

SUMMARY
Connexin43 (Cx43; gene name GJA1) is the most ubiquitously expressed gap junction protein, and understanding of its regulation largely falls under transcription and post-translational modification. In addition to Cx43, Gja1 mRNA encodes internally translated isoforms regulating gap junction formation, whose expression is modulated by TGF-β. Here, using RLM-RACE, we identify distinct Gja1 transcripts differing only in 5′ UTR length, of which two are upregulated during TGF-β exposure and hypoxia. Introduction of these transcripts into Gja1−/− cells phenocopies the response of Gja1 to TGF-β with reduced internal translation initiation. Inhibiting pathways downstream of TGF-β selectively regulates levels of Gja1 transcript isoforms and translation products. Reporter assays reveal enhanced translation of full-length Cx43 from shorter Gja1 5′ UTR isoforms. We also observe a correlation among UTR selection, translation, and reduced gap junction formation in aged heart tissue. These data elucidate a relationship between transcript isoform expression and translation initiation regulating intercellular communication.
In Brief
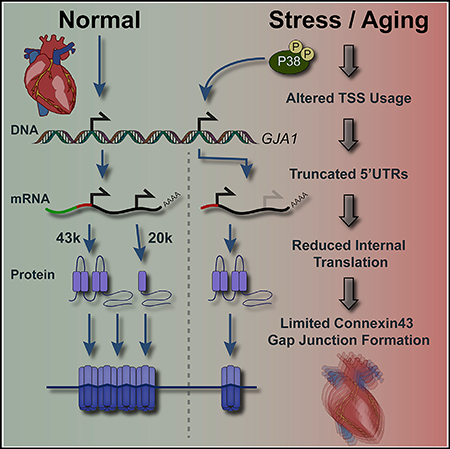
Connexin43 gap junctions enable direct intercellular communication facilitating action potential propagation. Internal translation of connexin43 mRNA generates the truncated isoform GJA1–20k, which promotes gap junction formation. During aging, Zeitz et al. find that activation of stress-response pathways shortens connexin43 mRNA UTRs to limit GJA1–20k translation coincident with gap junction loss.
Graphical Abstract
INTRODUCTION
Connexin43 (Cx43; gene name GJA1) (Fishman et al., 1990) is the most ubiquitously expressed gap junction protein, affecting direct intercellular communication crucial to organ function, such as in the heart where its expression and localization are tightly regulated to enable electrical coupling and maintain rhythmic contractions (Peters et al., 1997). Additional functions of Cx43 encompass cytoskeletal, metabolic, developmental, and cell cycle regulation, so it is unsurprising that alterations in Cx43 expression are implicated in a multitude of human pathologies (Chen et al., 2015; Clasadonte et al., 2017; Crespin et al., 2010; Meyer et al., 1997; Moorby and Patel, 2001). GJA1 mRNA encodes N-terminally truncated protein isoforms arising from alternative translation initiation within its coding sequence (Salat-Canela et al., 2014; Smyth and Shaw, 2013; Ul-Hussain et al., 2014). Tight regulation of GJA1 protein expression extends to its internally translated isoforms, as experimentally eliminating four of these isoforms, namely, GJA1–32k, GJA1–29k, GJA1–26k, and GJA1–20k, impairs gap junction formation reviewed in Basheer and Shaw (2016). Interestingly, reintroduction of GJA1–20k is able to rescue gap junction formation (Smyth and Shaw, 2013). We have subsequently identified changes in Gja1 translation initiation site selection in response to transforming growth factor β (TGF-β )-induced epithelial-mesenchymal transition (EMT), demonstrating the role of alternative translation in a physiological process that limits gap junction formation (James et al., 2018). Additionally, modulation of GJA1–20k expression has been reported during pathological stressors, including chemically induced hypoxia and cardiac ischemia (Basheer et al., 2018; Basheer et al., 2017; Ul-Hussain et al., 2014). The GJA1–20k fragment was also found in a model of fetal alcohol spectrum disorder (FASD) (Ramani et al., 2016). Although some signaling pathways have been implicated, the cellular mechanisms regulating changes in Gja1 translation are unknown.
Regulation of translation initiation can proceed through canonical and non-canonical mechanisms. Under normal physiological conditions, translation initiation typically involves eIF4F binding to the mRNA methyl 7-guanosine (m7G) cap, which is followed by formation and recruitment of the 43S pre-initiation complex (PIC). The PIC then scans for a start codon in a favorable context to initiate translation (Liu and Qian, 2014). Under conditions of cellular stress, traditional cap-dependent translation is suppressed, and many non-canonical modes of translation initiation are used to facilitate expression of key stress response and survival proteins (Holcik and Sonenberg, 2005). To date, it is unclear which mode of translation regulates the balance of Cx43 and its internally translated isoforms, but cap-dependency has been indicated (Salat-Canela et al., 2014). Regardless of the mechanism, a common feature of these modes of translation initiation is reliance on cis-acting elements within the 5′ UTR of mRNA.
Although the coding sequence of Gja1 is composed of an invariable single exon, one region of variability in Gja1 RNA composition is its 5′ UTR. Synthesis of this region was found to be regulated in a cell-type-specific manner through a combination of alternate promoter usage and splicing (Pfeifer et al., 2004). Gja1 5′ UTR variants were demonstrated to affect translation in vitro possibly through the use of an internal ribosome entry site (IRES) contained within the 5′ UTR (Pfeifer et al., 2004; Schiavi et al., 1999). Our previous work revealed upregulation of total Gja1 mRNA in response to TGF-β-induced EMT and identified downstream pathways responsible for subsequent translational changes (James et al., 2018). Here, we evaluate whether these signaling pathways play a role in Gja1 alternate promoter usage. Many of these pathways converge on activating protein 1 (AP-1), and this set of transcription factors has been demonstrated to regulate Gja1 expression (Geimonen et al., 1998; Geimonen et al., 1996; Hernandez et al., 2006). The presence of multiple transcription start sites (TSSs) for a given gene is common, and modulation of TSS selection by alternate promoter usage has been demonstrated to affect translational efficiency genome-wide (Cheng et al., 2018; Leenen et al., 2016; Rojas-Duran and Gilbert, 2012). This led us to hypothesize a role for TSS selection in regulating Gja1 alternative translation. We confirm this hypothesis in cell lines and in an in vivo model of cardiac aging, where remodeling of Cx43 at the intercalated disc has been demonstrated (Bonda et al., 2016). Here, we report the presence of stress responsive, dynamic alterations in TSS yielding Gja1 5′ UTR variants that regulate translation efficiency, alternative translation, and gap junction formation.
RESULTS
TGF-β Induces Translational and Transcriptional Changes to Gja1
In normal murine mammary gland (NMuMG) cells, we verify Cx43 induction with a decrease in the ratio of GJA1–20k to Cx43 by western blotting following stimulation with TGF-β (Figures 1A and 1B). Gja1 has been reported to express tissue-specific 5′ UTR isoforms capable of regulating translational efficiency (Pfeifer et al., 2004). We, therefore, performed RNA-ligase-mediated rapid amplification of cDNA ends (RLM-RACE) to determine whether TGF-β stimulation induces dynamic 5′ UTR alterations accompanying changes in alternative translation. RNA was isolated from NMuMG epithelial cells with or without 48 h of TGF-β treatment. Adaptor and gene-specific primers were used to amplify the entire Gja1 5′ UTR cDNA (Figure 1C). Interestingly, the prominent 5′ UTR isoforms from control and TGF-β stimulated RLM-RACE samples are unique, with the most intense bands from TGF-β-stimulated cells being shorter than those from untreated control cells (Figure 1D). DNA was gel extracted and Sanger sequenced with Gja1 5′ UTR sequences mapping to the most 5′ region of Gja1 exon 1, approximately 10.2 kb upstream of the start codon in the mouse genome (Figure 1E, green bar). These Gja1 5′ UTR isoforms labeled long (L), medium (M), and short (S) align with peaks of Cap analysis gene expression (CAGE) reads in the University of California, Santa Cruz (UCSC) genome browser, mouse genome build mm10 (Figure 1F), a readout from genome-wide cap analysis of gene expression representing TSSs from multiple cell types (Forrest et al., 2014; Lizio et al., 2015). This, therefore, suggests alternate promoter usage is responsible for generating truncated Gja1 5′ UTR isoforms arising from unique TSSs.
Figure 1. TGF-β Induces Translational and Transcriptional Changes to Gja1.

(A) Western blot of cell lysates probed with Cx43 C-terminal antibody to detect full-length Cx43 and internally translated GJA1–20k in control (C) and following 48 h of TGF-β treatment (T). α-tubulin is the loading control.
(B) Quantification of bands in (A).
(C) Schematic of primers in RACE procedure. Gene-specific reverse-transcription (RT) primer, primary RACE PCR (green arrows), and nested RACE PCR (orange arrows).
(D) Agarose gel electrophoresis of RACE PCR products in control (C), and following 48 h of TGF-β treatment (T).
(E) Location of RACE products in the Gja1 5′ UTR DNA.
(F) RACE products and CAGE reads mapped to mm10, UCSC genome browser.
See Table S2 for RLM-RACE primers. Statistical analysis performed using the Student’s t test (n = 3), **p ≤ 0.01.
Gja1 5′ UTR Isoforms Regulate Canonical and Alternative Translation
We next performed qRT-PCR to quantify the abundance of each 5′ UTR because RLM-RACE is not quantitative. Because truncated Gja1 5′ UTR isoforms share the same sequence as longer isoforms, absolute quantitation was performed using non-overlapping primers and by comparison to a standard curve of amplification products from a cloned Gja1 5′ UTR template. Once absolute copy numbers were calculated, overlapping transcripts were corrected by subtraction, and fold difference relative to L was plotted (Figure 2A). In untreated cells, S has the highest abundance, followed by M and L (Figures 2A and 2B).
Figure 2. UTR Isoforms Regulate Alternative Translation.

(A) qRT-PCR analysis of baseline levels of UTR isoforms.
(B) Schematic of UTR isoforms.
(C–E) qRT-PCR analysis of L (C), M (D), and S (E) UTR isoforms following treatment with vehicle (Veh; DMSO), TGF-β (T), SB 202190 (p), and SB431542 (SB).
(F) Western blot analysis of Cx43 following treatment as in graphs (C–E) with the addition of SP 600125 (G) and α-tubulin loading control.
(G) Quantification of bands from (F).
(H) Western blot of Gja1 translation products 24 h post-transfection into Gja1−/− cells with α-tubulin loading control.
(I) Quantification of bands from (H).
See Table S1 for primer sequences. Statistical analysis was performed using one-way ANOVA with Tukey’s multiple comparison test (n = 3). *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001.
The Gja1 5′ UTR encompasses multiple putative promoters (Pfeifer et al., 2004). Because alternate promoter usage is likely responsible for generating truncated Gja1 5′ UTRs in response to TGF-β (Figure 1F), we analyzed this genomic region using MatInspector software (Markoff, 2005) to identify transcription factor binding sites. We identified high-confidence transcription factor binding sites specific for AP-1, proximal to our putative TSSs within the 5′ UTR, consistent with published reports of its role in Gja1 expression (data not shown; Echetebu et al., 1999; Geimonen et al., 1998; Geimonen et al., 1996; Hernandez et al., 2006; Tacheau et al., 2008). TGF-β activates p38 mitogen-activated protein kinase (p38) through TAK1 upstream of AP-1 (Sorrentino et al., 2008; Tacheau et al., 2008; Watkins et al., 2006; Yamaguchi et al., 1995). To determine if this pathway contributes to the increase in TGF-β-induced Gja1 UTR isoforms, qRT-PCR was performed on RNA isolated from NMuMG cells following 24 h of TGF-β stimulation alone or in combination with the p38 inhibitor SB202190. TGF-β stimulation significantly increases the M and S Gja1-5′ UTRs with no change in expression of L (Figures 2C–2E). Inhibition of p38 prevents the increase in truncated Gja1 5′ UTRs M and S following TGF-β treatment, implicating this pathway in regulating alternate Gja1 promoter usage to increase M and S Gja1 5′ UTR isoform abundance in response to TGF-β.
To determine if the TGF-β-activated pathways p38 and/or c-Jun N-terminal kinase (JNK) contribute to TGF-β-induced increases in Cx43 protein, the p38 inhibitor SB202190 or JNK inhibitor SP600125 was applied 15 min prior to TGF-β treatment and protein was harvested after 24 h. Cx43 significantly increases following 24 h of exposure to TGF-β, whereas inhibition of p38 prevents the TGF-β-dependent increase in Cx43 (Figures 2F and 2G). JNK inhibition with TGF-β does not significantly reduce the increase in Cx43, and combining SB202190 and SP600125 does not result in a synergistic effect. This finding highlights the necessity for p38-activated alternate promoter usage for the TGF-β-induced increase in full-length Cx43 expression.
Alternative translation is regulated by cis and trans-acting factors (Faye and Holcik, 2015; Hellen and Sarnow, 2001; Stoneley and Willis, 2004), leading us to investigate whether the expression of truncated Gja1 5′ UTR isoforms alone affect alternative translation initiation. Isoforms L, M, and S were cloned upstream of the Gja1 coding sequence, and plasmid constructs were transfected into Gja1−/− cells, which were generated using CRISPR/Cas9 to excise the Cx43 coding sequence from 293FT and NMuMG cell lines (Figure S1). Protein was harvested 24 h post-transfection for western blot analysis (Figure 2H). When transfected into Gja1−/− NMuMG cells, both M and S isoforms result in significantly reduced GJA1–20k to Cx43 translation, compared with the L isoform (Figure 2I). This change in protein isoform ratio is the result of decreased GJA1–20k levels for the M isoform and increased Cx43 levels for the S isoform. When transfected into GJA1−/− 293FT cells, the M isoform has significantly reduced GJA1–20k to Cx43 translation compared with the L isoform (Figure S2). This indicates that UTR truncation alone is sufficient to phenocopy the effects on Gja1 translation following stimulation with TGF-β, resulting in reduced GJA1–20k relative to full-length Cx43 expression, which we had observed during EMT (James et al., 2018).
Previously, we found that inhibition of Erk signaling in NMuMG cells results in a robust increase in GJA1–20k to Cx43 ratios, opposite to the effect observed with TGF-β stimulation (Figures 3A and 3B) (James et al., 2018). We hypothesized that Erk inhibition would result in differential expression levels of L, M, or S Gja1 5′ UTR isoforms. Following 24 h of treatment with Erk inhibitor SCH772984 or vehicle, RNA was isolated and UTRs quantified by qRT-PCR. The L UTR increases significantly following Erk inhibition, whereas M and S are significantly downregulated (Figures 3C–3E). This further demonstrates the active regulatory effect of Gja1-UTR isoform expression in modulating alternative translation.
Figure 3. ERK Regulates Gja1 Transcript Isoforms and Alternative Translation.

(A) Western blot of cell lysates probed with Cx43 C-terminal antibody to detect full-length Cx43 and internally translated GJA1–20k following treatment with DMSO (Veh) or ERK inhibitor (ERKi) SCH772984 with loading control α-tubulin.
(B) Quantification of bands from (A).
(C–E) qRT-PCR analysis of L (C), M (D), and S (E) UTR isoforms following treatment with Veh (DMSO) or ERK inhibitor. Statistical analysis performed using the Student’s t test (n = 3), *p ≤ 0.05, **p ≤ 0.01.
GJA1 Stress Responsive Transcript Isoforms Are Conserved in Humans and Primary Mouse Tissue
To investigate whether alternate GJA1 5′ UTR usage occurs in human cells, RLM-RACE was performed in the human adult low calcium temperature (HaCaT) human keratinocyte cell line. RLM-RACE PCR products from control and TGF-β-stimulated HaCaT cells had a comparable banding pattern to NMuMG cells when analyzed by electrophoresis (Figure 4A). Also consistent with results from NMuMG cells, the second largest band also increases following 48 h of TGF-β. As was the case for the mouse UTRs, Sanger sequencing and mapping to human genome build hg38 reveal overlap with peaks of CAGE reads with both human and mouse UTRs mapping to homologous regions (Figure 4B). We next asked if alternate UTR usage occurs in vivo and turned to two primary cell types, astrocytes and neonatal mouse ventricular cardiomyocytes (NMVCMs), which are derived from tissues known to express high levels of Cx43. RLM-RACE products from both astrocytes and NMVCMs also had a similar banding pattern (Figures 4C and 4D). Gel-extracted and Sanger sequenced bands mapped proximal to L, M, and S UTR isoforms corresponding to blue, red, and black dotted lines, respectively (Figure 4E).
Figure 4. GJA1 Transcript Isoforms Are Conserved in Humans and Primary Mouse Tissue and Are Stress Responsive.

(A) Agarose gel electrophoresis of RACE PCR products in control (C) and following 48 h of TGF-β treatment (T).
(B) RACE products and CAGE reads mapped to hg18 in UCSC genome browser.
(C) Agarose gel electrophoresis of RACE PCR products from primary astrocytes (astros). Arrows indicate excised bands.
(D) Agarose gel electrophoresis of RACE PCR products from primary neonatal ventricular cardiomyocytes (NMVCMs). Arrows indicate excised bands.
(E) Astrocytes and NMVCM RACE products and CAGE reads mapped to mm10, UCSC genome browser. Vertical dotted lines indicate position of L (blue), M (red), and S (black) UTRs.
(F) Top panel: RACE PCR products in normoxia (N) and following 48 h of hypoxia (H). Bottom: western blot of iPSC-CM cell lysates probed with Cx43 C-terminal antibody to detect full-length Cx43 and internally translated GJA1–20k following 24 h of normoxic or hypoxic conditions (1% oxygen).
(G) Fixed cell confocal immunofluorescence (×100) of cells labeled with an antibody directed against the Cx43 C terminus (green) with cell borders detected using N-cadherin (red) in cells exposed to 48 h of normoxic or hypoxic conditions. Scale bars: 20 μm. Enlarged region of cell border shown below.
(H) Quantification of average fluorescence intensity profiles of 5-μm lines bisecting, and perpendicular to, cell-cell borders from confocal microscopy maximum intensity projections. n = 13.
(I) Western blot of NMuMG cell lysates probed with Cx43 C-terminal antibody to detect full-length Cx43 and internally translated GJA1–20k (20k) following 48 h of normoxic or hypoxic conditions (1% oxygen).
(J) Fixed cell confocal immunofluorescence (×60) of cells labeled with an antibody directed against the Cx43 C terminus (green) with cell borders detected using pan-cadherin (red) in cells exposed to 48 h of normoxic or hypoxic conditions. 4′,6-Diamidino-2-phenylindole (DAPI; blue). Enlarged region of cell border shown below.
(K) Quantification of NMuMG immunofluorescence as in (H).
n = 12. black line = normoxia, blue line = hypoxia.
Previous studies have shown that GJA1–20k is responsive to hypoxic conditions (Basheer et al., 2017; Kotini et al., 2018; Ul-Hussain et al., 2014). We, therefore, exposed human induced pluripotent stem cell-derived cardiomyocytes (iPSC-CMs) to 48 h of 1% oxygen to assess the effect on UTR selection. Like TGF-β, hypoxic stress results in an increase in truncated 5′ UTRs (Figure 4F, top). This was accompanied by a reduction in GJA1–20k relative to Cx43 (Figure 4F, bottom) and a reduction in membrane-localized Cx43 (Figures 4G and 4H). Levels of GJA1–20k relative to Cx43 were also reduced following 48 h of hypoxic stress in NMuMG cells, and Cx43 membrane localization was similarly limited (Figures 4I–4K; Figure S3). Finally, we found lower GJA1–20k to Cx43 ratios in A549 human lung adenocarcinoma cells exposed to 48 h of hypoxic conditions (Figure S4). These data highlight conservation of UTR variants in mammalian cells, demon-strating their involvement in a common response to physiological stress.
Gja1 5′ UTRs Have Distinct Translational Efficiency and Are TGF-β Responsive
The 5′ UTR of Gja1 has been reported to possess IRES activity (Pfeifer et al., 2004; Schiavi et al., 1999). To determine whether these Gja1 5′ UTR isoforms are capable of IRES-mediated translation, we performed luciferase assays with in-vitro-transcribed RNA transfected into NMuMG cells, avoiding the contribution of alternate promoter activity that could lead to false positives (Figure 5A). We first compared translation of m7G-capped and A-capped RNA with the L, M, or S UTR (Figures 5B–5D). The m7G-capped L UTR was translated less efficiently than the M and S isoforms. Cap-independent translation of the L UTR is a greater percentage of its capped counterpart, and yet cap-independent translation activity is similar for all isoforms. None of the UTR sequences cloned in between renilla luciferase (Rluc) and firefly luciferase (Fluc) on a bicistronic transcript had appreciable Fluc signal, which argues against the presence of an IRES in these 5′ UTR constructs (Figures 5B–5D). When these m7G-capped transcripts are transfected into NMuMG following 48 h of TGF-β treatment, L translation decreases, whereas M and S translation significantly increases (Figures 5E–5G). This demonstrates the capability of these UTRs to dynamically regulate translational efficiency and taken together suggests that truncated Gja1 5′ UTRs promote full-length Cx43 translation over alternative translation of the N-terminally truncated GJA1–20k isoform.
Figure 5. Gja1 5′ UTRs Regulate Translation Efficiency.

(A) Schematic of RNA constructs.
(B–D) Firefly luciferase (Fluc) reporter gene expression normalized to renilla luciferase (Rluc) for L (B), M (C), and S (D) 5′ UTR sequence when capped (m7G), uncapped or A-capped (A), and located internally in a bicistronic construct (dual).
(E–G) Fluc reporter gene expression normalized to Rluc for L (E), M (F), and S (G) m7G-capped 5′ UTR sequences following transfection into cells exposed to TGF-β for 48 h.
Statistical analysis was performed using the Student’s t test (n = 3), ***p ≤ 0.001, ****p ≤ 0.0001.
Truncation of the Gja1 mRNA-5′ UTR Occurs in Aged Hearts with Activation of p38 Correlating with Reduced Internal Translation
Remodeling of the intercalated disc occurs during aging and is associated with an age-related decline in cardiac function (Bonda et al., 2016). Loss of Cx43 enrichment at the intercalated disc is associated with arrhythmogenesis in cardiomyopathy (Peters et al., 1997). To assess Cx43 localization, we performed immunofluorescence labeling on cryosections from young (Y; 3.8 month) and old (O; 29 month) mouse hearts (Figure 6A). In aged hearts, there is a significant decline in Cx43 intensity at N-cadherin-labeled intercalated discs (Figure 6B).
Figure 6. Aging Hearts Have Increased p38, Altered UTRs, and Reduced Internal Translation.

(A) Immunofluorescence (×60) of mouse heart cryosections. Young (Y) = 3.8 months and old (O) = 29 months labeled with an antibody directed against the Cx43 C terminus (green), with intercalated discs labeled with N-cadherin (red) and DAPI DNA stain (blue). Scale bar: 10 μm.
(B) Quantification of average Cx43 fluorescence intensity within N-cadherin-labeled intercalated discs (n = 5 animals, 23 averaged intercalated disc intensities from young and n = 6 animals, 29 averaged intercalated disc intensities from old).
(C) Western blot of mouse heart lysates probed with antibodies directed against phosphorylated p38 (p38pT180/Y182), total p38, and a glyceraldehyde-3-phosphate dehydrogenase (GAPDH) loading control (n = 6 per group).
(D) Quantification of bands from (C).
(E–G) qRT-PCR analysis of L (E), M (F), and S (G) UTR isoforms from mouse heart RNA (n = 6 per group).
(H) Western blot of mouse heart lysates probed with a Cx43 C-terminal antibody to detect full-length Cx43 and internally translated isoform GJA1–20k, and a GAPDH loading control (n = 6 per group).
(I) Quantification of GJA1–20k relative to Cx43 from western blot in (H).
(J) Correlation between GJA1–20k relative to Cx43 and phosphorylated p38pT180/Y182; blue dotted lines represent 95% confidence interval. Statistical analysis performed using the Student’s t test, *p ≤ 0.05, ***p ≤ 0.001.
During aging, p38 protein kinase activity increases, which can result in a state of chronic stress signaling (Hsieh and Papaconstantinou, 2002; Hsieh et al., 2003). To determine levels of phosphorylated p38 (p38pT180/Y182) in cardiac ventricular tissue during aging, we performed western blots on protein lysates from young and old mouse hearts. There is a significant increase in p38pT180/Y182 at 29 months (Figures 6C and 6D). We next assessed whether increased p38pT180/Y182 correlates with the expression of shortened UTRs, as we found in cell lines (Figures 6E–6G). In agreement with these data, we find significantly elevated levels of M UTR isoform by qRT-PCR in aged hearts (Figure 6D). Importantly, our cell-based studies also identify the M isoform as least permissive to internal translation (Figures 2H and 2I). We performed western blotting for Cx43 on protein lysates from young and old mouse hearts (Figure 6H). Quantification by densitometry reveals significantly lower levels of Gja1 internal translation product GJA1–20k relative to Cx43 in aged hearts (Figure 6I). Importantly, this reduction in internal translation also correlates with increasing levels of p38pT180/Y182 independent of age (Figure 6J).
DISCUSSION
Pfeifer et al. (2004) reported the presence of a total of nine different Gja1 5′ UTR isoforms in mice. Subsets of these isoforms are expressed with tissue specificity and proposed to arise from a combination of alternate promoter usage and alternative splicing. Here, we find that alternate promoter usage gives rise to truncated Gja1 UTR isoforms, and yet we do not detect the presence of alternative splicing. This may be due to differences in the cell types used. Previously, we identified Smad and non-Smad signaling pathways responsible for TGF-β-induced alterations in Gja1 alternative translation (James et al., 2018). Here, we demonstrate that these signaling pathways act on alternate Gja1 promoters to dynamically regulate the level of UTR isoforms. Importantly, we reveal the activation of this mechanism in response to cellular stress and cardiac aging and demonstrate conservation of these UTR isoforms in humans.
Our data illustrate the complex regulation of Gja1 translation by UTR selection. We observe reduced ratios of GJA1–20k to Cx43 in response to both TGF-β and hypoxic stress. We reveal that Erk and p38, components of non-Smad signaling downstream of TGF-β, can modulate UTR selection, with Erk functioning to limit GJA1–20k translation, and p38 promoting translation of Cx43. The mechanism by which p38 and Erk modulate Gja1 translation may also involve their known role in activation of Mnk½, as it has been shown that inhibiting Mnk½ significantly increases levels of GJA1–20k (Salat-Canela et al., 2014). These findings are consistent with the fact that hypoxia also activates p38.
The Gja1 5′ UTR has been reported to possess IRES activity capable of enhancing translational efficiency (Schiavi et al., 1999). To assess IRES activity, we performed luciferase assays by using transfection of in-vitro-transcribed RNA as opposed to plasmid-based constructs to avoid confounding factors, such as cryptic promoter activity or splicing, as reviewed in Terenin et al. (2017). In our hands, we did not detect any IRES activity for these UTRs. This discrepancy may be due the lack of trans-acting nuclear factors binding to an in vitro-transcribed RNA. For instance, factors such as hnRNP A1 have been demonstrated to regulate IRES activity (James and Smyth, 2018; Jo et al., 2008).
Cx43 gap junction formation and maintenance consists of multiple levels of regulation from transcription to post-translational modifications. Previously, we revealed the necessity for the internal translated isoform GJA1–20k in Cx43 trafficking, with a suggested role in Cx43 hexamer formation, adding another layer of regulation to this process (James et al., 2018). We further demonstrate GJA1–20k’s role in Cx43 trafficking in response to hypoxic stress in epithelial cells and iPSC-CMs, where reduced GJA1–20k translation correlates with a reduction in levels of membrane-localized Cx43. Other functions have also been ascribed to GJA1–20k, including stabilization of actin filaments to guide Cx43 trafficking (Basheer et al., 2017) and promotion of mitochondrial transport (Fu et al., 2017). Recently, upregulation of GJA1–20k was shown to occur in response to ischemic injury, and experimental overexpression prior to ischemic injury therapeutically regulates cardiac mitochondrial function and biogenesis (Basheer et al., 2018). Here, we find that endogenous GJA1–20k levels are reduced in response to extended exposure to hypoxic conditions in a range of cells lines, including NMuMG, A549, and iPSC-CMs. Reported differences appear to stem from the duration of hypoxia, as we do detect a trend of rising GJA1–20k levels at earlier time points (4 h) before a reduction in expression at 24 h (Figure S3). Together, these data highlight the complexity of Cx43 versus GJA1–20k translation, and indeed our work herein uncovers independent pathways discretely regulating translation of each. Finally, another recent article has described a direct role for GJA1–20k in the transcriptional regulation of N-cadherin (Kotini et al., 2018), opening the possibility of feedback loops from products of alternative translation initiation influencing gene expression at a more global level.
Cardiac function declines with age independent of extrinsic factors (Bonda et al., 2016; Fannin et al., 2014; Sandstede et al., 2000). During aging, the heart undergoes pathological remodeling at multiple levels ranging from whole heart size to its molecular components (Bonda et al., 2016; Cheng et al., 2009; Cohn et al., 2000; Fannin et al., 2014). Gap junctions encompassing Cx43 facilitate well-orchestrated and rapid electrical coupling in the heart. Disruption of Cx43 localization at the intercalated disk of cardiomyocytes results in decreased gap junction intercellular communication (GJIC) and increased susceptibility to arrhythmias of sudden cardiac death (Peters et al., 1997; Poelzing and Rosenbaum, 2004; Smith et al., 1991). Here, we observe a role for alternate promoter usage and reduction of internal translation in limiting Cx43 gap junction formation during aging.
Variation in TSSs due to alternate promoter usage is widespread, with most genes having more than one TSS (Forrest et al., 2014). It is common for TSSs to cluster at multiple promoters within the same gene, resulting in multimodal peaks of transcription initiation (Carninci et al., 2006). This allows cell-type- and cell-state-specific transcription factors to modulate 5′ UTR composition and regulate translational efficiency (Tamarkin-Ben-Harush et al., 2017). Our transcript isoform data from cell lines and primary cells when combined with CAGE reads suggest a multimodal-based promoter region occupying the most 5′ exon of the Gja1 5′ UTR, which is dynamically regulated in response to stress signaling. Through transfection, we demonstrate the capability of cis-acting elements in the Gja1 5′ UTR to affect translation. Future work to dissect the mechanism of Gja1 alternative translation, including the contribution of trans-acting factors, will be necessary to fully understand the complex regulation of Cx43 gap junctions.
More broadly, altered translation initiation may also contribute to pathological phenotypes from genetic disorders involving Cx43, such as oculodentodigital dysplasia (Churko et al., 2011; Shibayama et al., 2005). It is interesting to speculate how mutations may not necessarily have deleterious impacts on the encoded full-length protein per se but rather alter RNA structure and/or affect trans-acting factor binding to modulate the translational readout from a given mRNA. Although this likely would not impact 5′ UTR usage, responsiveness to trans-acting factors may be altered and deserves further investigation in this context. Together, our data reveal dynamic regulation of Gja1 transcript isoforms and uncover a cellular mechanism to balance the translation of Cx43 and its alternative translation product GJA1–20k (Figure 7). This process is similar to the regulatory relationship between 5′ UTR splicing and N-terminally truncated isoform translation reported for the glucocorticoid receptor (Turner et al., 2014), suggesting it is a more widespread cellular mechanism of regulating alternative translation. Genome-wide mapping of translation initiation site activity identifies multiple upstream open reading frames (uORFs) in the Gja1 5′ UTR (Lee et al., 2012). Selective inclusion of these uORFs in 5′ UTR variants by alternate promoter usage is, therefore, a candidate mechanism to explain the differences in translation efficiency and initiation site selection. This may be analogous to examples of leaky ribosome scanning, where uORFs play a role in regulating translation under normal and stress conditions (Barbosa et al., 2013; Brubaker et al., 2014). Future work using deep sequencing is necessary to define the totality of Gja1 TSSs under conditions of stress to define the exact sequence elements regulating Gja1 translation.
Figure 7.

Model of Transcription Start Site Selection in the Gja1 5′ UTR and Transcript Isoform Effect on Alternative Translation and Gap Junction Formation
STAR★METHODS
Detailed methods are provided in the online version of this paper and include the following:
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, James W. Smyth, PhD. (smythj@vtc.vt.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell Culture
NMuMG cells, female, a kind gift from Prof. Rik Derynck (University of California San Francisco), and 293 FT cells, female, (Thermo Fisher Scientific) were grown as in James et al. (2018). Human induced pluripotent stem cell-derived cardiomyocytes (iPSC-CMs), male, and culture media were obtained from Axol BioSciences and maintained according to manufacturer’s instructions. Cells were maintained in a humidified atmosphere of 5% CO2 at 37°C, for hypoxic studies O2 was dropped to 1% in a tri-gas incubator. Cell treatments: TGF-β1 (2 ng/ml; Humanzyme), p38 inhibitor SB 202190 (20 μM; Cayman), JNK inhibitor SP 600125 (20 μM; Cayman), Erk½ inhibitor SCH 772984 (0.1 μM; Cayman), or vehicle (DMSO; Sigma).
Animals
All mice were C57BL/6N for primary cell isolation and adult aged (3.8 month old, male) were obtained from Charles River Laboratories with aged (29 month old, male) C57BL/6N obtained from the National Institute on Aging. All experiments were carried out under NIH guidelines and animal protocols were approved by the Virginia Tech Institutional Animal Care and Use Committee.
METHOD DETAILS
Primary cell isolation
Neonatal mouse ventricular myocytes (NMVCM) were isolated from C57BL/6 P7 mice as previously described (Smyth et al., 2012). Briefly, pups were decapitated and hearts removed and washed in Hank’s balanced salt solution without calcium or magnesium (HBSS; Thermo Fisher Scientific). Atria were removed and quartered ventricles placed in 5 mL HBSS with 1.5 mg/ml collagenase II (Worthington Biochemical) warmed to 37°C. Tissue was stirred for 5 min at 37°C and gently triturated 20 times with a transfer pipette prior to a 2 min rest allowing tissue to settle. Supernatant was removed and added to 5 mL ice-cold FBS (Thermo Fisher Scientific), and 5 mL warm HBSS with collagenase added to remaining tissue. This process was repeated three times, after which the cell/FBS suspension was centrifuged for 5 min at 300 × g. The supernatant was discarded and cell pellet resuspended in 10 mL F12/DMEM 50/50 (Thermo Fisher Scientific) supplemented with 5% FBS, insulin-transferrin sodium selenite media supplement (Thermo Fisher Scientific), and Mycozap-PR (Lonza). Cells were pipetted through a 70 μM cell strainer (BD Biosciences) and pre-plated for 30 min at 37°C on 100 mm dishes (Genesee Scientific). Cell suspension was removed and pre-plating repeated on a fresh 100 mm dish to further enrich for cardiomyocytes. Adherent cells were discarded and cell suspension centrifuged for 5 min at 500 × g. Supernatant was discarded and cell pellet snap frozen for RNA isolation.
Astrocyte isolation, Mice were decapitated, brains were removed and the cortical gray matter of eight C57BL/6 P7 mice was dissected in carbogenated (95% O2/ 5% CO2) ice cold artificial cerebral spinal fluid (ACSF, 120mM NaCl, 3.0 mM KCl, 2mM MgCl, 0.2 mM CaCl, 26.2 mM NaHCO3, 11.1 mM glucose, 5.0 mM HEPES). Meninges were removed. The tissue was minced into 1 mm3 pieces and dissociated into a single cell suspension using a Papain Dissociation kit (Worthington Biochemical). Magnetic cell sorting was used to remove myelin and microglia before positive selection for astrocytes. In short, a debris removal step using modified protocols from Miltenyi Biotec’s Myelin Removal Kit with anti-myelin Microbeads was performed first followed by removal of microglia using anti-Cd11b+ Microbeads. Astrocytes were selected using a modified protocol from Miltenyi Biotec’s Anti-ACSA-2 kit. A detailed protocol is provided in Holt and Olsen (2016).
RNA Extraction and RT-qPCR
RNA was extracted from NMuMG cells using Purelink RNA Mini Kit (Thermo Fisher Scientific) and homogenized by passage 10 times through an 18 gauge needle. Mouse hearts were homogenized in Trizol, and chloroform extracted prior to Purelink column purification. DNA was digested on column using PureLink DNase (Thermo Fisher Scientific). 1 μg of RNA was reverse transcribed with iScript Reverse Transcription Supermix for RT-qPCR (Bio-Rad). Quantitative PCR was performed using SYBR Select Master Mix for CFX (Thermo Fisher Scientific) on a Quant Studio 6 Flex cycler (Thermo Fisher Scientific) using PrimeTime qPCR primers (Integrated DNA technologies). UTR primers were designed using primer3 and ordered from Integrated DNA Technologies. GAPDH was used as an internal control. Cycling parameters consisted of 50°C 2 min; 95°C 2 min; 39 cycles of 95°C 15 s, 55°C 15 s, 72°C 1 min. To calculate baseline levels of UTR isoform abundance in untreated NMuMG cells, qPCR was performed on a dilution series made with plasmids containing cloned Gja1-UTR sequence, and a standard curve was used to calculate absolute quantity. Absolute quantities of overlapping UTRs were then subtracted from one another. Primer efficiencies were calculated from standard curves and were included in calculations of relative fold change by the Pfaffl equation (Pfaffl, 2001). For mouse experiments, primer efficiencies were calculated using RT miner software (Zhao and Fernald, 2005). Data were log transformed prior to statistical analysis.
Western blotting
Cells were lysed in RIPA buffer (50 mM Tris pH 7.4, 150 mM NaCl, 1 mM EDTA, 1% Triton X-100, 1% sodium deoxycholate, 2 mM NaF, 200 μM Na3VO4, 0.1% SDS, 5.6 mM NEM) supplemented with HALT Protease and Phosphatase Inhibitor Cocktail (Thermo Fisher Scientific) and western blotting conducted as previously described (Smyth et al., 2010; Smyth et al., 2012). Briefly, cells were scraped into RIPA buffer and sonicated prior to centrifugation at 10,000 × g for 20 min at 4°C. Protein concentration was quantified using the Bio-Rad DC Protein Assay and lysates normalized to the same concentration. 4X Bolt LDS sample buffer (Thermo Fisher Scientific) supplemented with DTT (400 mM, final concentration 100 mM) was added and samples heated for 10 min at 70°C prior to SDS-PAGE and western blotting with the following primary antibodies: rabbit anti-Cx43 (1:5000; Sigma), mouse anti-α-tubulin (1:5000; Sigma). Goat secondary antibodies conjugated to Alexa Fluor 555 and Alexa Fluor 647 (Thermo Fisher Scientific) were used at 1:1000, or secondary antibodies conjugated to HRP (1:5000) (Abcam) and detected with clarity western ECL substrate (BioRad), and membranes imaged on a ChemiDoc MP (Bio-Rad). Mouse heart tissue was homogenized in RIPA buffer with an omni tissue homogenizer (Omni international) and processed as above. Primary antibodies used were phospho-p38 MAPK Thr180/Tyr182 (1:1000, Cell Signaling Technology) and p38 MAPK (1:1000, Cell Signaling Technology). Mouse anti-GAPDH (1:5000, Fitzgerald) was used as loading control.
RLM-RACE
RLM-RACE was performed with the GeneRacer Kit (Thermo Fisher Scientific) according to manufacturer instructions. Briefly, RNA was isolated from NMUMG cells untreated or treated with TGF-β for 48 h. RNA integrity was verified by denaturing gel electrophoresis. 4 μg of RNA from each sample was dephosphorylated to prevent ligation of the RNA oligo adaptor to truncated mRNA and other non-mRNA. The 5′ cap was then removed and the GeneRacer RNA oligonucleotide was ligated to 5′ end of the RNA. Reverse transcription was performed with a Gja1 specific primer to generate cDNA. The FWD Adaptor specific GeneRacer alternative primer and a Gja1 specific Rev primer were used to amplify the entire 5′ UTR. A second round of nested PCR was performed. PCR products were run on an agarose gel followed by gel extraction of bands and cloning into the PCR 4 Blunt TOPO vector (Thermo Fisher Scientific) for sequencing. UTR sequences from RACE were aligned to the Gja1 5′UTR using Snapgene software. Sequences were then mapped to the appropriate genome (mouse: mm10; human: hg38) using the UCSC genome browser and correlated with Cap Analysis of Gene Expression (CAGE) reads from the FANTOM5 consortium, RIKEN. RACE was performed in HaCaT as above.
CRISPR/Cas9 gene editing
Generation of 293FT GJA1−/− cell line: Single guide RNA targeting 5′ and 3′ flanking regions of GJA1 exon 2, respectively were cloned into pSpCas9(BB)-2A-GFP (PX458) at Bbs1 site. PX458 was a gift from Feng Zhang (Addgene) (Ran et al., 2013). Cas9 transfected 293FT cells were selected for single cell colonies following serial dilution into 96 well plates. Clonal populations were screened by PCR using primers flanking GJA1 exon 2. Colonies positive for biallelic deletion of GJA1 exon 2 were further screened by Cx43 immunoblotting and immunofluorescence. Generation of NMuMG Gja1−/− cell line was done as above.
Cloning and expression of Gja1–5′UTR isoforms
The 5′UTR isoforms identified from RACE were cloned in front of Gja1 into expression vector pcDNA3.2/V5 -DEST using in-fusion HD cloning (TAKARA). Vector pcDNA3.2/V5-DEST was modified to exclude sequence downstream of the CMV promoter in order to exclude sequences that could influence translation. The ratio of GJA1–20k to Cx43 expression of each UTR isoform was analyzed by transfection of plasmids into GJA1−/− 239FT and Gja1−/− NMuMG cell lines. DNA Transfections were performed with Lipofectamine 3000 reagent (Thermo Fisher Scientific) and 500 ng of each UTR construct per well of a 24 well dish.
Luciferase assays
UTR isoforms were cloned into plasmid pcDNA3 RLUC POLIRES FLUC which was gift from Nahum Sonenberg (Addgene) (Poulin et al., 1998), in front of Fluc using infusion cloning. T7 tailed primers were used to PCR amplify luciferase constructs for in vitro transcription. The mMESSAGE mMACHINE T7 Transcription Kit (Thermo Fisher Scientific) was used for in vitro transcription according to manufacturer instructions with some modifications. RNA Cap mixtures included ribonucleotides with a 4:1 ratio of ARCA M7G cap (New England Biolabs), or an A cap analog (New England Biolabs). In vitro transcribed RNA was purified by lithium chloride precipitation. Poly (A) tailing of transcripts was performed with Poly (A) polymerase (New England Biolabs) at 37°C for 30 min. Luciferase assays were performed with the Dual-Luciferase Reporter Assay System (Promega) according to manufacturer’s instructions. Briefly, NMuMG cells in 24 well plates were transfected with in vitro transcribed RNA constructs consisting of 200 ng of UTR-Fluc and 10 ng of control Rluc, or 200 ng of an Rluc-UTR-Fluc bicistronic transcript, using Lipofectamine 3000. Cells were lysed in 100 μl of passive lysis buffer 3.5 hours post transfection. Luminescence measurements were recorded using an automated GloMax-Multi+ luminometer using luciferase Assay Reagent II and Stop & Glo.
Immunofluorescence
Cells were fixed in 37°C 4% paraformaldehyde for 20 min and stored in PBS at 4°C until immunostaining was conducted as previously described (Smyth et al., 2012) with the following primary antibodies: mouse anti–N-cadherin (BD Biosciences; 1:200), mouse anti-pan cadherin (Novus Biologicals; 1:500), rabbit anti-Cx43 (Sigma; 1:3000), and mouse anti–α-tubulin (Sigma; 1:3000). Secondary antibodies used were goat anti-rabbit Alexa Fluor 488 and anti-mouse Alexa Fluor 647 (Thermo Fisher Scientific; 1:500).
Heart tissue cryosections (10 μm) were prepared on glass slides and fixed in acetone for 1 minute followed by air drying. Sections were rehydrated for 10 minutes in PBS, followed by 1 h of blocking at room temperature with 5% NGS, 0.1% Triton X-100 in PBS. After blocking, sections were incubated overnight at 4°C with primary antibodies diluted in 5% NGS; rabbit anti-Cx43 (1:2500; Sigma) and mouse anti-N-cadherin (1:250; BD Biosciences). Washes included 10 quick rinses in PBS, two 10 min washes in 0.05% Tween 20 in PBS, two 5 min washes in PBS. Cells were then incubated for an additional hour at room temperature with goat secondary antibodies anti-rabbit Alexa Fluor 488 and anti-mouse Alexa Fluor 647 (Thermo Fisher Scientific; 1:500) in 5% NGS, 0.1% Triton X-100 in PBS with DAPI. Slides were then washed as above, and coverslips were mounted using ProLong gold antifade reagent.
Image analysis was performed with FIJI ImageJ software (NIH) (Schneider et al., 2012). For cell lines, average fluorescence intensity profiles of Cx43 were measured from 5 μm lines bisecting, and perpendicular to, N-cadherin labeled cell–cell borders from confocal microscopy maximum intensity projections. For cryosections, three N-cadherin labeled intercalated discs were randomly selected for each image. At least four images were analyzed per heart from which averaged intercalated disc values were obtained. The Cx43 containing channel was then merged with the N-cadherin channel and Cx43 fluorescence intensity measured along a 10 μm wide line covering the N-cadherin signal.
QUANTIFICATION AND STATISTICAL ANALYSIS
All quantification was performed on experiments repeated at least three times. Data are presented as mean ± SEM. Statistical analysis was conducted with GraphPad Prism 8.0.2 (GraphPad Software, Inc. La Jolla, CA). Data were analyzed for significance using Student’s t test or one-way ANOVA with Tukey’s post hoc tests. A value of p < 0.05 was considered statistically significant.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| rabbit anti-Connexin-43 | Sigma-Aldrich | Cat# C6219, RRID:AB_476857 |
| phospho-p38 MAPK (Thr180/Tyr182) (D3F9) XP® Rabbit mAb | Cell Signaling Technology | Cat# 4511, RRID:AB_2139682 |
| p38 MAPK (D13E1) XP® Rabbit | Cell Signaling Technology | Cat# 8690, RRID:AB_10999090 |
| mouse monoclonal anti-alpha-tubulin | Sigma-Aldrich | Cat# T6199, RRID:AB_477583 |
| Mouse Anti-Rabbit Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) Monoclonal Antibody, Unconjugated, Clone 6C5 | Fitzgerald Industries International | Cat# 10R-G109a, RRID:AB_1285808 |
| Mouse monoclonal anti-pan cadherin | Novus Biologicals | Novus Cat# NB 100–1907, RRID:AB_530742 |
| Mouse monoclonal anti–N-cadherin | BD Biosciences | Cat# 610921, RRID:AB_398236 |
| Goat anti-Rabbit IgG (H+L) Secondary Antibody, Alexa Fluor 488 conjugate | Thermo Fisher Scientific | Cat# A-11034, RRID:AB_2576217 |
| Goat anti-Rabbit IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor 647 | Thermo Fisher Scientific | Cat# A-21245, RRID:AB_2535813 |
| Goat anti-Mouse IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor 555 | Thermo Fisher Scientific | Cat# A-21424, RRID:AB_141780 |
| Goat Anti-Rabbit IgG - H&L Polyclonal antibody, Hrp Conjugated | Abcam | Abcam Cat# ab6721, RRID:AB_955447 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Human TGF-β1 | Humanzyme | HZ-1011 |
| SB 202190, p38 MAP kinase inhibitor | Cayman Chemical | Cat# 10010399, CAS:152121–30-7 |
| SP 600125, JNK inhibitor | Cayman Chemical | Cat# 10010466, CAS:129–56-6 |
| SCH 772984, ERK1/2 inhibitor | Cayman Chemical | Cat# 19166, CAS:942183–80-4 |
| PureLink DNase | Thermo Fisher Scientific | Cat# 12185010 |
| HALT Protease and Phosphatase Inhibitor Cocktail | Thermo Fisher Scientific | Cat# 78440 |
| 4X Bolt LDS sample buffer | Thermo Fisher Scientific | Cat# B0007 |
| Lipofectamine 3000 reagent | Thermo Fisher Scientific | Cat# L3000015 |
| Collagenase, Type 2 | Worthington Biochemical Corporation | Cat# LS004176 |
| Insulin-Transferrin-Selenium (ITS -G) (100X) | Thermo Fisher Scientific | Cat# 41400045 |
| ARCA M7G cap | New England BioLabs | Cat# S1411 |
| G(5′)ppp(5′)A RNA Cap Structure Analog | New England BioLabs | Cat# S1406 |
| Dimethyl sulfoxide | Sigma-Aldrich | Cat# D2438, CAS:67–68-5 |
| HBSS | Thermo Fisher Scientific | Cat# 14175095 |
| DMEM/F-12, GlutaMAX | Thermo Fisher Scientific | Cat# 10565018 |
| MycoZap Plus-CL | Thermo Fisher Scientific | Cat# VZA-2012 |
| Normal Goat Serum | Thermo Fisher Scientific | Cat# 01–6201 |
| DAPI (4′,6-diamidino-2-phenylindole) | Thermo Fisher Scientific | Cat# D1306 |
| ProLong Gold Antifade Mountant | Thermo Fisher Scientific | Cat# P36934 |
| TRIzol | Thermo Fisher Scientific | Cat# 15596026 |
| clarity western ECL substrate | BioRad | Cat# 1705060 |
| Critical Commercial Assays | ||
| GeneRacer Kit with SuperScript III RT and Zero Blunt TOPO PCR Cloning Kit for Sequencing | Thermo Fisher Scientific | Cat# L150202 |
| Dual-Luciferase Reporter Assay System | Promega | Cat# E1910 |
| mMESSAGE mMACHINE T7 Transcription Kit | Thermo Fisher Scientific | Cat# AM1344 |
| iScript Reverse Transcription Supermix for RT-qPCR | BioRad | Cat# 1708840 |
| Purelink RNA Mini Kit | Thermo Fisher Scientific | Cat# 12183018A |
| SYBR Select Master Mix for CFX | Thermo Fisher Scientific | Cat# 4472942 |
| DC Protein Assay Reagents | BioRad | Cat# 5000116 |
| In-fusion HD cloning | Takara | Cat# 638909 |
| Papain Dissociation kit | Worthington Biochemical Corporation | Cat# LK003153 |
| anti-myelin Microbeads | Miltenyi Biotec | Cat# 130096733 |
| anti-Cd11b+ Microbeads | Miltenyi Biotec | Cat# 130093634 |
| Experimental Models: Cell Lines | ||
| NMuMG cells | Laboratory of Rik Derynck | N/A |
| 293 FT cells | Thermo Fisher Scientific | Cat# R70007 |
| Human iPSC-derived cardiomyocytes | Axol Biosciences | Cat# ax2505 |
| NMuMG Gja1−/− clone6 | This paper | N/A |
| 293 FT GJA1−/ − clone 30 | This paper | N/A |
| Experimental Models: Organisms/Strains | ||
| C57BL/6N | Charles River Laboratories | N/A |
| C57BL/6N | National Institute on Aging | N/A |
| Oligonucleotides | ||
| GJA1 KO guide RNA: AGAAACAAAGAGACAATTCA | This paper | N/A |
| GJA1 KO guide RNA: TGGCTCTGCTTGAAGGTCGC | This paper | N/A |
| Gja1 KO guide RNA: AAGCCTACTCCACGGCCGGA | This paper | N/A |
| Gja1 KO guide RNA: CCACAATCGATTGGCAGCTT | This paper | N/A |
| CRISPR Screening primers human: GGTTTTGGAAAGAAGGTTATGGA | This paper | N/A |
| CRISPR Screening primers human: ATTACCACCCGCTCATTCAC | This paper | N/A |
| CRISPR Screening primers mouse: AGGTGGTGTCCAGAGCCTTA | This paper | N/A |
| CRISPR Screening primers mouse: GAGGACTTGGATTCCTGCTCA | This paper | N/A |
| Primers for RT-qPCR see Table S1 | This paper | N/A |
| Primers for RLM-RACE see Table S2 | This paper | N/A |
| Recombinant DNA | ||
| pSpCas9(BB)-2A-GFP (PX458) | Ran et al., 2013 | Addgene plasmid Cat# 48138 |
| pcDNA3.2/V5-DEST | Thermo Fisher Scientific | Cat# 12489019 |
| pcDNA3 RLUC POLIRES FLUC | Poulin et al., 1998 | Addgene plasmid Cat# 45642 |
| pcDNA3 RLUC-S-FLUC | This paper | N/A |
| pcDNA3 RLUC-M-FLUC | This paper | N/A |
| pcDNA3 RLUC-L-FLUC | This paper | N/A |
| Software and Algorithms | ||
| ImageJ | Schneider et al., 2012 | https://imagej.nih.gov/ij/ |
| SnapGene | GSL Biotech | https://www.snapgene.com/ |
| RT miner | Zhao and Fernald 2005 | http://www.ewindup.info/miner/ |
Highlights.
In aged hearts and during hypoxia or TGF-β treatment, the GJA1 mRNA 5′ UTR is truncated
Activation of P38 and ERK signal transduction pathways regulate GJA1 5′ UTR length
Truncated GJA1 5′ UTRs are sufficient to suppress internal translation of GJA1–20k
Reduced GJA1–20k expression is correlated with P38 activation in aged hearts
ACKNOWLEDGMENTS
The authors thank Dr. Allison N. Tegge (Fralin Biomedical Research Institute, Department of Statistics, Virginia Tech) for consultation on statistical methods used and Rachel Padget, M.S. (Fralin Biomedical Research Institute) and Darlon Jan (Virginia Tech Carilion School of Medicine) for critical review of this manuscript. This work was supported by an NIH NHLBI R01 grant (HL132236 to J.W.S.), NHLBI F31 grant (HL140909 to C.C.J.), NIA R01 grant (AG055545 to G.V.), NINDS R21 grant (NS106313 to G.V.), NINDS R01 grant (NS105807 to S.R.), NINDS R21 grant (NS107941 to S.R.), and the American Heart Association (18PRE33960573 to P.J.C.). Funding for open access charge: NIH.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2019.04.114.
REFERENCES
- Barbosa C, Peixeiro I, and Romão L (2013). Gene expression regulation by upstream open reading frames and human disease. PLoS Genet. 9, e1003529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basheer W, and Shaw R (2016). The “tail” of Connexin43: an unexpected journey from alternative translation to trafficking. Biochim. Biophys. Acta 1863, 1848–1856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basheer WA, Xiao S, Epifantseva I, Fu Y, Kleber AG, Hong T, and Shaw RM (2017). GJA1–20k Arranges Actin to Guide Cx43 Delivery to Cardiac Intercalated Discs. Circ. Res 121, 1069–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basheer WA, Fu Y, Shimura D, Xiao S, Agvanian S, Hernandez DM, Hitzeman TC, Hong T, and Shaw RM (2018). Stress response protein GJA1–20k promotes mitochondrial biogenesis, metabolic quiescence, and cardioprotection against ischemia/reperfusion injury. JCI Insight 3, 121900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonda TA, Szynaka B, Sokolowska M, Dziemidowicz M, Winnicka MM, Chyczewski L, and Kaminski KA (2016). Remodeling of the intercalated disc related to aging in the mouse heart. J. Cardiol 68, 261–268. [DOI] [PubMed] [Google Scholar]
- Brubaker SW, Gauthier AE, Mills EW, Ingolia NT, and Kagan JC (2014). A bicistronic MAVS transcript highlights a class of truncated variants in antiviral immunity. Cell 156, 800–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carninci P, Sandelin A, Lenhard B, Katayama S, Shimokawa K, Ponjavic J, Semple CA, Taylor MS, Engström PG, Frith MC, et al. (2006). Genome-wide analysis of mammalian promoter architecture and evolution. Nat. Genet 38, 626–635. [DOI] [PubMed] [Google Scholar]
- Chen CH, Mayo JN, Gourdie RG, Johnstone SR, Isakson BE, and Bearden SE (2015). The connexin 43/ZO-1 complex regulates cerebral endothelial F-actin architecture and migration. Am. J. Physiol. Cell Physiol 309, C600–C607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng S, Fernandes VR, Bluemke DA, McClelland RL, Kronmal RA, and Lima JA (2009). Age-related left ventricular remodeling and associated risk for cardiovascular outcomes: the Multi-Ethnic Study of Atherosclerosis. Circ Cardiovasc Imaging 2, 191–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Z, Otto GM, Powers EN, Keskin A, Mertins P, Carr SA, Jovanovic M, and Brar GA (2018). Pervasive, Coordinated Protein-Level Changes Driven by Transcript Isoform Switching during Meiosis. Cell 172, 910–923.e916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Churko JM, Shao Q, Gong XQ, Swoboda KJ, Bai D, Sampson J, and Laird DW (2011). Human dermal fibroblasts derived from oculodentodigital dysplasia patients suggest that patients may have wound-healing defects. Hum. Mutat 32, 456–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clasadonte J, Scemes E, Wang Z, Boison D, and Haydon PG (2017). Connexin 43-Mediated Astroglial Metabolic Networks Contribute to the Regulation of the Sleep-Wake Cycle. Neuron 95, 1365–1380.e1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohn JN, Ferrari R, and Sharpe N (2000). Cardiac remodeling–concepts and clinical implications: a consensus paper from an international forum on cardiac remodeling. Behalf of an International Forum on Cardiac Remodeling. J. Am. Coll. Cardiol 35, 569–582. [DOI] [PubMed] [Google Scholar]
- Crespin S, Bechberger J, Mesnil M, Naus CC, and Sin WC (2010). The carboxy-terminal tail of connexin43 gap junction protein is sufficient to mediate cytoskeleton changes in human glioma cells. J. Cell. Biochem 110, 589–597. [DOI] [PubMed] [Google Scholar]
- Echetebu CO, Ali M, Izban MG, MacKay L, and Garfield RE (1999). Localization of regulatory protein binding sites in the proximal region of human myometrial connexin 43 gene. Mol. Hum. Reprod 5, 757–766. [DOI] [PubMed] [Google Scholar]
- Fannin J, Rice KM, Thulluri S, Dornon L, Arvapalli RK, Wehner P, and Blough ER (2014). Age-associated alterations of cardiac structure and function in the female F344×BN rat heart. Age (Dordr.) 36, 9684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faye MD, and Holcik M (2015). The role of IRES trans-acting factors in carcinogenesis. Biochim. Biophys. Acta 1849, 887–897. [DOI] [PubMed] [Google Scholar]
- Fishman GI, Spray DC, and Leinwand LA (1990). Molecular characterization and functional expression of the human cardiac gap junction channel. J. Cell Biol 111, 589–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forrest AR, Kawaji H, Rehli M, Baillie JK, de Hoon MJ, Haberle V, Lassmann T, Kulakovskiy IV, Lizio M, Itoh M, et al. ; FANTOM Consortium and the RIKEN PMI and CLST (DGT) (2014). A promoter-level mammalian expression atlas. Nature 507, 462–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y, Zhang SS, Xiao S, Basheer WA, Baum R, Epifantseva I, Hong T, and Shaw RM (2017). Cx43 Isoform GJA1–20k Promotes Microtubule Dependent Mitochondrial Transport. Front. Physiol 8, 905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geimonen E, Jiang W, Ali M, Fishman GI, Garfield RE, and Andersen J (1996). Activation of protein kinase C in human uterine smooth muscle induces connexin-43 gene transcription through an AP-1 site in the promoter sequence. J. Biol. Chem 271, 23667–23674. [DOI] [PubMed] [Google Scholar]
- Geimonen E, Boylston E, Royek A, and Andersen J (1998). Elevated connexin-43 expression in term human myometrium correlates with elevated c-Jun expression and is independent of myometrial estrogen receptors. J. Clin. Endocrinol. Metab 83, 1177–1185. [DOI] [PubMed] [Google Scholar]
- Hellen CU, and Sarnow P (2001). Internal ribosome entry sites in eukaryotic mRNA molecules. Genes Dev. 15, 1593–1612. [DOI] [PubMed] [Google Scholar]
- Hernandez M, Shao Q, Yang XJ, Luh SP, Kandouz M, Batist G, Laird DW, and Alaoui-Jamali MA (2006). A histone deacetylation-dependent mechanism for transcriptional repression of the gap junction gene cx43 in prostate cancer cells. Prostate 66, 1151–1161. [DOI] [PubMed] [Google Scholar]
- Holcik M, and Sonenberg N (2005). Translational control in stress and apoptosis. Nat. Rev. Mol. Cell Biol 6, 318–327. [DOI] [PubMed] [Google Scholar]
- Holt LM, and Olsen ML (2016). Novel Applications of Magnetic Cell Sorting to Analyze Cell-Type Specific Gene and Protein Expression in the Central Nervous System. PLoS One 11, e0150290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh CC, and Papaconstantinou J (2002). The effect of aging on p38 signaling pathway activity in the mouse liver and in response to ROS generated by 3-nitropropionic acid. Mech. Ageing Dev 123, 1423–1435. [DOI] [PubMed] [Google Scholar]
- Hsieh CC, Rosenblatt JI, and Papaconstantinou J (2003). Age-associated changes in SAPK/JNK and p38 MAPK signaling in response to the generation of ROS by 3-nitropropionic acid. Mech. Ageing Dev 124, 733–746. [DOI] [PubMed] [Google Scholar]
- James CC, and Smyth JW (2018). Alternative mechanisms of translation initiation: An emerging dynamic regulator of the proteome in health and disease. Life Sci. 212, 138–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- James CC, Zeitz MJ, Calhoun PJ, Lamouille S, and Smyth JW (2018). Altered translation initiation of Gja1 limits gap junction formation during epithelial-mesenchymal transition. Mol. Biol. Cell 29, 797–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo OD, Martin J, Bernath A, Masri J, Lichtenstein A, and Gera J (2008). Heterogeneous nuclear ribonucleoprotein A1 regulates cyclin D1 and c-myc internal ribosome entry site function through Akt signaling. J. Biol. Chem 283, 23274–23287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotini M, Barriga EH, Leslie J, Gentzel M, Rauschenberger V, Schambony A, and Mayor R (2018). Gap junction protein Connexin-43 is a direct transcriptional regulator of N-cadherin in vivo. Nat. Commun 9, 3846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Liu B, Lee S, Huang SX, Shen B, and Qian SB (2012). Global mapping of translation initiation sites in mammalian cells at single-nucleotide resolution. Proc. Natl. Acad. Sci. USA 109, E2424–E2432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leenen FA, Vernocchi S, Hunewald OE, Schmitz S, Molitor AM, Muller CP, and Turner JD (2016). Where does transcription start? 5′-RACE adapted to next-generation sequencing. Nucleic Acids Res. 44, 2628–2645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B, and Qian SB (2014). Translational reprogramming in cellular stress response. Wiley Interdiscip. Rev. RNA 5, 301–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lizio M, Harshbarger J, Shimoji H, Severin J, Kasukawa T, Sahin S, Abugessaisa I, Fukuda S, Hori F, Ishikawa-Kato S, et al. ; FANTOM consortium (2015). Gateways to the FANTOM5 promoter level mammalian expression atlas. Genome Biol. 16, 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markoff A (2005). Analytical Tools for DNA, Genes and Genomes: Nuts & Bolts, First Edition (DNA Press; ). [Google Scholar]
- Meyer RA, Cohen MF, Recalde S, Zakany J, Bell SM, Scott WJ Jr., and Lo CW (1997). Developmental regulation and asymmetric expression of the gene encoding Cx43 gap junctions in the mouse limb bud. Dev. Genet 21, 290–300. [DOI] [PubMed] [Google Scholar]
- Moorby C, and Patel M (2001). Dual functions for connexins: Cx43 regulates growth independently of gap junction formation. Exp. Cell Res 271, 238–248. [DOI] [PubMed] [Google Scholar]
- Peters NS, Coromilas J, Severs NJ, and Wit AL (1997). Disturbed connexin43 gap junction distribution correlates with the location of reentrant circuits in the epicardial border zone of healing canine infarcts that cause ventricular tachycardia. Circulation 95, 988–996. [DOI] [PubMed] [Google Scholar]
- Pfaffl MW (2001). A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 29, e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeifer I, Anderson C, Werner R, and Oltra E (2004). Redefining the structure of the mouse connexin43 gene: selective promoter usage and alternative splicing mechanisms yield transcripts with different translational efficiencies. Nucleic Acids Res. 32, 4550–4562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poelzing S, and Rosenbaum DS (2004). Altered connexin43 expression produces arrhythmia substrate in heart failure. Am. J. Physiol. Heart Circ. Physiol 287, H1762–H1770. [DOI] [PubMed] [Google Scholar]
- Poulin F, Gingras AC, Olsen H, Chevalier S, and Sonenberg N (1998). 4E-BP3, a new member of the eukaryotic initiation factor 4E-binding protein family. J. Biol. Chem 273, 14002–14007. [DOI] [PubMed] [Google Scholar]
- Ramani M, Mylvaganam S, Krawczyk M, Wang L, Zoidl C, Brien J, Reynolds JN, Kapur B, Poulter MO, Zoidl G, and Carlen PL (2016). Differential expression of astrocytic connexins in a mouse model of prenatal alcohol exposure. Neurobiol. Dis 91, 83–93. [DOI] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, and Zhang F (2013). Genome engineering using the CRISPR-Cas9 system. Nat. Protoc 8, 2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rojas-Duran MF, and Gilbert WV (2012). Alternative transcription start site selection leads to large differences in translation activity in yeast. RNA 18, 2299–2305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salat-Canela C, Sesé M, Peula C, Ramón y Cajal S, and Aasen T (2014). Internal translation of the connexin 43 transcript. Cell Commun. Signal 12, 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandstede J, Lipke C, Beer M, Hofmann S, Pabst T, Kenn W, Neubauer S, and Hahn D (2000). Age- and gender-specific differences in left and right ventricular cardiac function and mass determined by cine magnetic resonance imaging. Eur. Radiol 10, 438–442. [DOI] [PubMed] [Google Scholar]
- Schiavi A, Hudder A, and Werner R (1999). Connexin43 mRNA contains a functional internal ribosome entry site. FEBS Lett. 464, 118–122. [DOI] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, and Eliceiri KW (2012). NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibayama J, Paznekas W, Seki A, Taffet S, Jabs EW, Delmar M, and Musa H (2005). Functional characterization of connexin43 mutations found in patients with oculodentodigital dysplasia. Circ. Res 96, e83–e91. [DOI] [PubMed] [Google Scholar]
- Smith JH, Green CR, Peters NS, Rothery S, and Severs NJ (1991). Altered patterns of gap junction distribution in ischemic heart disease. An immunohistochemical study of human myocardium using laser scanning confocal microscopy. Am. J. Pathol 139, 801–821. [PMC free article] [PubMed] [Google Scholar]
- Smyth JW, and Shaw RM (2013). Autoregulation of connexin43 gap junction formation by internally translated isoforms. Cell Rep. 5, 611–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smyth JW, Hong TT, Gao D, Vogan JM, Jensen BC, Fong TS, Simpson PC, Stainier DY, Chi NC, and Shaw RM (2010). Limited forward trafficking of connexin 43 reduces cell-cell coupling in stressed human and mouse myocardium. J. Clin. Invest 120, 266–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smyth JW, Vogan JM, Buch PJ, Zhang S-S, Fong TS, Hong T-T, and Shaw RM (2012). Actin cytoskeleton rest stops regulate anterograde traffic of connexin 43 vesicles to the plasma membrane. Circ. Res 110, 978–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorrentino A, Thakur N, Grimsby S, Marcusson A, von Bulow V, Schuster N, Zhang S, Heldin CH, and Landström M (2008). The type I TGF-beta receptor engages TRAF6 to activate TAK1 in a receptor kinase-independent manner. Nat. Cell Biol 10, 1199–1207. [DOI] [PubMed] [Google Scholar]
- Stoneley M, and Willis AE (2004). Cellular internal ribosome entry segments: structures, trans-acting factors and regulation of gene expression. Oncogene 23, 3200–3207. [DOI] [PubMed] [Google Scholar]
- Tacheau C, Fontaine J, Loy J, Mauviel A, and Verrecchia F (2008). TGF-beta induces connexin43 gene expression in normal murine mammary gland epithelial cells via activation of p38 and PI3K/AKT signaling pathways. J. Cell. Physiol 217, 759–768. [DOI] [PubMed] [Google Scholar]
- Tamarkin-Ben-Harush A, Vasseur JJ, Debart F, Ulitsky I, and Dikstein R (2017). Cap-proximal nucleotides via differential eIF4E binding and alternative promoter usage mediate translational response to energy stress. eLife 6, e21907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terenin IM, Smirnova VV, Andreev DE, Dmitriev SE, and Shatsky IN (2017). A researcher’s guide to the galaxy of IRESs. Cell. Mol. Life Sci 74, 1431–1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner JD, Vernocchi S, Schmitz S, and Muller CP (2014). Role of the 5′-untranslated regions in post-transcriptional regulation of the human glucocorticoid receptor. Biochim. Biophys. Acta 1839, 1051–1061. [DOI] [PubMed] [Google Scholar]
- Ul-Hussain M, Olk S, Schoenebeck B, Wasielewski B, Meier C, Prochnow N, May C, Galozzi S, Marcus K, Zoidl G, and Dermietzel R (2014). Internal ribosomal entry site (IRES) activity generates endogenous carboxyl-terminal domains of Cx43 and is responsive to hypoxic conditions. J. Biol. Chem 289, 20979–20990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watkins SJ, Jonker L, and Arthur HM (2006). A direct interaction between TGFbeta activated kinase 1 and the TGFbeta type II receptor: implications for TGFbeta signalling and cardiac hypertrophy. Cardiovasc. Res 69, 432–439. [DOI] [PubMed] [Google Scholar]
- Yamaguchi K, Shirakabe K, Shibuya H, Irie K, Oishi I, Ueno N, Taniguchi T, Nishida E, and Matsumoto K (1995). Identification of a member of the MAPKKK family as a potential mediator of TGF-beta signal transduction. Science 270, 2008–2011. [DOI] [PubMed] [Google Scholar]
- Zhao S, and Fernald RD (2005). Comprehensive algorithm for quantitative real-time polymerase chain reaction. J. Comput. Biol 12, 1047–1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.