines.
This hypothesis was tested by synthesizing the 8- and 6-regioisomers 15 and 16 respectively. Using variations of previously described chemical approaches,18, 20 compounds 15 and 16 could be prepared from the bromoketone building block 1425 (Scheme 2), in good yields over 5 synthetic steps.
Scheme 2.

Reagents and conditions: (i) 2-aminopyridine-3-methanol (for 15) or 2-aminopyridine-5-methanol (for 16), MeCN, NaHCO3, 90 °C, 18 h: 32–75%; (ii) MsCl, Et3N, THF, 0 °C, 4 h; (iii) Me2NH, THF, 0 °C – rt, 3 h, 60–89% for two steps; (iv) H2O2, Na2WO4·2H2O, AcOH, MeOH, rt, 3 h; (v) NH4OAc, melt, 120 °C, 3 h, 19–27% for two steps.
These two compounds showed reductions in their biochemical activity, as compared to 2 (Table 2), which were in line with predictions from the docking studies. Lipophilic ligand efficiency for the 8-substituted compound 15 was also higher than for 6-analogue 16, in part due to an interesting divergence in mLogD (values of 1.6 for 15, 2.3 for 16, as compared to 2.4 for 2). However, the key factor of lower synthetic accessibility for 8-position analogues emerged, which directed our efforts away from preparing further compounds of this kind. In contrast, the position of the two key acidic residues at the binding pocket mouth implied that re-design of the basic substituent into longer chain variants and appropriate conformationally constrained versions might be productive. This design hypothesis suggested that substituents of these new types at either the 6- or 7-positions should be evaluated.
Table 2.
| Compound | Substituent position | PfPKG pIC50 | LLE |
|---|---|---|---|
| 2 | 7 | 8.70 | 6.3 |
| 15 | 8 | 7.53 | 5.9 |
| 16 | 6 | 7.29 | 5.0 |
We tested this proposal by making compounds bearing such modifications to the dimethylaminomethyl side chain in 2, and chose to include adjustments in both expected pKa and conformation by varying the linking atom, chain length and ring size in new analogues. Preparation of key intermediates 17–19 in three steps, followed by palladium-catalysed aminations or microwave-mediated direct displacements provided the N-linked and O-linked examples 20–27 respectively (Scheme 3).26 The 7-C-linked analogue 29 was also prepared in four synthetic steps from intermediate 28, which was itself constructed by condensing bromoketone 14 with the appropriate building block 2-(2-aminopyridin-4-yl)ethan-1-ol.
Scheme 3.

Reagents and conditions: (i) 2-amino-4-bromopyridine or 2-amino-5-bromopyridine, EtOH, 4 Å sieves, 80 °C, 18 h, 20–36%; (ii) H2O2, Na2WO4·2H2O, AcOH, MeOH, rt – 50 °C, 18 h; (iii) For R1 = H: NH4OAc, melt, 130 °C, 18 h, 30–52% for two steps; for R1 = Me: Me2NH, THF, 70 °C, 18 h, 56% for two steps; (iv) for R2 = amine: Pd(OAc)2, JohnPhos, R2NH, NaOtBu, dioxane, 100 °C, 18 h, 3–29%; (v) for R2 = alcohol: KOtBu, ROH, NMP, microwave, 170 °C, 10 min, 6%; (vii) MsCl, Et3N, THF, 0 °C, 1 h; (vi) Me2NH, THF, 60 °C, 10 h, 49% for two steps.
These analogues (along with the earlier example 16) demonstrated that extending via a conformationally constrained basic group (as in 20) or via an open chain version (as in 21) at either the 7- or 6-position of the core could each provide good biochemical potency (Table 3). Neither of these compounds appeared to possess a particular advantage in any aspect of their in vitro profiles as compared to 2. Interestingly, the related pair of piperazine regioisomers 22 and 23 showed a subtle contrast in mLogD, where the 6-isomer 23 was found to possess the lower value. The cell activity of 23 was also slightly lower as compared to 22. Synthetic access to 6-substituted compounds was also found to be generally less efficient; considering this and other contributing factors,27 we decided to focus additional efforts on 7-linked analogues only.
Table 3.
| Compound | R1 | PfPKG pIC50 | Pf HXI pEC50a | LLE | mLogD |
|---|---|---|---|---|---|
| 2 | 7-CH2NMe2 | 8.70 | 6.44 | 6.3 | 2.4 |
| 16 | 6-CH2NMe2 | 7.29 | nt | 5.0 | 2.3 |
| 20 | 7-
|
8.07 | 6.07 | 6.0 | 2.1 |
| 21 | 6-NH(CH2)3NMe2 | 7.90 | nt | 5.7 | 2.2 |
| 22 | 7-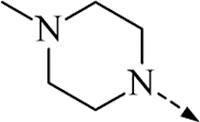
|
8.48 | 6.58 | 5.4 | 3.1 |
| 23 | 6-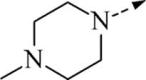
|
8.58 | 6.06 | 5.8 | 2.8 |
nt = not tested.
Using the same synthetic chemistry as shown in Scheme 3, a small additional set of 7-substituted analogues was prepared and evaluated (Table 4). As compared to 20, increasing the ring size and hence altering the conformational constraint in 24 gave modest improvements in biochemical potency and cell activity, though only a slight change to LLE. Microsomal stability was improved significantly, perhaps due to constraining the conformation in the basic side chain. For the open chain examples 25 and 29, in vitro ADME profiles very similar to 2 could be obtained, though both showed lower biochemical activity (and hence no further benefit in LLE) and microsomal stability had not improved. Both LLE and microsomal stability could be improved by returning to a nitrogen-linked design in the open chain analogue 26, for which the essentially unchanged mLogD (as compared to 25 and 29) was accompanied by better biochemical potency and LLE. Finally, positioning an additional carbon atom within the aminopyrimidine group gave 27; this notable compound showed an excellent balance of good biochemical potency, in vitro activity against the parasite and improved LLE and mLogD values. The effect of the secondary aminopyrimidine (in 27) on microsomal stability, relative to the primary aminopyrimidine (in 26), was also significant.
Table 4.
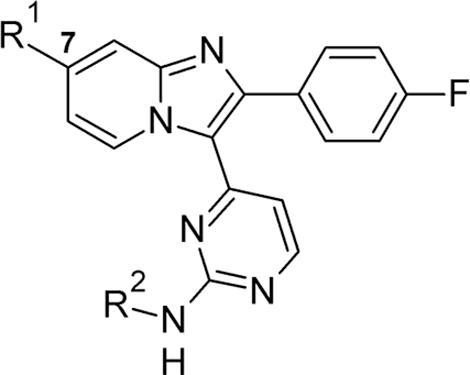
| Compound | R1 | R2 | PfPKG pIC50 | Pf HXI pEC50a | LLE | mLogD | MLM % remb |
|---|---|---|---|---|---|---|---|
| 2 | 7-CH2NMe2 | H | 8.70 | 6.44 | 6.3 | 2.4 | 52 |
| 24 | 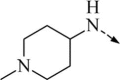 |
H | 8.44 | 6.46 | 6.2 | 2.2 | 88 |
| 25 | 7-O(CH2)2NMe2 | H | 8.13 | 6.09 | 5.9 | 2.2 | 54 |
| 26 | 7-NH(CH2)2NMe2 | H | 8.59 | nt | 6.5 | 2.1 | 80 |
| 27 | 7-NH(CH2)2NMe2 | Me | 8.56 | 6.28 | 6.4 | 2.2 | 93 |
| 29 | 7-(CH2)2NMe2 | H | 7.69 | 6.00 | 5.8 | 1.9 | 65 |
nt = not tested.
% remaining after 30 min incubation with mouse liver microsomes.
The two most promising compounds identified − 13 and 27 – were profiled and compared in vitro (Table 5). In addition to previously described improvements in mLogD and LLE, high kinetic solubility (measured using PBS at pH7.4 as buffer) was maintained in each case and both compounds were shown to be non-cytotoxic. Despite a significantly lower mLogD value, the mouse microsomal stability of 13 surprisingly remained at the same level as for 2, for which we have no clear explanation.28 In particular, 27 matched excellent biochemical and cell potency with significantly higher stability in mouse microsomes to give a highly promising and well-balanced overall profile. Selectivity was assessed by screening 13 and 27 against a human kinase panel29 at a single 1 µM concentration (Figure 3). As expected, the smaller cyclopropyl motif in 13 resulted in a decreased level of selectivity, whilst 27 showed an excellent selectivity profile against the kinases screened. We also tested compounds 2 and 27 against the two human orthologues of PKG; no activity was observed up to a top assay concentration of 1 µM,30 indicating a high level of selectivity for the malarial kinase.
Table 5.
Full in vitro profiles for compounds 2, 13 and 27; a % remaining after 30 min incubation with mouse liver microsomes; b kinetic solubility; cin vitro cytotoxicity assay measured in HepG2 human liver-derived cells – concentration at which half of cells remained viable at 48 h.16
| Compound | PfPKG pIC50 | Pf HXI pEC50 | LLE | mLogD | MLM % rema | Kin sol (µM)b | HepG2 pEC50c |
|---|---|---|---|---|---|---|---|
| 2 | 8.70 | 6.44 | 6.3 | 2.4 | 52 | 200 | < 4.7 |
| 13 | 8.35 | 6.70 | 6.8 | 1.5 | 53 | 189 | < 5 |
| 27 | 8.56 | 6.28 | 6.4 | 2.2 | 93 | 207 | < 4.7 |
Figure 3.

Kinase selectivity data for representative imidazopyridines 2, 13 and 27 on screening against a human kinase panel at 1 µM concentration; green < 50% inhibition; yellow 50–90% inhibition; red > 90% inhibition.29
We have reported here the results of our continuing effort to progress a series of imidazopyridines as inhibitors of PfPKG, focusing on alteration of the 4-fluorophenyl group and re-design of the basic substituent as key strategic aims. By concentrating on cell potency, lipophilic ligand efficiency and structural novelty in tandem, compounds such as 27 in particular were developed to populate a highly desirable and novel area of chemical space as potent, lower molecular weight, lipophilically efficient analogues with improved in vitro ADME and selectivity profiles. Studies towards the identification of additional analogues suitable for in vivo studies and further mechanistic considerations are ongoing and will be reported in due course.
Acknowledgments
This work was funded by MRC DPFS grant no G1000779. The authors thank David Tickle, Sadhia Khan, Katie Barnes and Hasina Mahmood (LifeArc) for compound purification and in vitro ADME data, Win Gutteridge and Simon Croft for many helpful discussions, and Jeremy Burrows and Sir Simon Campbell (Medicines for Malaria Venture) for their support of this work.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.bmcl.2019.08.014.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
References
- 1.World Health Organisation World Malaria Report. 2018 [Google Scholar]
- 2.Shepard D.S., Ettling M.B., Brinkmann U., Sauerborn R. Trop Med Parasitol. 1991;42:199. [PubMed] [Google Scholar]
- 3.Dondorp A.M., Fairhurst R.M., Slutsker L. New Engl J Med. 2011;365:1073. doi: 10.1056/NEJMp1108322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lucet I.S., Tobin A., Drewry D., Wilks A.F., Doerig C. Future Med Chem. 2012;4:2295. doi: 10.4155/fmc.12.183. [DOI] [PubMed] [Google Scholar]
- 5.Derbyshire E.R., Zuzarte-Luis V., Magalhaes A.D. Chem Bio Chem. 1920;2014:15. doi: 10.1002/cbic.201400025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Taylor H.M., McRobert L., Grainger M. Cell. 2010;9:37. doi: 10.1128/EC.00186-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Alam M.M., Solyakov L., Bottrill A.R. Nat Commun. 2015;6:7285. doi: 10.1038/ncomms8285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McRobert L., Taylor C.J., Deng W. PLoS Biol. 2008;6 doi: 10.1371/journal.pbio.0060139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Moon R.W., Taylor C.J., Bex C. PLoS Path. 2009;5 doi: 10.1371/journal.ppat.1000599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Govindasamy K., Jebiwott S., Jaijyan D.K. Mol Microbiol. 2016;102:349. doi: 10.1111/mmi.13466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Falae A., Combe A., Amaladoss A., Carvalho T., Menard R., Bhanot P. J Biol Chem. 2010;285:3282. doi: 10.1074/jbc.M109.070367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chapman T.M., Osborne S.A., Wallace C. J Med Chem. 2014;57:3570. doi: 10.1021/jm500342d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chapman T.M., Osborne S.A., Bouloc N. Bioorg Med Chem Lett. 2013;23:3064. doi: 10.1016/j.bmcl.2013.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Large J.M., Osborne S.A., Smiljanic-Hurley E. Bioorg Med Chem Lett. 2013;23:6019. doi: 10.1016/j.bmcl.2013.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bouloc N., Large J.M., Smiljanic E. Bioorg Med Chem Lett. 2008;18:5294. doi: 10.1016/j.bmcl.2008.08.043. [DOI] [PubMed] [Google Scholar]
- 16.Baker D.A., Stewart L.B., Large J.M. Nat Commun. 2017;8:430. doi: 10.1038/s41467-017-00572-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tsagris D.J., Birchall K., Bouloc N. Bioorg Med Chem Lett. 2018;28:3168. doi: 10.1016/j.bmcl.2018.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Biftu T., Feng D., Fisher M. Bioorg Med Chem Lett. 2006;16:2479. doi: 10.1016/j.bmcl.2006.01.092. [DOI] [PubMed] [Google Scholar]
- 19.Hopkins A.L., Keserü G.M., Leeson P.D., Rees D.C., Reynolds C.H. Nat Rev Drug Disc. 2014;13:105. doi: 10.1038/nrd4163. [DOI] [PubMed] [Google Scholar]
- 20.Large J.M., Birchall K., Bouloc N.S. Bioorg Med Chem Lett. 2019;29:509. doi: 10.1016/j.bmcl.2018.11.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Full details on biochemical assay and anti-malarial hypoxanthine incorporation (cell based) assay procedures can be found in reference 16.
- 22.Installing either a methyl group (7; PfPKG pIC50 7.24) or trifluoromethyl group (8; PfPKG pIC50 6.83) gave intermediate levels of biochemical potency.
- 23.For an improved synthetic approach to compound 13, see supplementary information.
- 24.Wernimont, A.K.; Tempel, W.; He, H.; Seitova, A.; Hills, T.; Neculai, A.M.; Baker, D.A.; Flueck, C.; Kettleborough, C.A.; Arrowsmith, C.H.; Edwards, A.M.; Bountra, C.; Hui, R.; Hutchinson, A.; El Bakkouri, M.; Crystal structure of PF3D7_1436600; Structural Genomics Consortium; to be published.
- 25.Scribner A., Dennis R., Hong J. Eur J Med Chem. 2007;42:1334. doi: 10.1016/j.ejmech.2007.02.006. [DOI] [PubMed] [Google Scholar]
- 26.Low yields for the final palladium-catalysed reactions could be improved by reversing the order of synthesis, such that this step was carried out before oxidation and displacement of the 2-thiomethylpyrimidine substituent. Full details are provided in the supplementary information.
- 27.It was found that 23 showed significant activity in an in vitro cytotoxicity assay16 whereas 22 did not. The same relationship between cell cytotoxicity and regiochemistry of the basic substituent on the bicyclic core was also observed for earlier compounds 16 and 2.
- 28.Kramer C., Ting A., Zheng H. an excellent recent review on correlating ADME properties, see. J Med Chem. 2018;61:3277. [Google Scholar]
- 29.Kinase selectivity profiling was carried out at the MRC Protein Phosphorylation Unit at the University of Dundee, U.K. at a single concentration of 1 µM for all three compounds. Kinases tested are listed below (reading from left to right in Figure 3) – the two kinases against which all three compounds showed some activity are in bold type: MKK1, ERK1, ERK2, JNK1, JNK2, p38a, MAPK, RSK1, RSK2, PDK1, PKBa, PKBb, SGK1, S6K1, PKA, ROCK2, PRK2, PKCa, PKCz, PKD1, MSK1, MNK1, MNK2, PRAK, CAMKKb, CAMK1, SmMLCK, PHK, CHK2, GSK3b, CDK2-Cyclin A, PLK1, Aurora A, Aurora B, AMPK, MARK3, BRSK2, MELK, CK1, CK2, DYRK1A, NEK2a, NEK6, IKKb, PIM1, SRPK1, MST2, EF2K, HIPK2, PAK4, Src, Lck, CSK, FGF-R1, IRR, MST4, SYK, YES1, IGF-1R, VEG-FR, BTK, EPH-B3, TBK1, IKKe, GCK, NUAK1, MLK1, MINK1, MLK3, LKB1, HER4, TTK, IR, RIPK2, TAK1, MEKK1, TrkA.
- 30.For additional details see supplementary information.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.