

Abstract
Objective
To determine the molecular and cellular bases of autoinflammatory syndromes in a multigenerational French family with Muckle‐Wells syndrome and in a patient originating from Portugal with familial cold autoinflammatory syndrome.
Methods
Sequencing of NLRP3 exon 3 was performed in all accessible patients. Microsatellite and whole‐genome single nucleotide polymorphism genotyping was used i) to test the intrafamilial segregation of the identified variant and ii) to look for a founder effect. Functional analyses included the study of i) apoptosis‐associated speck‐like protein containing a CARD (ASC) speck formation in HEK293T cells (stably expressing ASC–green fluorescent protein and pro‐caspase 1‐FLAG) transiently expressing the wild‐type or mutated NLRP3 protein, ii) levels of IL‐1β secreted from transfected THP‐1 cells, and iii) inflammasome‐related gene expression and cytokine secretion from monocytes isolated from patients in crisis (probands from the two families), related patients out of crisis, and from controls.
Results
The same heterozygous mutation (c.1322C>T, p.A441V) located in the NACHT domain, segregating with the disease within the first family, was identified in the two families. This mutation was found to be associated with different core haplotypes. NLRP3‐A441V led to increased ASC speck formation and high levels of secreted IL‐1β. Monocyte inflammasome‐related gene expression and cytokine secretion, which were within the normal range in patients out of crisis, were found to be differentially regulated between the two probands, correlating with their phenotypic status.
Conclusion
These molecular and cellular findings, which indicate a recurrent mutational event, clearly demonstrate the pathogenicity of the p.A441V missense mutation in NLRP3‐associated autoinflammatory disease and point to the interest of studying patients’ primary cells to assess disease activity.
Keywords: cryopyrin‐associated periodic syndromes, IL‐1β, inflammasome, NLRP3, NLRP3‐AID, variation
Introduction
NLRP3‐associated autoinflammatory disease (NLRP3‐AID), previously known as cryopyrin‐associated periodic syndromes (also known as cryopyrinopathies) 1, is a group of rare autoinflammatory diseases characterized by recurrent episodes of fever accompanied by localized sterile inflammation, mainly in the joints, skin, and serosal membranes (peritoneum, pleura, and pericardium). It encompasses a spectrum of three clinically overlapping syndromes with varying severity: the familial cold autoinflammatory syndrome (FCAS); the Muckle‐Wells syndrome (MWS); and the chronic infantile neurological, cutaneous, and articular syndrome 2. Heterozygous missense gain‐of‐function mutations in NLRP3 (also known as FCAS, CIAS1, NALP3, and PYPAF1), a gene located in the q44 region of chromosome 1, are causal for NLRP3‐AID 3, 4.
NLRP3 is mainly expressed in myeloid cells and encodes NLRP3 (also known as cryopyrin), a protein comprising an N‐terminal pyrin domain, a central NACHT (NAIP, CIITA, HET‐E, and TP1) domain, a NACHT‐associated domain (NAD), in addition to a C‐terminal leucine‐rich repeat (LRR) domain. NLRP3 is a member of a large family of intracellular receptors NLR (nucleotide‐binding and LRR receptor), which play major roles in host defense and innate immunity 5, 6. Following detection of a pathogen‐ or damage‐associated molecular pattern 7 or through the alteration of cellular homeostatic conditions 8, NLRP3 initiates inflammasome assembly. Inflammasome associates NLRP3 with pro‐caspase 1 through the adaptor protein apoptosis‐associated speck‐like protein containing a CARD (ASC) leading to caspase‐1 activation and subsequent maturation and secretion of the proinflammatory cytokines IL‐1β and IL‐18 5, 9.
In keeping with our knowledge on inflammasome biology, peripheral blood mononuclear cells (PBMCs) from NLRP3‐AID patients secrete high levels of IL‐1β, and, in most patients, biological therapies targeting IL‐1 significantly improve their clinical status 10, 11. Clinical suspicion of NLRP3‐AID is confirmed by molecular testing. However, given the heterogeneity of the clinical presentation, the existence of overlapping phenotypes, and the absence of pathognomonic clinical symptoms, NLRP3‐AID diagnosis remains a challenging issue. Despite the identification of more than 200 NLRP3 sequence variants (http://fmfighcnrsfr/ISSAID/infevers), their deleterious effects on NLRP3 function is difficult to assess, as the great majority are heterozygous missense variations, the functional consequences of which have not yet been studied.
Here, with the aim to identify the molecular bases of an autoinflammatory syndrome transmitted in a dominant manner in a large family originating from Brittany (France), we analyzed the NLRP3 gene and found a missense variation that perfectly segregates with the disease. The same variation was identified in a second family originating from Portugal. These data prompted us to test the pathogenicity of this sequence variant by means of different functional assays and to look for a founder effect in these two unrelated NLRP3‐AID families.
Patients and Methods
Patients
Clinical features were recorded on a standardized form. All adult patients were seen by the same clinician who provided access to complete medical files and results of basic biological tests (C‐reactive protein [CRP], serum amyloid A, hemogram, creatininemia proteinuria). Informed written consent for genetic studies was given by all participants.
Molecular analyses
Genomic DNA was extracted from peripheral blood leukocytes of affected members using standard procedures. NLRP3 exon 3 and its intronic junctions were amplified by polymerase chain reaction (PCR) and sequenced using the Big Dye Terminator sequencing kit (Applied Biosystems) on an ABI 3130XL automated capillary DNA sequencer (Applied Biosystems). Sequences were analyzed using the SeqScape software (Life Technologies) against the NM reference sequence (NM_004895).
Microsatellite markers (D1S2811, D1S1609, D1S3739, and D1S2682) were amplified by PCR using primers labeled with FAM‐ROX. The PCR products were analyzed on an ABI 3130XL sequencer (Applied Biosystems).
Whole‐genome single nucleotide polymorphism (SNP) genotyping was performed with the HumanOmniExpress BeadChip from Illumina, which contains more than 700 000 SNP markers with an average spacing of ~4 kb. The data were analyzed with the GenomeStudio software (Illumina). Logarithm of the odds (LOD) scores were calculated using the Merlin 1.1.2 software assuming full autosomal dominant inheritance as previously described 12.
Plasmid constructs
The generation of the NLRP3 expression vector (pNLRP3‐WT) was described previously 13. Site‐directed mutagenesis was performed to generate a plasmid carrying the missense variation identified in patients (pNLRP3‐A441V) using the “quick change site‐directed mutagenesis kit” (Stratagene). The resulting plasmid construct was confirmed by sequencing.
Cell culture and transfection
HEK293T cells stably expressing FLAG‐tagged pro‐caspase 1 (pro‐caspase 1‐FLAG) and green fluorescent protein (GFP)‐tagged–ASC (designated ASC‐GFP_C1‐FLAG)—as kindly provided by Emad Alnemri (Thomas Jefferson University, Philadelphia, PA)—were cultured in Dulbecco's modified Eagle's medium/Ham's F12 medium (Invitrogen) supplemented with 10% fetal calf serum, penicillin (100 IU/ml), and streptomycin (100 μg/ml). HEK293T (ASC‐GFP_C1‐FLAG) cells were transfected with either 375, 500, or 750 ng of pNLRP3‐WT or pNLRP3‐A441V or with the empty vector for 24 hours using FuGENE HD (Promega).
THP‐1 cells were cultured in Roswell Park Memorial Institute (RPMI) 1640 medium supplemented with glutamine, penicillin (100 IU/ml), and streptomycin (100 μg/ml). The cells were primed with 100 ng/ml phorbol myristate acetate (PMA) for 3 hours, then transfected either with 500 or 1000 ng of pNLRP3‐WT or pNLRP3‐A441V or with the empty vector using the FF‐100 program in the 4D‐Nucleofector, Amaxa (Lonza). Transfected THP‐1 cells were then distributed in six‐well plates and treated directly with 100 ng/ml lipopolysaccharide (LPS) at 37°C. Twenty‐four hours following transfection and treatment, supernatants were collected and stored at −80°C for cytokine measurement.
Speck quantification assay
The spontaneous formation of ASC aggregates (specks) was measured in HEK293T (ASC‐GFP_C1‐FLAG) cells 24 hours after transfection with pNLRP3‐WT or pNLRP3‐A441V plasmids or with the empty vector, which served as negative control. Manual counting of cells containing ASC‐GFP specks was performed in five representative randomly selected fields at ×20 magnification. The percentage of speck‐positive cells was calculated as the number of specks divided by the total number of counted cells. Cells were observed using a Nikon Eclipse TS100 inverted fluorescent microscope.
Human monocyte isolation
Monocytes were purified from human peripheral blood (EDTA tubes) of Proband I, patients out of crisis from the Family I, Proband II (Family II), and of apparently healthy volunteers provided from the Etablissement Francais du Sang. Briefly, PBMCs were isolated using Pancoll gradient tubes (PANbiotech). Following isolation, monocytes were selected from PBMCs by adherence for 1 hour as previously described 14. Cells were then cultured in the presence of RPMI 1640 complemented with glutamine, 10% pooled human serum, penicillin (100 IU/ml), and streptomycin (100 μg/ml).
Monocyte polarization
Adherent monocytes were treated for 24 hours either with 100 ng/ml interferon gamma (IFN‐γ) (to polarize the cells toward an M1 proinflammatory phenotype) or with 10 ng/ml IL‐4 + 10 ng/ml IL‐13 (to polarize the cells toward an M2 anti‐inflammatory phenotype) or with complete medium alone (control cells, M0) 14, 15. For the last 3 hours, cells were treated with 100 ng/ml LPS. Following treatments, cell lysates were collected for RNA extraction, and cell culture supernatants were used for ELISA. The messenger RNA (mRNA) expression of CXCL10 and ALOX 15 was used as a marker of polarization.
Reverse transcription and quantitative PCR
Following monocyte polarization, total RNA was isolated using the RNAeasy mini kit (Qiagen), including a DNase step (Qiagen). RNA was reversed transcribed in the presence of 2.5 mM oligo‐dT using the Transcriptor High Fidelity cDNA Synthesis Kit (Roche), following the manufacturer's instructions. Five nanograms of complementary DNA (cDNA) was amplified using the Mesa Blue qPCR MasterMix Plus for SYBR Assay (Eurogentec) in the Light Cycler LC480 (Roche). mRNA expression was normalized to the levels of ribosomal protein L13a, RPL13A (NM_012423.2), which was used as a housekeeping gene. The relative expression level of a gene between sample 1 (treated) and sample 2 (control) was calculated using the ΔΔCt formula: 2−(Ct1−Ct RPL13A 1)−(Ct2−Ct RPL13A 2). Normalized Ct values (mean of controls) were used to calculate the gene expression ratio between two samples. Primers were designed using the Probe Finder software (http://qpcr.probefinder.com/organism.jsp; Roche Life Sciences) as seen in Supplementary Table 1.
Cytokine measurements by ELISA
Supernatants of transfected THP‐1 cells or polarized monocytes isolated from healthy controls and patients were centrifuged for 10 minutes at 250g and kept at −80°C for cytokine measurements. Plasma from Proband I, patients out of crisis from Family I, and Proband II was isolated by blood centrifugation at 950g for 10 minutes and kept at −80°C for cytokine measurements. IL‐1α, IL‐1β, IL‐6, IL‐18, TNF‐α, and IL‐1Ra were quantified using the DuoSet ELISA kits (R&D, Biotechne), following the manufacturer's instructions.
Statistical analysis
Differences were analyzed statistically using the unpaired Student's t test and were plotted with GraphPad Prism software (GraphPad). A P value < 0.05 was considered to be statistically significant.
Results
Clinical phenotype
Proband I (Figure 1A, Patient III.3), born in 1982 (in the Brittany region of France), was referred to our laboratory for genetic confirmation of MWS. Her disease, which started during the neonatal period, was characterized by recurrent, spontaneously resolving attacks triggered by cold that lasted a few days, during which she presented with multiple chronic urticarial skin rashes, accompanied with arthralgia but with no ophthalmological, abdominal, or neurological manifestations (sensorineural hearing loss was not tested). Noteworthy, she was apyretic or had low‐grade fever (38°C or less) during the crises. Her CRP levels were 41 mg/L (at the time of monocyte isolation) and she did not wish to receive treatment (Table 1 and Supplementary Table 2 ). This patient, who was born as a result of a nonconsanguineous union, belongs to a large French family (Family I) spanning 4 generations with 10 identified living individuals with a clinical phenotype compatible with the diagnosis of MWS.
Figure 1.
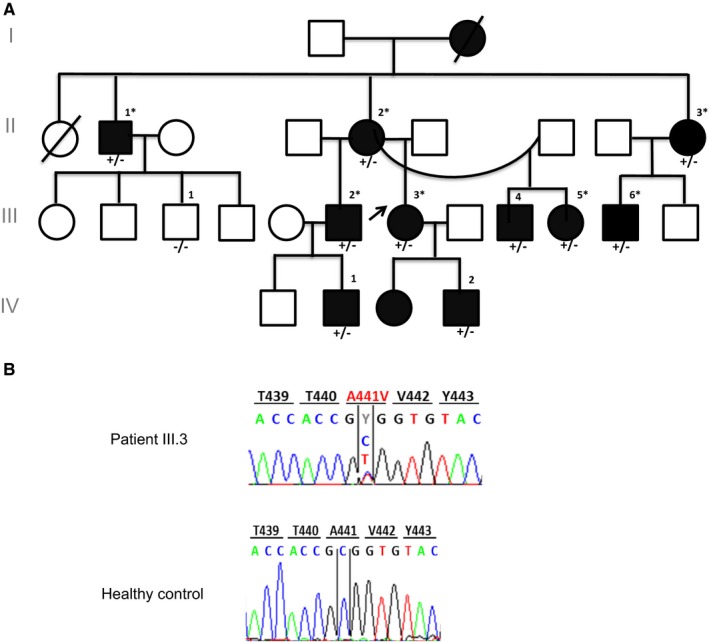
A, Genealogical tree of Family I with NLRP3‐AID and the identified NLRP3 mutation c.1322C>T (p.A441V). Filled symbols represent affected individuals, and empty symbols represent healthy subjects. The black arrow shows Proband I. The asterisk indicates that blood was available for monocyte isolation and ex vivo study. Nucleotide and amino acid coordinates are given assuming that the A of the first ATG codon is nucleotide #1. B, NLRP3 electropherograms corresponding to the direct Sanger sequencing of the polymerase chain reaction (PCR) products from one of the NLRP3‐AID patients (individual III.3, upper panel) showing the heterozygous mutation (c.1322C>T, p.A441V) and a control (lower panel).
Table 1.
Clinical diagnosis and crisis features of NLRP3‐AID members of Family I and Family II
| Family I | Family II | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Proband I (III.3) | II.I | II.2 | II.3 | III.2 | III.4 | III.5 | III.6 | IV.1 | IV.2 | Proband II | |
| Diagnosis | MWS | MWS | MWS | MWS | MWS | MWS | MWS | MWS | MWS | MWS | FCAS |
| Age at onset (years) | 0 | 0 | 1 | 0 | 0 | 1 | 1 | 0 | 0.5 | 2 | 3 |
| Duration of crises (days) | 3‐4 | 3‐4 | 3 | 2 | 3 | 3 | 2 | 1‐3 | 2‐3 | 2‐3 | 3 |
| Frequency | >1/wk | 2/mon | <1/mon | 1/wk | 1/wk | 1‐4/mon | 1/mon | 1/mon | >1/mon | >1/mon | >2/mon |
| >3 crises | + | + | + | + | + | + | + | + | + | + | + |
| Triggers (Cold) | + | + | + | + | + | + | + | + | − | − | + |
Abbreviation: FCAS, familial cold autoinflammatory syndrome; MWS, Muckle‐Wells syndrome.
We also had access to blood samples from nine more patients from the same family during a follow‐up visit. The clinical diagnosis and the features of crises in those patients are summarized in Table 1: the age of disease onset was variable among patients ranging from the neonatal period to childhood, with the majority of patients describing apyretic crises triggered by cold, lasting from 1 to 4 days, and characterized by chronic skin urticaria accompanied sometimes with macular erythema or aphthous ulcers. The majority of the patients presented with ophthalmological manifestations including conjunctivitis or uveitis, as well as articular involvement ranging from mild arthralgia to chronic arthritis. Some patients had sensorineural hearing loss. None of the patients had gastrointestinal manifestations (i.e. no abdominal pain, nausea, vomiting, or diarrhea). Those nine patients were out of crisis. The CRP levels were negative (less than 5 mg/L) for seven patients (II.1, II.2, II.3, III.2, III.5, IV.1 and IV.2, Figure 1). The CRP levels of the remaining two patients (III.4 and III.6, Figure 1), who were untreated and who did not feel to be in crisis, were elevated (28 and 40 mg/L, respectively).
Proband II, from a different family (Family II, originating from Portugal), was diagnosed with FCAS. As summarized in Table 1 and Supplementary Table 2, the patient presented with recurrent attacks triggered by cold and characterized by high‐grade fever (39°C) associated with arthralgia and arthritis, urticaria, and aphthous ulcers in addition to conjunctivitis, uveitis, and elevated CRP levels (recorded as 80 mg/L at time of monocyte isolation) since the age of 3 years. There was no report of neither abdominal or thoracic manifestations nor hearing loss or other neurological involvement. Each crisis lasted around 3 days with two or more crises per month.
Identification of a heterozygous variation in the NLRP3 gene
With the aim to identify the genetic defect underlying the disease in those patients, DNA samples were collected in Probands I and II as well as in nine affected members from Family I (Figures 1A and 1B). Sequencing of NLRP3 exon 3 revealed, in all affected individuals, a transition in exon 3 (c.1322C>T), which predicts a missense variation (p.A441V) in the NACHT domain of the protein (Figure 2A). Several lines of evidence argue for the involvement of this missense variation in NLRP3‐AID: i) it has not been described in sequence‐variant databases, such as the genome aggregation database (gnomAD); ii) it perfectly segregates with the disease in all tested members of Family I; iii) although the missense variation predicts the replacement of a hydrophobic residue (alanine) by another hydrophobic amino acid (valine), it is considered as probably damaging (score: 0.970), damaging (score: 0.04), or deleterious (score: −3.59) according to PolyPhen‐2, Sift, or Provean, respectively; iv) at first glance, this mutation could have an important effect on NLRP3 inflammasome activation, as another missense variation involving the same residue (c.1322A>G, p.A441T) has already been reported in a NLRP3‐AID patient 16. However, in the latter study, no functional study was performed to assess the pathogenicity of the p.A441T variation. In addition, A441 is not fully conserved throughout the evolution (Figure 2B, left panel); sequence alignment of the NACHT domains of the NLRP members with high conservation of their NACHT domain to NLRP3 NACHT domain—NLRP12, NLRP10, NLRP1 and NLRP6 17showed that A441 is only conserved in NLRP12 (Figure 2B, right panel).
Figure 2.
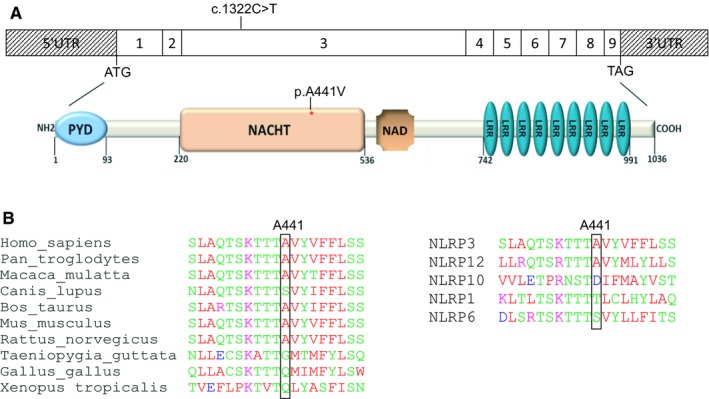
A, Impact of the NLRP3 c.1322C>T transition at the protein level. Exonic organization of the human NLRP3 cDNA and domain‐organization model of the corresponding protein. The identified mutation is indicated. The red asterisk shows the location of the identified mutation in the NACHT domain (top). PYD, pyrin domain; NACHT (NAIP,CIITA,HET‐E and TP1) domain; NAD, NACHT‐associated domain; LRR, leucine‐rich repeat domain. B, Partial interspecies alignment of NLRP3 showing the evolutionary conservation of the domain containing A441 (left panel), as well as the conservation of A441 in NLRP12, one of the human NLRP3 paralogs (right panel).
Microsatellite analysis and whole‐genome SNP genotyping in the two NLRP3‐AID families
The genotype of microsatellite markers flanking NLRP3 and exon 3 SNPs perfectly segregated with the disease phenotype in all tested members of Family I. Microsatellite analysis of all available DNA samples showed that the two unrelated families share the same disease haplotype, thereby suggesting a founder effect in these geographically unrelated NLRP3‐AID families (Figure S1). In order to further test whether the genotyping data obtained at the NLRP3 locus in the two families (i.e. possibly identical microsatellite genotypes) reflect a founder effect or occurred by chance, whole‐genome SNP genotyping was performed with high‐density chips. The results showed that the SNPs flanking NLRP3 perfectly segregated with the disease phenotype within Family I. However, somewhat unexpectedly, this haplotype was not shared with the second family (Proband II), thereby excluding that the two families share a common ancestral origin (Figure S2). More precisely, SNP genotyping showed that at least 88 (6%) among 1389 SNPs, flanking the NLRP3 region and spanning 5457 kb between rs10927034 and rs12135449—the closest SNPs to microsatellite markers D1S2811 and D1S3739, respectively—were found to be discordant between the two probands (Supplementary Table 3).
Impact of the p.A441V variation on ASC speck formations in HEK293T cells
In order to assess the pathogenicity of the p.A441V variation and test its effect on NLRP3 function, we first studied ASC speck formation, a common readout of inflammasome activation 18, in HEK293T (ASC‐GFP_C1‐FLAG) cells transfected with expression plasmids encoding NLRP3‐WT or NLRP3‐A441V. In agreement with the role of NLRP3 in inflammasome assembly and ASC speck formation, cells transfected with pNLRP3‐WT displayed a significantly higher percentage of ASC specks as compared with cells transfected with the empty vector alone (P = 0.0004, P < 0.0001, and P = 0.0002 for 375, 500, and 750 ng of plasmids, respectively). Importantly, cells transfected with the pNLRP3‐A441V construct showed a significantly higher percentage of ASC specks as compared with cells transfected with pNLRP3‐WT (Figure 3A) (P value = 0.0009, P value = 0.0113, and P value = 0.0031 for 375, 500, and 750 ng of plasmid, respectively).
Figure 3.
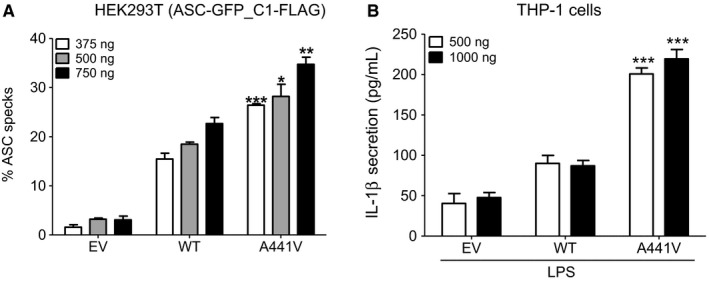
A, Impact of the c.1322C>T, (p.A441V) mutation on apoptosis‐associated speck‐like protein containing a CARD (ASC) speck formation. HEK293T (ASC‐GFP_C1‐FLAG) cells were transfected with different amounts (375, 500, or 750 ng) of the pNLRP3‐WT, pNLRP3‐A441V, or the empty vector (EV). The percentage of ASC specks was calculated as described in Materials and Methods. Results represent the means ± SD from three independent experiments. P values were calculated using unpaired student's t test. Asterisks indicates significant P value (<0.05) as compared with the wild‐type (***P value = 0.0009, *P value = 0.0113, **P value = 0.0031). B, IL‐1β secretion, as assessed by ELISA in cell culture supernatants of phorbol myristate acetate–primed THP‐1 cells transfected with either 500 or 1000 ng of pNLRP3‐WT, pNLRP3‐A441V, or the empty vector and treated with LPS, a known stimulus for NLRP3 inflammasome. Results represent the mean ± SD of 3 independent experiments performed in triplicates. P values were calculated using unpaired student's t test. Asterisks indicates significant P value (<0.05) as compared with the wild type (***P value = 0.0009 or P value = 0.0006).
Impact of the NLRP3 p.A441V variation on cytokine secretion from THP‐1 cells
To test the impact of the p.A441V variation on cytokine secretion, we used THP‐1 cells, which endogenously express NLRP3, ASC, pro‐caspase 1, and IL‐1β. Cells were primed with PMA that induces an adherent macrophage‐like phenotype of the monocytic THP‐1 cells 19 and IL1Β gene expression 20. Cells were then transfected with pNLRP3‐WT or pNLRP3‐A441V and treated with LPS. Cytokine secretion was assessed in the cell culture supernatants. IL‐18, IL‐1α, and IL‐6 levels were undetectable in all conditions tested, whereas no difference was observed in TNF‐α or IL‐1Ra (data not shown). However, as shown in Figure 3B, the levels of IL‐1β, a hallmark of inflammasome activation, were significantly higher in the supernatant of cells transfected with pNLRP3‐A441V as compared with cells transfected either with pNLRP3‐WT (P value = 0.0009 and 0.0006 for 500 and 1000 ng of plasmid, respectively) or with the empty vector (P value = 0.0004 and 0.0002 for 500 and 1000 ng of plasmid, respectively).
Cytokine levels in the plasma of NLRP3‐AID patients
For patients in whom plasma was available for further studies (Probands I and II, as well the following six affected members from Family I: II.1, II.2, II.3, III.2, III.5, and III.6), levels of cytokines IL‐1β, IL‐18, IL‐1α, IL‐6, TNF‐α, and IL‐1Ra were all within the normal range, thereby suggesting that the plasma cytokine levels are unhelpful for diagnosis or follow up of the patients (Supplementary Table 4).
NLRP3 inflammasome gene expression in polarized monocytes of NLRP3‐AID patients
NLRP3 inflammasome gene expression was tested in polarized M1 (proinflammatory phenotype), M2 (anti‐inflammatory phenotype), or nonpolarized monocytes (M0, basal conditions). Polarization markers CXCL10 for M1 and ALOX15 for M2 were upregulated in the respective phenotypes (Figure S3). Expression levels of NLRP3 transcripts at basal conditions (M0) were similar in Proband I; the same six available patients out of crisis from Family I, Proband II; and the healthy controls (Ct~31, mean Ct~31, Ct~29, and mean Ct~31.7, respectively). In accordance with previous studies 14, 21, 22, LPS induced NLRP3 expression in monocytes from healthy donors. A similar increase was observed in the two probands and in patients out of crisis from Family I (Figure 4). At basal conditions (M0), CASP1 mRNA expression was higher in both probands (Ct~25 and Ct~24 in Proband I and II, respectively) as compared with the patients out of crisis (mean Ct~27) or to healthy controls (mean Ct~27). Polarization toward an M1 phenotype increased CASP1 expression in all studied groups, with the highest expression levels found in Probands I and II. No difference was detected in ASC mRNA expression among all studied groups, whereas a minor decrease was seen in the presence of LPS. Basal gene expression of IL1Α and IL1Β was mildly increased in M0 cells of Proband I (IL1Α Ct~28 and Ct~23 for IL1Β) and strongly increased in Proband II (IL1Α Ct~21 and Ct~18 for IL1Β) as compared with the patients out of crisis (IL1Α mean Ct~31 and mean Ct~27 for IL1Β) or healthy controls (IL1Α mean Ct~31 and mean Ct~28 for IL1Β). LPS had a strong effect on the gene expression of IL1Α and IL1Β in healthy controls and patients out of crisis in all tested conditions (ie, M0, M1, M2), whereas its effect was milder in both probands (Figure 4). IL18 mRNA expression was found to be low at basal conditions in all studied groups and in all tested conditions. Interestingly, LPS induced IL18 mRNA only in the M2 cells of Proband II (Ct increased from ~30 to 26.8). Basal mRNA levels of TNF were slightly higher in Proband II as compared with all other groups. As expected, LPS significantly induced TNF expression in all tested conditions. Gene expression of IL6 was much higher in the M0 and M1 cells from Probands I and II (highest in Proband II) as compared with other groups. LPS significantly induced IL6 expression in all tested conditions. Noteworthy, IL1RN mRNA expression was much higher in Proband II in M0, M1, and M2 cells in the presence or absence of LPS (Figure 4).
Figure 4.
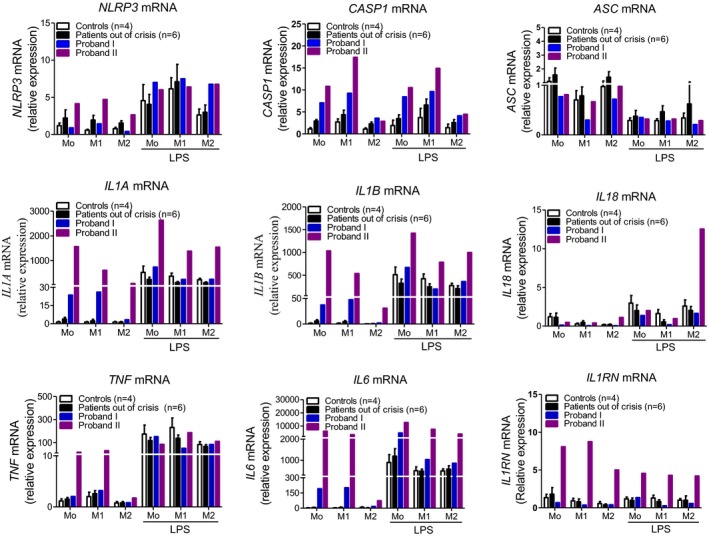
Expression of inflammasome‐related genes (NLRP3, CASP1, and ASC) and genes encoding cytokines in human polarized monocytes (M1: proinflammatory phenotype, M2: anti‐inflammatory phenotype, M0: basal conditions) in the presence or absence of LPS in Proband I, patients out of crisis (n = 6), and Proband II, as compared with apparently healthy donors (n = 4).
Overall, patients out of crisis and the healthy controls presented similar gene expression profiles. However, the expression of the inflammasome‐related genes in the two probands was different from that of patients out of crisis and healthy controls. In addition, this expression pattern was found to be differentially regulated between the two probands, correlating with their phenotypic status. The gene expression of proinflammatory cytokines was indeed found to be higher in Proband II, in whom disease activity was more important than in Proband I (Supplementary Table 2).
NLRP3 inflammasome activation in polarized monocytes of the NLRP3‐AID patients
We then assessed, in all study groups, the cytokine levels in the supernatant of polarized monocytes (M1 or M2 phenotype) following NLRP3 inflammasome activation with LPS. As shown in Figure 5, monocytes from Probands I and II secreted higher amounts of IL‐1β at basal conditions as compared with the patients out of crisis or the healthy controls; similar results were observed for TNF‐α and IL‐6. Noteworthy, levels of secreted IL‐1β, TNF‐α, and IL‐6, were higher in Proband II. As for IL‐1α and IL‐18, their secreted levels were much higher in Proband II as compared with Proband I, patients out of crisis, or healthy controls in all tested conditions (Figure 5). IL‐1Ra levels were higher in the two probands and in patients out of crisis as compared with healthy controls in all conditions. The presence of LPS for 3 hours did not significantly influence the secreted levels of all those cytokines in all conditions tested (Figure 5). Of note, cells from Probands I and II showed a maximal NLRP3 inflammasome activation in the presence of LPS. Treatment with Adenosine triphospshate (ATP) had no further effect on IL‐1β secretion (Figure S4).
Figure 5.
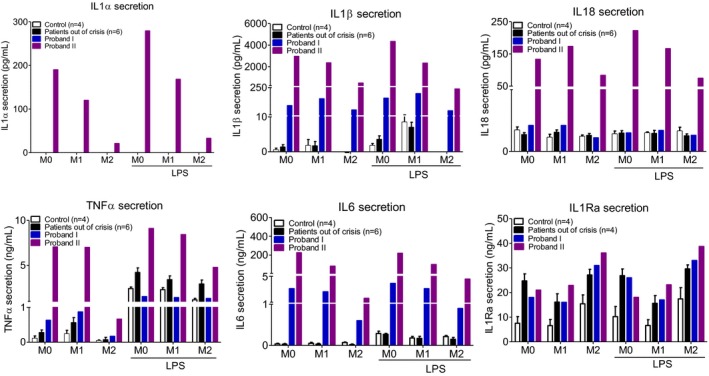
Cytokine levels (IL‐1β, IL‐1α, IL‐18, TNF‐α, IL‐6 and IL‐1Ra) in the supernatant of polarized monocytes (M1: proinflammatory phenotype, M2: anti‐inflammatory phenotype, M0: basal conditions) of Proband I, patients out of crisis (n = 6), and Proband II as compared with apparently healthy donors (n = 4) in the presence or absence of LPS.
Discussion
We studied one French family (Family I), in which several members presented a symptomatology consistent with a clinical diagnosis of MWS, and a patient from a second family originating from Portugal (Family II) who had a clinical suspicion of FCAS. All affected members to which we had access were shown to carry the same heterozygous c.1322C>T, (p.A441V) variation in exon 3 of NLRP3. Despite the fact that microsatellite and intragenic SNP analyses showed that both families may share the same disease haplotype, the results of whole‐genome SNP genotyping did not confirm this hypothesis. Rather, this much more informative approach revealed that the same mutation occurred independently in the two families, in keeping with a recurrent mutational event. Also consistent with a recurrent mutational event is the fact that the mutation is a C‐to‐T transition that occurred on a CpG dimer, a potential hot spot for mutations.
Proband I (Family I) presented with mild symptoms characterized by apyretic multiple urticarial skin lesions and mild arthralgia at time of blood sampling. In this family, to which we had blood sample access from nine other patients, there is a perfect segregation of the disease phenotype with the genotype at the NLRP3 locus, under the assumption of an autosomal dominant mode of inheritance of the disease. However, the family is not large enough to assess the involvement of the NLRP3 locus in the disease by the means of linkage analysis (LOD score at NLRP3 < 3) (Figure S5).
Proband II presented with acute febrile crisis and with highly elevated CRP levels (80 mg/L) at the time of blood sampling. Noteworthy, the heterogeneity of the clinical presentation associated with the c.1322C>T (p.A441V) variation, together with the conflicting data previously reported on this variation, prompted us to study its pathogenicity in vitro as well as ex vivo in patients’ cells. Indeed, on the one hand, the p.A441V variation was first described as a de novo mutation in a patient presenting a FCAS phenotype and in the two subsequent generations where it segregated with the disease haplotype 3; the same variation was also identified in another French family in which DNA samples from three affected siblings and from one of their children was available 23. However, on the other hand, Sobolewska et al. subsequently described a very large NLRP3‐AID family consisting of 94 members in which MWS spanned five generations. Noteworthy, although 15 members were mutation‐positive, up to 14 affected members were found to be mutation‐negative 24. From a genetic viewpoint, such observation, which corresponds to an exclusion of linkage of the NLRP3‐AID phenotype to the NLRP3 gene, seriously puts into question the involvement of the p.A441V missense variation in NLRP3‐AID.
Therefore, we decided to design functional studies in order to assess the pathogenicity of the p.A441V variation. We used the HEK293T (ASC‐GFP_C1‐FLAG) cell line, which we 25 and others 26 have previously used, to test the effect of NLRP3 variations on ASC speck formation. Our results clearly show that the expression of an NLRP3 isoform carrying the p.A441V variation was associated with a significant increase in ASC speck formation as compared with the wild‐type isoform. In accordance to what was previously shown by Duncan et al in a single experiment 27, we showed that transfected THP‐1 cells with pNLRP3‐A441V secrete significantly higher levels of IL‐1β as compared with cells transfected with the pNLRP3‐WT, a result in keeping with the gain‐of‐function effect of the p.A441V mutation. In transfected THP‐1 cells, IL‐1β was the only cytokine, among an array of cytokines studied, that was significantly associated with the expression of the mutated NLRP3. This latter observation, which is not surprising given the existing differences between THP‐1 cells and human primary monocytes 28, 29, underlines the importance of using both cell models in order to better understand the functional consequences of the identified mutation.
In order to understand the heterogeneity of the clinical presentation associated with the p.A441V mutation and to further demonstrate the impact of this mutation in NLRP3‐AID pathogenicity, we worked with patients’ primary monocytes in which we studied the inflammasome‐related gene expression and cytokine secretion. To the best of our knowledge, this is the first study in which patients’ primary cells are analyzed in order to assess the pathogenicity of this mutation. Most importantly, the current study shows that the phenotype of monocytes, which are the disease target cells, fits very well with the clinical presentation of the patients. Indeed, patients out of crisis show results similar to those found in healthy controls. Proband I, who had only chronic multiple urticarial skin rash and mild arthralgia at the time of blood sampling (CRP = 40 mg/L), showed a mild increase in inflammasome‐related gene expression and cytokine secretion as compared with Proband II, who presented with acute febrile crisis associated with urticarial rash, arthritis, and uveitis (CRP = 80 mg/L) at the time of blood sampling. Although the amounts of inflammasome‐related NLRP3, ASC, and IL18 transcripts were not different among the studied groups, levels of CASP1, IL1Α, IL1Β, and IL6 transcripts were mildly upregulated in Proband I (as compared with patients out of crisis or healthy controls) and strongly increased in Proband II, thereby correlating with the clinical status of the two probands. At the protein level, monocytes from Probands I and II secreted higher levels of IL‐1β, TNF‐α, and IL‐6 under basal conditions (the highest levels were observed in Proband II) as compared with patients out of crisis or healthy controls. Interestingly, in the supernatant of Proband II's monocytes, we could also detect IL‐1α and IL‐18, thereby suggesting a potential role for these two inflammatory cytokines in acute NLRP3‐AID crises.
In conclusion, our findings using in vitro cell lines (HEK293T and THP‐1 cells) and the patients’ primary cells clearly demonstrate the pathogenic role of the p.A441V missense mutation in the clinical phenotype of the affected members. Our study also shows that THP‐1 cells, a widely used cell line mimicking some characteristics of human monocytes 30, are useful as in vitro models to assess the pathogenicity and to establish the causality of NLRP3 mutations. When available, the study of monocytes from patients also represents a valuable model to explore and explain the heterogeneity of the disease phenotype.
Author Contributions
Awad, Assrawi, Giurgea, Karabina, and Amselem wrote the manuscript; Jumeau, Odent, Despert, Cam, Perdriger, Louvrier, Cobret, Copin, Chantot‐Bastaraud, Duquesnoy, Piterboth, Le Jeunne, Quenum‐Miraillet, Siffroi, Georgin‐Lavialle, Grateau, and Legendre reviewed the manuscript; and all authors approved the final manuscript.
Study conception and design
Giurgea, Karabina, Amselem.
Acquisition of data
Awad, Assrawi, Jumeau, Odent, Despert, Cam, Perdriger, Cobret, Chantot‐Bastaraud, Piterboth, Le Jeunne and Georgin‐Lavialle and Grateau.
Analysis and interpretation of data
Awad, Assrawi, Louvrier, Copin, Duquesnoy, Quenum‐Miraillet, Siffroi, Legendre, Giurgea, Karabina and Amselem.
Supporting information
Acknowledgments
We are grateful to the affected persons and their families whose cooperation made this study possible. We would like to thank Drs. Emmanuelle Génin and David‐Alexandre Trégouët for helpful discussions.
Dr. Awad's work was supported by a grant from the French government and Alquds University, Palestine, and from the Fondation pour la Recherche Médicale (FDT20130928419). Dr. Assrawi's work was supported by a grant from the French government and An‐Najah National University, Palestine. Dr. Jumeau's work was supported by a grant from the Doctoral School Complexité du Vivant.
Drs. Awad and Assrawi contributed equally to this article.
Prs. Grateau and Georgin‐Lavialle report honoraria as occasional consultants for SOBI, Bristol‐Myers Squibb, and Novartis laboratories unrelated to this study. No other disclosures relevant to this article were reported.
Contributor Information
Sonia‐Athina Karabina, Email: sonia.karabina@sorbonne-universite.fr.
Serge Amselem, Email: serge.amselem@inserm.fr.
References
- 1. Ben‐Chetrit E, Gattorno M, Gul A, Kastner DL, Lachmann HJ, Touitou I, et al. Consensus proposal for taxonomy and definition of the autoinflammatory diseases (AIDs): a Delphi study. Ann Rheum Dis 2018;77:1558–65. [DOI] [PubMed] [Google Scholar]
- 2. Kuemmerle‐Deschner JB. CAPS–pathogenesis, presentation and treatment of an autoinflammatory disease. Semin Immunopathol 2015;37:377–85. [DOI] [PubMed] [Google Scholar]
- 3. Hoffman HM, Mueller JL, Broide DH, Wanderer AA, Kolodner RD. Mutation of a new gene encoding a putative pyrin‐like protein causes familial cold autoinflammatory syndrome and Muckle‐Wells syndrome. Nat Genet 2001;29:301–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Feldmann J, Prieur AM, Quartier P, Berquin P, Certain S, Cortis E, et al. Chronic infantile neurological cutaneous and articular syndrome is caused by mutations in CIAS1, a gene highly expressed in polymorphonuclear cells and chondrocytes. Am J Hum Genet 2002;71:198–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Martinon F, Mayor A, Tschopp J. The inflammasomes: guardians of the body. Annu Rev Immunol 2009;27:229–65. [DOI] [PubMed] [Google Scholar]
- 6. Chen G, Pedra JH. The inflammasome in host defense. Sensors (Basel) 2010;10:97–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Martinon F. Detection of immune danger signals by NALP3. J Leukoc Biol 2008;83:507–11. [DOI] [PubMed] [Google Scholar]
- 8. Liston A, Masters SL. Homeostasis‐altering molecular processes as mechanisms of inflammasome activation. Nat Rev Immunol 2017;17:208–14. [DOI] [PubMed] [Google Scholar]
- 9. Schroder K, Tschopp J. The inflammasomes. Cell 2010;140:821–32. [DOI] [PubMed] [Google Scholar]
- 10. Agostini L, Martinon F, Burns K, McDermott MF, Hawkins PN, Tschopp J. NALP3 forms an IL‐1beta‐processing inflammasome with increased activity in Muckle‐Wells autoinflammatory disorder. Immunity 2004;20:319–25. [DOI] [PubMed] [Google Scholar]
- 11. Koné‐Paut I, Galeotti C. Anakinra for cryopyrin‐associated periodic syndrome. Expert Rev Clin Immunol 2014;10:7–18. [DOI] [PubMed] [Google Scholar]
- 12. Abecasis GR, Cherny SS, Cookson WO, Cardon LR. Merlin–rapid analysis of dense genetic maps using sparse gene flow trees. Nat Genet 2002;30:97–101. [DOI] [PubMed] [Google Scholar]
- 13. Jéru I, Hayrapetyan H, Duquesnoy P, Sarkisian T, Amselem S. PYPAF1 nonsense mutation in a patient with an unusual autoinflammatory syndrome: role of PYPAF1 in inflammation. Arthritis Rheum 2006;54:508–14. [DOI] [PubMed] [Google Scholar]
- 14. Awad F, Assrawi E, Jumeau C, Georgin‐Lavialle S, Cobret L, Duquesnoy P, et al. Impact of human monocyte and macrophage polarization on NLR expression and NLRP3 inflammasome activation. PLoS One 2017;12:e0175336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kittan NA, Allen RM, Dhaliwal A, Cavassani KA, Schaller M, Gallagher KA, et al. Cytokine induced phenotypic and epigenetic signatures are key to establishing specific macrophage phenotypes. PLoS One 2013;8:e78045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Dodé C, Le Dû N, Cuisset L, Letourneur F, Berthelot JM, Vaudour G, et al. New mutations of CIAS1 that are responsible for Muckle‐Wells syndrome and familial cold urticaria: a novel mutation underlies both syndromes. Am J Hum Genet 2002;70:1498–1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Tschopp J, Martinon F, Burns K. NALPs: a novel protein family involved in inflammation. Nat Rev Mol Cell Biol 2003;4:95–104. [DOI] [PubMed] [Google Scholar]
- 18. Man SM, Kanneganti TD. Regulation of inflammasome activation. Immunol Rev 265:6–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zeng C, Wang W, Yu X, Yang L, Chen S, Li Y. Pathways related to PMA‐differentiated THP1 human monocytic leukemia cells revealed by RNA‐Seq. Sci China Life Sci 2015;58:1282–7. [DOI] [PubMed] [Google Scholar]
- 20. Sharma B, McLeland CB, Potter TM, Stern ST, Adiseshaiah PP. Assessing NLRP3 inflammasome activation by nanoparticles. Methods Mol Biol 2018;1682:135–147. [DOI] [PubMed] [Google Scholar]
- 21. O'Connor W Jr, Harton JA, Zhu X, Linhoff MW, Ting JP. Cutting edge: CIAS1/cryopyrin/PYPAF1/NALP3/CATERPILLER 1.1 is an inducible inflammatory mediator with NF‐κB suppressive properties. J Immunol 2003;171:6329–33. [DOI] [PubMed] [Google Scholar]
- 22. Horstmann JP, Marzi I, Relja B. Adrenergic stimulation alters the expression of inflammasome components and interleukins in primary human monocytes. Exp Ther Med 2016;11:297–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hentgen V, Despert V, Leprêtre AC, Cuisset L, Chevrant‐Breton J, Jégo P, et al. Intrafamilial variable phenotypic expression of a CIAS1 mutation: from Muckle‐Wells to chronic infantile neurological cutaneous and articular syndrome. J Rheumatol 2005;32:747–51. [PubMed] [Google Scholar]
- 24. Sobolewska B, Angermair E, Deuter C, Doycheva D, Kuemmerle‐Deschner J, Zierhut M. NLRP3 A439V mutation in a large family with cryopyrin‐associated periodic syndrome: description of ophthalmologic symptoms in correlation with other organ symptoms. J Rheumatol 2016;43:1101–6. [DOI] [PubMed] [Google Scholar]
- 25. Jéru I, Marlin S, Le Borgne G, Cochet E, Normand S, Duquesnoy P, et al. Functional consequences of a germline mutation in the leucine‐rich repeat domain of NLRP3 identified in an atypical autoinflammatory disorder. Arthritis Rheum 2010;62:1176–85. [DOI] [PubMed] [Google Scholar]
- 26. Yu JW, Wu J, Zhang Z, Datta P, Ibrahimi I, Taniguchi S, et al. Cryopyrin and pyrin activate caspase‐1, but not NF‐κB, via ASC oligomerization. Cell Death Differ 2006;13:236–49. [DOI] [PubMed] [Google Scholar]
- 27. Duncan JA, Bergstralh DT, Wang Y, Willingham SB, Ye Z, Zimmermann AG, et al. Cryopyrin/NALP3 binds ATP/dATP, is an ATPase, and requires ATP binding to mediate inflammatory signaling. Proc Natl Acad Sci U S A 2007;104:8041–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gaffney EV, Stoner CR, Lingenfelter SE, Wagner LA. Demonstration of IL‐1α and IL‐1β secretion by the monocytic leukemia cell line, THP‐1. J Immunol Methods 1989;122:211–8. [DOI] [PubMed] [Google Scholar]
- 29. Schildberger A, Rossmanith E, Eichhorn T, Strassl K, Weber V. Monocytes, Peripheral Blood Mononuclear Cells, and THP‐1 Cells Exhibit Different Cytokine Expression Patterns following Stimulation with Lipopolysaccharide. Mediators Inflamm 2013;2013:697972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Auwerx J. The human leukemia cell line, THP‐1: a multifacetted model for the study of monocyte‐macrophage differentiation. Experientia 1991;47:22–31. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials