

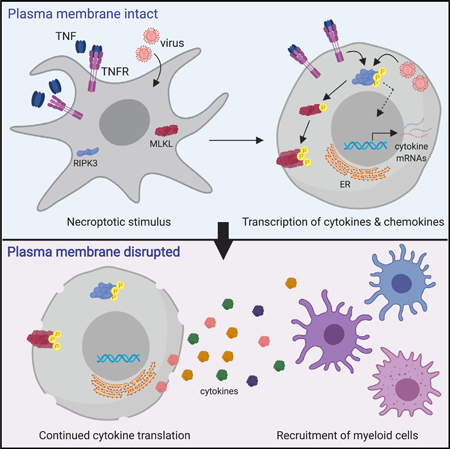
SUMMARY
Necroptosis is a form of programmed cell death that is defined by activation of the kinase RIPK3 and subsequent cell membrane permeabilization by the effector MLKL. RIPK3 activation can also promote immune responses via production of cytokines and chemokines. How active cytokine production is coordinated with the terminal process of necroptosis is unclear. Here, we report that cytokine production continues within necroptotic cells even after they have lost cell membrane integrity and irreversibly committed to death. This continued cytokine production is dependent on mRNA translation and requires maintenance of endoplasmic reticulum integrity that remains after plasma membrane integrity is lost. The continued translation of cytokines by cellular corpses contributes to necroptotic cell up-take by innate immune cells and priming of adaptive immune responses to antigens associated with necroptotic corpses. These findings imply that cell death and production of inflammatory mediators are coordinated to optimize the immunogenicity of necroptotic cells.
In Brief
Necroptotic cell death is associated with cytokine production. Orozco et al. show that necroptotic cell “corpses” continue to synthesize cytokines after they have lost membrane integrity and committed to cell death. This activity involves continued mRNA translation and requires ER function that continues after plasma membrane rupture.
Graphical Abstract
INTRODUCTION
Programmed cell death can occur via several pathways, and the way a cell dies influences subsequent immune responses (Yatim et al., 2017). Although apoptosis is generally considered immunologically silent, lytic forms of cell death, such as pyroptosis and necroptosis, can occur in response to pathogenic infection and are associated with inflammation and adaptive immunity (Green and Llambi, 2015). It is now appreciated that these cell death programs influence the immune system through the active generation of immunostimulatory signals during cell death. The activating cleavage of interleukin-1β (IL-1β) and IL-18 by caspase-1 that accompanies pyroptosis is a well-described example of this paradigm (de Vasconcelos et al., 2016; Vande Walle and Lamkanfi, 2016).
Necroptosis is a distinct cell death program, triggered in response to receptor ligation or viral infection through formation of a cytosolic complex containing the receptor-interacting protein kinases RIPK1 (Degterev et al., 2008; Lin et al., 2004) and RIPK3 (Cho et al., 2009; He et al., 2009; Zhang et al., 2009) and subsequent phosphorylation of the membrane-disrupting pseudokinase MLKL (Chen et al., 2013; Sun et al., 2012; Wu et al., 2013; Zhao et al., 2012). Several recent studies have highlighted additional roles for the RIP kinases in promoting nuclear factor κB (NF-κB)-dependent transcriptional responses, which in some cases occur simultaneously with necroptotic cell death (Snyder et al., 2019; Yatim et al., 2015). We have previously reported that this transcriptional signaling leads to an increase in cross-priming of T cells responsive to antigens derived from necroptotic cells. However, this finding raises the question of how a necroptotic cell is able to actively generate immunostimulatory cytokines while committing to the terminal process of cell death. Notably, an older report indicated that, although caspase activation associated with apoptosis actively suppresses protein translation by cleaving translation initiation factors, necroptotic cells retain the ability to translate mRNAs up to the point of death, as defined by loss of membrane integrity (Saelens et al., 2005).
Here, we report that cells undergoing necroptosis in response to direct RIPK3 activation or viral infection continue de novo synthesis of cytokines and chemokines for several hours after they have lost plasma membrane integrity and irreversibly committed to cell death. This process involves continued mRNA translation in cellular “corpses” and proceeds via an endoplasmic reticulum (ER)-dependent mechanism that reflects maintenance of ER integrity after MLKL-mediated plasma membrane (PM) permeabilization. This continued cytokine and chemokine synthesis enhances the uptake of necroptotic-cell-derived material and contributes to the immunogenicity of necroptotic cell-derived antigens in vivo. Together, these findings define an unexpected mechanism by which cells that have irreversibly committed to cell death continue to influence inflammatory and immune responses.
RESULTS
RIPK3 Activation Leads to Cytokine Synthesis that Continues after Loss of PM Integrity
To study the effects of RIPK3 activation, we employed a previously described system in which RIPK3 can be activated directly, independent of upstream receptor signaling (Orozco et al., 2014). Briefly, we created a chimeric form of RIPK3, composed of murine RIPK3 fused to tandem copies of the dimerizable domain FKBPF36V. We term the resulting chimeric, activatable RIPK3 construct “acRIPK3” (Figure 1A). Consistent with previous reports (Orozco et al., 2014; Yatim et al., 2015), clonal populations of NIH 3T3 cells expressing acRIPK3 underwent rapid and uniform necroptosis upon addition of the small-mole-cule dimerizer drug, as measured by the uptake of the cell-impermeable DNA-binding dye Sytox Green. We observed that >99% of cells were positive for Sytox Green 3 h after addition of dimerizer (Figure 1B). The robust induction of necroptosis by this system was further confirmed by kinetic imaging using an In-cuCyte system, release of lactate dehydrogenase (LDH), and CellTiter-Blue viability assay (Figure S1A).
Figure 1. RIPK3 Activation Leads to Transcriptional Changes and to Cytokine Synthesis that Continues after Loss of PM Integrity.

(A) Schematic representation of RIPK3 and the chimeric RIPK3 construct used in this study.
(B) NIH 3T3 fibroblasts expressing acRIPK3 were treated with dimerizer drug for the indicated times and analyzed for the uptake of the cell-impermeable dye Sytox Green by flow cytometry.
(C) NIH 3T3 cells expressing acRIPK3 weretreated with TNF-α or RIPK3 was activated by addition of dimerizer. 4 h later, poly-Aenrichment and RNA sequencing was performed and transcript abundance relative to untreated controls analyzed. Values reaching significance (fold change [FC] >1;p< 0.05) on x axis only are depicted in red, y axis only in green, and both axes in blue.
(D) ELISA analysis of indicated cytokines in the supernatant of NIH 3T3 cells induced to undergo necroptosis via activation of acRIPK3.(E and F) Kinetic comparison of Sytox Green uptake (black lines) versus de novo IL-6 production.
(E) NIH 3T3 cells expressing acRIPK3 were pre-treated with either actinomycin D (ActD) or cycloheximide (CHX) 30 min before dimerizer treatment.
(F) NIH 3T3 cells expressing acRIPK3 were induced to undergo nec-roptosis and were subsequently treated with ActD or CHX 3 h later. IL-6 in the supernatant was quantified by ELISA.
(G) Experimental schematic for data in (H).
(H) NIH 3T3 cellswere treated as in (G) and supernatants analyzed by ELISA. Values are plotted as fold increase in IL-6 afterSytox Green uptake (3 h). All data are representative of at least three independent experiments.Error bars indicate SD from the mean.
We next assessed the transcriptional changes associated with RIPK1-RIPK3 complex formation using poly-A enrichment and RNA sequencing. We found that direct activation of RIPK3 led to increased transcription of numerous genes encoding inflammatory mediators. This response closely mirrored the transcriptional signature observed upon treatment of these cells with tumor necrosis factor alpha (TNF-α), reflecting the common recruitment of RIPK1 to both activated RIPK3 and TNFR1 (Figure 1C). We next performed RNA sequencing on cells at various time points following RIPK3 activation. Increased transcription of inflammatory mediators was observable 30 min after RIPK3 activation (Figure S1B), and these continued to increase in samples collected 1 and 2 h after RIPK3 activation (Figure S1C). This transcriptional response plateaued by 3 h after RIPK3 activation, as samples collected at this time point or 1 h later showed few changes to the identity or abundance of mRNA species present (Figures S1D and S1E). As >99% of these cells lose PM integrity by 3 h after RIPK3 activation (Figure 1B), these observations are consistent with RIPK3 activation leading to a robust transcriptional program, which is curtailed upon cell lysis.
Given the concurrence of inflammatory transcription and cell death upon RIPK3 activation, we next wondered how the translation of these mRNA species into cytokines and chemokines was regulated during necroptosis. To assess this, we measured the accumulation of cytokines in cellular supernatants following RIPK3 activation, focusing on IL-6, CXCL1, CXCL2, and CCL2, each of which was upregulated by RIPK3 at the mRNA level. Consistent with RIPK3-dependent production of these mediators, we found that RIPK3 activation led to their accumulation in cellular supernatants, which was curtailed by addition of a small-molecule RIPK3 inhibitor (GSK’843), but not by the RIPK1 inhibitor necrostatin-1 (Figure S1F). Consistent with earlier reports of a scaffolding function for RIPK1 in this setting (Yatim et al., 2015), the use of a form of RIPK3 lacking a RHIM domain, which is unable to interact with RIPK1 (Orozco et al.,2014), induced robust cell death but failed to induce concurrent cytokine production (Figure S1G).
Given our observation that transcription of mRNAs following RIPK3 activation appeared to cease upon cell lysis, we next carried out kinetic analyses to see whether mRNA translation followed a similar pattern. In contrast to our observations of mRNA abundance, this analysis revealed that the quantities of inflammatory cytokines in cellular supernatants continued to increase up to 9 h after activation of RIPK3 (Figure 1D). This was unexpected, as these cells had uniformly lost PM integrity by 3 h after RIPK3 activation (Figures 1B and S1A).
These findings implied that necroptotic cells may continue to produce inflammatory cytokines even after the irreversible loss of cell membrane integrity and commitment to cell death. To test this idea, we assessed the effect of inhibiting transcription or translation, either before RIPK3 activation or after RIPK3-and MLKL-mediated loss of cell-membrane integrity, in each case measuring abundance of IL-6 protein as a marker of the RIPK3-dependent inflammatory response. We found that addition of either actinomycin D (ActD) or cycloheximide (CHX), which inhibit transcription and translation, respectively, to cells prior to RIPK3 activation abolished IL-6 production during necroptosis, consistent with de novo transcription and translation of IL-6 (Yatim et al., 2015; Figure 1E). We next activated RIPK3 and then waited 3 h, a time point at which >99% of cells have lost membrane integrity (Figure 1B). Addition of ActD to these cellular corpses did not alter continued accumulation of cyto-kines in the supernatant, consistent with our observation that de novo transcription has ceased at this time point (Figures S1D and S1E). However, addition of CHX at this time point abrogated further increase in cytokine levels in cellular supernatants (Figure 1F). This finding implies that protein translation, but not transcription, continues in necroptotic cell corpses following the loss of membrane integrity, allowing for continued production and accumulation of inflammatory cytokines.
A recent study demonstrated that the ESCRT-III machinery can act downstream of MLKL to regulate necroptotic death and sustain cell survival (Gong et al., 2017). We therefore sought to confirm that uptake of Sytox Green was an accurate marker of irreversible loss of cell-membrane integrity. To do this, we made use of a competitive inhibitor of the dimerizer drug, termed washout ligand (WL). We first added dimerizer drug to activate RIPK3 in our acRIPK3 cell line and then added WL in the presence of Sytox Green, waited 24 h, and then treated cells with another cell-impermeable dye, Yoyo-3 (Figure S1H). We hypothesized that, if cells transiently permeabilized their membranes and then recovered, we would detect a population of cells that became marked with Sytox Green and then regained membrane integrity sufficiently to exclude Yoyo-3. However, we did not observe this; all cells that lost membrane integrity upon RIPK3 activation (Sytox Green positive) remained permeable 24 h later (Yoyo-3-positive; Figure S1I). Consistent with these data, we found that, after 3 h of RIPK3 activity, the potential for clonogenic outgrowth was irrecoverably lost (Figure S1J). These data indicate that loss of cell-membrane integrity, as indicated by uptake of Sytox Green, is an irreversible event from which cells do not recover.
We next sought to understand whether the ongoing mRNA translation we observed occurred within necroptotic corpses; in an extracellular compartment, such as an exosome; or on free ribosomes released into the culture supernatant. To address this question, we fractionated necroptotic cells by low-speed centrifugation and analyzed further cytokine synthesis in either supernatant or cell corpses (Figure 1G). We observed that isolated corpses, but not their supernatants, had the same increase in IL-6 levels as unseparated necroptotic cell cultures. Addition of CHX to cell corpses following isolation from supernatants inhibited further cytokine synthesis (Figure 1H). These data suggest that ongoing translation following loss of PM integrity occurs within the necroptotic corpse. Together, these findings indicate that the translation of mRNA continues within necroptotic cell bodies even after PM integrity is irreversibly lost.
Ongoing Translation in Necroptotic Cell Corpses Is Associated with the ER
We performed transmission electron microscopy on necroptotic cells 3 h after RIPK3 activation. We observed that, although the PM was clearly disrupted at this time point, sections of ER studded with ribosomes remained intact (Figures 2Aand S2A, yellow and purple arrows, respectively). This observation led us to consider whether these or other membranous structures within cell corpses were required to support ongoing translation. To assess this, we treated cells with the detergent digitonin, using a concentration sufficient to permeabilize all cellular membranes. Although digitonin did not directly affect ribosome function in an in vitro assay (Figure S2B), adding digitonin to necroptotic corpses abolished ongoing translation (Figure 2B). To further investigate a possible role for preserved ER function in continued translation, we treated necroptotic cell corpses with brefeldin A (BfA), an inhibitor of protein transport from the ER to the Golgi apparatus (Figure 2C), or with thapsigargin, a disruptor of ER calcium homeostasis (Figure 2D). Each of these reagents abrogated ongoing production of IL-6 in necroptotic cell corpses. Together, these data implicate continued ER function within necroptotic corpses as a requirement for continued cytokine translation.
Figure 2. Ongoing Translation in Necroptotic Corpses Is Associated with the ER.

(A) NIH 3T3 cells expressing acRIPK3 were treated for 3 h with dimerizer and analyzed by TEM. Solid purple arrows indicate intact ER; yellow dashed arrows indicate disrupted plasma membrane; “N” indicates nucleus. Scale bar in left panel represents 2 μm. Scale bars in middle and right panels represent 400 nm.
(B-D) NIH 3T3 cells expressing acRIPK3 were induced to undergo necroptosis and then treated with eitherdigitonin (B), brefeldin A (BfA) (C), orthapsigargin (D) after 3 h (→). The concentration of cytokine in the supernatant was quantified at 9 h post-dimerizer addition.
(E) Experimental schematic for data in (F).
(F) Cellsweretreated as in (E), and intracellularCCL2 was analyzed by flow cytometry. The gating for both histogram (left panel) and geometric mean fluorescent intensity (gMFI; right panel) is all cells > singlets > Zombie Green+.
(G) Experimental schematic for data in (H).
(H) NIH 3T3 cells expressing acRIPK3 were treated as in (G) and analyzed by microscopy. Representative images for each condition are shown. Scale bars represent 10 μm. Data in (A) are representative of one independent experiment.
Data in (B)-(D), (F), and (H) are representative of at least three independent experiments. ****p < 0.0001; ***p < 0.001; **p < 0.01; *p < 0.05. nd, not detected; NS, not significant. Error bars indicate SD from the mean.
One possible explanation for the observation of continued protein translation within necroptotic cells would be the presence of an undetected population of viable cells in our cultures. Given our findings indicating that translation occurs within cellular corpses, we devised an assay that allowed us to quantify cytokine translation within individual necroptotic cells. To do this, we induced necroptosis in NIH 3T3 cells and then briefly pulsed them with a cell-impermeable dye (Zombie Green) to mark only cells that had lost membrane integrity at this time point. These cellular corpses were then incubated with BfA to allow the accumulation of cytokines within the ER and subjected to intracellular cytokine staining and flow cytometry (Figure 2E). By analyzing only those cells marked by the pulse of cell-impermeable dye, we were thereby able to quantify cytokine accumulation in individual cells that were confirmed to have lost PM integrity. We found that these corpses showed a significant increase in accumulation of CCL2 after loss of membrane integrity. Importantly, if CHX was added to these cells after PM integrity loss, this increase was abrogated (Figure 2F), consistent with continued mRNA translation in cells lacking PM integrity.
We next sought to directly visualize protein translation in necroptotic cell corpses using puromycilation, wherein puromycin is covalently incorporated into growing polypeptide chains by active ribosomes. These puromycilated polypeptides can then be detected using a monoclonal antibody to puromycin (Schmidt et al., 2009). We first pulsed necroptotic cell corpses with the cell-impermeable dye Zombie Green to confirm loss of PM integrity and then washed out excess dye before pulsing them with puromycin (Figure 2G). As expected, when this assay was applied to live cells, we observed no incorporation of Zombie Green but robust incorporation of puromycin, which was blocked by co-incubation with the translation inhibitor CHX (Figure 2H). In necroptotic cells, we observed uptake of Zombie Green consistent with loss of PM integrity but also clear incorporation of puromycin into newly synthesized poly-peptides, suggesting ongoing translation in these cells. Notably, puromycilated polypeptides co-localized with the ER marker ERp72 (Figure 2H), consistent with the idea that the ER is the site of ongoing translation in necroptotic cells. Also consistent with this, we did not observe puromycin incorporation (and thus ongoing translation) in cells in which intracellular membranes were disrupted by digitonin (Figure S2C). Together, these data implicate continued ER function within necroptotic corpses as a requirement for ongoing translation.
MLKL Fails to Completely Disrupt the ER during Necroptosis
Activated phosphorylated MLKL (pMLKL), the executioner molecule of necroptosis, has been shown to translocate to the PM and disrupt its integrity. Although imaging studies have indicated that most activated MLKL localizes to the PM, the ability of MLKL to disrupt other cellular membranes is less clear (Cai et al., 2014; Dondelinger et al., 2014; Gong et al., 2017; Quarato et al., 2016; Wang et al., 2014). Given our finding of ongoing translation following MLKL-mediated PM disruption and our implication of the ER in this process, we hypothesized that activated MLKL does not efficiently disrupt this organelle during necroptosis. Consistent with this idea, we observed limited co-localization of phosphorylated MLKL and the ER marker ERp72 following induction of necroptosis (Figures 3A and 3B).
Figure 3. Ongoing Translation Is Permitted by a Lack of ER Disruption by MLKL and Occurs during Necroptosis Induced by CMV, IAV, and TNF.

(A-C) NIH 43T3 + acRIPK3 cells expressing MLKL-ERCb5 or vector control were pre-treated with doxycycline for 4 h and then treated with dimerizer to induce necroptosis. Cells were pulsed with the cell-impermeable dye Zombie Green and then stained with antibodies against Pmlk Land ERp72. (A) Cells were analyzed by microscopy. Representative images for each condition are shown. Scale bars represent 10 μm. (B) Quantification of normalized colocalization ([red + blue signal]/[blue signal]) from (A). (C) IL-6 and CXCL2 levels in the supernatant were quantified by ELISA at 18 h post-dimerizer addition.
(D and E) SVEC4–10 cells with intact (scramble gRNA) or CRISPR/Cas9-disrupted RIPK3 (RIPK3 gRNA) were infected with MCMV M45DRHIM (MOI = 1), and death kinetics were analyzed via IncuCyte bioimaging (D) or imaged by microscopy (E). Representative images for each condition in (E) are shown. Scale bars represent 10 μm.
(F and G) SVEC4–10 cells with intact or disrupted RIPK3 were infected with X31 influenza (MOI = 1), and death kinetics were analyzed via IncuCyte bioimaging(F) or imaged by microscopy (G). Representative images for each condition in (G) are shown. Scale bars represent 10 μm.
(H) SVEC4–10 cells were subjected to TNF-α + zVAD treatment, and death kinetics were analyzed via IncuCyte imaging.
(I) SVEC4–10cellsweretreated with TNF-α + zVAD for 3h, followed by addition and wash-out of the cell-impermeable dye Zombie Green priorto incubation with puromycin (as illustrated in Figure 2G). All data are representative of at least two independent experiments. ***p < 0.001; ****p < 0.0001. Error bars indicate SD from the mean.
If a failure by MLKL to disrupt the ER during necroptosis is required to allow protein synthesis to continue, we reasoned that targeting MLKL to the ER might induce its disruption during necroptosis and thereby abolish this phenomenon. To test this, we engineered an MLKL construct targeted to the cytoplasmic surface of the ER, through addition of a sequence derived from the C terminus of cytochrome b5 (Wang et al., 2001). As expected, in corpses expressing both endogenous and ER-targeted MLKL, we observed increased co-localization of pMLKL and the ER following RIPK3 activation (Figures 3A and 3B). Notably, expression of this ER-targeted MLKL did not affect the kinetics of necroptosis, as cells also expressed endogenous MLKL to allow PM disruption (Figure S3A). However, in cells expressing ER-targeted MLKL, we observed a reduction in cytokine and chemokine production after PM disruption (Figure 3C). Taken together, these data suggest that, upon RIPK3 activation, MLKL selectively targets the PM, allowing for continued ER-dependent translation even after loss of PM integrity.
TNF- or Virus-Induced Necroptosis Leads to Continued Translation after Loss of PM Integrity
Several viruses have been shown to induce RIPK3-dependent death through activation of the nucleotide sensor DAI/ZBP1 (Guoetal., 2018; Koehler et al., 2017; Kuriakoseetal.,2016; Nogusa et al., 2016; Thapa et al., 2016; Upton et al., 2012), so we sought to determine whether translation occurs following necroptotic PM rupture induced by viral infection. To test this, we used a mutant strain of murine cytomegalovirus (MCMV M45ΔRHIM) that triggers robust necroptosis. Consistent with previous reports (Upton et al., 2012), SVEC4–10 endothelial cells expressing endogenous RIPK3 underwent RIPK3-dependent cell death upon infection with this virus (Figures 3D and S3B). To assess whether protein translation continued in these cells following PM lysis, we took advantage of the intracellular cytokine staining and puromycilation assays already described. Flow cytometric analysis indicated that CCL2 and IL-6 continued to accumulate in MCMV-killed cellular corpses after loss of PM integrity and that this effect was abrogated by blocking translation with CHX after PM rupture (Figures S3C and S3D). Consistently, imaging analysis of MCMV-killed cells following puromycin pulse treatment revealed that puromycin was incorporated into the proteome of cells that had lost PM integrity and that this puromycin incorporation could be blocked by inhibiting translation after loss of PM integrity (Figures 3E and S3E).
As influenza has also been shown to trigger RIPK3-dependent necroptosis (Nogusa et al., 2016; Thapa et al., 2016), we carried out analogous experiments following influenza infection. We first confirmed that SVEC4–10 cells died in response to infection with X31 influenza in a RIPK3-dependent manner (Figure 3F) and then subjected infected cells to the puromycilation assay. Analogous to our observations using MCMV M45DRHIM infection, we observed puromycin incorporation in cells that lost PM integrity due to X31 influenza infection (Figure 3G). Again, this translation could be blocked by treating infected cells with CHX (Figure S3F).
Treatment with TNF-α in combination with the caspase inhibitor zVAD is a canonical necroptosis-inducing stimulus, and we wondered whether mRNA translation continued following TNF-α-induced necroptosis. To test this, we carried out puromycilation experiments on SVEC cells treated with TNF-α + zVAD. Consistent with our findings using other stimuli, we found that translation continued after loss of PM integrity in these necroptotic cells (Figures 3H, 3I, and S3G). Together, these data indicate that continued translation after PM rupture is a feature of necroptosis induced by viral infection or TNF-α stimulation.
Cytokine and Chemokine mRNAs Are Associated with Translating Polyribosomes in Necroptotic Corpses
We next evaluated whether the program of ongoing translation observed in necroptotic cells was selective for specific mRNA species. To assess this, we subjected live cells and necroptotic corpses to polyribosome (polysome) analysis (Chassé et al., 2017). Briefly, NIH 3T3 cells with acRIPK3 were induced to undergo necroptosis using RIPK3 dimerization, and 4 h later (a time point at which >99% of cells had lost PM integrity, as in Figure 1B), lysates from these cells were fractionated by sucrose gradient ultracentrifugation, causing mRNAs associated with multiple ribosomes (polysomes, the translating fraction) to separate from free mRNAs or those associated with monosomes (non-translating fractions). Gradients were then fractionated while continuously monitoring absorbance at 254 nm, giving rise to polysome profiles (Figure 4A). In these experiments, CHX is added to freeze ribosomes to mRNA to allow fractionation, and EDTA, which dissociates ribosomes from mRNA, is used as a negative control. Using this approach, untreated cells or cells treated with TNF-α were found to display the expected profile of monosomes and polysomes (Figure S4A). Although necroptotic corpses had a reduced polysome peak (Figure S4A), we could nonetheless detect abundant cytokine and chemokine transcripts by qPCR in the corresponding fractions (Figure 4B). Interestingly, polysomes isolated from necroptotic corpses were enriched for cytokine and chemokine transcripts (relative to the housekeeping gene GAPDH), as compared to non-treated or TNF-α-treated cells (Figure 4B). For comparison, we also quantified the translation of CCL5, a gene we have observed to be induced by TNF-a treatment, but not RIPK3 activation (Figure 4C), and found that it was not associated with ribosomes in necroptotic corpses. Confirming that our fractionation was successful, we found that input lysates (taken prior to polysome fractionation) contained similar total transcript levels between samples that were treated with either CHX or EDTA (Figure S4B); however, EDTA treatment abolished the association of mRNAs with polysome fractions, as expected (Figure S4C). These data indicate that, upon RIPK3 activation, necroptotic cells upregulate cytokine and chemokine transcripts and that translation of these mRNAs is associated with polyribosomes even after loss of cell membrane integrity.
Figure 4. Cytokine and Chemokine mRNA Is Associated with Translating Polyribosomes in Necroptotic Corpses.

(A) Experimental schematic for polyribosome (polysome) analysis.
(B and C) RNA was extracted from the fractions corresponding to polysome peaks, and mRNA transcripts were analyzed by qPCR. Graphs represent the ratio of total cytokine transcripts for RIPK3-dependent (B) and -independent (C) genes to a housekeeping gene (GAPDH) being actively translated in each condition.
(D) mRNA from whole necroptotic corpses, or from polysome fractions isolated from these corpses, was analyzed by RNA sequencing. Fold change relative to untreated cells or polysomes isolated from untreated cells (respectively) is depicted. Values reaching significance (FC > 1; p < 0.05) on x axis only are depicted in red and those significant on both axes in blue.
(E) NIH 3T3 + acRIPK3 cells were induced to undergo necroptosis with dimerizer, and the amount of ATP present in cells at the indicated time points was assessed using CellTiter-Glo.
(F) NIH 3T3 + acRIPK3 were treated with dimerizer, digitonin, or treated with dimerizer followed by digitonin (→). 4 h later, ATP levels were quantified using CellTiter-Glo. All data are representative of three independent experiments. NT, no treatment.
Error bars indicate SD from the mean.
To test whether this reflected selective translation of cytokine and chemokine transcripts, we carried out unbiased RNA sequencing on polysome-associated mRNA fractions from necroptotic cells and compared the results to total RNA obtained by poly-A enrichment. This analysis revealed a strong correlation between these two pools of mRNA: the most abundant transcripts present in necroptotic corpses were also the most abundant in the actively translating fraction (Figure 4D). This finding indicates that ongoing protein translation in necroptotic cells is not selectively directed toward specific mRNA species. Notably, however, as shown in Figure 1C, the activation of RIPK3 leads to robust transcriptional upregulation of inflammatory mediators at the mRNA level, and these mediators therefore represent the most differentially expressed transcripts present in necroptotic corpses. These data therefore suggest that ongoing translation in necroptotic cells is not selective for specific mRNA transcripts but is biased toward production of inflammatory mediators by accumulation of mRNA encoding these mediators following RIPK3 activation.
Because protein translation is an energy-intensive process (Buttgereit and Brand, 1995), we sought to determine ATP levels in necroptotic corpses using CellTiter-Glo. Relative to their live-cell counterparts, necroptotic corpses lost much of their ATP by h after RIPK3 activation, a time point corresponding to completed loss of PM integrity; however, these corpses maintained low but detectible levels of ATP even up to 12 h post-dimerizer addition (Figure 4E). Given that global translation appears to be diminished in necroptotic corpses (Figure S4A) and that the ATP content of normal cells is several fold higher than the minimum required for protein translation (Jewett et al., 2009; Patel et al., 2017), we reasoned that perhaps this residual ATP is sufficient to maintain translation. We hypothesized that this residual ATP may be maintained by the architecture of organelle membranes present in necroptotic corpses, consistent with our finding that maintenance of ER function is required for ongoing translation. Consistent with the possibility, addition of digitonin to necroptotic corpses, which aspecifically disrupts cell membranes, diminished ATP to nearly undetectable levels (Figure 4F) and also halted translation (Figure 3B). These data suggest that necroptotic corpses that have lost cell membrane integrity maintain low levels of ATP and that this is sufficient to allow ongoing translation of cytokines and chemokines in corpses.
Translation of Inflammatory Mediators within Necroptotic Cells Influences Myeloid Cell Migration and Promotes CD8+ T Cell Responses to Necroptotic Cell-Derived Antigen
We next sought to evaluate the immunological consequences of continued translation of inflammatory mediators within necroptotic cell corpses. We first assessed whether blocking this translation program in necroptotic corpses affected phagocytosis of necroptotic cells in vitro by “feeding” necroptotic cells labeled with fluorescent nanoparticle FluoSpheres to bone marrow-derived macrophages (BMDM) cells (Figures 5A and S5A). To assess the effect of ongoing translation on this process, we also tested the effect of transiently pulsing necroptotic corpses with emetine, an irreversible translation inhibitor, to halt ongoing translation in these cells (Grollman, 1968; Figures S2B and S5B).
Figure 5. Preservation of Translation Capacity in Necroptotic Cell Corpses Influences Myeloid Cell Trafficking and Cross-Priming of Antigen Derived from Necroptotic Cells.

(A) Experimental schematic for (B)-(E). (B) Bone-marrow-derived macrophages (BMDM) were incubated with FluoSphere-labeled necroptotic cell corpses that had or had not been transiently treated with emetine. After 6 h, BMDMs that had taken up dead cell material were quantified by flow cytometry.
(C and D) Percentage and total number of FluoSphere-positive myeloid cells (CD3−B220−CD11b+; C) or migratory dendritic cells (CD11c+MHCIIhi; D) in the draining lymph node (popliteal) of mice, 24 h after injection in the footpad.
(E) NIH 3T3 cells expressing membrane-bound OVA (either actively dying in vivo [necroptosis in situ], corpses, or corpses pulse-treated with emetine) were injected into C57BL/6 mice. 9 days later, OVA-specific (SIINFEKL-H2Kbtetramer+) and activated (CD44+CD62L−) CD8+ T cell responses were measured by flow cytometry.
Data in (B) are representative of three independent experiments; data in (C)-(E) each represent two independent experiments combined. *p < 0.05; **p < 0.01; ****p < 0.0001. Error bars indicate SD from the mean.
We found that emetine treatment reduced the efficiency with which necroptotic cell material was taken up by phagocytes (Figure 5B), consistent with the possibility that chemokine production by necroptotic cells promotes their phagocytosis. We next assessed the activation status of phagocytes following uptake of necroptotic corpses in the presence of absence of ongoing translation. We found that phagocytes that had taken up necroptotic material (FluoSphere+) displayed increased expression of the activation markers MHC-II and CD-80 relative to phagocytes within the same culture that had not but that this phagocyte activation occurred irrespective of continued translation (Figure S5C), and importantly, the uptake of emetine-treated cells did not affect the number or viability of BMDM cells in vitro. Together, these findings imply that ongoing production of inflammatory mediators promotes localization and uptake of necroptotic cells by phagocytes but that other signals promote activation of these phagocytes following dead cell encounter.
We next used a similar experimental approach to assess the role of ongoing cytokine production in the uptake and trafficking material derived from necroptotic cell corpses in vivo. To do this, we injected FluoSphere-labeled corpses (verified >95% per-meabilized PMs by flow at time of injection; data not shown), with or without emetine pulse, into the footpads of mice and then analyzed FluoSphere-positive phagocytes in the draining lymph node 24 h later (Figure 5A). We observed a similar number of FluoSphere-positive myeloid cells in the draining lymph nodes (dLNs) of mice that received either actively dying cells (cells induced to undergo necroptosis in situ, as we have previously described; Yatim et al., 2015) or corpses; however, there were significantly fewer FluoSphere-positive cells in the dLNs of those animals that received emetine-pulsed corpses (Figure 5C). We also observed a decreased number of FluoSphere-positive migratory dendritic cells (DCs) (as defined by CD11c+ MHCIIhi myeloid cells) in the dLNs of animals that received emetine-treated corpses relative to animals that received either corpses in which continued cytokine synthesis was occurring or when cells underwent necroptosis in situ (Figure 5D). However, no difference was observed in the total number of myeloid cells present in the dLNs across treatment groups (Figure S5D). These data suggest that signals generated by translation of inflammatory mediators in necroptotic cells at peripheral sites can contribute to the efficient uptake and trafficking of material derived from necroptotic cells to the dLN.
Previously, we reported that NF-κ activity downstream of RIPK3 activation promotes the cross-priming of CD8+ T cell responses against antigen derived from necroptotic cells (Yatim et al., 2015). We hypothesized that ongoing translation of inflammatory mediators following PM rupture could contribute to the cross-priming potential of these corpses in vivo. To address this, we immunized C57BL/6 mice with cells containing membrane-bound ovalbumin (mOVA) and compared immunization with cells undergoing necroptosis in situ, with corpses in which translation was ongoing, or with emetine-pulsed corpses in which translation was abrogated. Nine days later, we enumerated OVA-specific CD8+ T cells as a measure of the cross-priming response elicited by each dying cell treatment. Mice that received either actively dying necroptotic cells or corpses showed efficient cross-priming and had no significant difference in the number of OVA-specific CD8+ T cells or activated CD8+ T cells; however, mice that received emetine-treated corpses had significantly fewer antigen-specific cells and activated CD8+T cells (Figure 5E). This finding suggests that the continued production of inflammatory mediators promotes a robust cross-priming response in vivo. Together, these data indicate that the continued production of inflammatory cytokines and chemo-kines by necroptotic cell corpses promotes the trafficking of necroptotic cell-derived antigens to draining lymph nodes and the initiation of T cell responses to dying cell-derived antigen.
DISCUSSION
Several recent reports have indicated that activation of the necroptotic pathway is associated with inflammatory transcription as well as with cell death (Orozco et al., 2014; Yatim et al.,2015). However, how the terminal process of cell death is coordinated with the production of cytokines and chemokines, two apparently opposing cellular processes, is poorly understood. Here, we report that necroptotic corpses can continue protein translation even after loss of PM integrity. We observed this phenomenon downstream of either direct RIPK3 activation or necroptosis in response to TNF-α or infection with MCMV or influenza A virus (IAV). This phenomenon contrasts with apoptotic cell death, during which protein synthesis is actively antagonized through caspase-mediated destruction of the translation machinery (Clemens et al., 2000). Indeed, our findings are consistent with a previous report, which indicated that direct disruption of the protein synthesis machinery is a feature of apoptosis that does not occur during necroptosis (Saelens et al., 2005). Although that report indicated that protein synthesis in necroptotic cells could continue up to the point of PM disruption, our data extend these observations by demonstrating that protein synthesis occurs even after the cell has irreversibly lost PM integrity and would be considered dead by commonly used measures of cell death. Our work supports the idea that MLKL leaves the ER network intact during necroptosis, thus allowing translation to continue; notably, we demonstrate here that forced targeting of MLKL to the ER disrupts translation without affecting the execution of MLKL-dependent cell death.
Protein translation is energy intensive, and necroptotic cells release ATP upon PM lysis. However, we found that necroptotic cell corpses sustained low levels of ATP up to 12 h after PM lysis. Although the physiological concentration of ATP in cells is quite high (Traut, 1994), a recent study suggested that this does not reflect the energetic needs of the cell but rather that high ATP concentration is required for proper protein folding and solubility (Patel et al., 2017), and protein synthesis can occur at much lower ATP concentrations (Jewett et al., 2009). We speculate that the residual ATP present in necroptotic corpses is sufficient to power the limited translation we observe in these cells.
Previous work has suggested that necroptosis can actually dampen inflammatory responses by eliminating cells that would otherwise continue cytokine synthesis in response to TNF-α or lipopolysaccharide (LPS) (Kearney et al., 2015). Our data do not contradict this idea, as the total magnitude of inflammatory mediators produced by a live cell in response to these stimuli would outstrip the post-death production we observe. Nonetheless, our data suggest that cytokines and chemokines produced after cell lysis are an important contributor to the inflammatory and immune response to necroptotic cells.
We speculate that the necroptotic program evolved as an antiviral mechanism, allowing elimination of the replicative niche by destroying the PM, while continuing cytokine and chemokine production to alert the immune system and recruit relevant immune cells to the site of infection (Daniels et al., 2017). A further, non-exclusive possibility is that viral proteins can also be translated in necroptotic corpses, thereby loading the necroptotic corpse with viral antigen. As we have previously shown that antigens derived from necroptotic cells more readily cross-prime CD8+ T cells (Yatim et al., 2015), this mechanism could contribute to the induction of antiviral immune responses. Together, our findings indicate that the necroptotic program involves the coordination of cell death with continued cytokine synthesis and that this property contributes to the inflammatory and immunogenic nature of necroptosis.
STAR★METHODS
Detailed methods are provided in the online version of this paper and include the following:
LEAD CONTACT AND MATERIALS AVAILABILITY
Plasmids and cell lines generated in this study will be made available under a standard material transfer agreement. Requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Andrew Oberst (oberst@uw.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
Wild-type C57BL/6J mice were obtained commercially (Jackson Laboratories) and were housed under specific-pathogen-free conditions at the University of Washington. All experiments were performed in female 6–8 week-old mice, in accordance with protocols approved by the University of Washington Animal Care and Use Committee (IACUC).
Cell Culture
NIH 3T3 cells (ATCC) and SVEC4–10 cells (ATCC), as well as derived cell lines (NIH 3T3 + acRIPK3, NIH 3T3 + acRIPK3 + MLKL-ERCb5, SVEC4–10 + scramble gRNA, SVEC4–10 + RIPK3 gRNA), were maintained in D-MEM supplemented with 10% FBS, 29.2 g/l glutamine, 10 000 U/ml penicillin and 10 000 mg/ml streptomycin, and grown at 37°C in 5% CO2.
METHOD DETAILS
NIH 3T3 + acRIPK3 Cell Line Generation
The acRIPK3 chimeric protein was constructed as previously described (Orozco et al., 2014; Yatim et al., 2015). Briefly, full-length murine RIPK3 was cloned upstream of 2 copies of FKBP carrying the F36V mutation. When two FKBPF36V domains were used, the first copy contained silent mutations to prevent DNA recombination. These RIPK3- FKBPF36V fusion proteins were cloned into pBabe-Puromycin retroviral vectors. This construct was transduced into NIH 3T3 cells (ATCC) using standard protocols for helper-dependent retroviral transduction. Transduced cells were selected for 5 days in 1 μg/ml puromycin, then single-cell cloned to obtain a homogeneous population.
NIH 3T3 + acRIPK3 + MLKL-ERcb5 Cell Line Generation
MLKL-ERcb5 was cloned into the previously described doxycycline-inducible pSLIK-Hygromycin backbone (Yatim et al., 2015). This construct was transduced into NIH 3T3 cells expressing acRIPK3. Transduced cells were selected for 7 days in 200 μg/ml hygromycin B.
Generation of CRISPR/Cas9 Gene-Edited SVEC4–10 Cell Lines
For CRISPR/Cas9-genome editing of SVEC4–10 cells (ATCC), guide RNA (gRNA) sequences were cloned into the lentiviral construct pRRL-Cas9-T2A-puromycin which encodes both a guide RNA (gRNA) (Gray et al., 2016) targeting eitherthe Ripk3 gene, or a murine non-targeting gRNA (Sanjana et al., 2014) as well as the Cas9 enzyme. Guide RNA sequences are presented in Table S1. Lentiviral particles were created using these constructs, and this virus was used to transduce SVEC4–10 cells using standard lentiviral transduction protocols. Transduced cells were selected for 5 days in 1 μg/ml puromycin, and validated via western blot analysis.
Cell death assays
Cell death was quantified by flow cytometry on an LSRII Flow Cytometer (BD) or using a 2-color IncuCyteZoom in-incubator imaging system (Essen Biosciences) as previously described (Orozco et al., 2014). Dead cells were detected by uptake of the cell-impermeable dye Sytox Green (Thermo Fisher). Cell death was also quantified by either CellTiter-Blue Cell Viability Assay (Promega) or LDH Cytotoxicity Assay (Thermo Fisher), both according to manufacturer’s instructions. To assess ATP levels in necroptotic corpses, CellTiter-Glo (Promega) was used according to the manufacturer’s instructions.
Electron microscopy
NIH 3T3 cells were induced to undergo necroptosis for 3 h; following treatment, samples were fixed in 2.5% glutaraldehyde, 2% para-formaldehyde in 0.1 M sodium cacodylate buffer pH 7.4, and postfixed for 1.5 h in reduced 2% osmium tetroxide with 1.5% potassium ferrocyanide in 0.1 M sodium cacodylate buffer. After rinsing in buffer, samples were dehydrated through a series of graded ethanol to propylene oxide solutions, infiltrated and embedded in epoxy resin, and polymerized at 70°C overnight. Semi-thin sections (0.5 micron) were stained with toluidine blue for light microscopic examination. Ultra-thin sections (80 nm) were cut and imaged using the Tecnai TF20 TEM with an AMT XR41 camera.
Polysome Analysis and qRT-PCR
Polysome analysis was performed as previously described (Chassé et al., 2017). Briefly, cells were treated for 4 h with either 20 ng/mL TNFα (Peprotech) or 500 nM dimerizer (Clontech), and then treated with 100 μg/mL of cycloheximide (Sigma-Aldrich) or 25 mM EDTA for 5 min at 37°C. Cells were then pelleted and lysed in hypotonic lysis buffer (5 mM Tris-HCl pH 7.5,2.5 mM MgCl2,1.5 mM KCl, and 1X protease inhibitor cocktail) followed by addition of 100 μg/mL cycloheximide, 2 mM DTT, 200 U/mL RNasin, 0.5% Triton-X, and 0.5% sodium deoxycholate. Lysates were clarified by spinning at max speed in a tabletop centrifuge for 5 min, and supernatants were loaded onto sucrose gradients (10%−50%). Samples were then ultracentrifuged using an SW40 Ti Rotor (Beckman Coulter) for 2 h at 4°C. Gradients were fractionated using an Auto-Densi Flow Pump (Labconco) and a Foxy R2 Fraction Collector (Teledyne Isco), while continuously monitoring absorbance at 254 nm using UA-6 UV detector and LoggerPro Software (Vernier).
RNA was extracted from the fractions corresponding to polysomes using RNA Stat-60 (Amsbio); RNA fractions from each sample were then pooled, and cDNA was made using SuperScript III Reverse Transcriptase (Thermo Fisher). Quantitative real-time PCR (qRT-PCR) analysis was performed using SYBR Green (Thermo Fisher) and ViiA7 Real-Time PCR System (Applied Biosystems). Cytokine and Gapdh transcripts were quantified against a standard curve of known amounts.
RNA sequencing
QC of total RNA was done to determine their quantity and quality. The concentration of RNA samples was determined using NanoDrop 8000 (Thermo Scientific) and the integrity of RNA was determined by Fragment Analyzer (Advanced Analytical Technologies). 0.1ug of total RNA was used as an input material for library preparation using TruSeq Stranded Total RNA Library Prep Kit (Illumina). Size of the libraries was confirmed using 4200 TapeStation and High Sensitivity D1K screen tape (Agilent Technologies) and their concentration was determined by qPCR based method using Library quantification kit (KAPA). The libraries were multiplexed and sequenced on Illumina HiSeq4000 (Illumina) to generate 30M of single end 50 base pair reads.
Immunocytochemistry
Fluorescent immunocytochemistry was performed as previously described (Daniels et al., 2017). Cells were fixed for 15 min with 4% paraformaldehyde (Fisher), permeabilized with 0.1% Triton-X, and blocked with 10% goat serum. Cells were stained using primary anti-puromycin (Clone 12D10, EMD Millipore) or anti-ERp72 (Cell Signaling Technologies) for 1 h at room temperature, followed by secondary goat anti-mouse AlexaFluor594 (Abcam) or goat anti-rabbit AlexaFluor405 (Abcam). Dead cells were visualized via staining with the fixable viability dye Zombie Green (BioLegend).
Cytokine Protein Quantification in Cell Supernatants
104 NIH 3T3 cells were plated per well in a 96-well tissue culture plate the day prior to treatment. Cells were then treated with 100 nM dimerizer and different combinations of inhibitors (as per figure legends) in a 200 μL volume. Supernatants were collected and clarified by centrifugation at various time points, and stored at −80°C. Cytokine concentrations in cell culture supernatants were measured using IL-6 (Thermo Fisher), CXCL2 (Peprotech), CXCL1 (Peprotech) or CCL2 (Thermo Fisher) ELISA kits, according to the manufacturer’s protocols.
Western Blots
Cells were lysed in NP-40 lysis buffer (150 mM NaCl, 20 mM tris-Cl, 1 mM EDTA, 1% NP-40 at pH 7.5) with 1X Halt Protease Inhibitor Cocktail (Thermo Fisher). 30 μg of protein was run on a 4%−20% Novex Tris-Glycine mini gel (Fisher) at 125 V for 2 h in WB running buffer (24 mM tris, 32 mM glycine, 3.5 mM SDS) and transferred onto PVDF membrane (Thermo Fisher) at 300 mAmpsfor 1 h in transfer buffer (6 mM tris, 8 mM glycine, 15% methanol). Membranes were blocked in 5% dry milkinTBS-Tween20 (1%) for 30 min at room temperature. Membranes were incubated overnight at 4°C with primary anti-RIPK3 (Novus Biologicals), anti-V5 (Thermo Fisher) or anti-actin (EMD Millipore), followed by staining with HRP-conjugated secondary antibodies (Santa Cruz). Membranes were developed using ECL Western Blotting Substrate (Pierce) and film.
In vitro transcription/translation
TnT® Quick Coupled Transcription/Translation System (Promega) was used to assess the effects of compounds on translation in a cell-free system. A construct template coding for V5-tagged TEV was used with the kit, according to manufacturer’s instructions. Lysates were analyzed via western blot.
In Vivo Migration Assays
NIH 3T3 cells were loaded with 106 particles/cell of FluoSpheres (Thermo Fisher) for one h at 37°C. Excess FluoSpheres were aspirated and cells were washed once with 1X PBS, then treated with 500 nM dimerizer for 3 h or pulsed for 15 min at 37°C as previously described (Yatim et al., 2015). Corpses were then treated with 25 μg/mL of emetine (EMD Millipore) or vehicle (water) for 15 min at 37°C, and washed 2x with 1X PBS. 5×105 cells or corpses were injected into each rear footpad of naive C57BL/6 mice. 24 h later, animals were sacrificed and the draining (popliteal) lymph nodes were harvested; single cell suspensions were made and stained for flow cytometric analysis.
In Vivo Cross-priming Assays
NIH 3T3 + acRIPK3 cells expressing membrane-bound ovalbumin (mOVA) were treated with 500 nM dimerizer for 3 h or pulsed for 15 min at 37°C as previously described (Yatim et al., 2015). Corpses were then treated with 25 μg/mL or emetine or vehicle (water) for 15 min at 37°C, and washed 2x with 1X PBS. One million cells were injected into the flank of naive C57BL/6 female mice, and nine days later, animals were sacrificed and spleen and draining (inguinal) lymph nodes were harvested and pooled. Single cell suspensions were made and cells were stained for OVA tetramer (SIINFEKL-H2Kb), and activation markers; cells were fixed and analyzed by flow cytometry.
Flow cytometric analysis
Cell viability was assessed by staining with Zombie Green (BioLegend) or Zombie Aqua (BioLegend), diluted 1:1000 in PBS and incubated at 4°C for 30 min. For surface staining, cells were first incubated with anti-CD16/CD32 receptor antibody (BD clone 2.4G2) diluted 1:100 in PBS, for 30 min at 4°C. Cells were then stained with fluorescently conjugated antibodies diluted 1:100 in PBS for 1 h at 4°C; cell suspensions were stained with antibodies to CD3 (BD clone 145–2C11), B220 (BD clone RA3–6B2), CD4 (BD clone RM4–5), CD8a (BD clone 53–6.7), CD44 (BD clone IM7), CD62L (BD clone MEL-14), CD11b (BD clone M1/70), CD11c (BD clone N418), MHCII I-A/E (BD clone M5/114.15.2), F4/80 (BD clone T45–2342), and SIINFEKL-H2Kb tetramer (NIH Tetramer Core).
For intracellular staining, cells were fixed with BD Perm/Fix for 20 min at 4°C, and then permeabilized with BD Perm/Wash for 10 min at4°C. Cells were stained intracellularly with fluorescently conjugated antibodies to CCL2 (BD clone 2H5) and IL-6 (BioLegend clone MP5–20F3), diluted 1:100 in BD Perm/Wash, for 1 h at 4°C.
All flow cytometry was performed on a BD LSRII flow cytometer. FlowJo (Tree Star, Inc.) software was used for all flow cytometric analysis.
QUANTIFICATION AND STATISTICAL ANALYSIS
Experimental replicate information is indicated in the figure legends. All results are given as mean ± SD and analyzed by using statistical tools in GraphPad Prism software (La Jolla, CA). For in vivo experiments, unpaired t test (for percentage) or Mann-Whitney test (for total cell numbers) were used. For protein quantification by ELISA, unpaired t tests were used. Differences with p < 0.05 were considered significant.
RNA Seq Differential analysis
FASTQ files were aligned to the mouse reference genome (mm9) using the GSNAP alignment tool. The count data from the alignment was normalized to rpkm and then filtered to remove lowly expressed genes. The resulting expression data were then adjusted by library size and corrected with voom precision weights. The differential expression analysis was performed using limma by defining the adequate contrasts and using the ebayes method. The resulting p values were adjusted by false discovery rate correction and the top deferentially expressed genes were ranked by fold change. All the analyses were conducted using R version 3.5.0.
DATA AND CODE AVAILABILITY
Complete RNA sequencing datasets are available via the NIH Gene Expression Omnibus (GEO) site (https://www.ncbi.nlm.nih.gov/geo), Series Record GSE134234.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-CD16/CD32 Receptor Antibody, clone 2.4G2 | BD Biosciences | Cat# 553142; RRID: AB_394657 |
| Anti-CD3e (clone 145-2C11) PerCP-Cy5.5 | BD Biosciences | Cat# 561108; RRID: AB_10562558 |
| Anti-B220 (RA3-6B2) AlexaFluor700 | BD Biosciences | Cat #557957; RRID: AB_396937 |
| Anti-CD4 (clone RM4-5) BV711 | BD Biosciences | Cat# 563726; RRID: AB_2738389 |
| Anti-CD8a (clone 53-6.7) BUV395 | 3D Biosciences | Cat# 563786; RRID: AB_2732919 |
| Anti-CD44 (clone IM7) PE-Cy7 | 3D Biosciences | Cat# 560569; RRID: AB_1727484 |
| Anti-CD62L (clone MEL-14) APC | BD Biosciences | Cat# 553152; RRID: AB_398533 |
| Anti-CD11b (clone M1/70) APC-R700 | BD Biosciences | Cat# 564985; RRID: AB_2739033 |
| Anti-CD11c (clone N418) BV421 | BD Biosciences | Cat# 565451; RRID: AB_2744278 |
| Anti-MHCII I-A/E (clone M5/114.15.2) BV711 | BD Biosciences | Cat# 563414; RRID: AB_2738191 |
| F4/80 (clone T45-2342) PE | BD Biosciences | Cat# 565410; RRID: AB_2687527 |
| Anti-CCL2 (clone 2H5) PE | BD Biosciences | Cat # 554443; RRID: AB_395394 |
| Anti-IL-6 (clone MP5-20F3) APC | BioLegend | Cat# 504507; RRID: AB_10694094 |
| OVA tetramer (SIINFEKL-H2Kb) PE | NIH Tetramer Core | N/A |
| Anti-MLKL (phospho S345) | Abcam | Cat# ab196436; RRID: AB_2687465 |
| Anti-RIPK3 | Novus Biological | Cat# NBP1-77299; RRID: AB_11040928 |
| Anti-V5 tag | Thermo Fisher | Cat# R960-25; RRID: AB_2556564 |
| Anti-actin, clone C4 | EMD Millipore | Cat# MAB1501; RRID: AB_2223041 |
| Mouse anti-rabbit IgG-HRP | Santa Cruz Biotechnology | Cat# sc-2357; RRID: AB_628497 |
| Goat anti-mouse IgG-HRP | Santa Cruz Biotechnology | Cat# sc-2005; RRID: AB_631736 |
| Anti-puromycin, clone 12D10 | EMD Millipore | Cat# MABE343; RRID: AB_2566826 |
| Anti-ERp72 | Cell Signaling Technology | Cat# 5033S; RRID: AB_10622112 |
| Goat anti-mouse IgG AlexaFluor594 | Abcam | Cat# ab150116; RRID: AB_2650601 |
| Goat anti-rabbit IgG AlexaFluor405 | Abcam | Cat# ab175652; RRID: AB_2687498 |
| Bacterial and Virus Strains | ||
| MCMV M45DRHIM | Upton et al., 2012 | N/A |
| Influenza A X31 | Thomas Lab, St. Jude Children’s Research Hospital | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Sytox Green | Thermo Fisher | Cat# S7020 |
| Syto Green 24 | Thermo Fisher | Cat# S7599 |
| Zombie Green Fixable Viability Dye | BioLegend | Cat# 423111 |
| Zombie Aqua Fixable Viability Dye | BioLegend | Cat# 423101 |
| Murine TNFα | Peprotech | Cat# 315-01A |
| zVAD | SM Biochemicals | Cat# SMFMK001 |
| GSK’843 | GlaxoSmithKline | N/A |
| Necrostatin-1 | Sigma-Aldrich | Cat# N9037 |
| Dimerizer (B/B homodimerizer) | Clontech | Cat# 635069 |
| B/B Washout Ligand | Clontech | Cat# 635088 |
| Actinomycin D | Sigma-Aldrich | Cat# A1410 |
| Cycloheximide | Sigma-Aldrich | Cat# C7698 |
| Gelonin | Enzo Life Sciences | Cat# ALX-350-150-M001 |
| Digitonin | EMD Millipore | Cat# 300410 |
| Thapsigargin | Thermo Fisher | Cat# T7458 |
| Brefeldin A | BioLegend | Cat# 420601 |
| Puromycin dihydrochloride | Thermo Fisher | Cat# A1113803 |
| Paraformaldehyde | Fisher | Cat# 50-980-487 |
| Emetine dihydrochloride | EMD Millipore | Cat# 324693 |
| Yellow-green FluoSpheres | Thermo Fisher | Cat #F8795 |
| HALT Protease Inhibitor Cocktail | Thermo Fisher | Cat# 78429 |
| DAPI | Thermo Fisher | Cat #62248 |
| Critical Commercial Assays | ||
| IL-6 Mouse ELISA Kit | Thermo Fisher | Cat# 88-7064-76 |
| MCP-1/CCL2 Mouse ELISA Kit | Thermo Fisher | Cat# 88-7391-76 |
| Murine KC (CXCL1) Standard ELISA Development Kit | Peprotech | Cat# 900-K127 |
| Murine MIP-2 (CXCL2) Standard ELISA Development Kit | Peprotech | Cat# 900-K152 |
| CellTiter-Blue Viability Assay | Promega | Cat# G8080 |
| Pierce LDH Cytotoxicity Assay Kit | Thermo Fisher | Cat# 88954 |
| CellTiter-Glo | Promega | Cat# G7572 |
| TnT® Quick Coupled Transcription/Translation System | Promega | Cat# L1171 |
| Deposited Data | ||
| RNA sequencing datasets | NIH GEO repository | GSE134234 |
| Experimental Models: Cell Lines | ||
| Mouse: NIH 3T3 + acRIPK3 | Orozco et al., 2014; Yatim et al., 2015 | N/A |
| Mouse: NIH 3T3 + acRIPK3 + mOVA-mCherry | This paper | N/A |
| Mouse: NIH 3T3 + acRIPK3 + MLKL-ERcb5 | This paper | N/A |
| Mouse: SVEC4-10 + scramble gRNA (CRISPR/Cas9) | This paper | N/A |
| Mouse: SVEC4-10 + RIPK3 gRNA (CRISPR/Cas9) | This paper | N/A |
| Experimental Models: Organisms/Strains | ||
| C57BL/6J | Jackson Laboratory | Stock# 000664 |
| Oligonucleotides | ||
| See Table S1 | N/A | N/A |
| Recombinant DNA | ||
| Plasmid: acRIPK3 in pBabe-puromycin | Orozco et al., 2014; Yatim et al., 2015 | N/A |
| Plasmid: Membrane-bound OVA (mOVA)-t2A-mCherry in pBabe-puromycin | This paper | N/A |
| Plasmid: MLKL-ERcb5 in pSLIK-Hygromycin | This paper | N/A |
| Plasmid: scramble (non-targeting) gRNA in pRRL-Cas9-t2a-puromycin | This paper | N/A |
| Plasmid: RIPK3 gRNA in pRRL-Cas9-t2a-puromycin | This paper | N/A |
| Software and Algorithms | ||
| Prism version 7 | GraphPad Software | https://www.graphpad.com |
| FlowJo version 10 | FlowJo, LLC | https://www.flowjo.com |
| LoggerPro Software | Vernier | https://www.vernier.com |
| ImageJ | NIH | https://imagej.nih.gov/ij |
| Other | ||
| 2-color IncuCyte Zoom in-incubator imaging system | Essen Bioscience | N/A |
| BD LSRII Flow Cytometer | BD Biosciences | N/A |
| SW40 Ti Rotor | Beckman Coulter | N/A |
| Auto-Densi Flow Pump | Labconco | N/A |
| Foxy R2 Fraction Collector | Teledyne Isco | N/A |
| UA-6 UV Detector | Teledyne Isco | N/A |
| ViiA7 Real-Time PCR System | Applied Biosystems | N/A |
Highlights.
RIPK3 activation induces both necroptotic cell death and cytokine production
Cytokine mRNA continues to be translated even after plasma membrane rupture
mRNA translation requires ER function, which remains after plasma membrane rupture
Continued cytokine synthesis contributes to the immune response to necroptotic cells
ACKNOWLEDGMENTS
We thank Drs. Ram Savan and Lomon So for helpful discussions and technical assistance with polysome experiments, Dr. William Kaiser for generously providing MCMV M45DRHIM virus, Dr. Jennifer Martinez for reagents, and Zora Modrusan for assistance with RNA sequencing. This work was supported by NIH grants R01AI132595 and R01CA228098 and a CRI Wade F. B. Thompson CLIP award (to A.O.) and an NIH NIAID R01 Diversity Supplement (to S.L.O.).
Footnotes
DECLARATION OF INTERESTS
A.O. is a co-founder, shareholder, and member of the Scientific Advisory Board of Walking Fish Therapeutics. A.G.S. acts as a consultant for and is a shareholder in Walking Fish Therapeutics. A.O. and D.R.G. hold a patent on applications of activatable RIPK3.
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2019.07.077.
REFERENCES
- Buttgereit F, and Brand MD (1995). A hierarchy of ATP-consuming processes in mammalian cells. Biochem. J. 312, 163–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai Z, Jitkaew S, Zhao J, Chiang HC, Choksi S, Liu J, Ward Y, Wu LG, and Liu ZG (2014). Plasma membrane translocation of trimerized MLKL protein is required for TNF-induced necroptosis. Nat. Cell Biol. 16, 55–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chassé H, Boulben S, Costache V, Cormier P, and Morales J (2017). Analysis of translation using polysome profiling. Nucleic Acids Res. 45, e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W, Zhou Z, Li L, Zhong CQ, Zheng X, Wu X, Zhang Y, Ma H, Huang D, Li W, et al. (2013). Diverse sequence determinants control human and mouse receptor interacting protein 3 (RIP3) and mixed lineage kinase domain-like (MLKL) interaction in necroptotic signaling. J. Biol. Chem. 288, 16247–16261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho YS, Challa S, Moquin D, Genga R, Ray TD, Guildford M, and Chan FK (2009). Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell 137, 1112–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clemens MJ, Bushell M, Jeffrey IW, Pain VM, and Morley SJ (2000). Translation initiation factor modifications and the regulation of protein synthesis in apoptotic cells. Cell Death Differ. 7, 603–615. [DOI] [PubMed] [Google Scholar]
- Daniels BP, Snyder AG, Olsen TM, Orozco S, Oguin TH 3rd, Tait SWG, Martinez J, Gale M Jr., Loo YM, and Oberst A (2017). RIPK3 restricts viral pathogenesis via cell death-independent neuroinflammation. Cell 169, 301–313.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Vasconcelos NM, Van Opdenbosch N, and Lamkanfi M (2016). Inflammasomes as polyvalent cell death platforms. Cell. Mol. Life Sci. 73, 2335–2347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degterev A, Hitomi J, Germscheid M, Ch’en IL, Korkina O, Teng X, Abbott D, Cuny GD, Yuan C, Wagner G, et al. (2008). Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat. Chem. Biol. 4,313–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dondelinger Y, Declercq W, Montessuit S, Roelandt R, Goncalves A, Bruggeman I, Hulpiau P, Weber K, Sehon CA, Marquis RW, et al. (2014). MLKL compromises plasma membrane integrity by binding to phosphatidylinositol phosphates. Cell Rep. 7, 971–981. [DOI] [PubMed] [Google Scholar]
- Gong YN, Guy C, Olauson H, Becker JU, Yang M, Fitzgerald P, Linkermann A, and Green DR (2017). ESCRT-III acts downstream of MLKL to regulate necroptotic cell death and its consequences. Cell 169, 286–300.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray EE, Winship D, Snyder JM, Child SJ, Geballe AP, and Stetson DB (2016). The AIM2-like receptors are dispensable for the interferon response to intracellular DNA. Immunity 45, 255–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green DR, and Llambi F (2015). Cell death signaling. Cold Spring Harb. Perspect. Biol. 7, a006080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grollman AP (1968). Inhibitors of protein biosynthesis. V. Effects of emetine on protein and nucleic acid biosynthesis in HeLa cells. J. Biol. Chem. 243, 4089–4094. [PubMed] [Google Scholar]
- Guo H, Gilley RP, Fisher A, Lane R, Landsteiner VJ, Ragan KB, Dovey CM, Carette JE, Upton JW, Mocarski ES, and Kaiser WJ (2018). Species-independent contribution of ZBP1/DAI/DLM-1-triggered necroptosis in host defense against HSV1. Cell Death Dis. 9, 816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He S, Wang L, Miao L, Wang T, Du F, Zhao L, and Wang X (2009). Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha. Cell 137,1100–1111. [DOI] [PubMed] [Google Scholar]
- Jewett MC, Miller ML, Chen Y, and Swartz JR (2009). Continued protein synthesis at low [ATP] and [GTP] enables cell adaptation during energy limitation. J. Bacteriol. 191, 1083–1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kearney CJ, Cullen SP, Tynan GA, Henry CM, Clancy D, Lavelle EC, and Martin SJ (2015). Necroptosis suppresses inflammation via termination of TNF- or LPS-induced cytokine and chemokine production. Cell Death Differ. 22, 1313–1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koehler H, Cotsmire S, Langland J, Kibler KV, Kalman D, Upton JW, Mocarski ES, and Jacobs BL (2017). Inhibition of DAI-dependent necroptosis by the Z-DNA binding domain of the vaccinia virus innate immune evasion protein, E3. Proc. Natl. Acad. Sci. USA 114, 11506–11511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuriakose T, Man SM, Malireddi RK, Karki R, Kesavardhana S, Place DE, Neale G, Vogel P, and Kanneganti TD (2016). ZBP1/DAI is an innate sensor of influenza virus triggering the NLRP3 inflammasome and programmed cell death pathways. Sci. Immunol. 1, aag2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y, Choksi S, Shen HM, Yang QF, Hur GM, Kim YS, Tran JH, Ne-dospasov SA,and Liu ZG (2004). Tumor necrosis factor-induced nonapoptotic cell death requires receptor-interacting protein-mediated cellular reactive oxygen species accumulation. J. Biol. Chem. 279, 10822–10828. [DOI] [PubMed] [Google Scholar]
- Nogusa S, Thapa RJ, Dillon CP, Liedmann S, Oguin TH 3rd, Ingram JP, Rodriguez DA, Kosoff R, Sharma S, Sturm O, et al. (2016). RIPK3 activates parallel pathways of MLKL-driven necroptosis and FADD-mediated apoptosis to protect against influenza A virus. Cell Host Microbe 20, 13–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orozco S, Yatim N, Werner MR, Tran H, Gunja SY, Tait SW, Albert ML, Green DR, and Oberst A (2014). RIPK1 both positively and negatively regulates RIPK3 oligomerization and necroptosis. Cell Death Differ. 21, 1511–1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel A, Malinovska L, Saha S, Wang J, Alberti S, Krishnan Y, and Hyman AA (2017). ATP as a biological hydrotrope. Science 356, 753–−756. [DOI] [PubMed] [Google Scholar]
- Quarato G, Guy CS, Grace CR, Llambi F, Nourse A, Rodriguez DA, Wakefield R, Frase S, Moldoveanu T, and Green DR (2016). Sequential engagement of distinct MLKL phosphatidylinositol-binding sites executes necroptosis. Mol. Cell 61, 589–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saelens X, Festjens N, Parthoens E, Vanoverberghe I, Kalai M, van Kuppeveld F, and Vandenabeele P (2005). Protein synthesis persists during necrotic cell death. J. Cell Biol. 168, 545–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanjana NE, Shalem O, and Zhang F (2014). Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods 11, 783–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt EK, Clavarino G, Ceppi M, and Pierre P (2009). SUnSET, a nonradioactive method to monitor protein synthesis. Nat. Methods 6, 275–277. [DOI] [PubMed] [Google Scholar]
- Snyder AG, Hubbard NW, Messmer MN, Kofman SB, Hagan CE, Orozco SL, Chiang K, Daniels BP, Baker D, and Oberst A (2019). Intratumoral activation of the necroptotic pathway components RIPK1 and RIPK3 potentiates antitumor immunity. Sci. Immunol. 4, eaaw2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L, Wang H, Wang Z, He S, Chen S, Liao D, Wang L, Yan J, Liu W, Lei X, and Wang X (2012). Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell 148, 213–227. [DOI] [PubMed] [Google Scholar]
- Thapa RJ, Ingram JP, Ragan KB, Nogusa S, Boyd DF, Benitez AA, Sridharan H, Kosoff R, Shubina M, Landsteiner VJ, et al. (2016). DAI senses influenza A virus genomic RNA and activates RIPK3-dependent cell death. Cell Host Microbe 20, 674–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traut TW (1994). Physiological concentrations of purines and pyrimidines. Mol. Cell. Biochem. 140, 1–22. [DOI] [PubMed] [Google Scholar]
- Upton JW, Kaiser WJ, and Mocarski ES (2012). DAI/ZBP1/DLM-1 complexes with RIP3 to mediate virus-induced programmed necrosis that is targeted by murine cytomegalovirus vIRA. Cell Host Microbe 11, 290–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vande Walle L, and Lamkanfi M (2016). Pyroptosis. Curr. Biol. 26, R568–R572. [DOI] [PubMed] [Google Scholar]
- Wang NS, Unkila MT, Reineks EZ, and Distelhorst CW (2001). Transient expression of wild-type or mitochondrially targeted Bcl-2 induces apoptosis, whereas transient expression of endoplasmic reticulum-targeted Bcl-2 is protective against Bax-induced cell death. J. Biol. Chem. 276, 44117–44128. [DOI] [PubMed] [Google Scholar]
- Wang H, Sun L, Su L, Rizo J, Liu L, Wang LF, Wang FS, and Wang X (2014). Mixed lineage kinase domain-like protein MLKL causes necrotic membrane disruption upon phosphorylation by RIP3. Mol. Cell 54, 133–146. [DOI] [PubMed] [Google Scholar]
- Wu J, Huang Z, Ren J, Zhang Z, He P, Li Y, Ma J, Chen W, Zhang Y, Zhou X, et al. (2013). Mlkl knockout mice demonstrate the indispensable role of Mlkl in necroptosis. Cell Res. 23, 994–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yatim N, Jusforgues-Saklani H, Orozco S, Schulz O, Barreira da Silva R, Reis e Sousa C, Green DR, Oberst A, and Albert ML (2015). RIPK1 and NF-κB signaling in dying cells determines cross-priming of CD8+ T cells. Science 350, 328–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yatim N, Cullen S, and Albert ML (2017). Dying cells actively regulate adaptive immune responses. Nat. Rev. Immunol. 17, 262–275. [DOI] [PubMed] [Google Scholar]
- Zhang DW, Shao J, Lin J, Zhang N, Lu BJ, Lin SC, Dong MQ, and Han J (2009). RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science 325, 332–336. [DOI] [PubMed] [Google Scholar]
- Zhao J, Jitkaew S, Cai Z, Choksi S, Li Q, Luo J, and Liu ZG (2012). Mixed lineage kinase domain-like is a key receptor interacting protein 3 down-stream component of TNF-induced necrosis. Proc. Natl. Acad. Sci. USA 109, 5322–5327. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Complete RNA sequencing datasets are available via the NIH Gene Expression Omnibus (GEO) site (https://www.ncbi.nlm.nih.gov/geo), Series Record GSE134234.