

Abstract
Objective
Type I interferon (IFN) is important to systemic lupus erythematosus (SLE) pathogenesis, but it is not clear how chronic elevations in IFN alter immune function. We compared cytokine responses after whole blood stimulation with Toll‐like receptor (TLR) agonists in high‐ and low‐IFN SLE patient subgroups.
Methods
SLE patients and nonautoimmune controls were recruited, and SLE patients were categorized as either high or low IFN. Whole blood was dispensed into tubes coated with lipopolysaccharide (LPS), oligonucleotides with cytosine‐guanine repeats, Resiquimod, IFN‐α, and IFN‐α + LPS. Cytokine production in patient sera and after whole blood TLR stimulation was measured by multiplex assay, and type I IFN was assessed using a functional assay.
Results
Circulating plasmacytoid dendritic cell numbers were specifically reduced in high‐IFN SLE patients and not in low‐IFN SLE patients. In serum, we observed that the correlations between cytokines in serum differed to a much greater degree between the high‐ and low‐IFN groups (P < 0.0001) than the absolute cytokine levels differed between these same groups. In stimulated conditions, the high‐IFN patients had less cytokine production in response to TLR ligation than the low‐IFN SLE patients. LPS produced the most diverse response, and a number of interactions between type I IFN and LPS were observed.
Conclusion
We find striking differences in resting and stimulated cytokine patterns in high‐ vs. low‐IFN SLE patients, which supports the biological importance of these patient subsets. These data could inform personalized treatment approaches and the pathogenesis of SLE flare following infection.
Introduction
Interferon (IFN)‐α has been implicated as a causal factor in human systemic lupus erythematosus (SLE) 1. Approximately half of SLE patients have high circulating IFN‐α levels in large patient cohorts 2, and elevated circulating IFN‐α has been associated with disease severity and activity 1, 3. Longitudinal studies of SLE cohorts have generally documented the stability of type I IFN levels over time, despite the association with disease activity 4, 5. These data taken together suggest that although the high‐IFN subgroup of patients generally has greater disease activity, the type I IFN levels are relatively stable, thus forming two large subgroups of patients that are largely distinct from each other. This is supported by the idea that both genetic factors and stable autoantibodies, such as anti‐Ro and anti‐ ribonucleoprotein, have demonstrated strong associations with circulating IFN‐α levels 2, 6, 7, 8.
High levels of IFN‐α have been associated with clinical features in SLE, such as renal disease, skin rash, and hematological manifestations 2, 9, 10. This would suggest underlying differences in immunopathogenesis between high‐ and low‐IFN SLE patients and that these differences contribute to the observed heterogeneity in clinical features between patients 11. Although it is clear that IFN‐α marks an important pathologic subgroup of SLE patients, the differences in the functional immune response between high‐ and low‐IFN SLE patients remains largely unknown. The impact of the chronic elevation of IFN‐α found in SLE upon the immune system would likely not be predicted well by short‐term in vitro experiments.
Some previous studies have examined differences in circulating cytokine levels in SLE patients, comparing those with high and low type I IFN levels 12, 13, 14. These have generally shown that B lymphocyte stimulator levels are correlated with type I IFN levels in patients and that tumor necrosis factor (TNF)‐α is not associated with type I IFN levels in circulation. In the present study, we used a standardized whole blood stimulation method to compare stimulated cytokine outputs after Toll‐like receptor (TLR) engagement, which allowed us to compare the functional immune system in SLE patients with high and low IFN levels. We found that stimulated cytokine outputs allows for greater differentiation between high and low IFN subjects than resting levels of cytokines in circulation. Interestingly, TLR4 stimulation demonstrated a great diversity of responses between high and low IFN groups, suggesting a particularly strong interaction between IFN and lipopolysaccharide (LPS) stimulation. This result may help to explain the immunopathogenesis of an SLE flare following gram negative bacterial infection.
Materials and Methods
Subjects and samples
We studied 32 female SLE patients in total between 18 and 55 years of age who met the American College of Rheumatology 1997 update to the 1982 criteria for the classification of lupus 15. Exclusion criteria were pregnancy, active acute or chronic infections, current daily prednisone dosage more than 10 mg, and current biologic or intravenous therapy. Ten healthy female controls were included after being screened for the absence of autoimmune, inflammatory, or infectious conditions, and they were not receiving any immunomodulatory or cancer treatments. Controls were recruited across a similar age range as the patients. Demographic and clinical data from the patients are shown in Table 1. There were 9 high‐IFN SLE patients and 23 low‐IFN SLE patients. High‐IFN SLE patients were defined as those having IFN levels more than 2SD above the mean of healthy controls, as we have done previously 2. For the overall analyses of plasmacytoid dendritic cells (pDCs) and serum cytokines, all patients were included (Figures 1, 2, 3). For the stimulated cytokine assays (Figures 4 and 5), all of the 9 high‐IFN patients were included, and 11 low‐IFN patients were included to balance the two groups. As has been reported in a number of previous studies, the high‐IFN SLE patients were younger and had lower complement C3 and C4 as compared with the low‐IFN SLE patients 2, 9, 16. Informed consent was obtained from all subjects, and the study was approved by the institutional review board (i14‐00487).
Table 1.
Patient demographics and characteristics
| Patient Characteristicsa | High IFN (n = 9) | Low IFN (n = 23) |
|---|---|---|
| Demographics | ||
| Age (mean in years)b | 27.3 | 54.3 |
| Ancestry (Euro‐American) | 88.8 | 100.0 |
| ACR SLE criteria | ||
| Malar rash | 55.5 | 56.5 |
| Discoid rash | 55.5 | 43.5 |
| Photosensitivity | 88.8 | 69.6 |
| Oral ulcers | 55.5 | 52.2 |
| Arthritis | 66.6 | 87.0 |
| Serositis | 22.2 | 42.9 |
| Renal disorder | 44.4 | 47.8 |
| Neurological disorder | 11.1 | 13.0 |
| Hematological disorder | 55.5 | 39.1 |
| Immunologic disorder | 55.5 | 86.4 |
| SLEDAI, mean (SD) | 4.11 (3.69) | 2.26 (2.51) |
| Laboratory values | ||
| low C3b | 28.6 | 4.4 |
| low C4b | 62.5 | 13.0 |
| Positive ANA | 100.0 | 95.4 |
| Positive anti‐dsDNA | 62.5 | 47.6 |
| Positive anti‐Smith | 28.6 | 26.3 |
| Medication use | ||
| Hydroxychloroquine | 77.0 | 82.6 |
| Mycophenolate | 44.4 | 17.4 |
| Azathioprine | 0.0 | 4.34 |
| Methotrexate | 11.1 | 8.70 |
| Leflunomide | 11.1 | 0.0 |
| Prednisone | 44.4 | 43.5 |
| Prednisone dose (mg), mean (SD) | 8.1 (19.6) | 2.0 (4.65) |
Abbreviation: ACR, American College of Rheumatology; ANA, antinuclear angibodies; dsDNA, double‐stranded DNA; IFN, interferon; SLE, systemic lupus erythematosus; SLEDAI, Systemic Lupus Erythematosus Disease Activity Index.
aAge is shown in years; other values represent the percentage of patients who have that finding or use the specific medication. bIndication of a significant difference between groups (P < 0.05 by Fisher's exact test or unpaired t test for age data). All demographic, laboratory, disease activity, and medication values reflect those at time of sampling.
Figure 1.
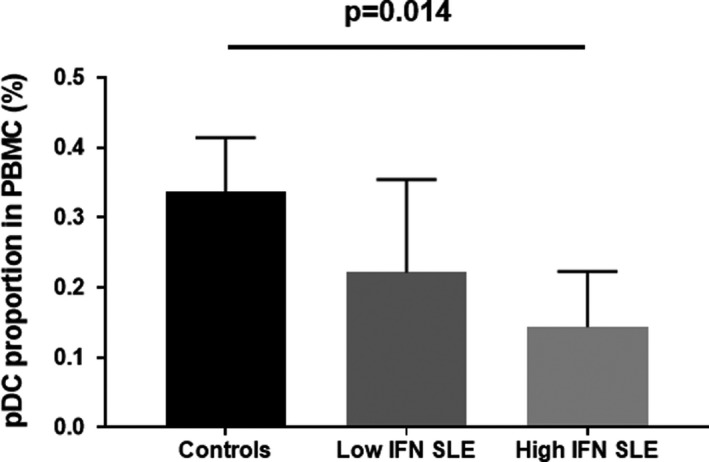
Circulating plasmacytoid dendritic cell (pDC) numbers in high‐ vs. low‐interferon (IFN) systemic lupus erythematosus (SLE) patients and controls. Proportion of pDCs in the total peripheral blood mononuclear cell (PBMC) fraction is shown on the y‐axis in percentage (0.5 = 0.5%). The P value by linear regression across the three categories, fit to a straight line of positive or negative slope with a horizontal line as the null hypothesis.
Figure 2.
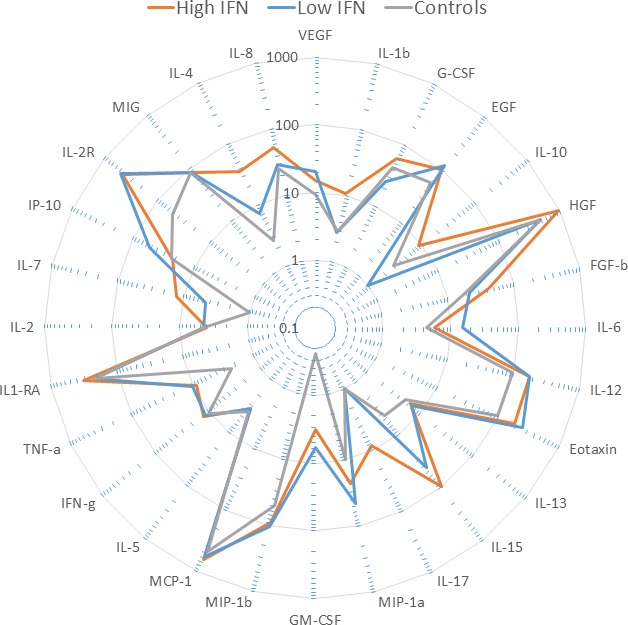
Radar plot of serum cytokine values in high‐ vs. low‐interferon (IFN) systemic lupus erythematosus (SLE) patients and controls. Each ray represents numeric values for one cytokine, presented as a median value in log scale for each subject group (see inset legend for color codes). Each cytokine was tested for statistical difference between high‐ and low‐IFN patient groups, and nominal significance (P < 0.05) was observed for increased interleukin (IL)‐1b and IL‐4 in the high IFN group.
Figure 3.
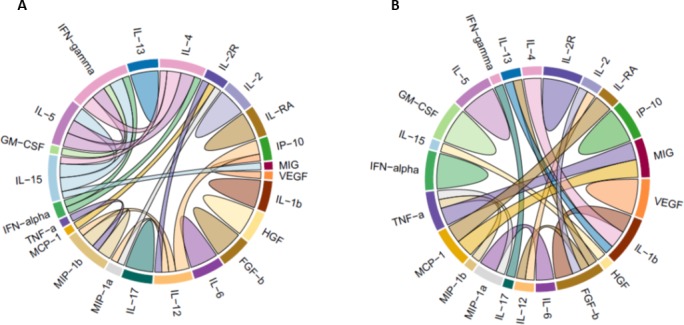
Chord diagrams demonstrating correlations between serum cytokines in high‐ vs. low‐ interferon (IFN) patient groups. The correlation pattern in low IFN systemic lupus erythematosus (SLE) patients (A) and the correlation pattern in high‐IFN SLE patients (B). Only correlations with a coefficient of 0.7 or greater are shown, and correlations of 0.7 to 0.8 were represented with thin chords, whereas correlation coefficients greater than 0.8 were represented with thick chords.
Figure 4.

Stimulated cytokine results from whole blood culture. Radar plots of stimulated cytokine outputs after whole blood culture are shown for low‐interferon (IFN) (A) and high‐IFN (B) systemic lupus erythematosus (SLE) patients. Each ray represents numeric values for one cytokine and is presented as a median value in log scale for each cytokine, with the tube type indicated by color of the line (see inset legend for color codes). See the text and supplemental tables for significant differences between patient and control groups. C, A summary of the lipopolysaccharide (LPS) stimulation data, with each line representing a cytokine or group of cytokines that shared the same pattern (color key in the inset legend). Elevated means a statistically significant elevation as compared to the null (no stimulus) tube for that patient group. Every condition shown is LPS stimulated, so “controls” = the control LPS stimulation tube as compared to the control null tube, and “High IFN SLE + IFN” = the high IFN SLE patient IFN‐α + LPS tube as compared to the high IFN SLE patient null tube. See supplemental tables for all individual comparison results.
Figure 5.
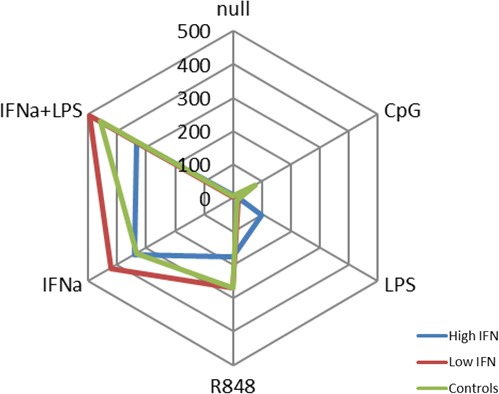
Radar plot of stimulated type I interferon (IFN) production in high‐ vs. low‐IFN systemic lupus erythematosus (SLE) patients and controls. Each ray represents numeric values for one cytokine and is presented as a median value in linear scale for each subject group (see inset legend for color codes).
Tru‐Culture system
Whole blood was dispensed into Tru‐Culture tubes (Myriad RBM) shortly following phlebotomy. The inside surface of the tubes were coated with a standardized amount of lyophilized stimulant to provide an exact concentration of stimulant in the whole blood sample. Stimulants in the tubes were as follows, with a separate tube for each: IFN‐α, oligonucleotides with cytosine‐guanine repeats (CpG), Resiquimod (R848), LPS, IFN‐α + LPS, and null (no stimulant). The tubes were incubated overnight at 37°C as per manufacturer protocol, and cytokines were measured in the samples from the tubes after an incubation period (22 hours) using the methods described below.
Multiplex cytokine assay
The following cytokines were measured in sera and supernatants from Tru‐Culture tubes using a human cytokine panel (Invitrogen) per manufacturer instructions: EGF, Exotaxin, fibroblast growth factor (FGF)‐β, hepatocyte growth factor (HGF), interleukin (IL)‐RA, IL‐1β, IL‐2, IL‐2r, IL‐4, IL‐5, IL‐6, IL‐7, IL‐8, IL‐10, IL‐12 (70), IL‐13, IL‐17, IP‐10, granulocyte‐colony stimulating factor (CSF), granulocyte‐macrophage–CSF, IFN‐γ, monocyte chemoattractant protein‐1 (MCP‐1; MCAF), monokine induced by gamma, macrophage inflammatory protein (MIP)‐1β, RANTES, vascular endothelial growth factor (VEGF), and TNF‐α.
WISH assay for type I IFN
Reporter cells were used to measure the ability of patient sera and Tru‐Culture supernatants to cause IFN‐induced gene expression. The reporter cells (WISH cells, American Type Culture Collection [ATCC] CCL‐25) are cultured in 50% patient serum or diluted supernatant for 6 hours. The cells are lysed and complementary DNA was made from total cellular messenger RNA. Canonical type I IFN‐induced gene expression (MX1, PKR and IFIT1) is measured using quantitative polymerase chain reaction. The relative expression of these three genes is standardized to healthy donors and summed to generate a score reflecting the ability of sera to cause IFN‐induced gene expression (serum type I IFN activity). For more detail, please see the references 17, 18. This assay has been highly sensitive and informative in a variety of human autoimmune disease cohorts 2, 19, 20.
Flow cytometry
Peripheral blood mononuclear cells (PBMCs) were isolated on Ficoll‐Paque gradients (Cederlane Laboratories). Cells were labeled with lineage (lin) antibody cocktail‐fluorescein isothiocyanate (CD3, CD14, CD16, CD19, CD20, and CD56; Biolegend Inc), anti‐HLADR‐eFluor 450 (clone LN3; Ebioscience Inc), and anti‐CD123‐PE‐Cy7 (clone 6H6). pDCs were identified as lineage negative cells that were HLADR+CD123+. Cells were analyzed using a BDTM LSR II flow cytometer using FACSDiva software (BD Biosciences) at the Flow Cytometry core facility. PBMCs were gated upon using light scatter properties. Isotype controls were used to determine nonspecific fluorescence.
Statistical analysis
Data were tested for normality using the D'Agostino‐Pearson normality test, and parametric vs. nonparametric analyses were used depending upon the result (unpaired t test vs. Mann‐Whitney U test). Most cytokine values did not conform to a normal distribution, and for multiple testing, such as multiple correlation or multiple t tests, nonparametric analyses were employed. Statistical correction for multiple comparisons was done on the multiple t tests for the stimulated cytokine results using the Benjamini, Kreiger, and Yekutieli method choosing a 5% false discovery rate as implemented in the GraphPad software program.
Results
pDCs are reduced in circulation in SLE patients with high IFN levels
We studied the frequency of circulating pDCs in the high‐ and low‐IFN SLE patients as well as controls, as the frequency of pDCs in circulation has been reported to be reduced in patients with SLE 21. Interestingly, the patients with high IFN had the lowest circulating pDC counts, which was significantly less than controls (Figure 1, P = 0.014). The low IFN SLE patients were intermediate between the controls and the high IFN SLE patients. Interestingly there were some low IFN SLE patients with pDC proportions that were similar to controls, whereas the distribution of pDC proportion in the high IFN SLE patients did not overlap that of the controls. There were no differences in the absolute lymphocyte count, CD19+CD20+ cell counts, or monocyte subset counts between high and low IFN patients (data not shown.)
Correlations between serum cytokines in high vs. low IFN SLE patients are strikingly different
We measured circulating cytokines in serum from patients and controls using a multiplex assay as outlined in the Methods section to assess 30 cytokines. Radar plots showing serum cytokine profiles in patients with high vs. low IFN are shown in Figure 2. IL‐1b and IL‐4 showed a trend toward being higher in serum in the high IFN patients as compared with the low IFN SLE patients (nominally significant P < 0.05 for both). We next examined correlation patterns between serum cytokines in the high‐ and low‐IFN patient groups. Interestingly, the correlations between cytokines were significantly different between the low‐ and high‐IFN patient groups (Figure 3). Of the 30 correlations observed with a correlation coefficient of 0.7 or greater, only 1 was common between the high and low‐IFN patient groups, whereas 29 were distinct and not shared with the other IFN subgroup. This differs significantly from a model in which half of the correlations are shared between the IFN subgroups (P < 0.0001 by the Fisher exact test). Notable differences include multiple correlations with IFN‐γ, IL‐4, IL‐5, IL‐12, IL‐15, and MIP‐1β in low‐IFN patients, and multiple correlations with IL‐1β, FGF‐β, and TNF‐α in the high‐IFN patients. The one shared cytokine correlation between high‐ and low‐IFN SLE patients was between TNF‐α and MIP‐1β (Figure 3). These data support the idea that the high‐ and low‐IFN subsets of SLE patients have immunological differences beyond the type I IFN system.
Stimulated cytokine patterns differ significantly between patients with high IFN and low IFN
Figure 4A and B shows all of the multiplex cytokine assay data from low‐ and high‐IFN SLE patients for each of the stimulated conditions. R848 induced significant increases in the production of VEGF, IL‐1b, IL‐10, MIP‐1a, MIP‐1b, TNF‐α, and IP‐10 in controls as well as in both low and high IFN SLE patient groups (Supplemental Table 1). FGF‐β, IL‐2R, IL‐6, IL‐7, IL‐8, and IL‐12 were significantly increased in both low‐ and high‐IFN SLE patient groups after R848 stimulation but were not increased significantly in controls. IFN‐γ, MCP‐1, and HGF were increased only in the low‐IFN SLE patients and not in high‐IFN SLE patients or controls after R848. In contrast, CpG stimulation did not induce any significant elevation in any of the cytokines tested in whole blood cultures from patients and controls. After finding this result, we began adding fresh CpG to the tubes in case the preloaded CpG had degraded, but we found similar results (data not shown). After IFN‐α stimulation, controls and low‐IFN SLE patients significantly increased production of IP‐10, whereas high‐IFN SLE patients did not (Supplemental Table 2).
Following LPS stimulation, VEGF was consistently elevated in all conditions: controls, low‐ and high‐IFN SLE patients, with and without the additional IFN‐α in the tube as a co‐stimulant (Figure 4C, Supplemental Table 3). The patterns observed after LPS stimulation were interesting and complex for many of the other cytokines, and different combinations of LPS‐induced cytokines were observed in the high vs. low endogenous IFN groups. The tubes that combine IFN‐α and LPS as co‐stimulants are interesting in this context as well (Figure 4C). TNF‐α was induced by LPS in all control and patient conditions except in the high IFN SLE group stimulated with LPS + IFN‐α. These data suggest that the high levels of IFN both endogenously in the patient sample and exogenously as a stimulant in the tube‐reduced TNF‐α production in response to LPS.
MIP‐1b was elevated in the high‐IFN SLE patients after LPS stimulation as well as the low IFN SLE patient IFN‐α + LPS tube (Figure 4C). These data suggest that IFN‐α is important to the induction of MIP‐1b after LPS stimulation in SLE patients. IL‐7 was induced by LPS in each category of SLE patient but not in controls treated with either LPS or IFN‐α + LPS. FGF‐β and IL‐2RA responses were interesting because the control IFN‐α + LPS tube resembled the SLE patient LPS alone and IFN‐α + LPS tubes, suggesting that the co‐stimulation of the control sample with IFN‐α resulted in a more lupus‐like response for these two cytokines. It is also notable that some of the cytokines that are induced in controls and low‐IFN SLE patients were not induced in the high‐IFN SLE patients. In the case of IL‐1b, MCP‐1, and IL‐1RA, the control subject LPS+IFN‐α tube mirrored the low‐IFN SLE LPS alone patient tube but not the high‐IFN SLE patient LPS alone tube. This could suggest that some priming of the IFN pathway in the controls via added IFN or low chronic levels of IFN in the low‐IFN SLE patients allows for induction of these cytokines, whereas the high chronic IFN levels in the high‐IFN SLE patients inhibited the production of the same cytokines. IP‐10 was induced in the IFN, LPS, and IFN‐α+LPS tubes in both the controls and low‐IFN SLE patients and was not induced in either of the high‐IFN SLE patient tubes, also suggesting that chronic high‐level IFN stimulation has suppressed the ability of the cells to induce IP‐10.
Stimulated type I IFN responses
We examined type I IFN production using a functional assay in supernatants from the stimulated tubes above, including both controls and patients with SLE, normalized to the pDC count in the blood sample. Interestingly, the high‐IFN patients produced less type I IFN than controls or low‐IFN patients in response to all TLR agonists, with the exception of LPS alone (Figure 5). In contrast, the low IFN patients demonstrated higher type I IFN production than controls after stimulation with IFN‐α and were similar to controls after stimulation with R848 and IFN‐α+LPS. These data suggest that type I IFN responses following TLR7/8 ligation are blunted in patients who already have high circulating type I IFN levels, whereas responses to LPS are stronger. This resembles the data from other cytokines above in which stimulated outputs differ significantly between high‐ and low‐IFN patients, and in general there are fewer and different cytokines induced in the high‐IFN patients as compared with the low‐IFN patients.
Discussion
Overall, we found many differences in circulating and stimulated cytokines when comparing the high‐ and low‐IFN SLE patients in this study. This suggests that the distinction between these groups is likely to be important with respect to the pathogenesis of disease features as well as response to therapy. Trials of anti–type I IFN drugs in SLE have largely supported the idea that patients with high vs. low IFN respond differently to treatment 1, 22. Although this may be expected because the target of the therapy is being measured prior to treatment, in this study we observed a number of different non‐IFN cytokine differences that segregate between the high‐ and low‐IFN patient groups. This suggests the possibility that circulating type I IFN levels could predict response to other therapies as well.
Finding a reduced proportion of circulating pDCs in high‐IFN SLE patients as compared with low‐IFN SLE patients and controls may seem paradoxical at first. However, it seems that these data support the model that pDCs migrate from the blood into inflamed tissues where type I IFNs are produced, and the type I IFN observed in the circulation is present as a “spill over” effect. This is supported by histology studies demonstrating pDCs infiltrating inflammatory lesions in SLE patients 23. Gene expression studies frequently find IFN‐induced gene expression to be elevated in peripheral circulating blood cells but do not observe type I IFN transcripts themselves to be elevated in these cells when comparing patients with controls 3, 24, 25, 26, 27. These peripheral blood data suggest that the although circulating cells are responding to type I IFN, they are not overproducing it in the blood. This study for the first time shows that the reduction in circulating pDC proportion is most prominent in the high‐IFN SLE patients, further supporting the idea that pDCs leave the circulation for the tissues to make type I IFN. Alternative explanations that cannot be excluded include increased apoptosis in pDCs, or that chronic type I IFN exposure suppresses the differentiation of cells into the pDC lineage.
Although we observed some differences in serum cytokine levels between high‐ vs. low‐IFN patients, more dramatic differences were found in the correlations between serum cytokines in high vs. low patients. These data suggest that although the absolute levels did not differ as greatly, the cytokine programs and overall coordination between cytokines was significantly different between the high‐ and low‐IFN groups. Interestingly IL‐4, IL‐5, IL‐12, and IL‐15 all demonstrated a large number of between‐cytokine correlations in the low‐IFN patients, whereas the inflammatory cytokines IL‐1b and TNF‐α demonstrated a large number of correlations in high‐IFN SLE patients with other cytokines in the same sample. Stimulated cytokine results also documented a greater array of differences between the high‐ and low‐IFN groups than did the serum cytokine comparison. Although CpG did not induce any differences in the whole blood cultures in cytokine levels, the TLR7/8 ligand R848 induced a number of changes. In general, and the low‐IFN patients were more responsive to R848 and IFN‐α than the high‐IFN patients. There were no instances of a cytokine that was induced in the high‐IFN patients after R848 or IFN‐α that was not also induced in the low‐IFN patients. In contrast, there were four cytokines that were induced in low‐IFN patients in response to either R848 or IFN‐α, which were not induced in the high‐IFN patient subset. One possibility to explain this is that the chronic high‐IFN state observed in the high‐IFN patients may result in a down‐regulation of endosomal TLR responses. In contrast, it is possible that the smaller amount of IFN present in the low‐IFN subjects may result in a “priming” phenomena, in which a small amount of IFN in the system results in a greater cytokine response after stimulation 28.
The responses to LPS showed the greatest divergence between subject groups, and the addition of IFN‐α in the IFN‐α + LPS tube provided some additional insights. The high‐IFN patients were notable for their MIP‐1b responses to LPS, and the low‐IFN SLE blood sample added to the IFN‐α + LPS tube also showed an increase in MIP‐1b, suggesting a synergy between LPS and IFN‐α in MIP‐1b induction. Although the high IFN group had the highest IL‐1b levels in serum, this group did not increase IL‐1b after LPS stimulation. In contrast, the low‐IFN SLE patients and the control IFN‐α + LPS condition showed increased IL‐1b after LPS stimulation. This pattern was also shared by IL‐1RA and MCP‐1. The pattern observed with these cytokines suggests some of the potential differences between acute IFN‐α stimulation and the chronic IFN‐α state present in the high‐IFN SLE patients. Notably, the high‐IFN SLE patients showed reduced responsiveness to LPS for a number of cytokines as compared with the low‐IFN SLE patients, and this difference was even more marked when a strong IFN‐α co‐stimulus was included in the high‐IFN SLE patient tube. IL‐1b, MCP‐1, IL‐1RA, and IP‐10 were all elevated in the low‐IFN SLE patients but not in the high‐IFN SLE patients after LPS, with or without IFN‐α as a co‐stimulant. IL‐6, IL‐8, IL‐12, and TNF‐α were all elevated in the low‐IFN SLE patients with and without IFN‐α co‐stimulation, but in the high IFN SLE patients, they were only elevated in the LPS alone tube and not in the IFN‐α + LPS tube. Type I IFN production was elevated in high‐IFN patients in the LPS alone tube but not the IFN‐α + LPS tube. Interestingly, the low‐IFN SLE patients produced more type I IFN after stimulation in all other conditions except LPS alone as compared with the high‐IFN SLE patients, also supporting the idea of a down‐regulated endosomal TLR response in the high‐IFN patients.
A limitation of this study is that the high‐IFN patients are significantly younger than the low‐IFN patients. This has been previously reported in large cohort studies 16. This trend is present in both women and men with SLE, making menopause a less likely explanation for the observed pattern 29. Previous studies have documented some age‐related differences in immune cell numbers and stimulated immune cell responses in healthy individuals 30, 31, and we cannot exclude that some of these age‐related effects could contribute to the patterns we observe in the SLE patients in this study. Our study does, however, reflect the patients we see in the clinic, in whom these variables will also be correlated and not separable.
These data provide a unique window into human SLE and the impact of type I IFN upon functional immune cell outputs. This type of information is critical as we consider blocking the type I IFN pathway in human patients, as we show that other distinct cytokine elevations coexist with the high vs. low type I IFN category. These differing cytokine patterns could mediate response vs. nonresponse to therapy, or persistent disease features despite efficient type I IFN blockade. The results presented here may also be informative in considering SLE patients who have an infection. LPS would be representative of a gram‐negative bacterial infection, whereas the CpG and R848 could simulate viral infections. The large differences we observe in the stimulated cytokine outputs following these pathogen‐associated stimuli could provide insight into the poorly understood and unpredictable phenomenon of SLE flare following infection. It is clear that in whole blood from SLE patients, responses to these canonical TLR pathways are very different depending upon type I IFN status.
Author Contributions
All authors were involved in drafting the article or revising it critically for important intellectual contact, and all authors approved the final version to be published. Dr. Niewold had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Study conception and design
Thanarajasingam, Muppirala, Jensen, Niewold.
Acquisition of data
Thanarajasingam, Muppirala, Jensen, Ghodke‐Puranik, Dorschner, Vsetecka, Amin, Makol, Ernste, Osborn, Moder, Chowdhary, Niewold.
Analysis and interpretation of data
Thanarajasingam, Muppirala, Jensen, Ghodke‐Puranik, Niewold.
Supporting information
Dr. Thanarajasingam is recipient of the Mayo Clinic Margaret Harvey Schering Clinician Career Development Award; Dr. Niewold is recipient of Colton Center for Autoimmunity, National Institutes of Health (NIH) grants AR060861, AR057781, AR065964, and AI071651); this study was supported by the Rheumatology Research Foundation, the Mayo Clinic Foundation, and the Lupus Foundation of Minnesota.
Uma Thanarajasingam, MD, PhD, Anoohya N. Muppirala, BS, Jessica M. Dorschner, BS, Danielle M. Vsetecka, MPH, Shreyasee Amin, MD, Ashima Makol, MD, Floranne Ernste, MD, Thomas Osborn, MD, Kevin Moder, MD, Vaidehi Chowdhary, MD: Mayo Clinic, Rochester, Minnesota; 2Mark A. Jensen, PhD, Yogita Ghodke‐Puranik, PhD, Timothy B. Niewold, MD: New York University School of Medicine.
Dr. Niewold has received research grants from EMD Serono, Janssen, and Microdrop unrelated to this study. No other disclosures relevant to this article were reported.
References
- 1. Muskardin TL, Niewold TB. Type I interferon in rheumatic diseases. Nat Rev Rheumatol 2018;14:214–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Weckerle CE, Franek BS, Kelly JA, Kumabe M, Mikolaitis RA, Green SL, et al. Network analysis of associations between serum interferon‐α activity, autoantibodies, and clinical features in systemic lupus erythematosus. Arthritis Rheum 2011;63:1044–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Baechler EC, Batliwalla FM, Karypis G, Gaffney PM, Ortmann WA, Espe KJ, et al. Interferon‐inducible gene expression signature in peripheral blood cells of patients with severe lupus. Proc Natl Acad Sci U S A 2003;100:2610–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Petri M, Singh S, Tesfasyone H, Dedrick R, Fry K, Lal P, et al. Longitudinal expression of type I interferon responsive genes in systemic lupus erythematosus. Lupus 2009;18:980–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bauer JW, Baechler EC, Petri M, Batliwalla FM, Crawford D, Ortmann WA, et al. Elevated serum levels of interferon‐regulated chemokines are biomarkers for active human systemic lupus erythematosus. PLoS Med 2006;3:e491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kariuki SN, Ghodke‐Puranik Y, Dorschner JM, Chrabot BS, Kelly JA, Tsao BP, et al. Genetic analysis of the pathogenic molecular sub‐phenotype interferon‐α identifies multiple novel loci involved in systemic lupus erythematosus. Genes Immun 2015;16:15–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Niewold TB. Advances in lupus genetics. Curr Opin Rheumatol 2015;27:440–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kariuki SN, Franek BS, Kumar AA, Arrington J, Mikolaitis RA, Utset TO, et al. Trait‐stratified genome‐wide association study identifies novel and diverse genetic associations with serologic and cytokine phenotypes in systemic lupus erythematosus. Arthritis Res Ther 2010;12:R151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kirou KA, Lee C, George S, Louca K, Peterson MG, Crow MK. Activation of the interferon‐α pathway identifies a subgroup of systemic lupus erythematosus patients with distinct serologic features and active disease. Arthritis Rheum 2005;52:1491–503. [DOI] [PubMed] [Google Scholar]
- 10. Dall'era MC, Cardarelli PM, Preston BT, Witte A, Davis JC Jr. Type I interferon correlates with serological and clinical manifestations of SLE. Ann Rheum Dis 2005;64:1692–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sinicato NA, Postal M, Appenzeller S, Niewold TB. Defining biological subsets in systemic lupus erythematosus: progress toward personalized therapy. Pharmaceut Med 2017;31:81–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Merrill JT, Immermann F, Whitley M, Zhou T, Hill A, O'Toole M, et al. The biomarkers of lupus disease study: a bold approach may mitigate interference of background immunosuppressants in clinical trials. Arthritis Rheumatol 2017;69:1257–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ritterhouse LL, Crowe SR, Niewold TB, Merrill JT, Roberts VC, Dedeke AB, et al. B lymphocyte stimulator levels in systemic lupus erythematosus: higher circulating levels in African American patients and increased production after influenza vaccination in patients with low baseline levels. Arthritis Rheum 2011;63:3931–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Weckerle CE, Mangale D, Franek BS, Kelly JA, Kumabe M, James JA, et al. Large‐scale analysis of tumor necrosis factor α levels in systemic lupus erythematosus. Arthritis Rheum 2012;64:2947–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hochberg MC. Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum 1997;40:1725. [DOI] [PubMed] [Google Scholar]
- 16. Niewold TB, Adler JE, Glenn SB, Lehman TJ, Harley JB, Crow MK. Age‐ and sex‐related patterns of serum interferon‐α activity in lupus families. Arthritis Rheum 2008;58:2113–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hua J, Kirou K, Lee C, Crow MK. Functional assay of type i interferon in systemic lupus erythematosus plasma and association with anti‐RNA binding protein autoantibodies. Arthritis Rheum 2006;54:1906–16. [DOI] [PubMed] [Google Scholar]
- 18. Niewold TB, Hua J, Lehman TJ, Harley JB, Crow MK. High serum IFN‐α activity is a heritable risk factor for systemic lupus erythematosus. Genes Immun 2007;8:492–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Feng X, Reder NP, Yanamandala M, Hill A, Franek BS, Niewold TB, et al. Type I interferon signature is high in lupus and neuromyelitis optica but low in multiple sclerosis. J Neurol Sci 2012;313:48–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Niewold TB, Wu SC, Smith M, Morgan GA, Pachman LM. Familial aggregation of autoimmune disease in juvenile dermatomyositis. Pediatrics 2011;127:e1239–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cederblad B, Blomberg S, Vallin H, Perers A, Alm GV, Rönnblom L. Patients with systemic lupus erythematosus have reduced numbers of circulating natural interferon‐α‐producing cells. J Autoimmun 1998;11:465–70. [DOI] [PubMed] [Google Scholar]
- 22. Niewold TB. Targeting type I interferon in systemic lupus erythematosus. Nat Rev Rheumatol 2016;12:377–8. [DOI] [PubMed] [Google Scholar]
- 23. Farkas L, Beiske K, Lund‐Johansen F, Brandtzaeg P, Jahnsen FL. Plasmacytoid dendritic cells (natural interferon‐ α/β‐producing cells) accumulate in cutaneous lupus erythematosus lesions. Am J Pathol 2001;159:237–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Crow MK, Kirou KA, Wohlgemuth J. Microarray analysis of interferon‐regulated genes in SLE. Autoimmunity 2003;36:481–90. [DOI] [PubMed] [Google Scholar]
- 25. Sharma S, Jin Z, Rosenzweig E, Rao S, Ko K, Niewold TB. Widely divergent transcriptional patterns between SLE patients of different ancestral backgrounds in sorted immune cell populations. J Autoimmun 2015;60:51–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ko K, Koldobskaya Y, Rosenzweig E, Niewold TB. Activation of the interferon pathway is dependent upon autoantibodies in African‐American SLE patients, but not in European‐American SLE patients. Front Immunol 2013;4:309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Jin Z, Fan W, Jensen MA, Dorschner JM, Bonadurer GF III, Vsetecka DM, et al. Single cell gene expression studies in lupus patient monocytes independently indicate disease activity, interferon, and therapy. Lupus Sci Med 2017;4:e000202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ivashkiv LB. Type I interferon modulation of cellular responses to cytokines and infectious pathogens: potential role in SLE pathogenesis. Autoimmunity 2003;36:473–9. [DOI] [PubMed] [Google Scholar]
- 29. Weckerle CE, Niewold TB. The unexplained female predominance of systemic lupus erythematosus: clues from genetic and cytokine studies. Clin Rev Allergy Immunol 2011;40:42–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Piasecka B, Duffy D, Urrutia A, Quach H, Patin E, Posseme C, et al. Distinctive roles of age, sex, and genetics in shaping transcriptional variation of human immune responses to microbial challenges. Proc Natl Acad Sci U S A 2018;115:E488–E97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Patin E, Hasan M, Bergstedt J, Rouilly V, Libri V, Urrutia A, et al. Natural variation in the parameters of innate immune cells is preferentially driven by genetic factors. Nat Immunol 2018;19:302–14. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials