

Abstract
We previously showed that classical 6-substituted pyrrolo[2,3-d]pyrimidine antifolates bind to folate receptor (FR) α and the target purine biosynthetic enzyme glycinamide ribonucleotide formyltransferase (GARFTase) with different cis and trans conformations. In this study, we designed novel analogs of this series with an amide moiety in the bridge region that can adopt both the cis and trans lowest energy conformations. This provides entropic benefit, by restricting the number of side-chain conformations of the unbound ligand to those most likely to promote binding to FRα and the target enzyme required for antitumor activity. NMR of the most active compound 7 showed both cis and trans amide bridge conformations in ~1:1 ratio. The bridge amide group in the best docked poses of 7 in the crystal structures of FRα and GARFTase adopted both cis and trans conformations, with the lowest energy conformations predicted by Maestro and evidenced by NMR within 1 kcal/mol. Compound 7 showed ~3-fold increased inhibition of FRα-expressing cells over its non-restricted parent analog 1 and was selectively internalized by FRα over the reduced folate carrier (RFC), resulting in significant in vitro antitumor activity toward FRα–expressing KB human tumor cells. Antitumor activity of 7 was abolished by treating cells with adenosine but was incompletely protected by 5-aminoimidazole-4-carboxamide (AICA) at higher drug concentrations, suggesting GARFTase and AICA ribonucleotide formyltransferase (AICARFTase) in de novo purine biosynthesis as the likely intracellular targets. GARFTase inhibition by compound 7 was confirmed by an in situ cell-based activity assay. Our results identify a “first-in-class” classical antifolate with a novel amide linkage between the scaffold and the side chain aryl l-glutamate that affords exclusive selectivity for transport via FRα over RFC and antitumor activity resulting from inhibition of GARFTase and likely AICARFTase. Compound 7 offers significant advantages over clinically used inhibitors of this class that are transported by the ubiquitous RFC, resulting in dose-limiting toxicities.
Keywords: classical antifolates; folate receptor; selective uptake; pyrrolo[2,3-d]pyrimidines; amide bridge
Graphical Abstract

1. INTRODUCTION
Antifolates that inhibit one-carbon biosynthetic enzymes such as dihydrofolate reductase and thymidylate synthase are important agents for anticancer chemotherapy.1,2 Clinically useful antifolates include methotrexate (MTX), pralatrexate (PDX), raltitrexed (RTX) and pemetrexed (PMX) (Figure 1) with applications for both hematologic malignancies and solid tumors.2 Unfortunately, dose-limiting toxicities plague the clinical utility of these drugs and are due, at least in part, to non-specific drug accumulation resulting in inhibition of critical enzyme targets and downstream pathways in both tumors and normal tissues without selectivity.
Figure 1.

Clinically used antifolate drugs for cancer.
The principal (anti)folate transport system in tissues and tumors is the ubiquitously expressed reduced folate carrier (RFC).3 Notably, all the aforementioned antifolates (Figure 1) are excellent substrates for transport by RFC and accumulate in normal tissues, as well as in tumor cells, precluding selectivity.1,3 Uptake systems with greater tumor specificity than RFC have been described and include the proton-coupled folate transporter (PCFT)4–7 and folate receptor (FR) α.8,9 PCFT is a proton symporter that is expressed in a wide range of solid tumors, including ovarian cancer, non-small cell lung cancer, and malignant pleural mesothelioma.10–13 Moreover, PCFT is active at acidic pH characterizing the microenvironments of solid tumors.6,14 Although certain normal tissues such as the proximal small intestine, liver, and kidney also express PCFT, the tissue microenvironments of most normal tissues are unlikely to be sufficiently acidic to support PCFT transport,7 except the jejunum where the pH is acidic and conducive of folate transport.
FRα is expressed on the surface of epithelial ovarian, non-small cell lung, kidney, endometrial, colorectal and breast cancers where it comes into contact with the systemic circulation.8,9,15 FRα is also expressed in a subset of normal tissues including kidney, lung, choroid plexus and placenta. However, in contrast to tumors, in normal tissues, FRα localizes to the luminal membranes without exposure to the systemic circulation. 8,9,15. Thus, there is a compelling rationale for developing FR- and PCFT-based therapies for cancer, particularly for solid tumors.
Clinically tested FRα-targeted therapies include a FRα monoclonal antibody [Farletuzumab (Morphotech)16,17], a FRα-targeting antibody-drug conjugate [IMGN853 (ImmunoGen)18], and cytotoxic folic acid (FA) conjugates [Vintafolide, EC1456 and EC1788 (Endocyte)] 9,19. Although an FR-targeted antifolate N-[4-[2-propyn-1-yl[(6S)-4,6,7,8-tetrahydro-2-(hydroxymethyl)-4-oxo-3H-cyclopenta [g]quinazolin-6-yl] amino] benzoyl]-l-γ-glutamyl-D-glutamic acid (ONX0801) was described20,21 and advanced to a phase 1 clinical trial,22 no FRα-targeted antifolate has yet been FDA-approved for cancer. We discovered novel 6-substituted pyrrolo[2,3-d]pyrimidine benzoyl L-glutamate antifolates with 3- or 4-bridge carbons (CH2) (1 and 2, respectively) (Figure 2) as selective transport substrates for FRs and/or PCFT over RFC.23,24 Following internalization by tumor cells, these compounds inhibited glycinamide ribonucleotide (GAR) formyltransferase (GARFTase), the first folate-dependent step in de novo purine biosynthesis. Side-chain replacement of the phenyl moiety of 1 with thienyl regioisomers (3–5) (Figure 2) increased the inhibitory effects toward FRα-expressing cells and with PCFT-expressing cells, as well.25,26 N-heteroatom substitution in the bridge region as in 6 (Figure 2) increased FRα-targeting at the expense of RFC and PCFT uptake; however, 6 did not show absolute selectivity for FR or PCFT over RFC.27
Figure 2.

6-Substituted pyrrolo[2,3-d]pyrimidine antifolates 1–6.
Cytotoxic drugs generally bind to different cellular targets in distinct conformations. For anionic compounds, a requisite step in drug activity involves mediated uptake into tumor cells (ideally via a tumor-selective uptake process), whereupon they inhibit intracellular target enzymes. In principle, these compounds could adopt distinct conformations, including one that strongly favors mediated uptake into cells and another favoring binding to an intracellular enzyme target(s) that ultimately effects a cytotoxic response. We previously reported that the pyrrolo[2,3-d]pyrimidine antifolate 3 adopts different poses when bound to FRα versus when bound to its target enzyme GARFTase (Figure 3).27,28 As an extension of this concept, since amides assume distinct conformations as cis and trans rotamers with the lowest energy conformers,29 we reason that an analog with a unique amide linkage in the bridge region would afford a somewhat restricted three atom bridge, compared to a more flexible three carbon (CH2) bridge. For the former, this could derive significant entropic benefit and increased target selectivity by restricting the number of side-chain conformations in both unbound and bound states. Thus, analogs that do not need to significantly alter their conformations to adopt the bound conformation at their cellular target would likely bind more effectively to target proteins than analogs that exhibit different low energy conformations compared to their bound conformations.30
Figure 3.

Compound 3 and its two different poses in FRα and GARFTase, as well as newly designed compounds 7–12.
To test this concept and to begin developing new targeted inhibitors as selective cytotoxic agents for cancer, we designed a novel series of amide-bridge 6-substituted pyrrolo[2,3-d]pyrimidine antifolates with different energy barriers between the cis and trans conformers. It was of interest to determine the effect of inserting an amide in the bridge region on biological activity and transport selectivity for FRα and/or PCFT over RFC for three C-atom-bridge compounds such as 1. In this report, we describe the synthesis and biological activities of the unique amide-substituted pyrrolo[2,3-d]pyrimidine compounds 7–12 (Figure 3). In designing this series, we also introduced an N-methyl group on the amide nitrogen to augment the metabolic stability of the amide linkage from possible enzymatic hydrolysis to make these more generally useful.31 Our results establish these compounds as potent and highly selective FR-targeted inhibitors of de novo purine biosynthesis at GARFTase, and likely at 5-aminoimidazole-4-carboxamide (AICA) ribonucleotide formyltransferase (AICARFTase) (Figure 3).
2. RESULTS AND DISCUSSION
2.1. Rationale for compound design
It was of interest to determine if introducing an amide into the 3-atom bridge of 6-substituted pyrrolo[2,3-d]pyrimidine compounds (7–12) would decrease the number of low energy accessible conformations, compared to the corresponding 3-atom carbon chain analogs (1, 3–5). By molecular modeling in Maestro,32 we determined that the amide bridge analogs afforded a reduced number of conformations compared to their corresponding 3-carbon atom bridge analogs (Table 1). This suggests that amide group insertion into the side-chain as in 7–12 should provide a distinct entropic advantage that should favor binding to cellular targets.
Table 1. Conformational search results using Maestro 11.8.
Target compounds (blue) and lead compounds (black)
| Compound (Ar) | Number of conformations within 5 kcal/mol of minimum energy conformation |
|---|---|
| 1 (1,4-phenyl) | 219 |
| 7 (1,4-phenyl) | 143 
|
| 8 (1,3-phenyl) | 152
|
| 12 (1,4-phenyl) | 121
|
| 3 (2,5-thienyl) | 192 |
| 9 (2,5-thienyl) | 134
|
| 4 (2,4-thienyl) | 148 |
| 10 (2,4-thienyl) | 97
|
| 5 (3,5-thienyl) | 131 |
| 11 (3,5-thienyl) | 83
|
From our previous studies, the side-chain aromatic ring is an important determinant of drug potency and transport selectivity of 6-substituted pyrrolo[2,3-d]pyrimidine inhibitors.25,33,34 Thus, different regioisomers involving a side-chain phenyl ring and its replacement with thienyl regioisomers profoundly impacted (generally improved) compound potency. Accordingly, we designed amide-bridge analogs, with side-chain 1,4- and 1,3-disubstituted phenyl (7 and 8), and 2,5-, 2,4- and 3,5-di-substituted thienyl moieties (9–11) (Figure 3), based on the parent molecules (1, 3–5), in order to determine the optimal side-chains for selective cellular uptake by FRα vis á vis RFC and for inhibition of intracellular target enzymes (i.e. GARFTase) resulting in antitumor activity. Finally, we designed a sulfonamide bridge compound 12 (Figure 3) as a bioisosteric replacement of the amide in 7.32
To determine the rotational barrier around the amide (N(CH3)-C(O)) bond, we performed a coordinate scan35 of target compounds 7 - 12 around the bridge amide bond, and of the parent compounds (1, 3–5) around the C-C bond (Maestro 11.8). The resulting plot of relative energy (∆E, kcal/mol) versus the dihedral angle (°) is shown in Figure 4. For the amide compounds (7–12), a coordinate scan predicted two distinct minimum energy conformations with trans and cis conformations of the amide bond and a high energy barrier (~8–20 kcal/mol) between them. In contrast, a torsional scan of the carbon bridge analogs (1, 3–5) predicted three distinct minimum energy conformations with a low energy barrier among them (~5 kcal/mol). This demonstrates the conformational restriction associated with the amide bridge. Based on these results, we propose that reducing the number of low energy conformations (Table 1) and increasing the energy barrier between bridge amide rotamers (Figure 4) in the target compounds (7–12) could enhance drug activity, reflecting uptake via FRα and inhibition of intracellular enzymes such as GARFTase.
Figure 4.

Plots of rotational barrier (relative energy, kcal/mol) around the carbon-carbon bridge of compounds 1, 3, 4 and 5 (left) and the bridged amide bond of compounds 7–12 (right). Coordinate Scan panel of Maestro 11.8 was used to determine and plot the rotational energy barrier for both the amide bridge and lead compounds; the settings were as follows: Force Field, OPLS3e; Solvent, water; Coordinate to Scan, dihedral angles of amide bridge for target compounds and carbon bridge for lead compounds (the rest of the settings were kept at default).32
We docked the amide-bridge compounds (Figure 3) in the FRα (PDB: 5IZQ27) and GARFTase (PDB: 4ZZ134) crystal structures. Figures 5 and 6 show the docked poses of compound 7 in FRα and GARFTase, respectively. The amide bridge in 7 adopts both cis and trans conformations within 1 kcal/mol (docking score) for both the FRα and GARFTase structures. The major binding interactions of 7 with the target proteins are similar to those for the co-crystallized ligands 6 (FRα)27 and 5 (GARFTase)34. The oxygen atom of the bridge amide group of 7 in both rotamers can form an ion-dipole interaction (distance: 2.78 Å and 2.67 Å, Figure 6) with the terminal amino group of the substrate GAR in the GARFTase binding site. Compound 7 shows better docking scores (FRα = −14.77 kcal/mol and GARFTase = −15.23 kcal/mol) than compound 1 (FRα = −13.33 kcal/mol and GARFTase = −14.43 kcal/mol), suggesting its more avid binding to these target proteins and predicting greater potency. The docked poses of 8 (FRα = −14.27 kcal/mol and GARFTase = −14.72 kcal/mol) and 9 (FRα = −15.23 kcal/mol and GARFTase = −14.75 kcal/mol) mimic those for 7 in FRα and GARFTase.
Figure 5.

Two alternate docked poses of 7 in FRα (PDB ID: 5IZQ). A) Cyan: compound 7, and pink: 6 (co-crystallized ligand). The amide linker in 7 adopted a trans conformation. Docking score: −14.77 kcal/mol. B) The amide linker in 7 adopted a cis conformation. Docking score: −13.89 kcal/mol. Maestro 11.8.
Figure 6.

Two alternate docked poses of 7 in GARFTase (PDB ID: 4ZZ1). A) Purple, compound 7, gray 5 (co-crystallized ligand), and red, GAR (endogenous substrate). The amide linker in 7 adopted a trans conformation. Docking score: −15.23 kcal/mol. B) Purple, compound 7. The amide linker in 7 adopted a cis conformation. Docking score: −14.79 kcal/mol. Maestro 11.8.
Compounds 10 (side-chain 2,4-thienyl) and 11 (side-chain 3,5-thienyl) were designed to vary the distance between the amide bridge and the glutamyl side-chain, and hence their conformations in comparison to the 2,5-thienyl side-chain analog 9.34 These changes introduce additional conformational restrictions and further reduce the number of possible low energy conformations even further (to 97 for 10 and 83 for 11, compared to the 134 possible conformations for the 2,5-thienyl side-chain analog 9; Table 1). Compound 12 is designed to explore the bioisosteric replacement of the amide group in compound 7. The sulfonamide moiety in compound 12 further reduces the number of possible low energy conformations compared to 7 (Table 1). Compounds 10 (FRα = −12.71 kcal/mol and GARFTase = −13.31 kcal/mol), 11 (FRα = −12.75 kcal/mol and GARFTase = −12.87 kcal/mol) and 12 (FRα = −13.42 kcal/mol and GARFTase = −13.53 kcal/mol) show comparable docking scores to compounds 1 (FRα = −13.33 kcal/mol and GARFTase = −14.43 kcal/mol) and 3 (FRα = −13.67 kcal/mol and GARFTase = −14.12 kcal/mol) for both FRα and GARFTase. Moderate docking scores of the compounds 10, 11 and 12 suggest that despite the conformational restriction, the nature of the aromatic side-chain and the bridge linker may dictate the binding affinity for the proposed compounds.
Based on our in silico studies, we predicted that compounds 7–12 with the amide group in the bridge should exhibit significant cellular uptake by FRα, resulting in GARFTase inhibition and antitumor efficacy. On the basis of the better docking scores in FRα and GARFTase, 7, 8 and 9 are predicted to be more potent inhibitors of tumor growth compared to 10–12 and the lead compounds 1–6.
3. Chemistry
There are two synthetic methods available for 6-substituted classical antifolates 15 (Scheme 1).34,36 The first method uses a Sonogashira coupling to install an alkyne linker on 6-halogenated pyrrolo[2,3-d]pyrimidines 13 and provides key intermediates 14. This approach is not suitable for heteroatom side chain compounds.36 The second method is to cyclize an α-bromoketone 17 with 2,6-diamino-4-hydroxy pyrimidine 18, which affords the pteroic ester precursors 19. In this approach, diazomethane is an indispensable, yet dangerous, starting material.37 Hence, a third synthetic method was devised for our proposed amide compounds to avoid the diazomethane reaction.
Scheme 1.

Two current synthesis methods for antifolate 15.
Our synthesis for the 6-aminomethyl pyrrolo[2,3-d]pyrimidines is shown in Scheme 2. Pivaloylation of the 2-amino moiety of the pyrrolo[2,3-d]pyrimidine 20 provided 21 (95% yield). The purpose of this step was to decrease the polarity and increase the solubility of 20. The product 21 was directly precipitated from the reaction mixture by adding a mixture of hexane and ethyl acetate (5:1). Subsequently, a Mannich reaction with 21 afforded 22 in 78% yield. Finally, Pd-catalyzed debenzylation of 22 provided 23 in 90% yield.
Scheme 2.

Synthesis of N-monosubstituted 6-amiomethyl pyrrolo[2,3-d]pyrimidines 23.
Since the Mannich reaction38–41 could afford 5- and/or 6- position substituted regioisomers of the pyrrolo[2,3-d]pyrimidine, we analyzed the regioselectivity of the reaction from 21 to 22 with compound 23 using NOESY42 and HMBC (Figure 7). A weak NOE signal of N7-H and 6’-H was observed. The weak NOE signal is explained by the quadrupole N7-H. In HMBC (Figure 7), the signal of 3J(C4-H5), as well as 3J(C5-H7 and C9-H3), confirmed the structure as 23 with the aminomethyl substitution at the 6-position. This demonstrated that under our Mannich reaction conditions, the 6-substituted regioisomer was the predominant product. In addition, our synthesis provided a secondary amine in 23, which was amenable to functionalization.
Figure 7.

HMBC spectrum of 23. a) HMBC spectrum and interpretation. The diagnostic 3JCH couplings are circled in red, and 2JCH couplings are circled in green; b) Highlighted diagnostic HMBC signals. 3J CH signals are highlighted with red arrows and 2J CH signals are highlighted with green arrows.
Amide or sulfonamide coupling of 23 (Scheme 3) with the corresponding acid 25a, 26b-c, 27a-b or methyl 4-(chlorosulfonyl)benzoate 27c afforded 24a-f in 69–82% yield. Compounds 26b-c were, in turn, synthesized by periodic acid oxidation of the corresponding aldehydes 25b-c in 56–63% yields.
Scheme 3.

Synthesis of 24a-f.
Depivaloylation and hydrolysis of the methyl esters in 24a-f (Scheme 4) with sodium carbonate afforded pteroic acids 28a-f in 78–91% yields. CDMT auxiliary amide coupling of 28a-f with dimethyl l-glutamate afforded 29a-f. Sodium carbonate hydrolysis of 29a-f provided the target classical antifolates 7–12 in 56–74% yields over two steps.
Scheme 4.

Synthesis of target compounds 7–12.
The 1H NMR of 7 (Figure 8) exhibited two sets of peaks of the NCH3, NCH2 and 5-H, for a mixture of the cis and trans rotamers, with all other protons at the same chemical shift. In each rotamer, the NCH3, NCH2 and 5-H have an integration ratio of 3 to 2 to 1, which was used to assign peaks to the two different sets. The chemical shifts of protons for the cis conformation of the amide oxo moiety are larger than those of the trans conformation.43 As a result, the red set (Figure 8) are assigned to the cis while the blue set are the peaks for the trans. This NMR result corroborates the two lowest energy rotamers of 7, as predicted by molecular modeling.
Figure 8.

1H NMR (DMSO-d6) of 7 at 400 MHz, the proton of NCH3, NCH2 and 5-H in the cis and trans rotamers are illustrated as red and blue, respectively. Red: δ 2.81 (1.82H, cis NCH3), 4.58 (1.15H, cis NCH2), 6.17 (0.53H, cis 5-H); Blue: δ 2.94 (1.3H, trans NCH3), 4.58 (0.9H, trans NCH2), 6.17 (0.48H, trans 5-H).
4. Biological evaluation
The bridge amide-substituted pyrrolo[2,3-d]pyrimidine compounds 7–12 were initially tested in cell proliferation assays with a unique panel of isogenic Chinese hamster ovary (CHO) cell lines engineered to individually express human RFC (PC43–10), PCFT (R2/PCFT4), or FRα (RT16).23,24,44 The results with the amide series were compared to previous 6-substituted pyrrolo[2,3-d]pyrimidine analogs with an alkyl bridge linked to side-chain phenyl (1)23 or thienyl (3–5)25,34 rings, as appropriate, and to standard antifolates (e.g., MTX) without transporter selectivity. The negative control for these experiments was the RFC-, FR-, and PCFT-null MTXRIIOuaR2–4 (R2) CHO cell line.45 For the FRα-expressing CHO cells, additional control involved treatment with excess folic acid (200 nM) to selectively block cellular uptake by FR (not shown). Proliferation experiments were also performed with KB human tumor cells that express highly elevated FRα, along with RFC and PCFT. These results are summarized in Table 2.
Table 2.
Structures of 1–12 and the IC50 values for inhibition of proliferation of transporter null (R2), FRα (RT16), PCFT (R2/PCFT4), and RFC (PC43–10)-expressing CHO cells, and KB human tumor cells (expresses FRα, RFC, and PCFT) in culture. Results are expressed as mean values (± standard errors) from 3–10 experiments. Abbreviations: MTX, methotrexate; PMX, pemetrexed; RTX, raltitrexed.
 | |||||||
|---|---|---|---|---|---|---|---|
| Compound | X | IC50 (nM) | Reference | ||||
| PC43–10 (RFC) | R2 | RT16 (FRα) | R2/PCFT4 (PCFT) | KB (FRα/RFC/PCFT) | |||
| 1* | - | 304 (89) | 448 (78) | 4.1(1.6) | 23.0(3.3) | 1.7(0.4) | 46 |
| 7 |  |
>1000 | >1000 | 1.4(0.24) | 48.60(16.40) | 1.40(0.24) | - |
| 8 |  |
>1000 | >1000 | 1.78 (0.17) | 747 (156) | 6.36 (1.00) | - |
| 12 |  |
>1000 | >1000 | >1000 | >1000 | 669 (112) | - |
| 3 | - | 101.0 (16.6) | 273.0(49.1) | 0.31 (0.14) | 3.34 (0.26) | 0.26 (0.03) | 25 |
| 9 |  |
>1000 | >1000 | 7.78(1.63) | >1000 | 4.23(0.57) | - |
| 4* | - | 189(51) | 290((8) | 0.61(0.11) | 6.51(1.30) | 0.09(0.02) | 34 |
| 10 | 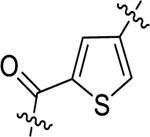 |
>1000 | >1000 | >1000 | >1000 | >1000 | - |
| 5* | - | 197(49) | 355(10) | 0.33(0.15) | 5.39(1.27) | 0.17(0.05) | 34 |
| 11 |  |
>1000 | >1000 | 867(147) | >1000 | 69.8(16.1) | - |
| MTX* | - | 12(1.1) | 216(8.7) | 114(31) | 121(17) | 6.0(0.6) | - |
| PMX* | - | 138(13) | 894(93) | 42(9) | 13.292.4) | 68(12) | - |
| RTX* | - | 6.3(1.3) | >1000 | 15(5) | 99.5(11.4) | 5.9(2.2) | - |
These data were previously published (see reference).
As previously reported, the non-amide pyrrolo[2,3-d]pyrimidine compounds (1, 3–5) were potent inhibitors of CHO cells expressing FRα or PCFT and are some of the most active analogs identified for this structural platform;4,23,25,34 however, this activity was not selective for FRα and/or PCFT as these compounds were taken up by a non-specific (i.e., non-mediated) process that results in equivalent growth inhibition toward RFC-expressing (PC43–10) and transporter-null (R2) cells23,25,34 (Table 2). Notably, the amide-substituted compounds, 7, 8 and 9 were inert toward RFC-expressing PC43–10 cells. Compounds 7, 8 and 9 all preserved FRα activities, reflected in growth inhibition of RT16 and KB tumor cells (Table 2). Thus, compounds 7, 8 and 9 were completely selective for FRα over RFC. For 7, inhibition of RT16 cells (FRα) significantly exceeded (~3-fold) that for the corresponding parent analog (1) (p<0.05), consistent with the notion that the presence of the amide bridge favors a conformation optimal for FR binding and internalization. While regioisomer 8 (meta substitution on the side chain phenyl ring) was equally potent as 7 toward FRα-expressing RT16 cells, 8 (meta-substituted on the side-chain phenyl) was ~4-fold less active than 7 (para substituted on the side chain phenyl ring) toward KB tumor cells (Table 2). Although these results may reflect factors unrelated to FRα internalization, it nonetheless suggests that that decreased distance between the scaffold and the l-glutamate moiety as in 8 compared to 7 is detrimental to antitumor activity, as previously described.27 Isosteric replacement of the phenyl side chain in 7 with a thiophene ring resulted in modestly decreased growth inhibition (for 9), or a substantial reduction (11) or complete loss (10) of FRα-targeted inhibition of RT16 and/or KB cells. The sulfonamide analog 12 was inactive regardless of the expressed transporter (Table 2).
While compounds 7–12 were all designed to inhibit GARFTase in de novo purine biosynthesis following internalization by FRα, this was directly tested for the compounds 7 and 9 with side-chain phenyl and 2,5-thiophene moieties analogous to parent compounds 1 and 3. For these experiments, we determined the extent of growth inhibition of KB tumor cells by these compounds in the presence of thymidine (10 µM) or adenosine (60 µM).25,27,33,34,36 We also tested the protective effects of glycine (130 μM) to explore the potential inhibition of mitochondrial C1 metabolism.47 Results for the nucleoside/glycine protection experiments are shown in Figure 9A. Adenosine completely reversed the drug effects of all the compounds, whereas thymidine and glycine were completely ineffective. Thus, neither thymidylate synthase nor mitochondrial C1 metabolism are involved in the mechanism of action of 7 or 9. The metabolite AICA (320 µM) is metabolized to AICA ribonucleotide (ZMP), the AICARFTase substrate which circumvents the GARFTase step. 27,28,33,34,36,44 AICA completely reversed the inhibitory effects of compounds 1 and 3 up to 1 μM; however, at higher concentrations of 3, AICA was less protective. By analogy with published results for related compounds,27,28,33,34,36,44,46 our results suggest that GARFTase is the primary intracellular target for this series, although at higher inhibitor concentrations, targeting of AICARFTase also contributes.
Figure 9.

Identification of the intracellular target by protection by nucleosides, glycine and AICA, and in situ GARFTase assays. A-D) KB cells were incubated with drugs in folate-, nucleoside-, and glycine-free RPMI 1640 medium with 10% dialyzed FBS, antibiotics, l-glutamine, and 2 nM leucovorin with a range of drug concentrations in the presence of folic acid (200 nM), adenosine (60 μM), thymidine (10 μM), glycine (130 μM) or AICA (320 μM). Cell proliferation was assayed with Cell Titer Blue (Promega) using a fluorescence plate reader. Data are representative of at least triplicate experiments. Error bars represent the standard errors. E) Inhibition of GARFTase activity in KB cells by the 6-substituted pyrrolo[2,3-d]pyrimidine compounds including compounds 1, 3, 7 and 9. KB cells were treated with the drugs under conditions approximating those for the cell outgrowth experiments (Table 2). Incorporation of [14C(U)]glycine into [14C]formyl GAR was used as an in situ measure of endogenous GARFTase activity. Experimental details are summarized in the Experimental Section. IC50 values (nM) were calculated as mean values +/− standard errors for triplicate experiments.
To further confirm GARFTase as the principal cellular target for our most potent analogs (compounds 7 and 9), GARFTase was directly assayed in situ in KB cells. For these experiments, KB cells were incubated with a range of concentrations of compounds 7 or 9, or of compounds 1 and 3 as controls, under conditions closely approximating those for the cell proliferation assays (Table 1). Cells were incubated with [14C]glycine, which is incorporated into [14C]GAR and [14C]FGAR (the GARFTase product). The latter accumulates in the presence of azaserine (inhibits FGAR amidotransferase) and is isolated by anion exchange fractionation for quantitation and calculation of IC50 values for GARFTase inhibition. All compounds inhibited GARFTase with IC50 values approximately one-order of magnitude higher than that for inhibition of KB proliferation. Collectively, these results establish that de novo purine biosynthesis is the principal targeted pathway by the active amide-substituted pyrrolo[2,3-d]pyrimidine compounds 7 and 9 at GARFTase, and likely AICARFTase, as well.
5. Conclusion
In this study, we used molecular modeling and docking studies to rationally design a series of amide-bridged pyrrolo[2,3-d]pyrimidine antifolates with cis and trans conformations as the two lowest energy conformations. These conformations were supported by 1H NMR analysis. We previously described carbon-bridge analogs of this series (1 – 6) that are not selective for FR and PCFT over other uptake mechanisms including RFC23,25,36 that allows the accumulation of the analogs in normal tissues and precludes tumor selectivity. Based on proliferation studies in isogenic CHO cell lines exclusively expressing human FRα, PCFT or RFC, we determined that the active amide compounds 7–9 are transported by FRα with or without PCFT, but not by RFC, and thus should possess absolute tumor selectivity. This indicates that the amide bridge is highly conducive to tumor transport selectivity for 7–9. Based on the inhibitory results toward KB human tumor cells, compounds 7 and 9 were tested to identify the likely targeted pathway and enzyme(s) in comparison to compounds 1 and 3, previously identified as exclusive inhibitors of GARFTase in de novo purine nucleotide biosynthesis.23,25 Whereas compounds 7 and 9 were confirmed as GARFTase inhibitors by nucleoside/AICA protection, a secondary target, most likely AICARFTase, was also implicated.
Our original hypothesis was that conformational restriction caused by the amide bridge in the analogs 7–12 would favor targeting FRα and possibly GARFTase by our analogs. Consistent with our hypothesis, compound 7 was ~3-fold more potent than 1 toward CHO cells exclusively expressing FRα, although 7 and 1 are equipotent toward KB cells that express RFC, FRα and PCFT. This suggests that the conformational restriction in 7 promotes FRα–targeting of our analogs, although at least for this analog, uptake by PCFT is preserved, as well, likely contributing to drug effects in KB cells. In the absence of crystal structures for RFC and PCFT, the information from this study can guide the design of conformationally restricted compounds that are exclusively transported by FRα-expressing cancer cells to afford highly selective, non-toxic cancer chemotherapeutic agents.
6. Experimental Section
Analytical samples were dried in vacuum (0.2 mmHg) in a CHEM-DRY drying apparatus over P2O5 at 50 °C. Melting points were determined on a digital MEL-TEMP II melting point apparatus with FLUKE 51K/J electronic thermometer and are uncorrected. Nuclear magnetic resonance spectra for protons (1H NMR, 13C NMR and HMBC) were recorded on a Bruker Avance II 400 (400 MHz) or on a 500 (500 MHz) NMR systems. The chemical shift values are expressed in ppm (parts per million) relative to tetramethylsilane as an internal standard: s, singlet; d, doublet; t, triplet; q, quartet; quint, quintet; m, multiplet; br, broad singlet. Thin-layer chromatography (TLC) was performed on Whatman Sil G/UV254 silica gel plates with a fluorescent indicator, and the spots were visualized under 254 and 365 nm illumination. Proportions of solvents used for TLC are by volume. Column chromatography was performed on a 230−400 mesh silica gel (Fisher Scientific) column. Elemental analyses were performed by Atlantic Microlab, Inc., Norcross, GA. Elemental compositions are within ±0.4% of the calculated values and indicate > 95% purity of the compounds. Fractional moles of water or organic solvents found in some analytical samples could not be prevented despite 24 h of drying in vacuum and were confirmed where possible by their presence in the 1H NMR spectra. UPLC-MS was analyzed on Acquity system. A linear gradient of 90% of 0.1% formic acid in water, 10% of 0.1% formic acid in acetonitrile (ACN) over 10 min, and then 100% ACN was used for 5 min. All final compounds were > 95% purity established by CHN, or UPLC-MS, or both CHN and UPLC-MS. All solvents and chemicals were purchased from Sigma-Aldrich Co. or Fisher Scientific Inc. and were used as received.
2-Pivalamido-4-oxo-4,7-dihydro-1H-pyrrolo[2,3-d]pyrimidine (21)
2-Amino-4-oxo-4,7-dihydro-1H-pyrrolo[2,3-d]pyrimidine (20) (3.0 g, 20 mmol) was added to pivalic anhydride (12 mL). The resulting suspension was stirring at 90 °C for 3 h. The reaction was cooled to rt and a mixture of hexane (40 mL) and EtOAc (8 mL) was added dropwise. The suspension was filtered, washed with acetone (10 mL), and dried at 50 °C under vacuum overnight to afford 4.44 g of pale yellow solid 21 in 95% yield. mp > 250 °C (lit48 295 °C); TLC Rf 0.55 (CHCl3: MeOH, 20:1); 1H NMR (DMSO-d6) δ 1.28 (s, 9H, Piv), 6.41 (d, J = 4.2 Hz, 1H, 5-H), 6.95 (d, J = 4.2 Hz, 1H, 6-H), 10.73 (br, 1H, exch, PivNH), 11.55 (br, 1H, exch, NH), 11.83 (br, 1H, exch, NH).
2-Pivalamido-4-oxo-6-(N-methyl-benzylaminomethyl)-4,7-dihydro-3H-pyrrolo[2,3-d]pyrimidine (22)
N-Methyl benzylamine (3.63 g, 30 mmol) and paraformaldehyde (1.05 g, 35 mmol) were added to water (20 mL). To this solution, acetic acid (20 mL) was added dropwise. The mixture was stirred at 80 °C for 3 h. Compound 21 (2.34 g, 10 mmol) and KOAc (2.94 g, 30 mmol) were added and the stirring was continued at 80 °C overnight. The solvent was removed by a rotary evaporator and acetone (40 mL) was added. The suspension was filtered and the filtrate was concentrated into a semisolid residue. This residue was purified by a silica column, which was flushed with CHCl3 and MeOH, to provide 2.86 g of a white solid, 22, in 78% yield. mp: 181.1 −182.5 °C; TLC Rf 0.37 (CHCl3: MeOH, 20:1); 1H NMR (DMSO-d6) δ 1.24 (s, 9H, Piv), 2.09 (s, 3H, NCH3), 3.48 (s, 2H, NCH2), 3.58 (s, 2H, NCH2), 6.31 (s, 1H, 5-H), 7.32 (m, 5H, Ph), 10.85 (br, 1H, exch, PivNH), 11.55 (br, 1H, exch, NH), 11.95 (br, 1H, exch, NH); Anal. calcd. for (C20H25N5O2·0.5H2O) C, H, N; UPLC-MS > 95%; m/z [M-MePhNH]+, calcd 247.12, found 247.25.
2-Pivalamido-4-oxo-6-(methylaminomethyl)-4,7-dihydro-3H-pyrrolo[2,3-d]pyrimidine hydrochloric salt (23)
Compound 22 (1.84 g, 5 mmol) was dissolved in methanol (40 mL) and chloroform (2 mL) in a Parr hydrogenation flask. Pd/C (10%, 200 mg) was added to the above solution under Ar atmosphere and placed on the Parr hydrogenation shaker under 50 psi hydrogen for 2 h. The catalyst was filtered through celite, and the solvent was evaporated. To the residue was added acetone (5 mL) and the suspension was filtered, and dried to afford 1.41 g of 23 as a white solid in 90% yield. mp > 250 °C; TLC Rf 0.13 (CHCl3: MeOH, 10:1); 1H NMR (DMSO-d6) δ 1.24 (s, 9H, Piv), 3.07 (s, 3H, NCH3), 4.17 (s, 2H, NCH2), 6.58 (s, 1H, 5-H), 9.32 (br, 2H, exch, NH2+), 10.95 (br, 1H, exch, PivNH), 11.86 (br, 2H, exch, NH); 13C NMR (DMSO-d6) δ 31.99, 44.48, 104.34, 105.09, 127.39, 148.67, 148.73, 157.11, 181.37; UPLC-MS > 95%; m/z [M-MeNH]–, calcd 247.12, found 247.18.
General procedures for the synthesis of methoxycarbonyl thiophene carboxylic acids 26b-c
To methoxycarbonyl thiophene carboxylaldehyde 25b-c in acetonitrile solution, periodic acid (2 eq.) was added and the reaction was stirred at rt for 1 h. To the above solution, water (10 mL) was added and mixture was extracted with EtOAc (3 X 10 mL). The organic layers were combined, dried, concentrated, and purified by a silica column with CHCl3 and MeOH, to provide a white solid 26b-c.
2-Methoxycarbonyl-thiophene-4-carboxylic acid (26b)
Yield: 59%. mp 136.9 – 137.8 °C; TLC Rf 0.22 (CHCl3: MeOH, 20:1); 1H NMR (CDCl3) δ 3.93 (s, 3H, OCH3), 8.28 (d, J = 1 Hz, 1H, ArH), 8.37 (d, J = 1 Hz, 1H, ArH); Anal. calcd. for (C7H6O4S·0.15H2O) C, H, S.
3-Methoxycarbonyl-thiophene-5-carboxylic acid (26c)
Yield: 63%. mp 189.1 – 189.5 °C; TLC Rf 0.22 (CHCl3: MeOH, 20:1); 1H NMR (CDCl3) δ 3.95 (s, 3H, OCH3), 8.24 (s, 1H, ArH), 8.40 (s, 1H, ArH); Anal. calcd. for (C7H6O4S) C, H, S.
Methyl 4-{[N-methyl-(2-pivalamido-4-oxo-4,7-dihydro-3H-pyrrolo[2,3-d]pyrimidin-6-yl)-methylamino]-carbonyl}-phenylcarboxylate (24a)
To a solution of 4-methoxycarbonyl phenyl carboxylic acid (180 mg, 1 mmol) in DMF (1 mL) was added N-methyl morpholine (310 mg, 3 mmol), EDCI (240 mg, 1.2 mmol) and HOAt (183 mg, 1.05 mmol). The mixture was stirred at rt for 1 h. Compound 23 (313 mg, 1 mmol) was added and stirring was continued overnight. The solvent was removed and the residue was purified by a silica column with CHCl3 and MeOH to provide 316 mg of 24a as a semi-solid in 72% yield. TLC Rf 0.16 (CHCl3/MeOH, 40:1); 1H NMR (DMSO-d6) δ 1.25 (s, 9H, Piv), 2.84 (s, 1.8H, cis NCH3), 2.99 (s, 1.2H, trans NCH3), 3.88 (s, 3H, OCH3), 4.44 (s, 0.8H, trans NCH2), 4.70 (s, 1.2H, cis NCH2), 6.24 (s, 0.4H, trans 5-H), 6.38 (s, 0.6H, cis 5-H), 7.60 (d, J = 6.8 Hz, 2H, Ph), 8.04 (d, J = 6.8 Hz, 2H, Ph), 10.79 (br, 1H, exch, PivNH), 11.62 (br, 1H, exch, NH), 11.87 (s, 1H, exch, NH).
Methyl 3-{[N-methyl-(2-pivalamido-4-oxo-4,7-dihydro-3H-pyrrolo[2,3-d]pyrimidin-6-yl)-methylamino]-carbonyl}-phenylcarboxylate (24b)
Compound 24b was synthesized following the procedure of 24a and was obtained as a semi-solid in 69% yield. TLC Rf 0.15 (CHCl3/MeOH, 40:1); 1H NMR (DMSO-d6) δ 1.25 (s, 9H, Piv), 2.73 (s, 1.2H, cis NCH3), 2.85 (s, 1.8H, trans NCH3), 3.98 (s, 3H, OCH3), 4.43 (s, 0.8H, trans NCH2), 4.69 (s, 1.2H, cis NCH2), 6.27 (s, 0.4H, trans 5-H), 6.39 (s, 0.6H, cis 5-H), 7.62 (m, 1H, Ph), 7.75 (m, 1H, Ph), 8.03 (m, 2H, Ph), 10.79 (s, 1H, exch, PivNH), 11.63 (s, 1H, exch, NH), 11.89 (s, 1H, exch, NH).
Methyl 5-{[N-methyl-(2-pivalamido-4-oxo-4,7-dihydro-3H-pyrrolo[2,3-d]pyrimidin-6-yl)-methylamino]-carbonyl}-thiophene-2-carboxylate (24c)
Compound 24c was synthesized following the procedure of 24a from 25a and was obtained as a semi-solid in 82% yield. TLC Rf 0.16 (CHCl3/MeOH, 40:1); 1H NMR (DMSO-d6) δ 1.24 (s, 9H, Piv), 3.05 (br, 3H, cis and trans NCH3), 3.85 (s, 3H, OCH3), 4.69 (s, 2H, cis and trans NCH2), 6.35 (s, 1H, cis and trans 5-H), 7.54 (s, 1H, thiophene), 7.78 (s, 1H, thiophene), 10.80 (s, 1H, exch, PivNH), 11.62 (s, 1H, exch, NH), 11.88 (s, 1H, exch, NH).
Methyl 4-{[N-methyl-(2-pivalamido-4-oxo-4,7-dihydro-3H-pyrrolo[2,3-d]pyrimidin-6-yl)-methylamino]-carbonyl}-thiophene-2-carboxylate (24d)
Compound 24d was synthesized following the procedure of 24a from 26b and was obtained as a semi-solid in 76% yield. TLC Rf 0.17 (CHCl3/MeOH, 40:1); 1H NMR (DMSO-d6) δ 1.25 (s, 9H, Piv), 2.89 (s, 1.5H, cis NCH3), 2.89 (s, 1.5H, trans NCH3), 3.80 (s, 3H, OCH3), 4.69 (s, 2H, cis and trans NCH2), 6.33 (s, 1H, cis and trans 5-H), 7.78 (s, 1H, thiophene), 8.54 (s, 1H, thiophene), 10.79 (s, 1H, exch, PivNH), 11.64 (s, 1H, exch, NH), 11.89 (br, 1H, exch, NH).
Methyl 5-{[N-methyl-(2-Pivalamido-4-oxo-4,7-dihydro-3H-pyrrolo[2,3-d]pyrimidin-6-yl)-methylamino]-carbonyl}-thiophene-3-carboxylate (24e)
Compound 24e was synthesized following the procedure of 24a from 26c and was obtained as a semi-solid in 79% yield. TLC Rf 0.16 (CHCl3/MeOH, 40:1); 1H NMR (DMSO-d6) δ 1.25 (s, 9H, Piv), 2.98 (s, 3H, cis and trans NCH3), 3.92 (s, 3H, OCH3), 4.65 (br, 2H, cis and trans NCH2), 6.37 (br, 1H, cis and trans 5-H), 7.91 (d, J = 2.8 Hz, 1H, thiophene), 8.34 (d, J = 3.0 Hz, 1H, thiophene), 10.78 (s, 1H, exch, PivNH), 11.55 (s, 1H, exch, NH), 11.86 (s, 1H, exch, NH).
Methyl 4-{[N-methyl-(2-pivalamido-4-oxo-4,7-dihydro-3H-pyrrolo[2,3-d]pyrimidin-6-yl)-methylamino]-sulfonyl}-phenylcarboxylate (24f)
To a solution of 23 (313 mg, 1 mmol) in DMF (1 mL), was added triethylamine (210 mg, 2.05 mmol), methyl 4-chlorosulfonyl benzoate 27c (235 mg, 1 mmol) and 4-dimethylaminopyridine (10 mg). The mixture was stirred at rt for 4 h. The solvent was removed and the residue was purified by a silica column with CHCl3 and MeOH to afford 356 mg of 24f as a white solid in 75% yield. TLC Rf 0.13 (CHCl3/MeOH, 40:1); 1H NMR (DMSO-d6) δ 1.25 (s, 9H, Piv), 2.61 (s, 3H, NCH3), 3.91 (s, 3H, OCH3), 4.21 (s, 2H, NCH2), 6.28 (s, 1H, 5-H), 7.95 (d, J = 8.0 Hz, 2H, Ph), 8.18 (d, J = 8.0 Hz, 2H, Ph), 10.85 (s, 1H, exch, PivNH), 11.66 (s, 1H, exch, NH), 11.88 (s, 1H, exch, NH).
4-{[N-Methyl-(2-amino-4-oxo-4,7-dihydro-3H-pyrrolo[2,3-d]pyrimidin-6-yl)-methylamino]-carbonyl}-phenylcarboxylic acid (28a)
To a solution of 24a (220 mg, 0.5 mmol) in water (10 mL) and methanol (10 mL) was added Na2CO3 (0.5 g). The mixture was stirred at 80 °C for 2h. The methanol was removed and the pH of the solution was adjusted to 3 with 1N HCl. The suspension was filtered and the filter cake was washed with deionized water and acetone to facilitate drying, and dried to afford 152 mg white solid 28a in 90% yield. mp > 250 °C; TLC Rf 0.53 (CHCl3: MeOH: AcOH, 10:1:0.5); 1H NMR (DMSO-d6) δ 2.78 (s, 1.8H, cis NCH3), 2.93 (s, 1.2H, trans NCH3), 4.30 (s, 0.8H, trans NCH2), 4.58 (s, 1.2H, cis NCH2), 6.01 (s, 0.4H, trans 5-H), 6.07 (s, 2H, exch, 2-NH2), 6.16 (s, 0.6H, cis 5-H), 7.58 (d, J = 6.8 Hz, 2H, Ph), 8.01 (d, J = 6.8 Hz, 2H), 10.25 (s, 1H, exch), 10.33 (s, 1H, exch), 13.14 (s, 1H, exch); Anal. calcd. for (C16H15N5O4 · 1.40H2O) C, H, N; UPLC-MS > 95%; m/z [M+H]+, calcd 342.11, found 342.25.
3-{[N-methyl-(2-amino-4-oxo-4,7-dihydro-3H-pyrrolo[2,3-d]pyrimidin-6-yl)-methylamino]-carbonyl}-phenylcarboxylic acid (28b)
Compound 28b was synthesized in 83% yield following the procedure of 28a. mp > 250 °C; TLC Rf 0.51 (CHCl3: MeOH: AcOH, 10:1:0.5); 1H NMR (DMSO-d6) δ 2.79 (s, 1.8H, cis NCH3), 2.92 (s, 1.2H, trans NCH3), 4.32 (s, 0.8H, trans NCH2), 4.58 (s, 1.2H, cis NCH2), 6.02 (s, 0.4H, trans 5-H), 6.08 (s, 2H, exch, 2-NH2), 6.16 (s, 0.6H, cis 5-H), 7.58 (m, 1H, Ph), 7.72 (m, 1H, Ph), 8.18 (m, 2H, Ph), 10.26 (s, 1H, exch, NH), 11.06 (s, 1H, exch, NH), 13.20 (s, 1H, exch, COOH); Anal. calcd. for (C16H15N5O4 · 0.4Acetone·1.8H2O) C, H, N; UPLC-MS > 95%; m/z [M+H]+, calcd 342.11, found 342.28.
5-{[N-methyl-(2-amino-4-oxo-4,7-dihydro-3H-pyrrolo[2,3-d]pyrimidin-6-yl)-methylamino]-carbonyl}-thiophene-2-carboxylic acid (28c)
Compound 28c was synthesized in 82% yield following the procedure of 28a. mp > 250 °C; TLC Rf 0.47 (CHCl3: MeOH: AcOH, 10:1:0.5); 1H NMR (DMSO-d6) δ 2.95 (br, 3H, cis and trans NCH3), 4.58 (s, 2H, cis and trans NCH2), 6.07 (s, 2H, exch, 2-NH2), 6.12 (s, 1H, cis and trans 5-H), 7.52 (s, 1H, thiophene), 7.67 (s, 1H, thiophene), 10.34 (s, 1H, exch, NH), 11.08 (s, 1H, exch, NH), 13.20 (s, 1H, exch, COOH); Anal. calcd. for (C14H13N5O4S · 0.1Acetone·0.95H2O) C, H, N, S; UPLC-MS > 95%; m/z [M+H]+, calcd 348.07, found 348.20.
4-{[N-methyl-(2-amino-4-oxo-4,7-dihydro-3H-pyrrolo[2,3-d]pyrimidin-6-yl)-methylamino]-carbonyl}-thiophene-2-carboxylic acid (28d)
Compound 28d was synthesized in 90% yield following the procedure of 28a. mp > 250 °C; TLC Rf 0.50 (CHCl3: MeOH: AcOH, 10:1:0.5); 1H NMR (DMSO-d6) δ 3.12 (br, 3H, cis and trans NCH3), 4.59 (s, 2H, cis and trans NCH2), 6.12 (br, 3H, 2-NH2(exch), cis and trans 5-H), 7.71 (s, 1H, thiophene), 8.43 (s, 1H, thiophene), 10.28 (s, 1H, exch, NH), 11.10 (s, 1H, exch, NH), 12.95 (s, 1H, exch, COOH); Anal. calcd. For (C14H13N5O4S· 0.1Acetone · 2.55H2O) C, H, N, S; UPLC-MS > 95%; m/z [M+H]+, calcd 348.07, found 348.21.
5-{[N-methyl-(2-amino-4-oxo-4,7-dihydro-3H-pyrrolo[2,3-d]pyrimidin-6-yl)-methylamino]-carbonyl}-thiophene-3-carboxylic acid (28e)
Compound 28e was synthesized in 78% yield following the procedure of 28a. mp > 250 °C; TLC Rf 0.51 (CHCl3: MeOH: AcOH, 10:1:0.5); 1H NMR (DMSO-d6) δ 2.95 (s, 3H, cis and trans NCH3), 4.45–4.54 (d, 2H, cis and trans NCH2), 6.07 (s, 2H, exch, 2-NH2), 6.13 (s, 1H, cis and trans 5-H), 7.81 (s, 1H, thiophene), 8.17 (s, 1H, thiophene), 10.24 (s, 1H, exch, NH), 10.98 (s, 1H, exch, NH), 13.35 (s, 1H, exch, COOH); Anal. calcd. for (C14H13N5O4S · 1.06H2O) C, H, N, S; UPLC-MS > 95%; m/z [M+H]+, calcd 348.07, found 348.24.
4-{[N-methyl-(2-amino-4-oxo-4,7-dihydro-3H-pyrrolo[2,3-d]pyrimidin-6-yl)-methylamino]-sulfonyl}-phenylcarboxylic acid (28f)
Compound 28f was synthesized in 91% yield following the procedure of 28a. mp > 250 °C; TLC Rf 0.45 (CHCl3: MeOH: AcOH, 10:1:0.5); 1H NMR (DMSO-d6) δ 2.57 (s, 3H, NCH3), 4.12 (s, 2H, NCH2), 6.69 (s, 1H, 5-H), 6.78(s, 2H, exch, 2-NH2), 7.92 (d, J = 7.6 Hz, 2H, Ph), 8.15 (d, J = 7.6 Hz, 2H, Ph), 10.87 (s, 1H, exch, NH), 11.48 (s, 1H, exch, NH), 13.50 (br, 1H, exch, COOH); UPLC-MS > 95%; m/z [M+H]+, calcd 378.08, found 378.26.
General procedures for the synthesis of 29a-f
To a solution of 28a-f (1 eq.) in DMF (1 mL) were added N-methyl morpholine (3 eq.) and 2-chloro-4,6-dimethoxy-1,3,5-triazine (1.05 eq.). The mixture was stirred at rt for 1 h. Dimethyl l-glutamate (1.1 eq.) was added and the mixture was stirred overnight. The solvent was evaporated and the residue was purified by a silica column with chloroform and methanol to give 29a-f. Compounds 29a-f were directly used in the next step reaction without identification.
General procedures for the synthesis of 7–12
To a solution of 29a-f (1.0 eq.) in water (10 mL) and methanol (10 mL) was added Na2CO3 (0.5 g). The mixture was stirred at 80 °C rt. Methanol was removed and the mixture was adjusted to pH 3 with 1N HCl. The suspension was filtered and the filter cake was washed with deionized water, and acetone to facilitate the drying process. The resulting solid was dried at 50 °C in vacuum overnight to afford a white solid 7–12.
(S)-2-(4-{[N-Methyl-(2-amino-4-oxo-4,7-dihydro-3H-pyrrolo[2,3-d]pyrimidin-6-yl)-methylamino]-carbonyl}-phenyl-1-carbonyl)amino pentanedioic acid (7)
Yield: 64%. TLC Rf 0.12 (CHCl3/MeOH/AcOH, 10:1:0.1); mp > 250 °C; 1H NMR (DMSO-d6) δ 1.94 (m, 1H, CH2), 2.09 (m, 1H, CH2), 2.35 (t, J = 7.6 Hz, 2H, CH2), 2.81 (s, 1.8H, cis NCH3), 2.94 (s, 1.2H, trans NCH3), 4.31 (s, 0.8H, trans NCH2), 4.41 (s, 1.2H, cis NCH2), 4.59 (m, 1H, CH), 6.02 (s, 0.4H, trans 5-H), 6.08 (s, 2H, exch, 2-NH2), 6.17 (s, 0.6H, cis 5-H), 7.55 (d, J = 6.4 Hz, 2H, Ph), 7.93 (d, J = 6.4 Hz, 2H, Ph), 8.71 (s, 1H, exch, CONH), 10.23 (s, 1H, exch, NH), 11.02 (s, 1H, exch, NH), 12.59 (br, 2H, exch, COOH); Anal. calcd. for (C21H22N6O7 · 0.95H2O) C, H, N; UPLC-MS > 95%; m/z [M+H]+, calcd 471.16, found 471.26.
(S)-2-(3-{[N-methyl-(2-amino-4-oxo-4,7-dihydro-3H-pyrrolo[2,3-d]pyrimidin-6-yl)-methylamino]-carbonyl}phenylcarbonyl)amino pentanedioic acid (8)
Yield: 56%. TLC Rf 0.18 (CHCl3/MeOH/AcOH, 10:1:0.1); mp > 250 °C; 1H NMR (DMSO-d6) δ 1.99 (m, 1H, CH2), 2.09 (m, 1H, CH2), 2.33 (m, 2H, CH2), 2.81 (s, 1.5H, cis NCH3), 2.93 (s, 1.5H, trans NCH3), 4.32 (s, 1.0H, trans NCH2), 4.32 (s, 1.0H, cis NCH2), 4.60 (m, 1H, CH), 5.93 (s, 0.5H, trans 5-H), 6.06 (s, 2H, exch, 2-NH2), 6.17 (s, 0.5H, cis 5-H), 7.54 (m, 1H, Ph), 7.61 (m, 1H, Ph), 7.96 (m, 2H), 8.61 (s, 1H, exch, CONH), 10.09 (s, 1H, exch, NH), 10.23 (s, 1H, exch, NH), 11.05 (br, 1H, exch, COOH), 11.13 (br, 1H, exch, COOH); UPLC-MS> 95%; m/z [M+H]+, calcd 471.16, found 471.26.
(S)-2-(5-{[N-methyl-(2-amino-4-oxo-4,7-dihydro-3H-pyrrolo[2,3-d]pyrimidin-6-yl)-methylamino]-carbonyl}-thiophene-2-carbonyl)amino pentanedioic acid (9)
Yield: 61%. TLC Rf 0.19 (CHCl3/MeOH/AcOH, 10:1:0.5); mp > 250 °C; 1H NMR (DMSO-d6) δ 1.91 (q, J = 7.2 Hz, 2H, CH2), 2.35 (t, J = 6.8 Hz, 2H, CH2), 3.11 (br, 3H, cis and trans NCH3), 4.40 (q, J = 9.6 Hz, 1H, CH), 4.58 (s, 2H, cis and trans NCH2), 6.09 (s, 2H, exch, 2-NH2), 6.12 (s, 1H, cis and trans 5-H), 7.51 (d, J = 7.2 Hz, 1H, thiophene), 7.82 (d, J = 6.4 Hz, 1H, thiophene), 8.81 (d, J = 2.0 Hz, 1H, exch, CONH), 10.25 (s, 1H, exch, NH), 11.04 (s, 1H, exch, NH), 12.27 (s, 1H, exch, COOH), 12.82 (s, 1H, exch, COOH); Anal. calcd. for (C19H20N6O7S · 0.2Acetone·1.16H2O) C, H, N, S; UPLC-MS > 95%; m/z [M+H]+, calcd 477.11, found 477.16.
(S)-2-(4-{[N-methyl-(2-amino-4-oxo-4,7-dihydro-3H-pyrrolo[2,3-d]pyrimidin-6-yl)-methylamino]-carbonyl}-thiophene-2-carbonyl) amino pentanedioic acid (10)
Yield: 70%. TLC Rf 0.15 (CHCl3/MeOH/AcOH, 10:1:0.5); mp > 250 °C; 1H NMR (DMSO-d6) δ 1.90 (m, 1H, CH2), 2.07 (m, 1H, CH2), 2.34 (t, J = 7.6 Hz, 2H, CH2), 3.12 (s, 3H, cis and trans NCH3), 4.36 (q, J = 7.6 Hz, 1H, CH), 4.54 (s, 2H, cis and trans NCH2), 6.08 (s, 2H, exch, 2-NH2), 6.12 (s, 1H, cis and trans 5-H), 7.86 (s, 1H, thiophene), 8.38 (m, 1H, thiophene), 8.56 (d, J = 7.6 Hz, 1H, exch, CONH), 10.24 (s, 1H, exch, NH), 11.04 (s, 1H, exch, NH), 12.27 (br, 1H, exch, COOH), 12.70 (br, 1H, exch, COOH); Anal. calcd. for (C19H20N6O7S · 0.45Acetone·1.83H2O) C, H, N, S; UPLC-MS > 95%; m/z [M+H]+, calcd 477.11, found 477.22.
(S)-2-(5-{[N-methyl-(2-amino-4-oxo-4,7-dihydro-3H-pyrrolo[2,3-d]pyrimidin-6-yl)-methylamino]-carbonyl}-thiophene-3-carbonyl)amino pentanedioic acid (11)
Yield: 74%. TLC Rf 0.19 (CHCl3/MeOH/AcOH, 10:1:0.5); mp > 250 °C; 1H NMR (DMSO-d6) δ 1.92 (m, 1H, CH2), 2.09 (m, 1H, CH2), 2.35 (m, 2H, CH2), 3.01 (s, 3H, cis and trans NCH3), 4.35 (m, 1H, CH), 4.54 (s, 2H, cis and trans NCH2), 6.08 (s, 2H, exch, 2-NH2), 6.12 (s, 1H, cis and trans 5-H), 7.96 (s, 1H, thiophene), 8.07 (s, 1H, thiophene), 8.79 (d, J = 1.2 Hz, 1H, exch, CONH), 10.24 (s, 1H, exch, NH), 11.00 (s, 1H, exch, NH), 12.27 (br, 1H, exch, COOH), 12.69 (s, 1H, exch, COOH); Anal. calcd. for (C19H20N6O7S·1.77H2O) C, H, N, S; UPLC-MS > 95%; m/z [M+H]+, calcd 477.11, found 477.26.
(S)-2-(4-{[N-methyl-(2-amino-4-oxo-4,7-dihydro-3H-pyrrolo[2,3-d]pyrimidin-6-yl)-methylamino]-sulfonyl}-phenylcarbonyl)amino pentanedioic acid (12)
Yield: 60%. TLC Rf 0.17 (CHCl3/MeOH/AcOH, 10:1:0.5); mp 239.3 – 241.7 °C; 1H NMR (DMSO-d6) δ 1.98 (m, 1H, CH2), 1.99 (m, 1H, CH2), 2.38 (t, J=8.0 Hz, 2H, CH2), 3.38 (s, 3H, NCH3), 4.06 (s, 2H, NCH2), 4.44 (m, 1H, CH), 6.11 (s, 2H, exch, 2-NH2), 6.12 (s, 1H, 5-H), 7.95 (d, J = 8.0 Hz, 2H, Ph), 8.13 (d, J = 8.0 Hz, 2H, Ph), 8.92 (d, J = 1.2 Hz, 1H, exch, CONH), 10.24 (s, 1H, exch, NH), 11.11 (s, 1H, exch, NH), 12.16 (br, 1H, exch, COOH), 12.69 (br, 1H, exch, COOH); Anal. calcd. for (C20H22N6O8S· 0.5Acetone·0.92H2O) C, H, N, S; UPLC-MS > 95%; m/z [M+H]+, calcd 507.12, found 507.20.
Molecular modeling and computational studies
Molecular modeling was performed for all analogs with the FRα (PDB: 5IZQ) and GARFTase (PDB: 4ZZ1) crystal structures using the induced fit docking protocol of Maestro.49 The ligands were prepared using the Ligprep application of Maestro. The docking protocol was validated by re-docking the co-crystallized ligands 6 (FRα) and 3 (GARFTase) into the crystal structures with RMSD values of 0.15 Å and 0.23 Å, respectively. The centroid around the ligands were defined as the ligand binding site. The OPLS3e force field was used and amino acid residues within 5 Å from the docked poses were allowed to be optimized using prime refinement. Using prepared ligands from the Ligprep files, we used Conformational Search (Macromodel) to determine the number of low energy conformations within 5 kcal/mol from the minimum energy conformation; the settings were as follows: Force Field, OPLS3e; Solvent, water; Conformation Search Method, Mixed Torsional/Low-Mode sampling (the remaining settings were set default).
Reagents for biological studies.
[14C(U)]Glycine (87 mCi/mmol) was purchased from Moravek Biochemicals (Brea, CA). Unlabeled folic acid was purchased from the Sigma Chemical Co. (St. Louis, MO). Leucovorin [(6R,S) 5-formyl tetrahydrofolate] was provided by the Drug Development Branch, National Cancer Institute, Bethesda, MD. The sources of the classical antifolate drugs were as follows: MTX, Drug Development Branch, National Cancer Institute (Bethesda, MD); RTX [N-(5-[N-(3,4-dihydro-2-methyl-4-oxyquinazolin-6-ylmethyl)-N-methyl-amino]-2-thienoyl)- l-glutamic acid], AstraZeneca Pharmaceuticals (Maccesfield, Cheshire, England); and PMX [N-{4-[2-(2-amino-3,4-dihydro-4-oxo-7H-pyrrolo[2,3-d]pyrimidin-5-yl)ethyl]benzoyl}- l-glutamic acid] (Alimta) (LC Laboratories, Woburn, MA). Other chemicals were obtained from commercial sources in the highest available purity.
Cell culture.
MTXRIIOuaR2–4 (hereafter, referred to as R2) is a RFC-, PCFT-and FRα-null CHO cell line45 that was a gift from Dr. Wayne Flintoff (University of Western Ontario). From this parental cell line, RFC, PCFT and FRα were transfected to generate isogenic CHO cell lines designated PC43–10 (expresses human RFC but not PCFT or FRα), R2/PCFT4 (expresses human PCFT), and RT16 (expresses human FRα).23,24,44 The CHO sublines were maintained in α-minimal essential medium (α-MEM) supplemented with 1% penicillin-streptomycin solution, 2 mM l-glutamine and 10% heat-treated bovine calf serum (Invitrogen). Transfected cell lines were cultured with 1 mg/ml G418. FR-expressing CHO cell lines were cultured for 72 hours prior to cell viability experiments in folate-free RPMI 1640 (FF-RPMI) (Invitrogen) with 10% dialyzed fetal bovine serum (dFBS) (Invitrogen), supplemented with 1% penicillin-streptomycin and l-glutamine. Human KB carcinoma cells were purchased from ATCC (Manassas, VA) continuously maintained in complete FF-RPMI with 10% fetal bovine serum and 1% penicillin-streptomycin and l-glutamine.
Cell proliferation assays were performed exactly as previously described.23,25,27,28,33,34,36,44 Cell lines were treated with a range of concentrations of standard or novel inhibitors (0–1000 nM) in a 96 well plate with FF-RPMI complete and 10% dFBS, supplemented with 2 nM (RT-16, D4, KB) or 25 nM (R2/PCFT4, R2, PC43–10) leucovorin, 1% penicillin–streptomycin and l-glutamine (2 mM), over a 96 h incubation period at 37 °C with 5% CO2. To confirm FR-mediated drug uptake and growth inhibition, 200 nM folic acid was added to parallel incubations with drug. To quantitate viable cells, the media was removed and Cell Titer-blue™ reagent (Promega) was added. Relative cell numbers were determined by fluorescence measurements with a fluorescence plate reader (Molecular Devices) at 590 nm emission and 560 nm excitation. IC50 values, corresponding to drug concentrations that resulted in 50% loss of cell growth, were calculated from dose-response curves using Sigma Plot (v.12) software.
In order to identify the targeted pathways/folate-dependent enzymes by the classic and novel pyrrolo[2,3-d]pyrimidine antifolates, proliferation assays were performed with KB cell cultured in complete glycine free FF-RPMI with dFBC in the presence of adenosine (60 μM), thymidine (10 μM), glycine (130 μM) or AICA hydrochloride (320 μM); results were compared to incubations in parallel without nucleoside/AICA or drug additions.
In situ GARFTase inhibition assays.
To measure the extent of intracellular GARFTase inhibition in the presence of antifolate inhibitors, we measured the metabolic incorporation of [14C(U)] glycine into [14C]formyl GAR in the presence of azaserine.23–25,27,28,33,34,36,44,50 KB cells were seeded in complete media (above) and allowed to adhere to the substratum for 24 h. Cells were washed with FF-RPMI, L-glutamine free with 10% dFBS, 1% penicillin-streptomycin and 2 nM LCV. Cells were incubated at 37°C with 5% CO2 for 1 h with folate- and l-glutamine-depleted media containing 2 nM LCV with the antifolate compounds over a range of concentrations. Control cells were treated with an equal volume of DMSO. Azaserine (4 µM final concentration) was added to the cells and cells were incubated for 30 minutes. This was followed by the addition of l-glutamine (2 mM) and [14C(U)]glycine (final specific activity 0.1 mCi/L).
Cells were incubated for an additional 16 h, then washed with ice-cold DPBS, trypsinized and collected. Cells were treated with 5% trichloroacetic acid (TCA) at 0°C. Samples were centrifuged (4°C, 14,000 rpm) and the protein precipitants were solubilized with 0.5 N NaOH for quantitation of protein concentrations with the Folin-phenol protein method.51 The TCA supernatants were extracted with cold ether. After evaporation of ether, the remaining aqueous layer was gravity filtered through a 1 cm chromatography column (Bio-rad) of AG1×8 (chloride form). The columns were washed with 10 mL each of 0.5 N formic acid, followed by 4 N formic acid and finally eluted with 8 ml of 1 N HCl collected in 8 one ml fractions. The eluate was measured for radioactivity and the percentages of radioactivity in the [14C]formyl GAR and non-specific [14C]fractions determined. The results were calculated as pmol [14C]formyl GAR/mg protein. The data were plotted using Sigmaplot (v.12) and the IC50s were calculated for the drug-treated samples, compared to the untreated controls.
ACKNOWLEDGMENTS
This work was supported in part by grants from the National Institutes of Health R01 CA53535 (LHM and ZH), R01 CA125153 (AG), R01 CA152316 (LHM and AG), and R01 CA166711 (AG, LHM and CED), the Eunice and Milton Ring Endowed Chair for Cancer Research (LHM), and the Duquesne University Adrian Van Kaam Chair in Scholarly Excellence (AG).
Abbreviations used:
- FR
Folate Receptor
- GAR
Glycinamide Ribonucleotide
- GARFTase
Glycinamide Ribonucleotide Formyltransferase
- RFC
Reduced Folate Carrier
- AICA
5-Aminoimidazole-4-Carboxamide
- AICARFTase
5-Aminoimidazole-4-Carboxamide Ribonucleotide Formyltransferase
- MTX
Methotrexate
- PDX
Pralatrexate
- RTX
Raltitrexed
- PMX
Pemetrexed
- PCFT
Proton-Coupled Folate Transporter
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Matherly LH, Wilson MR, Hou Z. The major facilitative folate transporters solute carrier 19A1 and solute carrier 46A1: biology and role in antifolate chemotherapy of cancer. Drug Metab Dispos. 2014;42:632–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Visentin M, Zhao R, Goldman ID. The Antifolates. Hematol Oncol Clin N. 2012;26(3):629–ix. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Matherly LH, Hou Z, Deng Y. Human reduced folate carrier: translation of basic biology to cancer etiology and therapy. Cancer Metastasis Rev. 2007;26:111–128. [DOI] [PubMed] [Google Scholar]
- 4.Kugel Desmoulin S, Wang L, Polin L, et al. Functional loss of the reduced folate carrier enhances the antitumor activities of novel antifolates with selective uptake by the proton-coupled folate transporter. Mol Pharmacol. 2012;82:591–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Matherly LH, Hou Z, Gangjee A. The promise and challenges of exploiting the proton-coupled folate transporter for selective therapeutic targeting of cancer. Cancer Chemother Pharmacol. 2018;81:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhao R, Min SH, Qiu A, et al. The spectrum of mutations in the PCFT gene, coding for an intestinal folate transporter, that are the basis for hereditary folate malabsorption. Blood. 2007;110:1147–1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhao R, Goldman ID. The proton-coupled folate transporter: physiological and pharmacological roles. Curr Opin Pharmacol. 2013;13(6):875–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Elnakat H, Ratnam M. Distribution, functionality and gene regulation of folate receptor isoforms: implications in targeted therapy. Adv Drug Deliv Rev. 2004;56:1067–1084. [DOI] [PubMed] [Google Scholar]
- 9.Xia W, Low PS. Folate-targeted therapies for cancer. J Med Chem. 2010;53(19):6811–6824. [DOI] [PubMed] [Google Scholar]
- 10.Cherian C, Kugel Desmoulin S, Wang L, et al. Therapeutic targeting malignant mesothelioma with a novel 6-substituted pyrrolo[2,3-d]pyrimidine thienoyl antifolate via its selective uptake by the proton-coupled folate transporter. Cancer Chemother Pharmacol. 2013;71:999–1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hou Z, Gattoc L, O’Connor C, et al. Dual targeting of epithelial ovarian cancer via folate receptor alpha and the proton-coupled folate transporter with 6-substituted pyrrolo[2,3-d]pyrimidine antifolates. Mol Cancer Ther. 2017;16:819–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wilson MR, Hou Z, Yang S, et al. Targeting Nonsquamous Nonsmall Cell Lung Cancer via the Proton-Coupled Folate Transporter with 6-Substituted Pyrrolo[2,3-d]Pyrimidine Thienoyl Antifolates. Mol Pharmacol. 2016;89:425–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Giovannetti E, Zucali PA, Assaraf YG, et al. Role of proton-coupled folate transporter in pemetrexed-resistance of mesothelioma: clinical evidence and new pharmacological tools. Ann Oncol. 2017;28:2725–2732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Desmoulin SK, Hou Z, Gangjee A, Matherly LH. The human proton-coupled folate transporter: Biology and therapeutic applications to cancer. Cancer Biol Ther. 2012;13:1355–1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Parker N, Turk MJ, Westrick E, Lewis JD, Low PS, Leamon CP. Folate receptor expression in carcinomas and normal tissues determined by a quantitative radioligand binding assay. Anal Biochem. 2005;338:284–293. [DOI] [PubMed] [Google Scholar]
- 16.Kamen BA, Smith AK. Farletuzumab, an anti-folate receptor alpha antibody, does not block binding of folate or anti-folates to receptor nor does it alter the potency of anti-folates in vitro. Cancer Chemother Pharmacol. 2012;70(1):113–120. [DOI] [PubMed] [Google Scholar]
- 17.Armstrong DK, White AJ, Weil SC, Phillips M, Coleman RL. Farletuzumab (a monoclonal antibody against folate receptor alpha) in relapsed platinum-sensitive ovarian cancer. Gynecologic oncology. 2013;129(3):452–458. [DOI] [PubMed] [Google Scholar]
- 18.Kurkjian C, LoRusso P, Sankhala KK, Birrer MJ, Kirby M, Ladd S, Hawes S,Running KL, O’Leary JJ, Moore KN. A phase I, first-in-human studyto evaluate the safety, pharmacokinetics (PK), and pharmacodynamics (PD) ofIMGN853 in patients (Pts) with epithelial ovarian cancer (EOC) and other FOLR1-positive solid tumors. J Clin Oncol 2013;31 (15 Suppl.):2573. [Google Scholar]
- 19.Assaraf YG, Leamon CP, Reddy JA. The folate receptor as a rational therapeutic target for personalized cancer treatment. Drug Resist Updat. 2014;17(4–6):89–95. [DOI] [PubMed] [Google Scholar]
- 20.Gibbs DD, Theti DS, Wood N, et al. BGC 945, a novel tumor-selective thymidylate synthase inhibitor targeted to alpha-folate receptor-overexpressing tumors. Cancer Res. 2005;65(24):11721–11728. [DOI] [PubMed] [Google Scholar]
- 21.Leamon CP, Jackman AL. Exploitation of the folate receptor in the management of cancer and inflammatory disease. Vitam Horm. 2008;79:203–233. [DOI] [PubMed] [Google Scholar]
- 22.Udai Banerji AHIG, Michalarea Vasiliki, Ruddle Ruth, Raynaud Florence I., Riisnaes Ruth, Rodrigues Daniel Nava, Tunariu Nina, Porter Joanna C, Ward Sarah Emily, Parmar Mona, Turner Alison Joanne, Seeramreddi Satyanarayana, Hall Emma, Dean Emma Jane, Basu Bristi, George Angela, Kaye Stan B, Banerjee Susana N., De Bono Johann S. An investigator-initiated phase I study of ONX-0801, a first-in-class alpha folate receptor targeted, small molecule thymidylate synthase inhibitor in solid tumors. J Clin Oncol. 2017; 35 (15 suppl):2503. [Google Scholar]
- 23.Deng Y, Wang Y, Cherian C, et al. Synthesis and discovery of high affinity folate receptor-specific glycinamide ribonucleotide formyltransferase inhibitors with antitumor activity. J Med Chem. 2008;51:5052–5063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kugel Desmoulin S, Wang Y, Wu J, et al. Targeting the proton-coupled folate transporter for selective delivery of 6-substituted pyrrolo[2,3-d]pyrimidine antifolate inhibitors of de novo purine biosynthesis in the chemotherapy of solid tumors. Mol Pharmacol. 2010;78:577–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang L, Kugel Desmoulin S, Cherian C, et al. Synthesis, biological, and antitumor activity of a highly potent 6-substituted pyrrolo[2,3-d]pyrimidine thienoyl antifolate inhibitor with proton-coupled folate transporter and folate receptor selectivity over the reduced folate carrier that inhibits beta-glycinamide ribonucleotide formyltransferase. J Med Chem. 2011;54:7150–7164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang L, Wallace A, Raghavan S, et al. 6-Substituted Pyrrolo[2,3-d]pyrimidine Thienoyl Regioisomers as Targeted Antifolates for Folate Receptor α and the Proton-Coupled Folate Transporter in Human Tumors. J Med Chem. 2015;58(17):6938–6959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Golani LK, Wallace-Povirk A, Deis SM, et al. Tumor Targeting with Novel 6-Substituted Pyrrolo [2,3-d] Pyrimidine Antifolates with Heteroatom Bridge Substitutions via Cellular Uptake by Folate Receptor alpha and the Proton-Coupled Folate Transporter and Inhibition of de Novo Purine Nucleotide Biosynthesis. J Med Chem. 2016;59(17):7856–7876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Golani LK, George C, Zhao S, et al. Structure-activity profiles of novel 6-substituted pyrrolo[2,3-d]pyrimidine thienoyl antifolates with modified amino acids for cellular uptake by folate receptors alpha and beta and the proton-coupled folate transporter. J Med Chem. 2014;57(19):8152–8166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Laursen JS, Engel-Andreasen J, Fristrup P, Harris P, Olsen CA. Cis–Trans Amide Bond Rotamers in β-Peptoids and Peptoids: Evaluation of Stereoelectronic Effects in Backbone and Side Chains. J Am Chem Soc. 2013;135(7):2835–2844. [DOI] [PubMed] [Google Scholar]
- 30.Sliwoski G, Kothiwale S, Meiler J, Lowe EW. Computational Methods in Drug Discovery. Pharmacol Rev. 2014;66(1):334–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Szostak M, Aubé J. The Chemistry of Bridged Lactams and Related Heterocycles. Chem Rev. 2013;113(8):5701–5765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen P, Feng D, Qian X, et al. Structure-activity-relationship of amide and sulfonamide analogs of omarigliptin. Bioorganic & medicinal chemistry letters. 2015;25(24):5767–5771. [DOI] [PubMed] [Google Scholar]
- 33.Wang L, Cherian C, Kugel Desmoulin S, et al. Synthesis and antitumor activity of a novel series of 6-substituted pyrrolo[2,3-d]pyrimidine thienoyl antifolate inhibitors of purine biosynthesis with selectivity for high affinity folate receptors and the proton-coupled folate transporter over the reduced folate carrier for cellular entry. J Med Chem. 2010;53:1306–1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang L, Wallace A, Raghavan S, et al. 6-Substituted Pyrrolo[2,3-d]pyrimidine Thienoyl Regioisomers as Targeted Antifolates for Folate Receptor alpha and the Proton-Coupled Folate Transporter in Human Tumors. J Med Chem. 2015;58(17):6938–6959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li X, Liu S, Huang H, et al. Gambogic Acid Is a Tissue-Specific Proteasome Inhibitor In Vitro and In Vivo. Cell Reports. 2013;3(1):211–222. [DOI] [PubMed] [Google Scholar]
- 36.Wang L, Cherian C, Kugel Desmoulin S, et al. Synthesis and Biological Activity of 6-Substituted Pyrrolo[2,3-d]pyrimidine Thienoyl Regioisomers as Inhibitors of de Novo Purine Biosynthesis with Selectivity for Cellular Uptake by High Affinity Folate Receptors and the Proton-Coupled Folate Transporter over the Reduced Folate Carrier. J Med Chem. 2012;55(4):1758–1770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Proctor LD, Warr AJ. Development of a Continuous Process for the Industrial Generation of Diazomethane1. Org Process Res Dev. 2002;6(6):884–892. [Google Scholar]
- 38.Akimoto H, Imamiya E, Hitaka T, Nomura H, Nishimura S. Synthesis of queuine, the base of naturally occurring hypermodified nucleoside (queuosine), and its analogues. Journal of the Chemical Society, Perkin Transactions 1. 1988(7):1637–1644. [Google Scholar]
- 39.Gangjee A, Mavandadi F, Kisliuk RL, Queener SF. Synthesis of Classical and a Nonclassical 2-Amino-4-oxo-6-methyl-5-substituted Pyrrolo[2,3-d]pyrimidine Antifolate Inhibitors of Thymidylate Synthase. J Med Chem. 1999;42(12):2272–2279. [DOI] [PubMed] [Google Scholar]
- 40.Linz S, Troschuetz R. Synthesis of 5-[(4-phenylpiperazin-1-yl)methyl]pyrrolo[2,3-d]pyrimidine derivatives as potential dopamine D4 receptor ligands. J Heterocycl Chem. 2007;44(2):349–354. [Google Scholar]
- 41.Seela F, Chen Y, Zulauf M. Regioselectivity of the Mannich reaction on pyrrolo[2,3-d]pyrimidine nucleosides related to 7-deaza-2’-deoxyadenosine or 7-deaza-2’-deoxyguanosine. Synthese. 1997(9):1067–1072. [Google Scholar]
- 42.Xiang W, Choudhary S, Hamel E, Mooberry SL, Gangjee A. Structure based drug design and in vitro metabolism study: Discovery of N-(4-methylthiophenyl)-N,2-dimethyl-cyclopenta[d]pyrimidine as a potent microtubule targeting agent. Bioorganic & medicinal chemistry. 2018;26(9):2437–2451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Quintanilla-Licea R, Colunga-Valladares J, Caballero-Quintero A, et al. NMR Detection of Isomers Arising from Restricted Rotation of the C-N Amide Bond of N-Formyl-o-toluidine and N,N’-bis-Formyl-o-tolidine. Molecules. 2002;7(8):662. [Google Scholar]
- 44.Deng Y, Zhou X, Kugel Desmoulin S, et al. Synthesis and biological activity of a novel series of 6-substituted thieno[2,3-d]pyrimidine antifolate inhibitors of purine biosynthesis with selectivity for high affinity folate receptors over the reduced folate carrier and proton-coupled folate transporter for cellular entry. J Med Chem. 2009;52:2940–2951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Flintoff WF, Nagainis CR. Transport of methotrexate in Chinese hamster ovary cells: a mutant defective in methotrexate uptake and cell binding. Arch Biochem Biophys. 1983;223:433–440. [DOI] [PubMed] [Google Scholar]
- 46.Mitchell-Ryan S, Wang Y, Raghavan S, et al. Discovery of 5-Substituted Pyrrolo[2,3-d]pyrimidine Antifolates as Dual-Acting Inhibitors of Glycinamide Ribonucleotide Formyltransferase and 5-Aminoimidazole-4-carboxamide Ribonucleotide Formyltransferase in De Novo Purine Nucleotide Biosynthesis: Implications of Inhibiting 5-Aminoimidazole-4-carboxamide Ribonucleotide Formyltransferase to AMPK Activation and Antitumor Activity. J Med Chem. 2013;56(24):10016–10032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ducker GS, Ghergurovich JM, Mainolfi N, et al. Human SHMT inhibitors reveal defective glycine import as a targetable metabolic vulnerability of diffuse large B-cell lymphoma. Proc Natl Acad Sci U S A. 2017;114(43):11404–11409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Taylor EC, Kuhnt D, Shih C, et al. A dideazatetrahydrofolate analog lacking a chiral center at C-6: N-[4-[2-(2-amino-3,4-dihydro-4-oxo-7H-pyrrolo[2,3-d]pyrimidin-5yl)ethyl[benzoyl]-L-glutamic acid is an inhibitor of thymidylate synthase. J Med Chem. 1992;35(23):4450–4454. [DOI] [PubMed] [Google Scholar]
- 49.Saez-Calvo G, Sharma A, Balaguer FA, et al. Triazolopyrimidines Are Microtubule-Stabilizing Agents that Bind the Vinca Inhibitor Site of Tubulin. Cell Chem Biol. 2017;24(6):737–750 e736. [DOI] [PubMed] [Google Scholar]
- 50.Kugel Desmoulin S, Wang L, Hales E, et al. Therapeutic targeting of a novel 6-substituted pyrrolo [2,3-d]pyrimidine thienoyl antifolate to human solid tumors based on selective uptake by the proton-coupled folate transporter. Mol Pharmacol. 2011;80:1096–1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with the Folin phenol reagent. The Journal of biological chemistry. 1951;193(1):265–275. [PubMed] [Google Scholar]