

Abstract
Modification of proteins by O-linked β-N-acetylglucosamine (O-GlcNAc) promotes tumor cell survival, proliferation, epigenetic changes, angiogenesis, invasion, and metastasis. Here we demonstrate that in conditions of elevated glucose, there is increased expression of key drug resistance proteins (ABCB1, ABCG2, ERCC1, and XRCC1), all of which are regulated by the Hedgehog pathway. In elevated glucose conditions, we determined that the Hedgehog pathway transcription factors, GLI1 and GLI2, are modified by O-GlcNAcylation. This modification functionally enhanced their transcriptional activity. The activity of GLI was enhanced when O-GlcNAcase was inhibited, while inhibiting O-GlcNAc Transferase caused a decrease in GLI activity. The metabolic impact of hyperglycemic conditions impinges on maintaining PKM2 in the less active state that facilitates the availability of glycolytic intermediates for biosynthetic pathways. Interestingly, under elevated glucose conditions, PKM2 directly influenced GLI activity. Specifically, abrogating PKM2 expression caused a significant decline in GLI activity and expression of drug resistance proteins. Cumulatively, our results suggest that elevated glucose conditions upregulate chemoresistance through elevated transcriptional activity of the Hedgehog/GLI pathway. Interfering in O-GlcNAcylation of the GLI transcription factors may be a novel target in controlling cancer progression and drug resistance of breast cancer.
Keywords: breast cancer, drug resistance, GLI, Hedgehog pathway, O-GlcNAcylation
Introduction
Breast cancer is the most commonly diagnosed cancer among women in the United States. Efforts are being made to pinpoint factors that determine and dictate breast tumor malignancy and metastasis. Complications such as diabetes provide increased mortality risk due to the hyperinsulinemic and hyperglycemic state feeding a tumor (1–4). Energy metabolism in cancer involves a shift away from mitochondrial oxidative phosphorylation to less efficient glycolysis, necessitating greatly increased uptake of glucose. Increased glucose uptake by cancer cells enables increased flux through the Hexosamine Biosynthesis Pathway (HBP). Increased HBP flux and elevated UDP-GlcNAc is a general feature of cancer cells that contributes to O-GlcNAcylation events (5, 6). The O-linked β-N-acetylglucosamine (O-GlcNAc) monosaccharide is commonly found on Serine or Threonine residues in proteins. O-GlcNAc may take part in a variety of important cellular functions such as aiding stem cells in switching from totipotency to pluripotency, countering post-translational modifications such as phosphorylation, aiding in the adjustment of permeability of the nuclear pore, as well as many forms of disease-relevant signaling and the regulation of enzymes. The addition of O-GlcNAc to proteins is catalyzed by O-GlcNAc transferase (OGT) that transfers the GlcNAc moiety from the high-energy donor UDP-GlcNAc to substrate proteins while O-GlcNAcase (OGA) catalyzes its removal (5). These processes are restricted to the nuclear, mitochondrial, and cytoplasmic compartments of the cell. Also, both OGT and OGA are necessary for cell survival, which further highlights the importance of O-GlcNAcylation and the roles that these enzymes play in this process (7–9).
Increased O-GlcNAcylation is also known to be associated with cancer cells; elevated O-GlcNAcylation is seen in several human malignancies including breast (10, 11), prostate (12), lung (13), colorectal (13, 14), liver (15), and nonsolid cancers such as chronic lymphocytic leukemia (16). Knockdown of OGT reduces cancer hyper-O-GlcNAcylation and inhibits transformed phenotypes, indicating that elevated O-GlcNAc contributes to cancer progression (17). Cancer cell hyper-O-GlcNAcylation appears, in part, to contribute to excessive growth through up-regulation of key proteins that drive cell cycle progression such as Cyclin D1 and down-regulation of cell cycle inhibitory proteins such as p27Kip1 (17). Suppression of GlcNAcylation in breast cancer cells leads to reversal of the Warburg Effect by regulation of HIF-1α (18).
Breast cancer tissues and cell lines bear alterations in the levels of O-GlcNAcylated proteins and levels of OGT and OGA. Immunohistochemical assessments have demonstrated characteristically elevated O-GlcNAc modification in human breast tumor tissue compared with adjacent normal tissue (10, 11, 19). Krzeslak et al., (20) demonstrated an increase in OGT transcript levels with concomitant decrease in the OGA transcript levels in tumor tissues relative to normal samples. This was confirmed in a separate study showing that breast tumor tissues bear elevated levels of OGT and O-GlcNAc when compared with normal patient samples (21). Moreover, the increase in O-GlcNAc profile was concordant with the histological grade of breast tumors. These attributes of O-GlcNAc modification and elevated OGT levels are maintained in breast tumor cell lines (10). While a separate study with a reduced sample size reported an increase in OGA activity with breast cancer tumor grade (22), there is a cumulatively large body of data that supports increased O-GlcNAcylation in breast cancer.
The Hedgehog (Hh) pathway, in mammalian cells, is classically initiated with the binding of soluble, lipid-modified morphogens, Sonic Hedgehog (SHH), Indian Hedgehog (IHH), or Desert Hedgehog (DHH) to the Patched (PTCH) receptors on the cell membrane. This relieves the inhibitory effect of PTCH on Smoothened (SMO) causing activation of GLI transcription factors. Targeting the Hh pathway has been one of the recent promising strategies to combat breast cancer (23–26). In the present study, we report novel findings that in conditions of elevated glucose, breast cancer cells demonstrate O-GlcNAc modification of GLI1 and GLI2 proteins that augments their transcriptional activity. The implications of our study have important bearings on understanding the detrimental effect of hyperglycemic conditions in cancer progression and drug resistance.
Materials and Methods
Cell Culture:
SUM1315 and SUM159 breast cancer cells (Asterand, Detroit, MI) were cultured in DMEM/F12 (Life Technologies, Carlsbad, CA) supplemented with 5% fetal bovine serum (FBS; Life Technologies), 10 μg/ml insulin (Sigma-Aldrich; St. Louis, MO), and either 10 ng/ml EGF (Sigma-Aldrich) or 1 μg/ml hydrocortisone, respectively. MDA-MB-468 breast cancer cells were cultured in DMEM/F12 supplemented with 10% FBS. MDA-MB-231 and MDA-MB-435 cells were cultured in DMEM/F12 supplemented with 5% FBS. All cells were maintained in a humidified 5% CO2 environment. For normal and high glucose experiments, the cells were conditioned in either ~5mM or ~25 mM glucose containing medium, respectively, for 5 passages.
Transient Transfectants:
MDA-MB-468 and MDA-MB-435 cells were transfected with Fugene6 (Promega; Madison, WI) following the manufacturer’s protocol. The various plasmids used for OGT/OGA knock-in and knock-down were CMV-OGT+1042 for OGT knock-in (27), CMV-GFP-OGT-DN-1433 for OGT knockdown (27), CMV-OGA+995 for OGA knock-in (28), and CMV-OGA-DN-997 for OGA knockdown (28). CMV-GFP-979 was the control vector for all these transient transfections. The plasmids were a kind gift from Prof. John C. Chatham and Dr. Andrew Patterson at UAB. PKM2 silencing was achieved using 50nM non-targeting control or PKM siRNA (Dharmacon ON-TARGETplus Human PKM siRNA SMARTpool, cat # 5315) transiently transfected using Lipofectamine2000. Where applicable, twenty-four hours after transfection, media was changed to media containing DMSO or 10μM GANT61 or 25μM PUGNAc. Forty-eight hours post treatment, RNA was isolated. RTQ was done for PKM2 to validate knockdown, and then RTQ was done for ABCB1, ABCG2, ERCC1, and XRCC1 (TaqMan, ThermoFisher).
Small molecule inhibitors:
PUGNAc (Sigma-Aldrich; A7229), OSMI-1 (GLIXX Lab; Southborough, MA; GLXC-06077) and Thiamet-G (TOCRIS; Bristol, UK; 4390) were used in the study.
Western Blotting Analysis:
Whole cell lysates were collected in Nonidet P-40 lysis buffer (150 mM NaCl, 50 mM Tris, 1% Nonidet P-40). Isolation of cytosolic and nuclear fractions was done with an NE-PER kit (Pierce; Rockford, IL) following the manufacturer’s protocol. Protein (30 μg) was resolved by SDS-PAGE, transferred to polyvinylidene difluoride membranes, and immunoblotted overnight at 4 °C with primary antibodies. The various primary antibodies used were CTD110.6 (IgM, UAB), GLI1 (Cell Signaling; 2643), GLI2 (Boster Biological Technology; Pleasanton, CA; PA1941), Histone H3 (Cell Signaling; 4499), OGA (Santa Cruz Biotechnology; Dallas, TX; sc-376429), OGT (Sigma-Aldrich; O6264), PKM2 (Cell Signaling; 4053), phospho-Y105–PKM2 (Cell Signaling; 3827), and RL2 (Abcam; Cambridge, MA; ab2739). Alpha-tubulin (Cell Signaling; 12351) or beta-actin (Sigma-Aldrich; A3854) was used to confirm equal loading. Anti-rabbit or anti-mouse HRP-conjugated secondary antibody (GE Healthcare; Chicago, IL) was used for detection, and blots were developed with either ECL or SuperSignal substrate (Pierce) and imaged on films or Amersham Imager 600. The purity of cytosolic and nuclear fractions was confirmed with beta-tubulin (2146; Cell Signaling) or histone deacetylase 1 (Cell Signaling; 2062) antibodies, respectively. The depicted images are representative of experiments repeated at least three times.
Co-immunoprecipitation:
Whole cell lysates (500μg) from SUM159 cells grown in normo- or hyperglycemic conditions were immunoprecipitated with RL2 antibody and immunoblotted with GLI1 or GLI2. In a reverse co-immunoprecipitation experiment, the lysate was immunoprecipitated with GLI1 antibody and immunoblotted with the RL2 antibody. MDA-MB-468 cells conditioned in normo or hyperglycemic conditions were transfected with pLNCX-myc-GLI1 (26). The cell lysates (500μg) were immunoprecipitated with GLI1 and immunoblotted for O-GlcNAc detection. The results depicted are representative of experiments performed at least three times.
Luciferase Assays:
Cells (25 X 103) were transfected with an 8X-GLI construct in pGL3 promoter plasmid as described previously (29). Similarly, SUM1315 and SUM159 cells (25 × 103) were transiently co-transfected with plasmids encoding a non-targeting (NT) control or shPKM2 and 8X-GLI construct. Cells were cultured in either high (25mM) or normal (5mM) glucose containing medium for 24 hours. Luciferase readings were normalized to total protein content. Each parameter was studied in triplicate and the experiment repeated at least three times. The data are represented as percent luciferase activity, which is derived as a percent of the relative light units in treated groups compared with the untreated groups.
Drug/Inhibitor Treatments:
Cells (104) conditioned in high (25mM) or normal glucose (5mM) media were seeded per well of a 96-well plate. Twenty-four hours after plating, cells were treated with PUGNAc in the presence or absence of GANT61 (Tocris) or doxorubicin (Calbiochem, Billerica, MA) or cisplatin (Sigma-Aldrich) as indicated. After treating for 48 hours, the medium was removed, and fresh, serum-free medium containing MTS reagent (CellTiter 96 AQueous One Solution Cell Proliferation Assay; Promega; Madison, WI) was added. The cells were placed at 37⁰C/5%CO2 for 3 hours to allow for color development. Spectrophotometric readings were taken at 490 nm in a BioTek Eon plate reader. All assays were done in triplicate and the experiment was repeated at least three times.
Quantitative RT-PCR (qRT-PCR):
RNA was harvested from cells grown in 10-cm plates using QIAGEN RNAeasy Mini Kit (Hilden, Germany). cDNA was generated using High Capacity Reverse Transcriptase Kit (Applied Biosystems; Foster City, CA). Real time PCR was performed using a StepOnePlus Real-Time PCR System (Applied Biosystems). All reactions were done as three independent replicates and repeated at least once. All assays were done using the TaqMan Gene Expression Assays from Applied Biosystems. The genes queried include GLI1, GLI2, PTCH1, ABCB1, ABCG2, ERCC1 AND XRCC1. Transcript levels were normalized to endorse control gene beta-actin, using change in the CT values (ΔCT) to calculate changes in respective expression (ΔΔCT).
Statistical Analysis:
Statistical differences between groups were assessed after applying the test per the mandates of the experimental design using GraphPad Prism 4 software. Statistical significance was determined if the analysis reached 95% confidence. The precise p values are listed in the corresponding figure legends.
Results
Hyperglycemic conditions activate Hh/GLI signaling and enhance drug resistance.
Relative to normoglycemic conditions, high glucose levels significantly impact tumor cell behavior, associating with increased cancer cell proliferation, invasion, and metastasis. We conditioned MDA-MB-468 and SUM159 breast cancer cells in media containing normoglycemic levels of glucose (5.5mM glucose) or hyperglycemic conditions (25mM glucose). When these cells were treated with cisplatin or doxorubicin, the cells conditioned to hyperglycemic conditions (HG) demonstrated a notable resistance to these drugs (Figures 1A-B). Both cells demonstrated significantly elevated levels of the two drug resistance proteins, ABCB1 and ABCG2 in conditions of elevated glucose. The levels of DNA damage repair genes, ERCC1 and XRCC1, were also upregulated in hyperglycemic conditions (Figure 1C). A similar result was also evident in MDA-MB-231 breast cancer cells which showed significantly increased expression of ABCG2, ERCC1, and XRCC1 (Supplementary Figure S1A).
Figure 1. Breast cancer cells demonstrate drug resistance and dysregulated Hh signaling in hyperglycemic conditions.
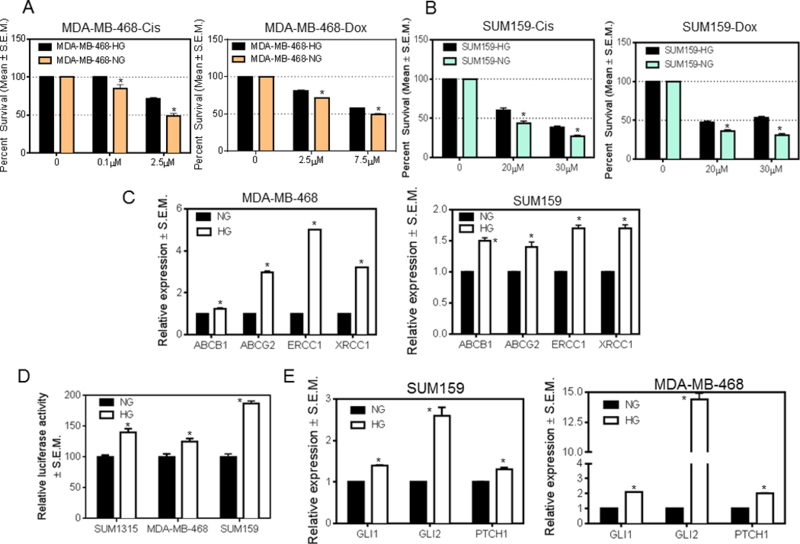
A, MDA-MB-468 cells show a decreased sensitivity to cisplatin (p=0.04 at 0.1μM; p=0.037 at 2.5μM) and doxorubicin (p=0.002 at 2.5μM; p=0.023 at 7.5μM) when conditioned in high glucose as compared to normoglycemic media. B, SUM159 cells also show a decreased sensitivity to cisplatin (p=0.017 for 20μM; p=0.009 for 30μM) and doxorubicin (p=0.01 for 20μM; p=0.001 for 30μM) when grown in high glucose versus normoglycemic media. C, multi-drug-resistance (ABCB1 and ABCG2) and DNA-damage-repair (ERCC1 and XRCC1) genes show increased expression in MDA-MB-468 (p<0.0001) and SUM159 (p=0.007 (ABCB1); p=0.01 (ABCG2); p=0.002 (ERCC1); p=0.003 (XRCC1)) cells grown in high versus normal glucose. D, GLI activity is significantly upregulated in cells grown in hyperglycemic medium relative to normoglycemic medium (p=0.004 (SUM1315); p=0.02 (468); p=0.0002 (SUM159)). E, Expression of GLI1, GLI2, and PTCH1 is increased in cells grown in hyperglycemic versus normoglycemic conditions (p<0.01 for all comparisons). The unpaired t-test was used for analysis.
We have previously demonstrated that Hh signaling functionally enhances drug resistance (30, 31); whereas inhibiting Hh activity with the Smoothened inhibitor cyclopamine or with RNA silencing significantly reduced expression of drug resistance and DNA repair proteins. We assessed the activity of GLI in SUM1315, MDA-MB-468, and SUM159 cells. Relative to normoglycemic conditions, cells conditioned in hyperglycemic conditions showed increased GLI transcriptional activity (read as luciferase activity) (Figure 1D). Increased GLI transcriptional activity is reflected in significantly increased levels of the bonafide GLI transcriptional targets, GLI1, GLI2, and PTCH (Figure 1E and Supplementary Figure S1B) in breast cancer cells.
O-GlcNAcylation of GLI proteins augments their activity.
We evaluated the levels and sub-cellular localization of the GLI proteins as a possible explanation for their increased activity. In hyperglycemic conditions, we detected increased nuclear presence of GLI1 and GLI2 in SUM159 and SUM1315 cells (Figure 2A). Moreover, the protein levels of GLI1 and GLI2 were notably increased in SUM159 and SUM1315 cells cultured in hyperglycemic conditions (Figure 2B). The increase in GLI levels corresponded with an elevated expression of OGT and decreased expression of OGA in high glucose conditions. This suggested a likelihood for higher cellular O-GlcNAcylation. Thus, we assessed the cellular O-GlcNAc landscape and determined that in elevated glucose culture conditions, breast cancer cells demonstrate an overall increase in O-GlcNAcylation (Figure 2C). In order to assess the possibility that GLI proteins may be O-GlcNAcylated, we immunoprecipitated the lysate of SUM159 breast cancer cells with RL2 antibody (recognizes O-GlcNAcylated residues) and immunoblotted for GLI1 and GLI2. In hyperglycemic conditions, GLI1 and GLI2 demonstrate evidence of being modified by O-GlcNAcylation (Figure 2D; Supplementary Figure S2A). We also confirmed this by immunoprecipitating endogenous GLI1 from SUM159 cells and assessed O-GlcNAc modification with RL2 antibody (Figure 2E). In an independent approach, we transfected a GLI1 expressing construct in MDA-MB-468 cells. When enriched by immunoprecipitation and immunoblotted with RL2, GLI1 showed evidence of being O-GlcNAc-ylated in hyperglycemic conditions (Figure 2E). In order to directly ascribe a role for O-GlcNAc-modification, we treated SUM159 cells with OGA inhibitor Thiamet-G and enriched for GLI1 and GLI2. Thiamet-G treatment resulted in greater signal intensity of O-GlcNAc-ylated GLI1 and GLI2 affirming direct O-GlcNAc-ylation of GLI proteins (Supplementary Figure S2B).
Figure 2. Hyperglycemic conditions increase cellular O-GlcNAcylation and promote nuclear accumulation of GLI transcription factor.
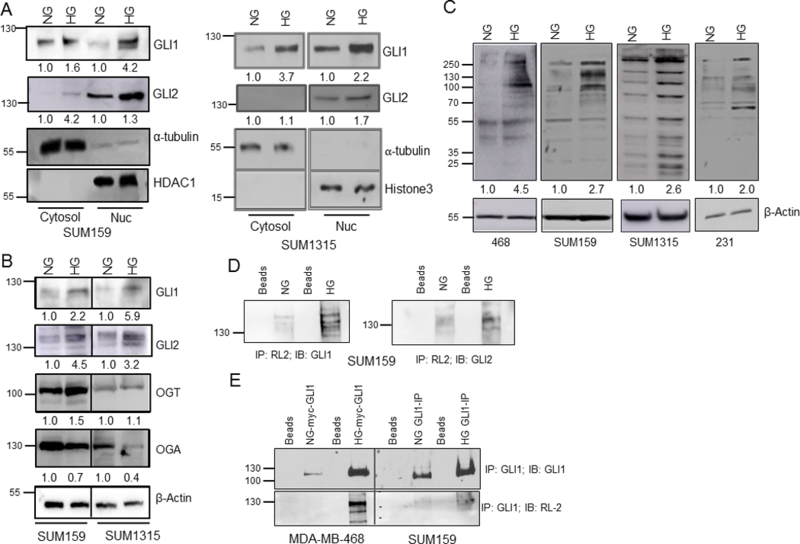
A, the activity of GLI1 and GLI2 are increased in high glucose versus normal glucose conditioned cells as evidenced by their increased nuclear presence in high glucose conditioned cells. Alpha-tubulin was used as a control for the cytosolic fraction, and HDAC1 or Histone H3 was used as a control for the nuclear fraction. B, levels of GLI1, GLI2, and OGT are elevated in cells grown in high versus normal glucose while levels of OGA are decreased in those cells. C, O-GlcNAcylation is increased in cells grown in high verses normal glucose media across several cell lines. D, the levels of O-GlcNAcylated GLI1 and GLI2 are increased in cells grown in high glucose. Lysates of SUM159 cells grown in normal or high glucose conditions were immunoprecipitated (IP’d) with RL2 antibody which detects O-GlcNAc residues. The IP’d proteins were immunoblotted for GLI1 or GLI2. E, the signal for O-GlcNAcylated GLI1 is increased in cells cultured in high glucose versus normal glucose conditions. Cell lysates were immunoprecipitated for GLI1, then immunoblotted with RL2. Endogenous GLI1 was IP’d from SUM159 cells; whereas in MDA-MB-468 cells, myc-GLI1 was transfected and IP’d. Both kinds of enrichments were immunoblotted with RL2 antibody. The numbers beneath each panel in panels A, B, C represent relative signal intensity (NG versus HG) normalized to endogenous control for each panel.
The protein sequences of GLI1 and GLI2 bear several potential sites of O-GlcNAc modifications (http://www.cbs.dtu.dk/services/YinOYang/) at Serine and Threonine residues; many of these sites are predicted to be at high threshold levels (Figure 3A-D). In order to functionally assign a role for O-GlcNAc modifications of GLI, we used a plasmid-based approach to regulate cellular O-GlcNAcylation. Introduction of the OGT-expressing and the OGA-dominant negative (DN) constructs resulted in elevated cellular O-GlcNAcylation, while expression of the OGA and OGT-DN constructs tempered the modifications in MDA-MB-468 cells (Supplementary Figure S2C). Expression of OGT and OGA-DN resulted in a significant increase in the transcriptional activity of GLI indicating that an increase in O-GlcNAcylation significantly impacts GLI activity while expression of OGA had modest effects on reduction of GLI activity (Figure 3E).
Figure 3. GLI transcription factors bear signatures of O-GlcNAcylation.
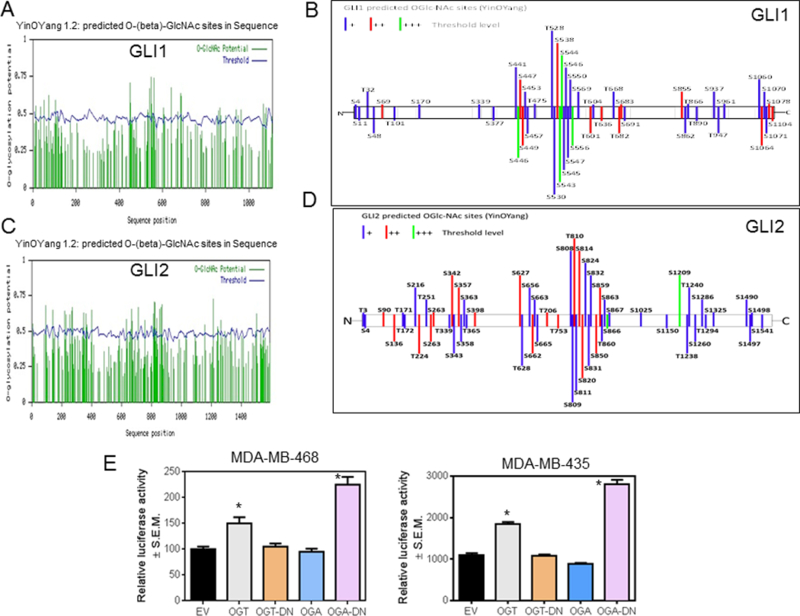
A and C, O-GlcNAcylated sites on GLI1 and GLI2 as predicted by YinOYang 1.2 Server. Potential sites are indicated by the green lines. Green lines that cross the threshold, as indicated by the wavy blue line, are predicted sites. B and D, the YinOYang prediction algorithm suggests that these proteins will be notably O-GlcNAc modified as shown in the schematics (15 of the 49 predicted O-GlcNAc sites in GLI1 and 19 out of 55 predicted O-GlcNAc sites in GLI2 have a projected threshold of ++ or +++). E, 8X-GLI activity is increased when OGT is knocked-in and OGA is knocked-out (p=0.03 (OGT) and p<0.0001 (OGA-DN)). The unpaired t-test was used for analysis.
Inhibiting OGT and OGA have opposing effects on GLI activity.
Thus far, our data demonstrate that GLI transcription factors are modified by O-GlcNAcylation in hyperglycemic conditions; further enhanced cellular O-GlcNAc-ylation significantly upregulates Hh/GLI activity. In order to assign mechanistic roles for OGT and OGA in regulating GLI activity, we used small molecule inhibitors. OSMI-1 is a small molecule OGT inhibitor developed from a high-throughput screening hit (32). Inhibition of OGT with OSMI-1 (Supplementary Figure S3A) in MDA-MB-468 and SUM1315 cells decreased GLI activity in a dose dependent manner indicating the importance of O-GlcNAcylation mediated by OGT in the transcriptional activity of GLI proteins (Figure 4A). Similar results were evident in SUM159 cells (Supplementary Figure 4A). Thiamet-G and PUGNAc, both inhibitors of OGA (Supplementary Figures S3B and C), increased the activity of GLI (Figure 4B and4C, Supplementary Figure 4B and4C). Elevated O-GlcNAcylation supports enhanced cell division (33, 34). As such, we assessed the effect of inhibiting OGA on cell proliferation. Treatment with PUGNAc significantly increased proliferation; this effect was notably reduced with the GLI inhibitor, GANT61 (Figure 4D). In order to test the role of O-GlcNAcylation in impacting Hh/GLI activity leading to augmentation of drug resistance, we evaluated SUM1315 cells in which we stably abrogated GLI1 expression. These cells show significantly downregulated expression of drug resistance proteins (Figure 4E) (30). Thiamet-G caused a notable upregulation in the expression of all drug resistance proteins; however, GLI1-silenced cells were hindered in their ability to upregulate expression of drug resistance proteins when treated with Thiamet-G (Figure 4E). This suggests that Hh/GLI activity is critical for mediating the effects of enhanced O-GlcNAcylaton on cell proliferation and manifestation of multidrug resistance.
Figure 4. O-GlcNAcylation functionally regulates the activity of GLI.
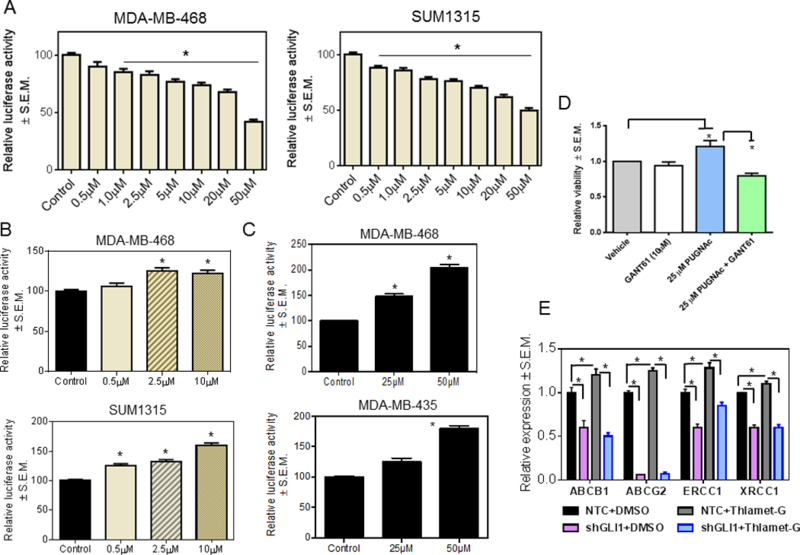
A, 8X-GLI luciferase activity is decreased in a dose-dependent manner by treatment with OSMI-1, an OGT inhibitor. DMSO was used as a control (p<0.0001 for all comparisons). B and C, inhibiting OGA with Thiamet-G (p<0.0001 for all comparisons) or PUGNAc ((MDA-MB-468: p=0.003 (25μM); p<0.0001 (50μM). MDA-MB-435: p=0.004 (25μM); p<0.0001 (50μM)) increases 8X-GLI activity in a dose dependent manner. DMSO was used as a control for Thiamet-G and ethanol for PUGNAc. D, MDA-MB-468 cells treated with PUGNAc show increased viability (p=0.04 for 25μM PUGNAc). This is reversed when the cells are co-treated with PUGNAc and GANT61 (p=0.0087 for PUGNAc+GANT61). E, SUM1315 cells stably transfected with a non-targeting shRNA control (NTC) or a GLI1-silencing plasmid (shGLI1), were treated with DMSO (vehicle control) or Thiamet-G. Thiamet-G upregulates transcript levels of the drug resistance proteins. GLI1 silencing renders cells unable to respond to Thiamet-G as evidenced by decreased expression of drug resistance proteins (*p<0.05). ANOVA was applied for data depicted in A and B and the unpaired t-test was used for analysis of data in C-E.
O-GlcNAcylation overcomes the inhibitory effects of limiting PKM2 on regulating GLI activity.
The M2 isoform of pyruvate kinase enzyme (PKM2) is enriched in cancer cells and functionally drives glycolysis. Post-translational modification of PKM2 including acetylation, phosphorylation, and O-GlcNAcylation inhibits its activity enabling upstream glycolytic intermediates to accumulate and thus contribute to the shift of metabolism towards biosynthetic pathways, including HBP (35–37). In liver cancer, ablating PKM led to a decrease in GLI expression in hepatocellular carcinoma cells (38). O-GlcNAc modifications on PKM2 protein resulted in elevated cellular levels of PKM2 but decreased PKM2-specific activity in colorectal carcinoma cells (Supplementary Figure S5A) (39). With these compelling data, we evaluated the effects of normo and hyperglycemic culture conditions on expression of PKM2 in breast cancer cells. While we did not register obvious changes in the levels of PKM2 protein in cells cultured in hyperglycemic conditions, there was a stark upregulation of PKM2 phosphorylated at Tyr105, a signature of inactive PKM2 (Figure 5A). As demonstrated earlier (Figure 2), these hyperglycemic conditions elicit upregulated Hh/GLI transcriptional activity. In order to evaluate the effects of PKM2 on GLI activity in hyperglycemic conditions, we silenced expression of endogenous PKM2. This resulted in a significant reduction in protein levels of GLI1 (Figure 5B) concomitant with a decrease in GLI transcriptional activity (Figure 5C) and expression of markers of drug resistance (Figure 5D), without impacting total cellular O-GlcNacylation (Supplementary Figure S5B). In conditions of elevated glucose, PUGNAc caused an elevation in GLI activity; PUGNAc was also able to partly overcome the effects of silencing PKM on GLI activity (Figure 5E). This suggests that PKM2 and O-GlcNAcylation have significant effects on regulating GLI activity. Enhanced O-GlcNAcylation overcomes the inhibitory effects of limiting PKM2 on regulating GLI activity. Cumulatively, these data suggest that PKM2 and O-GlcNAcylation jointly regulate the activity of Hh/GLI signaling.
Figure 5. PKM2 regulates Hh/Gli activity.
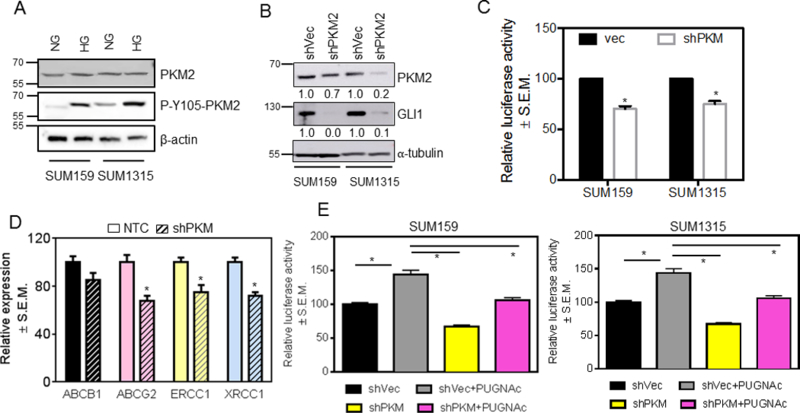
A, under high glucose conditions, the inactive form of PKM2, P-Y105, is increased. B, knocking-down PKM2 causes a decrease in protein levels of GLI1. The numbers beneath each panel represent relative signal intensity (shVec versus shPKM2) normalized to respective loading control. C, knocking-down PKM2 also inhibits the activity of GLI as shown by a decrease in activity of the 8X-GLI luciferase reporter (p=0.002 for SUM159; p=0.004 for SUM1315). D, knocking down PKM2 in SUM159 cells decreases expression of multi-drug-resistance (ABCB1 and ABCG2 (p=0.007)) and DNA-damage-repair (ERCC1 (p=0.04) and XRCC1 (p=0.005)) genes relative to control cells (transfected with non-targeting control siRNA, NTC). E, the effect on GLI1 activity of knocking-down PKM2 is reversed by the inhibition of OGA with PUGNAc (p<0.005 for all comparisons). The unpaired t-test was used for analysis.
Discussion
O-GlcNAcylation is considered a “rheostat” that controls the intensity of the signals traveling through different pathways according to the nutritional status of the cell (40). O-GlcNAc is a dynamic and ubiquitous posttranslational modification of serine and threonine residues and is emerging as an important regulator of cellular processes such as cell signaling, transcription, translation, apoptosis, cell proliferation and differentiation, among others. Cancer cells are known to be highly glycolytic to meet the cancer cells increased energy demands. In addition to increased glycolysis, elevated glucose uptake can also facilitate increased glucose flux through the HBP resulting in increased levels of protein O-GlcNAcylation. In addition an increase in OGT levels resulting from the PI3K-mTOR/MYC pathway facilitates the addition of more O-GlcNAc residues onto target proteins (11, 17). Cells also tend to see an increase of glucose going through the HBP when oncogenes are activated, and this trend is vital for cancer cell survival (12). This increased expression of O-GlcNAc residues may serve as a marker in cancer cells and also may promote cancer cell invasion and metastasis. Specifically, in breast cancer cells, the increase in OGT and GlcNAc levels was shown to occur with AKT and mTOR activation (10). OGT also functions to increase metastasis and cancer cell survival through decreasing SIRT1 levels thereby enabling the activity of FOXM1 on tumor cell invasion and metastasis (41). Several proteins that are known to be O-GlcNAcylated in breast cancer include but are not limited to: Akt, C-Myc, E-cadherin, p27(kip1), and PKM2 (39). Importantly, O-GlcNAcylation also has been demonstrated to enhance the activity of transcription factors in breast cancer including p53, NF-κB p65, c-Rel, FOXM1 (10), HIF-1α, and Sp1 (8, 18, 42, 43).
In this study, we have demonstrated that GLI1 and GLI2 transcription factors of the Hh pathway, are modified by O-GlcNAcylation. Elevated cellular O-GlcNAcylation, either by upregulating OGT or by abrogating OGA, led to an increase in the activity of GLI. Further, OSMI-1, a small molecule inhibitor of OGT, decreased GLI activity in stark contrast to inhibition of OGA (with Thiamet-G or PUGNAc). Thus, GLI activity is profoundly affected by O-GlcNAcylation. Under conditions of elevated glucose, the levels of GLI transcriptional targets, ABCB1, ABCG2, ERCC1, and XRCC1, were significantly elevated and associated with increased tumor cell survival in the presence of chemotherapeutic drugs. We focused the current study on cells representing the triple-negative type of breast cancer, which are characterized by exceptionally activated Hh signaling, lack of targeted therapy, and frequent emergence of drug resistance (44, 45). Due to the lack of targeted therapy, triple-negative breast cancer is usually administered cytotoxic chemotherapy including doxorubicin, docetaxel, or cisplatin. Disease relapse is due to the evolution and selection of a drug resistant tumor cell population. Resistance to doxorubicin predominantly involves the ABC proteins ABCB1, ABCC, and ABCG2 that act as ATP-dependent efflux pumps. Cisplatin resistance is largely conferred by activity of the ERCC1-XPF heterodimer that repairs DNA double-strand breaks. In breast cancer, increased ERCC1/XRCC1 expression indicates worse prognosis (46–49).
Hyper-O-GlcNAcylation is reported to induce tolerance to cisplatin, tamoxifen, and the PI-3-Kinase inhibitor Pictilisib (50–52). Modulation of cellular O-GlcNAc levels has been linked to altered cellular development, mitotic progression, growth, and survival patterns (53). Interestingly, we see that the increase in cell proliferation brought about by PUGNAc is blunted when the activity of Hh/GLI is inhibited, suggesting that GLI activity is an important determinant of the effects of cellular O-GlcNAc levels on cell growth. In this process, we also discovered an essential role for PKM2 in regulating the functions of GLI. PKM2 expression is critical in determining the glycolytic phenotype of cancer cells. PKM2 is expressed in tissues with anabolic functions, including proliferating cells and cancer cells, and is subject to complex allosteric regulation. In the majority of cancer cells, the expression of PKM2 is increased (32). Increased cellular O-GlcNAcylation is associated with a decrease in pyruvate kinase activity (39). It has been proposed that hyper-O-GlcNAcylation in cancer cells likely decreases PKM2 activity, which contributes to directing glycolytic intermediates toward biosynthetic pathways (54). In addition to being an important control point in glycolysis, PKM2 also modulates gene expression. PKM2 functions as a transcription co-activator of HIF-1α, Oct-4, and β-catenin and vitally impacts epigenetic regulation of gene transcription by phosphorylating Histone H3 (55, 56). While Hh/GLI signaling upregulates the expression of PKM2 (57), PKM2 is reported to transcriptionally activate GLI1 gene expression (38), thereby presenting a metabolically important feed-forward loop. Our data reveal that in conditions of hyperglycemia, PKM2 is phosphorylated at Y105, which is a signature of its existence in the inactive dimeric form. Phosphorylation of PKM2 at Y105 is regulated by FGFR1 activity (36). Thus, less active PKM2 channels the metabolic flux to HBP, which increases O-GlcNAcylation. This is in agreement with the overall increase in cellular O-GlcNAcylation that we see in conditions of elevated glucose. Consistent with prior reports, abrogating cellular PKM2 significantly blunted the transcriptional activity of GLI (38) and decreased the expression of drug resistance proteins. While increased cellular O-GlcNAcylation by PUGNAc culminated in increased GLI activity, the activity of PUGNAc was tempered when PKM was silenced, indicating that PKM2 and O-GlcNAcylation jointly regulate the activity of Hh/GLI signaling.
The body of data presented in the current work indicates that under high glucose condition there is an increase in O-GlcNAcylation which is likely sustained by the upregulation of the mTOR pathway (with the removal of AMPK inhibition) (Supplementary Figure S5C). Transcription factors GLI1 and GLI2 are O-GlcNAcylated in these conditions and this modification functionally enhances their transcriptional activity. It is likely that simultaneous nuclear localization of PKM2 in conditions of elevated glucose, as evidenced by its phosphorylation at S37, not only sustains O-GlcNAcylation by increasing flux through HBP, but also activates the GLI transcription factors (Supplementary Figure S5D).
Hh signaling has been demonstrated by us and others to promote epithelial-mesenchymal-transition (EMT) characterized by molecular alterations and enhanced invasion, migration, and metastasis (26, 29, 58, 59). Many of the signature EMT proteins are modified by glycans containing GlcNAc. This includes SNAI1, β-catenin, NF-κB, c-Myc, c/EBPβ, E-cadherin, N-cadherin, Wnt ligands, Hh ligands, and FOXO1 (60). Inhibition of glucose uptake and metabolism disabled the HBP pathway and suppressed known oncogenic pathways resulting in phenotypic reversion in breast cancer cells in 3D assays (61). Thus, inhibiting OGT and O-GlcNAcylation in cancer cells is likely to have beneficial outcomes, particularly in cancer patients with diabetes or metabolic disease (62). Interfering in O-GlcNAcylation of Hh transcription factors may be a novel target in controlling cancer metabolism and progression and/or drug resistance of breast cancer.
Supplementary Material
Acknowledgements
We would like to acknowledge funding from NCI R01CA169202, the Department of Defense (W81XWH-14–1-0516 and W81XWH-18–1-0036), The Breast Cancer Research Foundation of Alabama (BCRFA), AMC21 funds from UAB, Diabetes Research Center Pilot Grant, and The George G. and Amelia G. Tapper Foundation to L.A.S.
Abbreviations:
- ABCB1
ATP Binding Cassette Subfamily B Member 1
- ABCG2
ATP Binding Cassette Subfamily G Member 2
- AMPK
Adenosine monophosphate-activated protein kinase
- DHH
Desert Hedgehog
- EMT
epithelial-mesenchymal-transition
- ERCC1
ERCC Excision Repair 1, Endonuclease Non-Catalytic Subunit
- FGFR1
Fibroblast Growth Factor Receptor 1
- FOXM1
Forkhead Box M1
- GLI1
Glioma-Associated Oncogene Family Zinc Finger 1
- GLI2
Glioma-Associated Oncogene Family Zinc Finger 2
- HBP
Hexosamine Biosynthesis Pathway
- HG
hyperglycemic
- Hh
Hedgehog
- HIF-1α
Hypoxia Inducible Factor 1 Alpha Subunit
- IHH
Indian Hedgehog
- MDR
multi-drug resistance
- mTOR
Mammalian Target of Rapamycin
- NF-κB
Nuclear Factor Kappa B
- NG
normoglycemic
- OGA
O-GlcNAcase
- O-GlcNAc
O-linked β-N-actylglucosamine
- OGT
O-GlcNAc transferase
- PI3K
Phosphatidylinositol-3-Kinase
- PKM2
Pyruvate kinase muscle isozyme M2
- PTB
polypyrimidine-tract binding protein
- PTCH1
Patched 1
- SHH
Sonic Hedgehog
- SIRT1
Sirtuin 1
- SMO
Smoothened
- SNAI1
Snail Family Transcriptional Repressor 1
- XRCC1
X-Ray Repair Cross Complementing 1
Footnotes
Disclosure/Conflict of Interest
None of the authors have any conflict of interest that could be perceived to bias their work.
Author Contributions
All authors have read and approved the manuscript. S.D, S.K.B, D.C.H, B.J.M., R.S.S., J.C.C., A.F., and L.A.S. contributed to the conception and design of this work. S.D., S.K.B., and L.A.S. were involved in the development of methodology. S.D., S.K.B. B.J.M., D.C.H., A.H., M.M. were responsible for acquisition of data. S.D., S.K.B., B.J.M., A.H., R.S.S., J.C.C., L.A.S. analyzed and interpreted the data. All authors were involved in writing the manuscript.
REFERENCES
- 1.Boyle P, Boniol M, Koechlin A, et al. Diabetes and breast cancer risk: a meta-analysis. Br J Cancer 2012;107(9):1608–1617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lipscombe LL, Goodwin PJ, Zinman B, McLaughlin JR, Hux JE. Diabetes mellitus and breast cancer: a retrospective population-based cohort study. Breast Cancer Res Treat 2006;98(3):349–356. [DOI] [PubMed] [Google Scholar]
- 3.Lipscombe LL, Goodwin PJ, Zinman B, McLaughlin JR, Hux JE. The impact of diabetes on survival following breast cancer. Breast Cancer Res Treat 2008;109(2):389–395. [DOI] [PubMed] [Google Scholar]
- 4.Ryu TY, Park J, Scherer PE. Hyperglycemia as a risk factor for cancer progression. Diabetes Metab J 2014;38(5):330–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hart GW, Akimoto Y. The O-GlcNAc Modification In: Varki A, Cummings RD, Esko JD, et al. editors. Essentials of Glycobiology, 2nd edn Cold Spring Harbor (NY), 2009. [Google Scholar]
- 6.Slawson C, Hart GW. O-GlcNAc signalling: implications for cancer cell biology. Nat Rev Cancer 2011;11(9):678–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.West CM, Hart GW. Nucleocytoplasmic Glycosylation In: Varki A, Cummings RD, Esko JD, et al. editors. Essentials of Glycobiology, 3rd edn Cold Spring Harbor (NY), 2015. [Google Scholar]
- 8.Zachara N, Akimoto Y, Hart GW. The O-GlcNAc Modification In: Varki A, Cummings RD, Esko JD, et al. editors. Essentials of Glycobiology, 3rd edn Cold Spring Harbor (NY), 2015. [Google Scholar]
- 9.Banerjee PS, Lagerlof O, Hart GW. Roles of O-GlcNAc in chronic diseases of aging. Mol Aspects Med 2016;51:1–15. [DOI] [PubMed] [Google Scholar]
- 10.Caldwell SA, Jackson SR, Shahriari KS, et al. Nutrient sensor O-GlcNAc transferase regulates breast cancer tumorigenesis through targeting of the oncogenic transcription factor FoxM1. Oncogene 2010;29(19):2831–2842. [DOI] [PubMed] [Google Scholar]
- 11.Gu Y, Mi W, Ge Y, et al. GlcNAcylation plays an essential role in breast cancer metastasis. Cancer Res 2010;70(15):6344–6351. [DOI] [PubMed] [Google Scholar]
- 12.Lynch TP, Ferrer CM, Jackson SR, Shahriari KS, Vosseller K, Reginato MJ. Critical role of O-Linked beta-N-acetylglucosamine transferase in prostate cancer invasion, angiogenesis, and metastasis. J Biol Chem 2012;287(14):11070–11081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mi W, Gu Y, Han C, et al. O-GlcNAcylation is a novel regulator of lung and colon cancer malignancy. Biochim Biophys Acta 2011;1812(4):514–519. [DOI] [PubMed] [Google Scholar]
- 14.Yehezkel G, Cohen L, Kliger A, Manor E, Khalaila I. O-linked beta-N-acetylglucosaminylation (O-GlcNAcylation) in primary and metastatic colorectal cancer clones and effect of N-acetyl-beta-D-glucosaminidase silencing on cell phenotype and transcriptome. J Biol Chem 2012;287(34):28755–28769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhu Q, Zhou L, Yang Z, et al. O-GlcNAcylation plays a role in tumor recurrence of hepatocellular carcinoma following liver transplantation. Medical oncology 2012;29(2):985–993. [DOI] [PubMed] [Google Scholar]
- 16.Shi Y, Tomic J, Wen F, et al. Aberrant O-GlcNAcylation characterizes chronic lymphocytic leukemia. Leukemia 2010;24(9):1588–1598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ma Z, Vosseller K. Cancer metabolism and elevated O-GlcNAc in oncogenic signaling. J Biol Chem 2014;289(50):34457–34465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ferrer CM, Lynch TP, Sodi VL, et al. O-GlcNAcylation regulates cancer metabolism and survival stress signaling via regulation of the HIF-1 pathway. Mol Cell 2014;54(5):820–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Champattanachai V, Netsirisawan P, Chaiyawat P, et al. Proteomic analysis and abrogated expression of O-GlcNAcylated proteins associated with primary breast cancer. Proteomics 2013;13(14):2088–2099. [DOI] [PubMed] [Google Scholar]
- 20.Krzeslak A, Forma E, Bernaciak M, Romanowicz H, Brys M. Gene expression of O-GlcNAc cycling enzymes in human breast cancers. Clinical and experimental medicine 2012;12(1):61–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chaiyawat P, Netsirisawan P, Svasti J, Champattanachai V. Aberrant O-GlcNAcylated Proteins: New Perspectives in Breast and Colorectal Cancer . Frontiers in endocrinology 2014;5:193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Slawson C, Pidala J, Potter R. Increased N-acetyl-beta-glucosaminidase activity in primary breast carcinomas corresponds to a decrease in N-acetylglucosamine containing proteins. Biochim Biophys Acta 2001;1537(2):147–157. [DOI] [PubMed] [Google Scholar]
- 23.Shevde LA, Samant RS. Nonclassical hedgehog-GLI signaling and its clinical implications. Int J Cancer 2014;135(1):1–6. [DOI] [PubMed] [Google Scholar]
- 24.Hanna A, Shevde LA. Hedgehog signaling: modulation of cancer properies and tumor mircroenvironment. Mol Cancer 2016;15(1):24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Das S, Samant RS, Shevde LA. The hedgehog pathway conditions the bone microenvironment for osteolytic metastasis of breast cancer. International journal of breast cancer 2012;2012:298623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Das S, Harris LG, Metge BJ, et al. The hedgehog pathway transcription factor GLI1 promotes malignant behavior of cancer cells by up-regulating osteopontin. J Biol Chem 2009;284(34):22888–22897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yang X, Su K, Roos MD, Chang Q, Paterson AJ, Kudlow JE. O-linkage of N-acetylglucosamine to Sp1 activation domain inhibits its transcriptional capability. Proc Natl Acad Sci U S A 2001;98(12):6611–6616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Toleman C, Paterson AJ, Whisenhunt TR, Kudlow JE. Characterization of the histone acetyltransferase (HAT) domain of a bifunctional protein with activable O-GlcNAcase and HAT activities. J Biol Chem 2004;279(51):53665–53673. [DOI] [PubMed] [Google Scholar]
- 29.Harris LG, Pannell LK, Singh S, Samant RS, Shevde LA. Increased vascularity and spontaneous metastasis of breast cancer by hedgehog signaling mediated upregulation of cyr61. Oncogene 2012;31(28):3370–3380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Das S, Samant RS, Shevde LA. Nonclassical activation of Hedgehog signaling enhances multidrug resistance and makes cancer cells refractory to Smoothened-targeting Hedgehog inhibition. J Biol Chem 2013;288(17):11824–11833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kudo K, Gavin E, Das S, Amable L, Shevde LA, Reed E. Inhibition of Gli1 results in altered c-Jun activation, inhibition of cisplatin-induced upregulation of ERCC1, XPD and XRCC1, and inhibition of platinum-DNA adduct repair. Oncogene 2012. [DOI] [PubMed] [Google Scholar]
- 32.Ortiz-Meoz RF, Jiang J, Lazarus MB, et al. A small molecule that inhibits OGT activity in cells. ACS Chem Biol 2015;10(6):1392–1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zachara NE, Hart GW. Cell signaling, the essential role of O-GlcNAc! Biochim Biophys Acta 2006;1761(5–6):599–617. [DOI] [PubMed] [Google Scholar]
- 34.Zachara NE, Hart GW. The emerging significance of O-GlcNAc in cellular regulation. Chemical reviews 2002;102(2):431–438. [DOI] [PubMed] [Google Scholar]
- 35.Wong N, De Melo J, Tang D. PKM2, a Central Point of Regulation in Cancer Metabolism. Int J Cell Biol 2013;2013:242513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hitosugi T, Kang S, Vander Heiden MG, et al. Tyrosine phosphorylation inhibits PKM2 to promote the Warburg effect and tumor growth. Science signaling 2009;2(97):ra73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wellen KE, Thompson CB. A two-way street: reciprocal regulation of metabolism and signalling. Nature reviews Molecular cell biology 2012;13(4):270–276. [DOI] [PubMed] [Google Scholar]
- 38.Xu Q, Liu X, Zheng X, Yao Y, Liu Q. PKM2 regulates Gli1 expression in hepatocellular carcinoma. Oncology letters 2014;8(5):1973–1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chaiyawat P, Chokchaichamnankit D, Lirdprapamongkol K, Srisomsap C, Svasti J, Champattanachai V. Alteration of O-GlcNAcylation affects serine phosphorylation and regulates gene expression and activity of pyruvate kinase M2 in colorectal cancer cells. Oncol Rep 2015;34(4):1933–1942. [DOI] [PubMed] [Google Scholar]
- 40.Issad T, Masson E, Pagesy P. O-GlcNAc modification, insulin signaling and diabetic complications. Diabetes & metabolism 2010;36(6 Pt 1):423–435. [DOI] [PubMed] [Google Scholar]
- 41.Ferrer CM, Lu TY, Bacigalupa ZA, Katsetos CD, Sinclair DA, Reginato MJ. O-GlcNAcylation regulates breast cancer metastasis via SIRT1 modulation of FOXM1 pathway. Oncogene 2017;36(4):559–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hart GW. Nutrient regulation of immunity: O-GlcNAcylation regulates stimulus-specific NF-kappaB-dependent transcription. Science signaling 2013;6(290):pe26. [DOI] [PubMed] [Google Scholar]
- 43.Dias WB, Cheung WD, Hart GW. O-GlcNAcylation of kinases. Biochem Biophys Res Commun 2012;422(2):224–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tao Y, Mao J, Zhang Q, Li L. Overexpression of Hedgehog signaling molecules and its involvement in triple-negative breast cancer. Oncology letters 2011;2(5):995–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhao J, Chen G, Cao D, et al. Expression of Gli1 correlates with the transition of breast cancer cells to estrogen-independent growth. Breast Cancer Res Treat 2010;119(1):39–51. [DOI] [PubMed] [Google Scholar]
- 46.Bewick MA, Lafrenie RM, Conlon MS. Nucleotide excision repair polymorphisms and survival outcome for patients with metastatic breast cancer. Journal of cancer research and clinical oncology 2011;137(3):543–550. [DOI] [PubMed] [Google Scholar]
- 47.Gerhard R, Carvalho A, Carneiro V, et al. Clinicopathological significance of ERCC1 expression in breast cancer. Pathology, research and practice 2013;209(6):331–336. [DOI] [PubMed] [Google Scholar]
- 48.Ozkan C, Gumuskaya B, Yaman S, Aksoy S, Guler G, Altundag K. ERCC1 expression in triple negative breast cancer. Journal of BUON : official journal of the Balkan Union of Oncology 2012;17(2):271–276. [PubMed] [Google Scholar]
- 49.Pei XH, Yang Z, Lv XQ, Li HX. Genetic variation in ERCC1 and XPF genes and breast cancer risk. Genetics and molecular research : GMR 2014;13(1):2259–2267. [DOI] [PubMed] [Google Scholar]
- 50.Luanpitpong S, Angsutararux P, Samart P, Chanthra N, Chanvorachote P, Issaragrisil S. Hyper-O-GlcNAcylation induces cisplatin resistance via regulation of p53 and c-Myc in human lung carcinoma. Scientific reports 2017;7(1):10607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kanwal S, Fardini Y, Pagesy P, et al. O-GlcNAcylation-inducing treatments inhibit estrogen receptor alpha expression and confer resistance to 4-OH-tamoxifen in human breast cancer-derived MCF-7 cells. PloS one 2013;8(7):e69150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kwei KA, Baker JB, Pelham RJ. Modulators of sensitivity and resistance to inhibition of PI3K identified in a pharmacogenomic screen of the NCI-60 human tumor cell line collection. PloS one 2012;7(9):e46518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang S, Huang X, Sun D, et al. Extensive crosstalk between O-GlcNAcylation and phosphorylation regulates Akt signaling. PloS one 2012;7(5):e37427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fardini Y, Dehennaut V, Lefebvre T, Issad T. O-GlcNAcylation: A New Cancer Hallmark? Frontiers in endocrinology 2013;4:99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yang W, Xia Y, Ji H, et al. Nuclear PKM2 regulates beta-catenin transactivation upon EGFR activation. Nature 2011;480(7375):118–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Macintyre AN, Rathmell JC. PKM2 and the tricky balance of growth and energy in cancer. Mol Cell 2011;42(6):713–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Di Magno L, Manzi D, D’Amico D, et al. Druggable glycolytic requirement for Hedgehog-dependent neuronal and medulloblastoma growth. Cell cycle 2014;13(21):3404–3413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Thayer SP, di Magliano MP, Heiser PW, et al. Hedgehog is an early and late mediator of pancreatic cancer tumorigenesis. Nature 2003;425(6960):851–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ramaswamy B, Lu Y, Teng KY, et al. Hedgehog signaling is a novel therapeutic target in tamoxifen-resistant breast cancer aberrantly activated by PI3K/AKT pathway. Cancer Res 2012;72(19):5048–5059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Taparra K, Tran PT, Zachara NE. Hijacking the Hexosamine Biosynthetic Pathway to Promote EMT-Mediated Neoplastic Phenotypes. Front Oncol 2016;6:85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Onodera Y, Nam JM, Bissell MJ. Increased sugar uptake promotes oncogenesis via EPAC/RAP1 and O-GlcNAc pathways. The Journal of clinical investigation 2014;124(1):367–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Itkonen HM, Gorad SS, Duveau DY, et al. Inhibition of O-GlcNAc transferase activity reprograms prostate cancer cell metabolism. Oncotarget 2016;7(11):12464–12476. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.