

Abstract
As synucleinopathies, Parkinson’s disease (PD) and multiple system atrophy (MSA) are neurodegenerative diseases that involve the spread of pathogenic alpha-synuclein (αSyn) throughout the brain. Recent studies have suggested a role for αSyn as an antimicrobial peptide in response to PD- and MSA-related infections of peripheral tissues, including those in the respiratory, gastrointestinal, and urogenital systems. In this chapter, we examine epidemiological and experimental evidence for a role of peripheral microbial infections in triggering alpha-synucleinopathies. We propose a model of how infectious triggers, in conjunction with inflammatory, environmental, and genetic facilitators, may result in transfer of pathogenic αSyn strains from the periphery to the brain, where they propagate and spread. Finally, we discuss future research challenges and programs necessary to clarify the role of infections as triggers of PD and MSA and, ultimately, to prevent the onset of these diseases by infectious triggers.
1. Introduction
1.1. Alpha-synucleinopathies
Neurological protein misfolding diseases include common and pernicious neurodegenerative diseases such as Alzheimer’s disease (AD), tauopathies, Amyotrophic Lateral Sclerosis, Creutzfeldt-Jakob disease, prion diseases, and Huntington’s disease. Some neurological protein misfolding diseases are characterized by intracellular inclusions of misfolded alpha-synuclein (αSyn) and are collectively termed alpha-synucleinopathies. They include Parkinson’s disease (PD), dementia with Lewy Bodies (DLB), and multiple system atrophy (MSA). Although the proteins implicated across the spectrum of neurodegenerative diseases and the clinical manifestations thereof vary, the molecular mechanisms underlying protein misfolding share several characteristics, including the accumulation and progressive spread of protein aggregates in the brain. The triggers of protein misfolding and propagation and the means by which misfolded proteins exert their toxic effects are still hotly debated. Furthermore, there are no known cures or effective treatments for these progressive disorders, which means that, in total, protein misfolding neurological diseases are associated with very high societal costs and represent a major unmet medical need.
In order to develop preventive strategies or disease-modifying therapies, it is imperative to define and understand the mechanisms that underlie disease progression and to identify genetic and environmental factors that play roles in triggering disease. In this chapter, we first introduce the two alpha-synucleinopathies, PD and MSA, describing the clinical neurological features and molecular neuropathologies. We then discuss a new potential triggering event, arising from the impact of microbial infection, on the initial steps of alpha-synucleinopathies. Finally, we discuss if unraveling how pathogens trigger αSyn expression and, potentially, its misfolding could lead to the future development of preventive or disease-modifying therapies.
1.2. Parkinson’s disease
Parkinson’s disease is the second most common neurodegenerative disease, affecting 7–10 million people worldwide. With the growing size of the aging population and advances in health care and life expectancy, these numbers are expected to increase.1 The rate of growth of the prevalence of PD has surpassed that of other neurodegenerative diseases, such as AD, resulting in some researchers labeling PD a “pandemic.”2
In the PD prodromal stage, people destined to develop PD often exhibit non-motor abnormalities, including Rapid Eye Movement (REM) sleep behavior disorder (RBD), hyposmia, autonomic dysfunction, depression, and constipation. Currently, the diagnosis of PD cannot formally be established until the characteristic motor features, namely, resting tremor, bradykinesia, and rigidity, are apparent.3 These motor symptoms are believed to be largely due to progressive degeneration of dopaminergic neurons in the substantia nigra pars compacta (SNpc) and of their axon terminals in the striatum.4 The loss of nigral dopamine neurons is one of the major histopathological hallmarks of PD. The second key neuropathological feature of PD is the accumulation of αSyn protein into intraneuronal aggregates, known as Lewy bodies and Lewy neurites. The vast majority of PD cases are sporadic. Only an estimated 5–10% of patients exhibit monogenic inherited forms of the disease.5 While a review of inherited forms of PD is beyond the scope of the current review and has been covered in detail elsewhere,6 a number of PD genes are noteworthy. The most common autosomal dominant monogenic form of PD (with a penetrance estimated at 40–80%) involves mutations in the gene encoding the kinase LRRK2. This kinase has been implicated in endosomal cycling and has been suggested to play a role in several cellular functions including autophagy, possibly in neurons or immune cells.7,8 In other very rare forms of autosomal dominant PD and closely related neurological disorders, point mutations or multiplications of the αSyn gene SNCA occur,9 supporting the idea that the formation of Lewy pathology is likely a key part of the pathogenesis in sporadic disease as well. Other inherited mutations are strong risk factors but still exhibit very low penetrance. For example, heterozygous mutations in the gene encoding the lysosomal enzyme glucocerebrosidase (GBA), which is deficient in Gaucher’s disease, also elevate apparent PD risk to about four times that of controls10 and are particularly common among Ashkenazi Jews. Additionally, several genome-wide association studies (GWAS) have identified genetic loci associated with altered PD risk. The most recent GWAS lists 90 polymorphisms in 78 genetic loci associated with PD risk.11 While the functional implications of these polymorphisms in non-coding DNA have not yet been defined, some are located close to established PD risk genes (e.g., LRRK2, SNCA, and GBA) and a large proportion are located close to genes implicated in immune functions and the lysosomal-autophagy pathway.12–16 A recent study suggests that the PD genetic risk loci exert their risk-modifying effects in non-neuronal cells in the central nervous system and cells and tissues outside the nervous system.16 This latter observation suggests that the triggering of the PD process might involve peripheral tissues. Despite the large number of genes and non-coding genetic loci implicated in PD, it is important to note that genetic risk factors have been estimated to collectively account for 26–36% of total PD risk.11 This modest genetic link has been further corroborated by a twin study of PD concordance between mono- and dizygotic twins, with proband-wise concordance at 0.20 for monozygotic and 0.13 for dizygotic twins, and heritability estimates at 0.27.17 Furthermore, if estimates from the recent GWAS are accurate, this means that 64–74% of PD risk can be attributed to environmental factors, a large category that might include infections.11
Epidemiological and experimental studies have identified strong associations between certain environmental factors, such as exposure to MPTP, the precursor to neurotoxin MPP+, or to pesticides, and elevated PD risk.18 This risk may be further elevated both by genetic impairment in handling of and by opportunity for passive or occupational exposure to environmental toxins.19,20 Nevertheless, the role of many other environmental factors and their interactions with genetic risk in PD are relatively unexplored areas of research.21 The identification of factors that trigger PD, especially in idiopathic cases without clear exposure to toxins known to reliably induce the disease, has been particularly challenging because the disease process is believed to begin several years or decades before the presentation of motor symptoms.22 This makes it particularly difficult to associate exposure to a particular environmental risk factor with subsequent PD development.
The anatomical distribution of Lewy pathology in PD has provided a new insight into the possible triggers of PD pathology. Almost two decades ago, Braak and colleagues suggested that Lewy pathology first appears in the dorsal motor nucleus of the vagal nerve—the source of enteric nerves controlling gut motility and other organs in the digestive tract—as well as in the brainstem and the olfactory bulbs.23 They proposed that this pathology is present prior to the appearance of classic motor symptoms in PD24,25 and could contribute to the constipation and hyposmia that are frequently present in the PD prodrome.3,26 They further suggested that the Lewy pathology then gradually spreads throughout the brain, among different anatomical regions, in a stereotypic fashion, only reaching the nigral dopaminergic neurons after several years.23,24,27 Thus, the death of dopamine neurons and concomitant development of motor symptoms are suggested to develop decades after the initial trigger of the disease.28 During the past decade, it has been demonstrated in experimental models that misfolded αSyn can be secreted from neurons and then internalized by neighboring neurons.29–33 Once inside the new cell, it can act as a seed and recruit endogenous native αSyn from the new cell, causing it to misfold and aggregate in a manner reminiscent of prions. Small aggregates of αSyn can be transported in both directions along axons.34 Based on all these observations, it has been proposed that αSyn exhibits prion-like behavior which underlies the protracted and gradual spread of Lewy pathology from one brain region to another.35 The model is also consistent with the idea that an initial accumulation of misfolded αSyn in peripheral tissue (e.g., the gut) or the olfactory bulbs could be the first pathogenic event in PD. As the digestive and respiratory systems are exposed to the exterior environment, it is tempting to suggest that external triggers use these systems as entry points to gain access to the central nervous system.
1.3. Multiple system atrophy
Multiple system atrophy (MSA) is a rapidly progressing alpha-synucleinopathy characterized by a breakdown of basic and vital functions including breathing, digestion, urination, blood pressure control, and motor functions. It is a rare neurodegenerative disease with an estimated prevalence of 1 per 20,000 individuals.36 MSA is sub-divided into cerebellar (MSA-C) and parkinsonian (MSA-P) forms, although during later disease stages, patients usually exhibit both cerebellar and parkinsonian features.37 It is not known how MSA arises, and no environmental or genetic contributors have been identified to date. Similar to PD, several disease mechanisms have been suggested, including brain inflammation, metabolic failure, and αSyn accumulation.38 An accurate identification of genetic risk factors has been hampered by the fact that the disease is both heterogeneous and rare. Some studies suggest that there are certain genetic loci that affect both PD and MSA risk. A study with autopsy-proven cases of MSA suggested that specific GBA variants are more frequent in MSA patients in comparison to AD patients or normal subjects.39 Another study of 900 MSA patients found similar results and furthermore showed that GBA variants were much more frequent in patients that had the MSA-C subtype.40
RBD also seems to be a clinical feature that predicts future development of not only PD and DLB, but also MSA.41 In contrast to PD, however, hyposmia is not clearly associated with MSA.42–44 By contrast, what is particularly striking in MSA is that symptoms from the genitourinary tract are prominent and can occur many years before the earliest motor deficits appear.45,46 This is notable considering that the parasympathetic pudendal nerve innervates the urogenital system and directly synapses with different regions within the spinal cord and brainstem that are affected in MSA, particularly the MSA-C subtype. In fact, a recent report suggests that αSyn pathology is prominent in the nerves innervating muscles of the urinary tract of MSA patients, but not PD patients.47
2. Infections as potential triggers of alpha-synucleinopathies
In this section, we discuss whether microbial or viral infections can initiate alpha-synucleinopathy-related disease processes. We propose that, following pathogen infection and a simultaneous inflammatory response, αSyn expression in peripheral neurons will increase, leading to accumulation of the protein, which will misfold and form high molecular weight assemblies. Under normal conditions, and hence, for the majority of people exposed to pathogens that are potential triggers, these αSyn seeds will not propagate as pathological aggregates but will instead be cleared by normal cellular homeostatic mechanisms. Therefore, in most cases, we hypothesize that exposure to pathogens alone is not sufficient to trigger central αSyn pathology. We suggest that only under certain conditions (genetic predisposition, metabolic perturbations, cellular aging, etc.) will the cellular milieu facilitate the accumulation and spread of αSyn aggregates, eventually leading to alpha-synucleinopathies such as PD or MSA. We have divided the evidence supporting this model into two parts, one that discusses it extensively in the context of PD, and a shorter section that considers how the model might apply to MSA.
2.1. Evidence of infections in Parkinson’s disease patients and animal models
Both the nose and the gut, along with other visceral organs innervated by the vagus nerve, including the lungs and liver, are continuously exposed to environmental factors that could potentially trigger αSyn aggregation. Infectious diseases have historically been proposed as contributing factors to the onset of neurodegenerative diseases. Early evidence exhibited a link between the 1918 influenza pandemic known as the Spanish flu and an increase in the number of PD cases in survivors of the flu decades later (in the 1940s and 1950s).48 Gamboa and colleagues further studied the association between the Spanish influenza outbreak and encephalitis lethargica and post-encephalitic parkinsonism, and found influenza antigens in the brains of these patients,49 although subsequent retroactive epidemiological studies found no link.50 Nevertheless, these observations were the first to link viral infections and parkinsonism.
Associations between brain penetrant viral infections—in particular herpes simplex virus (HSV), Influenza-A, West Nile, and norovirus—and neurodegenerative diseases have been demonstrated in experimental models and epidemiological and case-control studies.51 Chronic infections like those caused by HSV have the potential to produce prolonged or recurring inflammatory responses. Even transient infections like Influenza-A (e.g., H5N1, H1N1) may be triggers of αSyn production through their point of entry. For respiratory infections, the nasal cavity, innervated by the olfactory nerves from the olfactory bulbs, and the lungs, innervated by the vagal nerve, may be the starting point (Fig. 1A). After a triggering event in the respiratory system, alpha-synucleinopathy could spread outward through olfactory structures or along the vagus nerve to the brainstem.52,53
Fig. 1.
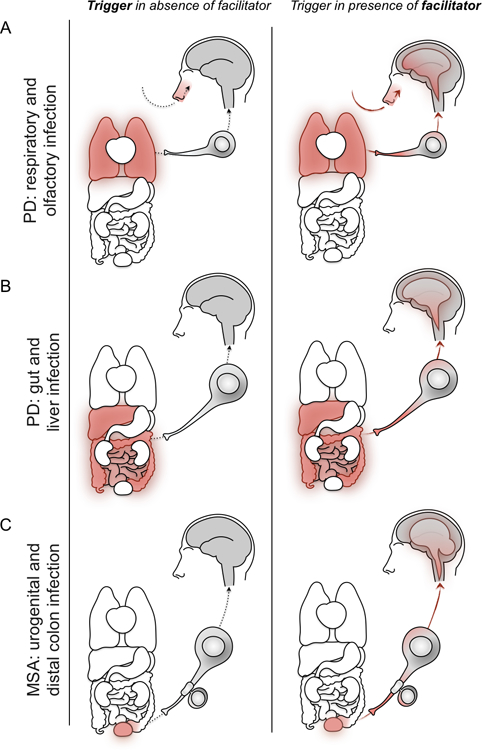
Potential routes of entry for infectious triggers of alpha-synucleinopathies. (A) Infectious triggers of Parkinson’s disease (PD) may infect respiratory system tissues, including the nasal passage, innervated by the olfactory nerves of the olfactory bulbs, and the lungs, innervated by the vagus nerve (left). In the presence of facilitators, alpha-synuclein (αSyn) deposition occurs in neurons before propagating interneuronally along these nerves to the olfactory bulbs and brainstem before spreading through the brain (right). (B) Infectious triggers of PD may also infect gastrointestinal system tissues, including the liver and intestines, which are innervated by the vagus nerve (left). In the presence of facilitators, αSyn propagates interneuronally along the vagus nerve to the brainstem before spreading through the brain (right). (C) Infectious triggers of multiple system atrophy (MSA) may enter the body and infect tissues of the urogenital system or distal colon (left). In the presence of facilitators, αSyn deposition occurs in Schwann cells and oligodendrocytes. αSyn transfers between these cells and neurons to the brainstem, from which alpha-synucleinopathy spreads in the brain, including the cerebellum (right).
Proteins implicated in protein misfolding diseases that can form oligomers and fibrils, such as αSyn, appear to be part of the first line of defense against pathogens acting as natural antimicrobial peptides (AMPs).54 It has been suggested that AMPs can act as potent antibiotics by the destabilization of biological membranes, or as immunomodulators in tissues with limited adaptive immune response, such as the neural system. It is conceivable that the immune response initiates αSyn production as part of the defense mechanism to combat infections. Before eradication, the infectious agent may be escorted by αSyn, possibly in the form of small, potentially seeding-competent assemblies, as it spreads along the vagus nerve before penetrating the brain, inducing central αSyn pathology or aggravating neuroinflammation (Fig. 2). The soluble form of αSyn has broad spectrum antimicrobial properties at concentrations as low as 0.2 μM, which is two magnitudes lower than the concentration of αSyn found in neuronal synapses.55 At these low concentrations, αSyn was shown to dramatically impact the survival of various types of bacteria and fungi in vitro.56 Furthermore, αSyn expression was upregulated in response to viral infection.57 There is support for this concept in other neurodegenerative diseases as well. In brains of AD patients with Candida infections, accumulation of amyloid β-protein (Aβ) aggregates was observed around infections. This observation was hypothesized to represent a neuroprotective sequestration of the infection for attack by a special type of neuroimmune response.58,59 Indeed, Candida infections induced activation of proinflammatory transcription factor NF-κB and production of the cytokines IL-1β and TNFα,60 tying together aggregation of Aβ around Candida with neuroinflammation. In a similar way, it has been suggested that Aβ1–42 exerts an antibacterial effect against Porphyromonas gingivalis. This pathogen has been found in the brain of AD patients and was proposed to act as a trigger of the disease.61 There is evidence that the protease gingipain R1, which is produced by P. gingivalis, is present in blood clots of PD patients but not controls, suggesting this bacteria may be associated with PD risk as well.62
Fig. 2.
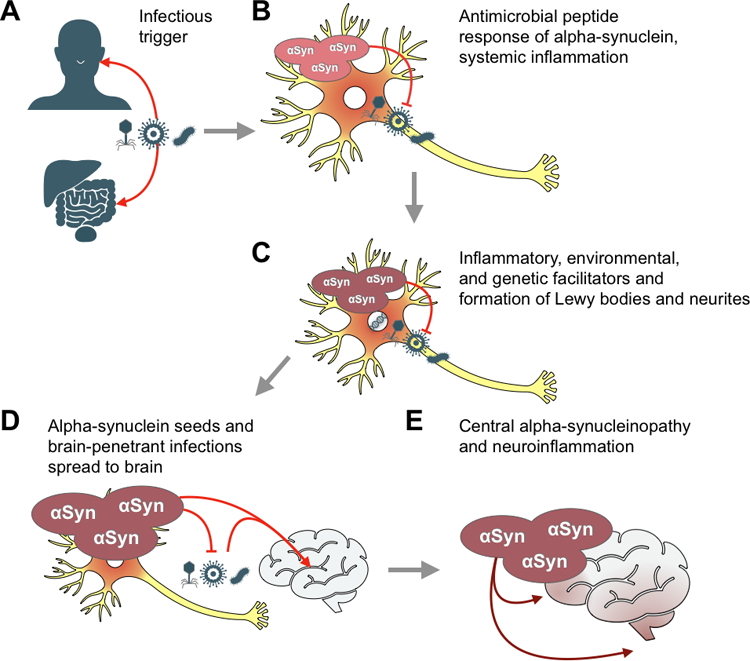
Model of peripheral infections as triggers of alpha-synucleinopathies in the brain. (A) Tissues of the respiratory, gastrointestinal, or urogenital systems are infected by microbial triggers including viruses, fungi, or bacteria. (B) Cells of these tissues, including neurons in PD and oligodendrocytes in MSA, experience an inflammatory response to the infection and produce alpha-synuclein (αSyn) as an antimicrobial peptide. (C) A second hit of environmental, genetic, or inflammatory facilitators leads to aggregation of potentially seeding-competent assemblies of alpha-synuclein, which form Lewy bodies and neurites. (D) αSyn and brain penetrant infections are transferred retrogradely along the nervous tissues that innervate the infected peripheral organs, ultimately reaching the brain. (E) Pathogenic αSyn seeds lead to neuroinflammation and propagation of alpha-synucleinopathy throughout the brain over decades.
Studies in animal models using intranasal administration of the influenza H5N1 and H1N1 viruses showed that both viruses induce microglial activation and associated neuroinflammation in the SNpc of mice63,64 (Table 1). While the brain-penetrant H5N1 induced alpha-synucleinopathy, neuroinflammation, and a temporary period of dopaminergic neuron atrophy and loss of function after which the neurons recovered,63,69 the non-penetrant H1N1 exacerbated dopaminergic neuron death induced by the parkinsonian toxin MPTP65 (Table 1). This demonstrates that viral infections, whether they penetrate the CNS or not, can induce neuroinflammatory responses that under certain conditions can lead to accumulation and spread of αSyn, and ultimately to neurodegeneration. Another brain-penetrant virus, the Japanese encephalitis virus, also induces dopaminergic neuron loss and motor symptoms in rats66 (Table 1), although changes in αSyn were not monitored because its connection to PD had not been discovered at the time when the study was conducted.
Table 1.
Functional contribution of infections to PD phenotypes in animal models.
| Virus | Animal | Manipulation | Outcome | References |
|---|---|---|---|---|
| H5N1 | C57BL/6J mice | Intranasal inoculation | Microglial activation, alpha-synuclein phosphorylation and aggregation. Nigral dopaminergic neuron loss | Jang et al.63 |
| H1N1 | C57BL/6J mice | Intranasal inoculation | Microglial activation in the SNpc. Reduced levels of neurotrophic factors and increased neuroinflammation markers | Sadasivan et al.64 |
| H1N1 | C57BL/6J mice+ MPTP | Intranasal inoculation | Increased loss of nigral dopaminergic neurons | Sadasivan et al.65 |
| Japanese encephalitis | Fischer rats | Intracerebral inoculation | Nigral dopaminergic neuron loss and gliosis. L-DOPA-responsive bradykinesia | Ogata et al.66 |
| Bacteria | Animal | Manipulation | Outcome | References |
| Curli producing E. coli | Aged Fischer 344 rats | Oral administration | Microglial and astroglial activation. Increased alpha-synuclein deposition | Chen et al.67 |
| Other | Animal | Manipulation | Outcome | References |
| PD patients microbiota | Germ-free Th1-αSyn mice | Oral administration | Increased motor dysfunction | Sampson et al.68 |
An intriguing potential link between viral infections and processes linked to PD pathogenesis has been revealed in studies of systemic West Nile virus infections. In mouse models of West Nile virus infection, a major cause of viral encephalitis transferred by mosquitos, expression of endogenous αSyn was upregulated in response to the viral infection in cell culture.57 Furthermore, when αSyn knockout mice were infected, there was an increased viral particle load in the brain and mortality due to encephalitis was greater, supporting the novel idea that αSyn modulates viral infections. Because the presence or absence of αSyn did not affect the host response when West Nile virus was inoculated directly into the brain, as opposed to systemically, the investigators proposed that αSyn restricts the transfer of the viral infection from the PNS to the CNS.57 Furthermore, other neurodegenerative disorders, such as AD, have been associated with brain penetrant viral infections—brains of AD patients have shown higher levels of HSV antigen than the brains of HSV-positive, non-AD controls,70–72 and HSV accelerated deposition of Aβ in a manner that entrapped viral particles in 3D neural cell cultures.71
Other common infections have also been associated with altered PD risk. One case-control study utilizing 110 newly diagnosed PD cases and 220 controls identified various infectious diseases, including mumps, scarlet fever, influenza, whooping cough, and HSV, as risk factors for PD.73
Visceral organs innervated by the vagal nerve, including the gut and the liver, could constitute other peripheral sites that may be impacted by infections, subsequently triggering PD. In this case, infections in visceral organs could potentially first affect neurons in the enteric nervous system (ENS) and spread to the brain by traveling up the vagus nerve, ultimately triggering central PD pathology (Fig. 1B). In line with this, epidemiological studies have shown that both vagotomy and appendectomy are associated with decreased risk of developing PD, in particular if the surgery takes place early in life.74–76
Hepatitis C virus (HCV), which infects the liver, was associated with increased PD risk in two large Taiwanese cohorts (62,276 cases, and 49,967 cases and 199,868 controls, respectively; adjusted Odds Ratios for both ≈ 1.3).77,78 The former study also found 60% dopamine neuron death, levels similar to exposure to 100 nM of the PD-inducing neurotoxin MPP+, and inflammatory cytokine release in a human HCV-exposed neuron-glia coculture rat model.78 Clinical treatment of HCV with interferon increases the incidence of comorbid PD,79 making it difficult to distinguish whether HCV itself or its treatment is associated with PD. Furthermore, a study of a separate cohort from the aforementioned two studies corroborated the finding that HCV confers an elevated risk for PD, as does Hepatitis B.80
The metabolic kynurenine pathway is activated during infection and has been shown to produce large amounts of picolinic acid in response to HCV infection.81 Interestingly, one genetic risk factor for PD is polymorphisms in a locus close to the ACMSD gene,82 the enzyme responsible for the production of picolinic acid at the expense of the excitotoxin quinolinic acid. The combination of genetic risk factors involving such metabolic mechanisms and an infection triggering αSyn production could represent a “perfect storm” of cellular events necessary to facilitate αSyn propagation and spreading of pathology. With the recent development of a mouse model of HCV,83 it may be possible to unravel the mechanisms that underlie the association between HCV infection and PD.
Disturbances in the neuronal signaling between the gut and the brain may underlie some of the preclinical/prodromal PD symptoms, in particular gastrointestinal issues, including constipation as well as other syndromes and diseases associated with PD, such as peptic ulcers, inflammatory bowel disease (IBD), or Crohn’s disease. Chronic infection with the gastric pathogen Helicobacter pylori, a potential causative agent of peptic ulcers, is associated with elevated risk of developing PD.84
The scope of potential insults that may act as triggers of PD pathogenesis extends beyond infections and may also include alterations (dysbiosis) of the resident gut microbiota. Even though this is a relatively new field of study in PD, there are several human case-control studies addressing the taxonomic changes in gut microbiota. These studies invariably suffer from the short-coming that it is not clear whether any observed changes in gut microbiota are due to the constipation that commonly occurs in PD or due to anti-PD medications. Furthermore, changes in gut microbiota that might act as triggers of PD pathogenic processes are likely to have occurred several years prior to the diagnosis of PD. Notwithstanding these significant caveats, the information available suggests that the composition of the PD microbiome is characterized by increased representation of Lactobacillus, Bifidobacterium, and Verrucomicrobiaceae Akkermansia and Enterobacteriaceae, and decreased representation of Faecalibacterium, Coprococcus, Blautia, and Prevotella.85 Interestingly, the gut microbiota also comprises a large variety of viruses, fungi, and archaea that have not hitherto been the subject of extensive study. Bacteriophages, a viral component of the gut microbiota, can alter gut bacterial composition and impact intestinal permeability as well as local inflammation. A recent study reported that PD patients’ microbiome is enriched in lytic lactococcal phages.86 An altered gut microbiota composition can also lead to changes in the levels of metabolic products from gut bacteria, including short-chain fatty acids (SCFAs), which are major players in the maintenance of immune homeostasis and neuroprotective or neurotoxic kynurenine metabolites.87 Therefore, bacterial metabolites can have profound effects on the inflammatory milieu both in the periphery and the brain. A lower abundance of Coprococcus, Blautia, and Prevotella has been associated with decreases in SCFAs and could contribute to the increased intestinal permeability observed in PD patients.88,89 As mentioned above, most studies have been performed in patients already diagnosed with PD, and it is known that disease duration and medication can alter microbiota composition. Therefore, in the future it will be important to perform prospective longitudinal studies that include people with prodromal PD to identify specific dysbiosis signatures that might be triggers of the disease.
Few functional studies have been performed in animal models to address the role of the microbiome in the initial stages of PD. Fecal microbiota transplantation from PD patients into germ-free transgenic mice that overexpress αSyn worsened the motor deficits in this PD model68 (Table 1). Even when it has been shown that PD patients display decreased absolute concentrations of SCFAs,90 SCFA supplementation promoted microglial activation and motor dysfunction in the αSyn overexpressing mouse.68 These results in animal models contradict the notion that SCFAs are anti-inflammatory and protective against pathological conditions, and again reveal a need for more studies in PD patients and animal models to better understand the role of the gut microbiota and its metabolites.
Aside from changes in SFCAs, there are additional possible mechanisms through which gut infections or dysbiosis could trigger the disease process, including bacterial amyloid cross-seeding. This notion is supported in an aged rat model of αSyn accumulation in the GI tract, in which it was observed that the inoculation of an E. coli strain that produces the bacterial amyloid protein Curli increases glial activation and αSyn deposition67 (Table 1). The most noteworthy mechanism, however, proposes that gut dysbiosis and/or infections increase intestinal permeability and peripheral inflammatory response, triggering pathological αSyn deposition and propagation, cytokine release, and glial activation.91,92
2.2. Evidence of infections in multiple system atrophy
To date, no epidemiological studies have strongly associated environmental factors or infectious diseases to MSA. A recent study established an association between increased MSA risk and chronic inflammation in the gut and found that peripheral inflammation might be a risk factor for MSA.93 Furthermore, the gut microbiome in MSA patients differs from that of control subjects and is consistent with gut inflammatory activation.94 While PD and MSA exhibit some intriguing similarities, their clinical presentations are often remarkably distinct. The question, then, is why are MSA and PD so different?
In contrast to PD, where neuronal inclusions are the key defining histopathological lesion, MSA is characterized by αSyn deposits in oligodendrocytes and Schwann cells in the central and peripheral nervous system, respectively.95 Smaller aggregates of αSyn that are difficult to detect with conventional staining methods are found in neurons.96 When comparing neuron-derived aggregated αSyn with αSyn isolated from oligodendrocytes, both the structures and biological activities of αSyn are different.97 Furthermore, under given assembly conditions, monomeric αSyn can misfold and aggregate into specific high molecular weight structures that are ultrastructurally and biochemically unique, and can be considered distinct strains of αSyn aggregates.98 Such αSyn strains can have differential pathological effects in experimental models.99 Hence, this might provide a clue as to why alpha-synucleinopathies are so heterogeneous. How αSyn can form various types of assemblies (strains) in different diseases, however, remains a mystery.
Central to the idea of a distinct type of αSyn strain being present in MSA is that the formation and maturation of αSyn occurs in oligodendrocytes. αSyn inclusions are found in differentiated, myelinating oligodendrocytes that do not express αSyn under normal conditions in adulthood. Immature oligodendrocytes express αSyn but downregulate its expression during maturation, and once oligodendrocytes start to myelinate, αSyn becomes undetectable.100–102 One possibility could be that exogenous aggregates of αSyn are generated in the periphery, outside of the nervous system, and trigger aggregation of αSyn in oligodendrocytes. Notably, erectile dysfunction and incontinence are some of the earliest symptoms in MSA. Therefore, one can hypothesize that bacterial, viral, or fungal infections of the urinary tract or distal colon trigger the formation of unique, pathological αSyn seeds in peripheral nerves innervating these organs. These nerves would provide an entry route into the CNS via sympathetic and parasympathetic innervations of the urinary tract and distal colon (Fig. 1C). MSA patients were indeed shown to have abundant Lewy pathology in the nerves innervating the bladder, which was not the case for PD patients in the same study.47 Furthermore, when seeding-potent fibrils of synthetic αSyn were injected into the lower urinary tract of hemizygous TgM83+/— mice, which overexpress human A53T mutant αSyn, they were found to spread from the bladder to the spinal cord where it gave rise to neuronal and sparse glial pathology.47
A more speculative hypothesis that could explain why αSyn aggregates are present in the cytoplasm of glia in MSA is that mature oligodendrocytes could conceivably use αSyn to combat infections. Given the purported antimicrobial properties of αSyn,56,103,104 it is possible that mature oligodendrocytes defend themselves against invading pathogens by expressing αSyn. Intracellular accumulation of αSyn in response to pathogens could provide oligodendrocytes with a mechanism to defend themselves against intracellular microbes before they cause irreversible damage and demyelination. Over 15% of MSA patients experience repeated urinary tract infections as an initial symptom before an MSA diagnosis is made. After diagnosis, recurring urinary tract infections are suffered by more than half of MSA patients, of which a significant number die due to related complications.105 In light of recurrent urinary tract infections being common in MSA,106 recurring bacterial (uropathogenic E. coli) or fungal (Candida) infections in the urinary tract might trigger the formation of distinct αSyn strains in neurons, Schwann cells and oligodendrocytes, eventually leading to prion-like propagation of αSyn pathology in MSA. Urinary tract infections, however, are common,107 and urinary tract infections alone cannot explain why an individual would develop MSA. One has to posit that a facilitator that is specific for the disease increases a person’s susceptibility to develop MSA.
3. Research challenges regarding infections as triggers of Parkinson’s disease
The aforementioned epidemiological, case-control, and animal studies support the notion that infections, using different entry points in the body, could trigger alpha-synucleinopathy and inflammatory processes that ultimately lead to PD and related synucleinopathies. We recently proposed a model for PD pathogenesis in which different factors relevant to the pathogenic process are active during different temporal phases of the disease28 (Fig. 1). In the past it has been suggested that inflammatory processes may be triggers of PD.108 However, in the present model of pathogen-induced PD and MSA, we suggest that inflammation acts as a facilitator necessary for development of pathology that is initiated by other triggers, such as infectious diseases. We further propose that there is a complex interaction between genetic and environmental factor(s) that determine an individual’s susceptibility to alpha-synucleinopathies. For example, the susceptibility to infections by the intracellular bacterial pathogen Mycobacterium tuberculosis has been postulated to be governed by the autosomal recessive PD gene parkin.109 GBA has been demonstrated to be necessary for H1N1 endocytosis.110 LRRK2 also affects maturation of phagosomes containing Mycobacterium tuberculosis.111
We hypothesize that neurons in PD, or oligodendrocytes in MSA, accumulate assemblies of αSyn that act as AMPs in the defense against infections. We further suggest that this accumulation only happens when the affected person is genetically predisposed or if there is a second environmental or inflammatory insult (or some combination of facilitators) (Fig. 2). This hypothesis implies that αSyn aggregates have a useful normal function in combating the initial trigger before the process goes awry. Despite all the evidence implicating microbes and infections as triggers of PD and MSA, the mechanisms by which these lead to αSyn pathology are unknown. As mentioned above, αSyn exhibits antibacterial and antifungal activities, in addition to restricting viral infections in the brain.56,103,104 It is possible that, depending on the infection, the mechanism of action of αSyn could vary. While the ability of αSyn oligomers to destabilize lipid membranes could be one endogenous function in cases of bacterial and fungal infections, its function in ER-to-Golgi vesicular transport, which some viruses manipulate to sustain self-replication, could affect viral spread.103 It is also possible that loss of normal αSyn function due to misfolding and oligomerization causes disruptions to secretory pathways, inducing an overload of endolysosomal-autophagy pathways and subsequent accumulation of the vesicular structures and dysmorphic organelles identified as major components of Lewy bodies.112 Related to this is the finding that the oligomer forms of αSyn target and disrupt intracellular membranes, leading to accumulation of lipids within αSyn-positive inclusions, as observed in PD patient brains.112 αSyn has also been demonstrated to have an immunomodulatory role in acute inflammatory conditions of the gut, including IBD and norovirus infections in pediatric patients. In the former study’s patients, αSyn aggregates were observed in the colon and in macrophages.113 In the latter study’s patients, αSyn expression positively correlated with the degree of inflammation, as various unfolded and high molecular weight assemblies of αSyn manifest a chemoattractant activity for phagocytic cells.114
Our model suggests that public health measures and treatments to reduce the infections implicated in PD and MSA might reduce the incidence of these disorders. Interventions could include different methods for microbiota modulation, ranging from dietary changes and transfer of fecal microbiota to the use of antibiotics or even phagotherapies. Because pathogen-induced inflammation might constitute a final common pathway, one can speculate that anti-inflammatory treatments might reduce the risk for triggering PD and MSA, regardless of which pathogens sparked the initial molecular cascade. Intriguingly, patients with IBD, which is associated with higher incidence of PD, have a reduced risk of developing PD if they have had early anti-inflammatory interventions with anti-TNF treatment.115 However, caution should be taken when designing PD-modifying interventions. If αSyn indeed is part of an important first-line defense mechanism, αSyn-targeted immuno-therapies, which currently are being tested against PD and MSA,116,117 could potentially be harmful if the treated individual is infected by certain pathogens. Therefore, identifying early, low-risk targets such as potential infectious triggers is imperative to modify disease course and prevent initiation of alpha-synucleinopathy.
Because of the apparently long incubation period, which one can assume based on the PD prodrome extending over years or even decades, it is challenging to identify which potential infectious triggers are most important. It is also unclear whether only specific pathogens can trigger alpha-synucleinopathies or whether any pathogen may contribute inflammatory responses that initiate disease. While we highlight infections as potential triggers of alpha-synucleinopathies in this chapter, they are most likely not the only triggers of PD or MSA. Recent reports have suggested different clusters of PD based on development of symptoms,118 and the existence of different triggers might explain some of this clinical variability. Another possibility is that the site of primary infection (e.g., respiratory, gastrointestinal, urogenital, or other peripheral tissues) significantly impacts the eventual pattern of neuropathology.
In conclusion, we believe that there is sufficient and compelling evidence that infections might act as triggers of alpha-synucleinopathies to warrant expanded research efforts designed to test the hypothesis. These efforts could provide us with new therapeutic targets for prevention of PD and MSA, but even if they do not, they will help to both clarify the relationship between infections and alpha-synucleinopathies and to elucidate the role of αSyn in immunity.
Acknowledgments
We acknowledge the Van Andel Research Institute and the many individuals and corporations that supported financially the neurodegenerative research at Van Andel Research Institute. We would like to acknowledge Farmer Family Foundation for funding related to the topic of this review. P.B. is supported by grants from the National Institutes of Health (1R01DC016519-03) which supports G.M. and the Office of the Assistant Secretary of Defense for Health Affairs (Parkinson’s Research Program, Award No. W81XWH-17-1-0534) which supports C.T. L.B. is supported by grant 14939 from the Michael J. Fox Foundation. W.P. is supported by a Fulbright Fellowship and FWO.
P.B. has received commercial support as a consultant from Renovo Neural, Inc., Roche, Teva Inc., Lundbeck A/S, AbbVie, Neuroderm, Fujifilm-Cellular Dynamics International, Axial Biotherapeutics, ClearView Healthcare, FCB Health, IOS Press Partners and Capital Technologies, Inc. He has received commercial support for grants/research from Renovo and Roche. He has ownership interests in Acousort AB and is on the steering committee of the NILO-PD trial.
Footnotes
Competing interests
The authors declare no additional competing financial interests.
References
- 1.Dorsey ER, Bloem BR. The Parkinson Pandemic-A call to action. JAMA Neurol 2018;75(1):9–10. [DOI] [PubMed] [Google Scholar]
- 2.Dorsey ER, Sherer T, Okun MS, Bloem BR. The emerging evidence of the Parkinson Pandemic. J Park Dis 2018;8(s1):S3–S8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Postuma RB, Berg D, Stern M, et al. MDS clinical diagnostic criteria for Parkinson’s disease. Mov Disord 2015;30(12):1591–1601. [DOI] [PubMed] [Google Scholar]
- 4.Tagliaferro P, Burke RE. Retrograde axonal degeneration in parkinson disease. J Park Dis 2016;6(1):1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Klein C, Westenberger A. Genetics of Parkinson’s disease. Cold Spring Harb Perspect Med 2012;2(1):a008888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Deng H, Wang P, Jankovic J. The genetics of Parkinson disease. Ageing Res Rev 2018;42:72–85. [DOI] [PubMed] [Google Scholar]
- 7.Roosen DA, Cookson MR. LRRK2 at the interface of autophagosomes, endosomes and lysosomes. Mol Neurodegener 2016;11(1):73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Manzoni C, Mamais A, Dihanich S, et al. Pathogenic Parkinson’s disease mutations across the functional domains of LRRK2 alter the autophagic/lysosomal response to starvation. Biochem Biophys Res Commun 2013;441(4):862–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xu W, Tan L, Yu JT. Link between the SNCA gene and parkinsonism. Neurobiol Aging 2015;36(3):1505–1518. [DOI] [PubMed] [Google Scholar]
- 10.Sidransky E, Lopez G. The link between the GBA gene and parkinsonism. Lancet Neurol 2012;11(11):986–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nalls MA, Blauwendraat C, Vallerga CL, et al. Expanding Parkinson’s disease genetics: novel risk loci, genomic context, causal insights and heritable risk. [published online ahead of print March 4, 2019]. bioRxiv. 2019;388165 10.1101/388165. [DOI]
- 12.Mullin S, Schapira A. The genetics of Parkinson’s disease. Br Med Bull 2015; 114(1):39–52. [DOI] [PubMed] [Google Scholar]
- 13.Hardy J, Cai H, Cookson MR, Gwinn-Hardy K, Singleton A. Genetics of Parkinson’s disease and parkinsonism. Ann Neurol 2006;60(4):389–398. [DOI] [PubMed] [Google Scholar]
- 14.Coetzee SG, Pierce S, Brundin P, Brundin L, Hazelett DJ, Coetzee GA. Enrichment of risk SNPs in regulatory regions implicate diverse tissues in Parkinson’s disease etiology. Sci Rep 2016;6:30509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gagliano SA, Pouget JG, Hardy J, et al. Genomics implicates adaptive and innate immunity in Alzheimer’s and Parkinson’s diseases. Ann Clin Transl Neurol 2016; 3(12):924–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Reynolds RH, Botia J, Nalls MA, et al. Moving beyond neurons: the role of cell type-specific gene regulation in Parkinson’s disease heritability. NPJ Parkinsons Dis 2019;5:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Goldman SM, Marek K, Ottman R, et al. Concordance for Parkinson’s disease in twins: a 20-year update. Ann Neurol 2019;85(4):600–605. [DOI] [PubMed] [Google Scholar]
- 18.Nandipati S, Litvan I. Environmental exposures and Parkinson’s disease. Int J Environ Res Public Health 2016;13(9):881–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang A, Costello S, Cockburn M, Zhang X, Bronstein J, Ritz B. Parkinson’s disease risk from ambient exposure to pesticides. Eur J Epidemiol 2011;26(7):547–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tanner CM, Goldman SM, Ross GW, Grate SJ. The disease intersection of susceptibility and exposure: chemical exposures and neurodegenerative disease risk. Alzheimers Dement 2014;10(3 Suppl):S213–S225. [DOI] [PubMed] [Google Scholar]
- 21.Chen H, Ritz B. The search for environmental causes of Parkinson’s disease: moving forward. J Park Dis 2018;8(s1):S9–S17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kalia LV, Lang AE. Parkinson’s disease. Lancet 2015;386(9996):896–912. [DOI] [PubMed] [Google Scholar]
- 23.Del Tredici K, Rub U, De Vos RA, Bohl JR, Braak H. Where does parkinson disease pathology begin in the brain? J Neuropathol Exp Neurol 2002;61(5):413–426. [DOI] [PubMed] [Google Scholar]
- 24.Braak H, Del Tredici K, Rub U, de Vos RA, Jansen Steur EN, Braak E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging 2003;24(2):197–211. [DOI] [PubMed] [Google Scholar]
- 25.Braak H, Del Tredici K. Invited article: nervous system pathology in sporadic Parkinson disease. Neurology 2008;70(20):1916–1925. [DOI] [PubMed] [Google Scholar]
- 26.Postuma RB, Berg D. Advances in markers of prodromal Parkinson disease. Nat Rev Neurol 2016;12(11):622–634. [DOI] [PubMed] [Google Scholar]
- 27.Braak H, Del Tredici K. Neuropathological staging of brain pathology in sporadic Parkinson’s disease: separating the wheat from the chaff. J Park Dis 2017;7(s1):S71–S85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Johnson ME, Stecher B, Labrie V, Brundin L, Brundin P. Triggers, facilitators, and aggravators: redefining Parkinson’s disease pathogenesis. Trends Neurosci 2019; 42(1):4–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Steiner JA, Quansah E, Brundin P. The concept of alpha-synuclein as a prion-like protein: ten years after. Cell Tissue Res 2018;373(1):161–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Del Tredici K, Braak H. Review: sporadic Parkinson’s disease: development and distribution of alpha-synuclein pathology. Neuropathol Appl Neurobiol 2016;42(1):33–50. [DOI] [PubMed] [Google Scholar]
- 31.Desplats P, Lee HJ, Bae EJ, et al. Inclusion formation and neuronal cell death through neuron-to-neuron transmission of alpha-synuclein. Proc Natl Acad Sci U S A 2009;106(31):13010–13015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lee HJ, Bae EJ, Lee SJ. Extracellular alpha—synuclein-a novel and crucial factor in Lewy body diseases. Nat Rev Neurol 2014;10(2):92–98. [DOI] [PubMed] [Google Scholar]
- 33.Hansen C, Angot E, Bergstrom AL, et al. alpha-Synuclein propagates from mouse brain to grafted dopaminergic neurons and seeds aggregation in cultured human cells. J Clin Invest 2011;121(2):715–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Brahic M, Bousset L, Bieri G, Melki R, Gitler AD. Axonal transport and secretion of fibrillar forms of alpha-synuclein, Abeta42 peptide and HTTExon 1. Acta Neuropathol 2016;131(4):539–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Brundin P, Melki R. Prying into the prion hypothesis for Parkinson’s disease. J Neurosci 2017;37(41):9808–9818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Glasmacher SA, Leigh PN, Saha RA. Predictors of survival in progressive supranuclear palsy and multiple system atrophy: a systematic review and meta-analysis. J Neurol Neurosurg Psychiatry 2017;88(5):402–411. [DOI] [PubMed] [Google Scholar]
- 37.Gilman S, Wenning GK, Low PA, et al. Second consensus statement on the diagnosis of multiple system atrophy. Neurology 2008;71(9):670–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Walsh RR, Krismer F, Galpern WR, et al. Recommendations of the global multiple system atrophy research roadmap meeting. Neurology 2018;90(2):74–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sklerov M, Kang UJ, Liong C, et al. Frequency of GBA variants in autopsy-proven multiple system atrophy. Mov Disord Clin Pract 2017;4(4):574–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mitsui J, Matsukawa T, Sasaki H, et al. Variants associated with Gaucher disease in multiple system atrophy. Ann Clin Transl Neurol 2015;2(4):417–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Palma JA, Fernandez-Cordon C, Coon EA, et al. Prevalence of REM sleep behavior disorder in multiple system atrophy: a multicenter study and meta-analysis. Clin Auton Res 2015;25(1):69–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Garland EM, Raj SR, Peltier AC, Robertson D, Biaggioni I. A cross-sectional study contrasting olfactory function in autonomic disorders. Neurology 2011;76(5):456–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Muller A, Mungersdorf M, Reichmann H, Strehle G, Hummel T. Olfactory function in Parkinsonian syndromes. J Clin Neurosci 2002;9(5):521–524. [DOI] [PubMed] [Google Scholar]
- 44.Rey NL, Wesson DW, Brundin P. The olfactory bulb as the entry site for prion-like propagation in neurodegenerative diseases. Neurobiol Dis 2018;109(pt B):226–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sakakibara R, Panicker J, Simeoni S, et al. Bladder dysfunction as the initial presentation of multiple system atrophy: a prospective cohort study. Clin Auton Res 2018. 10.1007/s10286-018-0550-y. [DOI] [PubMed]
- 46.McKay JH, Cheshire WP. First symptoms in multiple system atrophy. Clin Auton Res 2018;28(2):215–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ding X, Wang X, Ma M, et al. Spread of pathological alpha-synuclein from urogenital nerves initiates multiple system atrophy-like symptoms. [published online ahead of print January 24, 2019]. bioRxiv. 2019;529594 10.1101/529594. [DOI]
- 48.Poskanzer DC, Schwab RS. Cohort analysis of Parkinson’s syndrome: evidence for a single etiology related to subclinical infection about 1920. J Chronic Dis 1963; 16:961–973. [DOI] [PubMed] [Google Scholar]
- 49.Gamboa ET, Wolf A, Yahr MD, et al. Influenza virus antigen in postencephalitic parkinsonism brain. Detection by immunofluorescence. Arch Neurol 1974;31(4):228–232. [DOI] [PubMed] [Google Scholar]
- 50.McCall S, Henry JM, Reid AH, Taubenberger JK. Influenza RNA not detected in archival brain tissues from acute encephalitis lethargica cases or in postencephalitic Parkinson cases. J Neuropathol Exp Neurol 2001;60(7):696–704. [DOI] [PubMed] [Google Scholar]
- 51.Jang H, Boltz DA, Webster RG, Smeyne RJ. Viral parkinsonism. Biochim Biophys Acta 2009;1792(7):714–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Doty RL. The olfactory vector hypothesis of neurodegenerative disease: is it viable? Ann Neurol 2008;63(1):7–15. [DOI] [PubMed] [Google Scholar]
- 53.Lema Tome CM, Tyson T, Rey NL, Grathwohl S, Britschgi M, Brundin P. Inflammation and alpha-synuclein’s prion-like behavior in Parkinson’s disease—is there a link? Mol Neurobiol 2013;47(2):561–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wiesner J, Vilcinskas A. Antimicrobial peptides: the ancient arm of the human immune system. Virulence 2010;1(5):440–464. [DOI] [PubMed] [Google Scholar]
- 55.Wilhelm BG, Mandad S, Truckenbrodt S, et al. Composition of isolated synaptic boutons reveals the amounts of vesicle trafficking proteins. Science 2014;344(6187): 1023–1028. [DOI] [PubMed] [Google Scholar]
- 56.Park SC, Moon JC, Shin SY, et al. Functional characterization of alpha-synuclein protein with antimicrobial activity. Biochem Biophys Res Commun 2016;478(2):924–928. [DOI] [PubMed] [Google Scholar]
- 57.Beatman EL, Massey A, Shives KD, et al. Alpha-synuclein expression restricts RNA viral infections in the brain. J Virol 2015;90(6):2767–2782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Alonso R, Pisa D, Marina AI, Morato E, Rabano A, Carrasco L. Fungal infection in patients with Alzheimer’s disease. J Alzheimers Dis 2014;41(1):301–311. [DOI] [PubMed] [Google Scholar]
- 59.Pisa D, Alonso R, Juarranz A, Rabano A, Carrasco L. Direct visualization of fungal infection in brains from patients with Alzheimer’s disease. J Alzheimers Dis 2015; 43(2):613–624. [DOI] [PubMed] [Google Scholar]
- 60.Wu Y, Du S, Johnson JL, et al. Microglia and amyloid precursor protein coordinate control of transient Candida cerebritis with memory deficits. Nat Commun 2019; 10(1):58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dominy SS, Lynch C, Ermini F, et al. Porphyromonas gingivalis in Alzheimer’s disease brains: evidence for disease causation and treatment with small-molecule inhibitors. Sci Adv 2019;5(1):eaau3333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Adams B, Nunes JM, Page MJ, et al. Parkinson’s disease: a systemic inflammatory disease accompanied by bacterial inflammagens. [published online ahead of print May 24, 2019]. bioRxiv. 2019;646307 10.1101/646307. [DOI] [PMC free article] [PubMed]
- 63.Jang H, Boltz D, Sturm-Ramirez K, et al. Highly pathogenic H5N1 influenza virus can enter the central nervous system and induce neuroinflammation and neurodegeneration. Proc Natl Acad Sci U S A 2009;106(33):14063–14068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sadasivan S, Zanin M, O’Brien K, Schultz-Cherry S, Smeyne RJ. Induction of microglia activation after infection with the non-neurotropic A/CA/04/2009 H1N1 influenza virus. PLoS One 2015;10(4):e0124047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sadasivan S, Sharp B, Schultz-Cherry S, Smeyne RJ. Synergistic effects of influenza and 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) can be eliminated by the use of influenza therapeutics: experimental evidence for the multi-hit hypothesis. NPJ Parkinsons Dis 2017;3:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ogata A, Tashiro K, Nukuzuma S, Nagashima K, Hall WW. A rat model of Parkinson’s disease induced by Japanese encephalitis virus. J Neurovirol 1997;3(2):141–147. [DOI] [PubMed] [Google Scholar]
- 67.Chen SG, Stribinskis V, Rane MJ, et al. Exposure to the functional bacterial amyloid protein curli enhances alpha-synuclein aggregation in aged fischer 344 rats and caenorhabditis elegans. Sci Rep 2016;6:34477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sampson TR, Debelius JW, Thron T, et al. Gut microbiota regulate motor deficits and neuroinflammation in a model of Parkinson’s disease. Cell 2016;167(6): 1469–1480.e1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Jang H, Boltz D, McClaren J, et al. Inflammatory effects of highly pathogenic H5N1 influenza virus infection in the CNS of mice. J Neurosci 2012;32(5): 1545–1559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Readhead B, Haure-Mirande JV, Funk CC, et al. Multiscale analysis of independent Alzheimer’s cohorts finds disruption of molecular, genetic, and clinical networks by human herpesvirus. Neuron 2018;99(1):64–82.e67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Eimer WA, Vijaya Kumar DK, Navalpur Shanmugam NK, et al. Alzheimer’s disease-associated beta-amyloid is rapidly seeded by herpesviridae to protect against brain infection. Neuron 2018;99(1):56–63.e53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kumar DK, Choi SH, Washicosky KJ, et al. Amyloid-beta peptide protects against microbial infection in mouse and worm models of Alzheimer’s disease. Sci Transl Med 2016;8(340):340ra372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Vlajinac H, Dzoljic E, Maksimovic J, Marinkovic J, Sipetic S, Kostic V. Infections as a risk factor for Parkinson’s disease: a case-control study. Int J Neurosci 2013;123(5): 329–332. [DOI] [PubMed] [Google Scholar]
- 74.Killinger BA, Madaj Z, Sikora JW, et al. The vermiform appendix impacts the risk of developing Parkinson’s disease. Sci Transl Med 2018;10(465). 10.1126/scitranslmed.aar5280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Liu B, Fang F, Pedersen NL, et al. Vagotomy and Parkinson disease: a Swedish register-based matched-cohort study. Neurology 2017;88(21):1996–2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Svensson E, Horvath-Puho E, Thomsen RW, et al. Vagotomy and subsequent risk of Parkinson’s disease. Ann Neurol 2015;78(4):522–529. [DOI] [PubMed] [Google Scholar]
- 77.Tsai HH, Liou HH, Muo CH, Lee CZ, Yen RF, Kao CH. Hepatitis C virus infection as a risk factor for Parkinson disease: a nationwide cohort study. Neurology 2016; 86(9):840–846. [DOI] [PubMed] [Google Scholar]
- 78.Wu WY, Kang KH, Chen SL, et al. Hepatitis C virus infection: a risk factor for Parkinson’s disease. J Viral Hepat 2015;22(10):784–791. [DOI] [PubMed] [Google Scholar]
- 79.Wangensteen KJ, Krawitt EL, Hamill RW, Boyd JT. Parkinsonism in patients with chronic Hepatitis C treated with interferons: case reports and review of the literature. Clin Neuropharmacol 2016;39(1):1–5. [DOI] [PubMed] [Google Scholar]
- 80.Pakpoor J, Noyce A, Goldacre R, et al. Viral hepatitis and Parkinson disease: a national record-linkage study. Neurology 2017;88(17):1630–1633. [DOI] [PubMed] [Google Scholar]
- 81.Zuwala-Jagiello J, Pazgan-Simon M, Simon K, Warwas M. Picolinic acid in patients with chronic hepatitis C infection: a preliminary report. Mediators Inflamm 2012; 2012:762863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lim CK, Fernandez-Gomez FJ, Braidy N, et al. Involvement of the kynurenine pathway in the pathogenesis of Parkinson’s disease. Prog Neurobiol 2017;155:76–95. [DOI] [PubMed] [Google Scholar]
- 83.Billerbeck E, Wolfisberg R, Fahnoe U, et al. Mouse models of acute and chronic hepacivirus infection. Science 2017;357(6347):204–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Weller C, Oxlade N, Dobbs SM, Dobbs RJ, Charlett A, Bjarnason IT. Role of inflammation in gastrointestinal tract in aetiology and pathogenesis of idiopathic parkinsonism. FEMS Immunol Med Microbiol 2005;44(2):129–135. [DOI] [PubMed] [Google Scholar]
- 85.Gerhardt S, Mohajeri MH. Changes of colonic bacterial composition in Parkinson’s disease and other neurodegenerative diseases. Nutrients 2018;10(6):708–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Tetz G, Brown SM, Hao Y, Tetz V. Parkinson’s disease and bacteriophages as its over-looked contributors. Sci Rep 2018;8(1):10812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Rothhammer V, Borucki DM, Tjon EC, et al. Microglial control of astrocytes in response to microbial metabolites. Nature 2018;557(7707):724–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Forsyth CB, Shannon KM, Kordower JH, et al. Increased intestinal permeability correlates with sigmoid mucosa alpha-synuclein staining and endotoxin exposure markers in early Parkinson’s disease. PLoS One 2011;6(12):e28032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Keshavarzian A, Green SJ, Engen PA, et al. Colonic bacterial composition in Parkinson’s disease. Mov Disord 2015;30(10):1351–1360. [DOI] [PubMed] [Google Scholar]
- 90.Unger MM, Spiegel J, Dillmann KU, et al. Short chain fatty acids and gut microbiota differ between patients with Parkinson’s disease and age-matched controls. Parkinsonism Relat Disord 2016;32:66–72. [DOI] [PubMed] [Google Scholar]
- 91.Caggiu E, Arru G, Hosseini S, et al. Inflammation, infectious triggers, and Parkinson’s disease. Front Neurol 2019;10:122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Perez-Pardo P, Hartog M, Garssen J, Kraneveld AD. Microbes tickling your tummy: the importance of the gut-brain axis in Parkinson’s disease. Curr Behav Neurosci Rep 2017;4(4):361–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Villumsen M, Aznar S, Pakkenberg B, Jess T, Brudek T. Inflammatory bowel disease increases the risk of Parkinson’s disease: a Danish nationwide cohort study 1977–2014. Gut 2019;68(1):18–24. [DOI] [PubMed] [Google Scholar]
- 94.Engen PA, Dodiya HB, Naqib A, et al. The potential role of gut-derived inflammation in multiple system atrophy. J Park Dis 2017;7(2):331–346. [DOI] [PubMed] [Google Scholar]
- 95.Nakamura K, Mori F, Kon T, et al. Filamentous aggregations of phosphorylated alpha-synuclein in Schwann cells (Schwann cell cytoplasmic inclusions) in multiple system atrophy. Acta Neuropathol Commun 2015;3:29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Sekiya H, Kowa H, Koga H, et al. Wide distribution of alpha-synuclein oligomers in multiple system atrophy brain detected by proximity ligation. Acta Neuropathol 2019;137(3):455–466. [DOI] [PubMed] [Google Scholar]
- 97.Peng C, Gathagan RJ, Covell DJ, et al. Cellular milieu imparts distinct pathological alpha-synuclein strains in alpha-synucleinopathies. Nature 2018;557(7706): 558–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Bousset L, Pieri L, Ruiz-Arlandis G, et al. Structural and functional characterization of two alpha-synuclein strains. Nat Commun 2013;4:2575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Peelaerts W, Bousset L, Van der Perren A, et al. alpha-Synuclein strains cause distinct synucleinopathies after local and systemic administration. Nature 2015;522(7556): 340–344. [DOI] [PubMed] [Google Scholar]
- 100.Djelloul M, Holmqvist S, Boza-Serrano A, et al. Alpha-synuclein expression in the oligodendrocyte lineage: an in vitro and in vivo study using rodent and human models. Stem Cell Rep 2015;5(2):174–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Richter-Landsberg C, Gorath M, Trojanowski JQ, Lee VM. alpha-synuclein is developmentally expressed in cultured rat brain oligodendrocytes. J Neurosci Res 2000;62(1):9–14. [DOI] [PubMed] [Google Scholar]
- 102.Asi YT, Simpson JE, Heath PR, et al. Alpha-synuclein mRNA expression in oligodendrocytes in MSA. Glia 2014;62(6):964–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Lesteberg KE, Beckham JD. Immunology of West Nile virus infection and the role of alpha-synuclein as a viral restriction factor. Viral Immunol 2019;32(1):38–47. [DOI] [PubMed] [Google Scholar]
- 104.Berstad K, Berstad JER. Parkinson’s disease; the hibernating spore hypothesis. Med Hypotheses 2017;104:48–53. [DOI] [PubMed] [Google Scholar]
- 105.Papatsoris AG, Papapetropoulos S, Singer C, Deliveliotis C. Urinary and erectile dysfunction in multiple system atrophy (MSA). NeurourolUrodyn 2008;27(1):22–27. [DOI] [PubMed] [Google Scholar]
- 106.Papapetropoulos S, Tuchman A, Laufer D, Papatsoris AG, Papapetropoulos N, Mash DC. Causes of death in multiple system atrophy. J Neurol Neurosurg Psychiatry 2007;78(3):327–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Flores-Mireles AL, Walker JN, Caparon M, Hultgren SJ. Urinary tract infections: epidemiology, mechanisms of infection and treatment options. Nat Rev Microbiol 2015;13(5):269–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Tansey MG, McCoy MK, Frank-Cannon TC. Neuroinflammatory mechanisms in Parkinson’s disease: potential environmental triggers, pathways, and targets for early therapeutic intervention. Exp Neurol 2007;208(1):1–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Manzanillo PS, Ayres JS, Watson RO, et al. The ubiquitin ligase parkin mediates resistance to intracellular pathogens. Nature 2013;501(7468):512–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Drews K, Calgi MP, Harrison WC, et al. Glucosylceramidase maintains influenza infection by regulating endocytosis. J Virol 2019;93 10.1128/JVI.00017-19. [DOI] [PMC free article] [PubMed]
- 111.Hartlova A, Herbst S, Peltier J, et al. LRRK2 is a negative regulator of Mycobacterium tuberculosis phagosome maturation in macrophages. EMBO J 2018;37(12). 10.15252/embj.201798694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Shahmoradian SH, Lewis AJ, Cenoud C, et al. Lewy pathology in Parkinson’s disease consists of crowded organellar, membranous medley. [published online ahead of print February 13, 2019]. bioRxiv. 2019;137976 10.1101/137976. [DOI] [PubMed]
- 113.Grathwohl S, Quansah E, Maroof N, et al. Experimental colitis drives enteric alpha-synuclein accumulation and Parkinson-like brain pathology. [published online ahead of print April 28, 2019] bioRxiv. 2019;505164 10.1101/505164. [DOI] [PMC free article] [PubMed]
- 114.Stolzenberg E, Berry D, Yang, et al. A role for neuronal alpha-synuclein in gastrointestinal immunity. J Innate Immun 2017;9(5):456–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Peter I, Dubinsky M, Bressman S, et al. Anti-tumor necrosis factor therapy and incidence of Parkinson disease among patients with inflammatory bowel disease. JAMA Neurol 2018;75(8):939–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Sardi SP, Cedarbaum JM, Brundin P. Targeted therapies for Parkinson’s disease: from genetics to the clinic. Mov Disord 2018;33(5):684–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Merchant KM, Cedarbaum JM, Brundin P, et al. A proposed roadmap for Parkinson’s disease proof of concept clinical trials investigating compounds targeting alpha-synuclein. J Park Dis 2019;9(1):31–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Lawton M, Ben-Shlomo Y, May MT, et al. Developing and validating Parkinson’s disease subtypes and their motor and cognitive progression. J Neurol Neurosurg Psychiatry 2018;89(12):1279–1287. [DOI] [PMC free article] [PubMed] [Google Scholar]