

Abstract
Objective
We sought to determine if any histopathologic component of the pulmonary microcirculation can distinguish systemic sclerosis (SSc)‐related pulmonary fibrosis (PF) with and without pulmonary hypertension (PH).
Methods
Two pulmonary pathologists blindly evaluated 360 histologic slides from lungs of 31 SSc‐PF explants or autopsies with (n = 22) and without (n = 9) PH. The presence of abnormal small arteries, veins, and capillaries (pulmonary microcirculation) was semiquantitatively assessed in areas of preserved lung architecture. Capillary proliferation (CP) within the alveolar walls was measured by its distribution, extent (CP % involvement), and maximum number of layers (maximum CP). These measures were then evaluated to determine the strength of their association with right heart catheterization–proven PH.
Results
Using consensus measures, all measures of CP were significantly associated with PH. Maximum CP had the strongest association with PH (P = 0.013; C statistic 0.869). Maximum CP 2 or more layers and CP % involvement 10% or greater were the optimal thresholds that predicted PH, both with a sensitivity of 56% and specificity of 91%. The CP was typically multifocal rather than focal or diffuse and was associated with a background pattern of usual interstitial pneumonia. There was a significant but weaker relationship between the presence of abnormal small arteries and veins and PH.
Conclusion
In the setting of advanced SSc‐PF, the histopathologic feature of the pulmonary microcirculation best associated with PH was capillary proliferation in architecturally preserved lung areas.
Introduction
Pulmonary hypertension (PH) is an independent risk factor for mortality in the setting of systemic sclerosis (SSc)‐related pulmonary fibrosis (PF). A prior UK Registry reported a dismal 3‐year survival of 28% for SSc‐PF‐PH in the modern treatment era 1. Furthermore, the presence of PH, as compared to no PH, has been associated with a 5‐fold increased mortality in SSc‐PF 2. Interestingly, although our group has reported improved transplant‐free survival for SSc‐PF‐PH compared with historical cohorts 3, the results of pulmonary arterial hypertension (PAH)‐specific therapy in SSc‐PF‐PH have been largely disappointing 4.
Although we know that the entire pulmonary microcirculation (small arteries, veins, and capillaries) may be affected in SSc‐PAH (without PF) 5, 6, 7, the pulmonary microcirculation has not been systematically contrasted between SSc‐PF with, as compared to without, PH. From our University of California, Los Angeles (UCLA) lung explant pathology experience, we have incidentally noted capillary proliferation (CP) in areas of lung specifically without significant fibrosis in our SSc‐PF cohort, a finding also previously reported in idiopathic PF (IPF). To our knowledge, CP has not been evaluated as a possible association with right heart catheterization (RHC)‐proven PH, complicating PF. As such, and to better understand the PH phenotype complicating SSc‐PF, we elected to evaluate the pathology of the pulmonary microcirculation with a focus on the capillary bed. For purposes of this study, the pulmonary microcirculation was exclusively evaluated in areas of relatively nonfibrotic lung. In contrast to nonfibrotic lung, the vascular changes in areas of fibrosis are generally predictable and homogeneous 8, 9. In addition, a focus on nonfibrotic lung allows for better recognition of vascular abnormalities that may otherwise be obscured by interstitial pneumonia.
We took advantage of the SSc‐PF lung tissue repository at UCLA and sought to systematically characterize the pulmonary microcirculation, dichotomized by those with and without RHC‐proven PH. We hypothesized that CP in architecturally preserved areas of advanced SSc‐PF lung tissue would be the element of the pulmonary microcirculation most likely to differentiate the PH and non‐PH subgroups.
Materials and Methods
The UCLA Institutional Review Board approved the study (IRB#11‐003042‐CR‐00002). We identified 31 cases of SSc‐PF with available explanted (n = 22) or autopsy (n = 9) lung tissue between January 2003 and December 2012.
Data collection
Patient charts were retrospectively analyzed and data were collected, including demographics, pulmonary hemodynamics, and pulmonary function testing.
Definition of CP
We defined CP as an abnormal proliferation of alveolar capillaries at least two layers thick (as previously characterized by Wagenvoort and colleagues; 10), in contrast to the normal alveolar‐capillary architecture that consists of a single capillary lumen lined by endothelial cells. However, this definition of CP is broad and does not account for confounding factors that may include underlying structural abnormalities, artifacts of processing, and/or tangential sectioning. Therefore, we only evaluated areas of nonfibrotic lung that were appropriately sectioned and free of artifact, as determined by the judgement of the expert pulmonary pathologists. Areas deemed to be distorted by crush artifact, poor tissue processing, insufficient inflation, and vascular congestion were excluded. Areas of CP in other portions of the lung, such as airway walls and pleural tissue, were not included for analysis.
Definition of PH
PH was defined using the following hemodynamic criteria at RHC: a resting mean pulmonary artery pressure (mPAP) ≥25 mmHg, pulmonary artery wedge pressure <15 mmHg, and pulmonary vascular resistance >3 Wood units. The “SSc‐PF without PH” classification required a normal mPAP (<25 mmHg) at both the screening RHC and the RHC done on the day of lung transplantation to ensure that PH had not developed during the period of active listing as previously reported 11. The reported pulmonary hemodynamics in Table 1 were obtained from the screening RHC for both patient groups.
Table 1.
Baseline demographics, pulmonary and systemic hemodynamics, and pulmonary function of a systemic sclerosis–related pulmonary fibrosis cohort divided into subgroups either with pulmonary hypertension or without pulmonary hypertension
| Total (n = 31) | With PH (n = 22) | No PH (n = 9) | P Value | |
|---|---|---|---|---|
| Tissue Type | 0.029 | |||
| Autopsy | 9 (30.0%) | 9 (40.9%) | 0 | |
| Transplant | 22 (70.0%) | 13 (59.1%) | 9 (100%) | |
| Age at tissue acquisition (y) | 0.071 | |||
| Mean (SD) | 53.6 (11.5) | 56.0 (10.3) | 47.8 (12.6) | |
| Median (Q1‐Q3) | 55.0 (48.5‐60.0) | 55.0 (49.0‐63.8) | 50.0 (33.0‐58.0) | |
| Gender | 0.042 | |||
| Female | 19 (61.3%) | 12 (54.5%) | 7 (77.8%) | |
| Male | 12 (38.7%) | 10 (45.5%) | 2 (22.2%) | |
| Time from right heart catheterization to tissue acquisition (d) | 0.056 | |||
| Mean (SD) | 379.7 (428.1) | 286.5 (391.0) | 607.6 (451.1) | |
| Median (Q1‐Q3) | 168.0 (39.0‐718.5) | 152.0 (33.5‐232.0) | 771.0 (90.0‐956.0) | |
| Body mass index (kg/m 2 ) | 0.886 | |||
| Mean (SD) | 23.3 (3.7) | 23.2 (4.0) | 23.4 (3.1) | |
| Median (Q1‐Q3) | 23.0 (21.0‐25.5) | 23.0 (21.0‐24.8) | 22.0 (21.026.0) | |
| Systemic mean arterial pressure (mmHg) | 0.612 | |||
| Mean (SD) | 86.4 (13.1) | 87.1 (13.2) | 84.4 (13.6) | |
| Median (Q1‐Q3) | 84.0 (76.5‐93.0) | 85.0 (76.2‐97.5) | 79.0 (77.0‐90.0) | |
| Heart rate (beats/min) | 0.237 | |||
| Mean (SD) | 88.4 (14.4) | 86.4 (14.7) | 93.2 (13.2) | |
| Median (Q1‐Q3) | 88.0 (77.0‐96.0) | 82.5 (77.0‐94.5) | 91.0 (88.0‐102.0) | |
| Right atrial pressure (mmHg) | 0.007 | |||
| Mean (SD) | 9.9 (6.6) | 11.9 (6.8) | 5.1 (2.2) | |
| Median (Q1‐Q3) | 8.0 (5.5‐12.5) | 11.5 (7.2‐14.5) | 5.0 (5.0‐7.0) | |
| Mean pulmonary artery pressure (mmHg) | <0.001 | |||
| Mean (SD) | 39.1 (15.6) | 47.9 (8.1) | 17.7 (3.0) | |
| Median (Q1‐Q3) | 43.3 (22.5‐51.0) | 47.5 (42.6‐53.2) | 17.3 (15.7‐17.7) | |
| Pulmonary artery wedge pressure (mmHg) | 0.125 | |||
| Mean (SD) | 10.1 (4.0) | 10.8 (3.8) | 8.3 (4.3) | |
| Median (Q1‐Q3) | 10.0 (7.0‐13.5) | 10.5 (7.2‐13.8) | 6.0 (5.0‐11.0) | |
| Cardiac index (L/min/m 2 ) | 0.003 | |||
| Mean (SD) | 2.6 (0.8) | 2.4 (0.8) | 3.3 (0.6) | |
| Median (Q1‐Q3) | 2.4 (2.1‐3.0) | 2.2 (1.8‐2.5) | 3.2 (3.0‐3.7) | |
| Pulmonary vascular resistance (Wood units) | <0.001 | |||
| Mean (SD) | 7.4 (5.0) | 9.8 (3.9) | 1.7 (0.6) | |
| Median (Q1‐Q3) | 7.8 (2.3‐11.6) | 9.1 (7.1‐12.7) | 1.7 (1.5‐1.9) | |
| Forced vital capacity (L) | 0.019 | |||
| Mean (SD) | 1.7 (0.6) | 1.9 (0.6) | 1.3 (0.4) | |
| Median (Q1‐Q3) | 1.5 (1.3‐2.0) | 1.8 (1.4‐2.1) | 1.2 (1.2‐1.3) | |
| Missing | 1 | 1 | 0 | |
| Forced vital capacity (% predicted) | 0.04 | |||
| Mean (SD) | 45.5 (15.9) | 49.3 (15.9) | 36.4 (12.5) | |
| Median (Q1‐Q3) | 41.5 (34.5‐51.0) | 43.0 (38.0‐55.0) | 34.0 (28.0‐40.0) | |
| Missing | 1 | 1 | 0 | |
| Forced expiratory volume, 1 s (L) | 0.004 | |||
| Mean (SD) | 1.4 (0.6) | 1.6 (0.6) | 0.9 (0.2) | |
| Median (Q1‐Q3) | 1.2 (1.0‐1.6) | 1.5 (1.2‐1.8) | 1.0 (0.9‐1.1) | |
| Missing | 1 | 1 | 0 | |
| Forced expiratory volume, 1 s (% predicted) | 0.003 | |||
| Mean (SD) | 46.8 (18.4) | 53.0 (18.1) | 32.3 (8.9) | |
| Median (Q1‐Q3) | 45.5 (32.5‐54.5) | 50.0 (41.0‐56.0) | 32.0(26.0‐36.0) | |
| Missing | 1 | 1 | 0 | |
| Diffusing capacity (ml/min per mmHg) | 0.518 | |||
| Mean (SD) | 6.5 (4.1) | 6.9 (4.7) | 5.7 (2.3) | |
| Median (Q1‐Q3) | 5.7 (4.7‐7.4) | 5.8 (4.9‐6.9) | 4.8 (4.0‐7.6) | |
| Missing | 3 | 2 | 1 | |
| Diffusing capacity (% predicted) | 0.337 | |||
| Mean (SD) | 24.9 (10.4) | 23.7 (7.9) | 27.8 (15.0) | |
| Median (Q1‐Q3) | 21.0 (20.0‐30.8) | 21.0 (20.0‐28.0) | 21.0 (17.0‐36.0) | |
| Missing | 1 | 1 | 0 | |
| Supplemental oxygen at tissue acquisition (L/min) | 0.53 | |||
| Mean (SD) | 4.6 (3.2) | 4.8 (3.2) | 4.0 (3.3) | |
| Median (Q1‐Q3) | 4.0 (2.0‐6.0) | 4.0 (3.2‐6.0) | 2.0 (2.0‐5.0) | |
Study design
Histopathologic evaluation
As per standard UCLA protocol for surgical and autopsy specimens, all lungs were inflated through the bronchi with 10% neutral buffered formalin. After fixation in formalin for at least 24 hours prior to sampling, at least two sections per lobe were sampled from central (hilar) and peripheral parenchyma. As per protocol, lung samples were taken from both macroscopically fibrotic and normal‐appearing lung. After paraffin embedding, all tissue sections were stained with hematoxylin and eosin (H&E), and one to two representative blocks for each case were stained with combined Masson's trichrome/elastin van Gieson (TRI‐EVG) and CD31 immunohistochemistry stains. The H&E stain provides standard light microscopy evaluation; the TRI‐EVG stain is used for evaluation of arterial and venous intimal fibrosis (small‐vessel vasculopathy); and CD31 stains vascular endothelium. For purposes of this study, a case report form (Supplemental Figure E1) was developed to systematically document the histologic features of the pulmonary microcirculation in our SSc‐PF cohort. CP is also a reported pathologic finding from severely congested lungs 12, usually in the setting of long‐standing clinical left‐sided congestive heart failure, which, importantly, was not a feature in our SSc‐PF cohort. A 1‐hour comprehensive training session including the pathologists and pulmonologist (Rajan Saggar) allowed for consistent methodologic consensus in reporting the presence and degree of abnormal alveolar capillary pathology.
A blinded evaluation was then performed by two senior pulmonary pathologists (MCF, WDW) with extensive surgical and autopsy pathology experience. Each pathologist evaluated 360 stained slides (324 histology slides [H&E or TRI‐EVG] and 36 immunohistochemistry slides [CD31]). Each patient had between 6 and 18 slides, with a median of 11 slides per patient. The pathology evaluation was limited to only those areas of lung with preserved architecture (eg, areas of parenchyma where the architecture was not obscured or effaced by fibrosis. CP was assessed as a binary variable (yes/no) after review of all histology slides for a given patient. If present, the distribution of CP was noted as focal, multifocal, or diffuse. The maximum CP (ie, maximum number of layers) was reported for each slide relevant to each patient. Similarly, the extent of CP was reported for each slide and reflected the percentage of architecturally preserved lung area (nearest decile; Supplemental Figure E1) estimated to be occupied by CP.
Given that all subjects had a clinical diagnosis of PF, the blinded pathologists reported the major histologic fibrosis subtype 13 after review of all slides relevant to each patient as one of the following: usual interstitial pneumonia (UIP); nonspecific interstitial pneumonia (NSIP); a combination of UIP and NSIP; pulmonary veno‐occlusive disease (PVOD); or other.
We attempted to distinguish arteries from veins based on anatomic location in accordance with topographic guidelines described by the Development and Pathology Working group 14 (see supplement: Histopathologic evaluation). However, despite adhering to these guidelines and focusing in areas of nonfibrotic lung, there was poor interpathologist reliability for the presence and extent of abnormal small arteries and veins (Supplemental Tables E2‐E5). In light of this finding, we elected to analyze and report abnormal small arteries and veins collectively as a single category termed “small‐vessel vasculopathy.”
Finally, the blinded pathologists were asked to comment on the presence or absence of hemosiderin‐laden macrophages after reviewing all slides relevant to a particular patient.
Scanning electron microscopy evaluation
After the study period, one SSc‐PF‐PH subject underwent a limited thoracic autopsy 15 hours after sudden death. The lung specimen was prepared for scanning electron microscopy (SEM) and light microscopy. A previously published SEM image 15 demonstrating normal pulmonary microvasculature and capillary bed is shown for comparison.
Sequential SSc‐PF histopathology associated with the interval development of pulmonary hypertension
A surgical lung biopsy specimen was accessible from a single patient who had developed PH in the time between the surgical lung biopsy and lung transplantation. The pathology at the two time points was then compared specifically for CP in architecturally preserved areas.
Statistical Analysis
Categorical variables were summarized using frequencies and percentages, whereas continuous variables were summarized using means, standard deviations, and quartiles. Analysis of PH as an outcome was formulated as a logistic regression model. Consensus predictors were constructed to reconcile the different assessments by the two pathologists. For binary assessments, we compared consensus “yes” with consensus “no” valuations. After initial analysis of the data, we found poor interpathologist correlations for identifying the presence and extent of abnormal small arteries and veins (Supplemental Tables E2‐E5). As a result, we generated a single group termed “small‐vessel vasculopathy” (representing abnormal small arteries and veins) and analyzed the ability of the binary assessment to discriminate the PH and non‐PH subgroups within the following patient‐level score construct between the two pathologists: −1 points for consensus normal artery/vein; +1 points for consensus abnormal artery/vein; and 0 points for discrepant artery/vein. As a result, the final patient‐level score could range from −2 (consensus normal artery and vein) to +2 (consensus abnormal artery and vein). For median assessments, we used the median of the two pathologists’ median assessments. For maximum assessments, we used the maximum of the two pathologists’ maximum assessments. Prediction of PH was also evaluated by adjusting for forced vital capacity (FVC) in order to evaluate pathologist assessment value‐added. Other outcomes were analyzed using mixed effects logistic regression models. These models included random patient and pathologist effects in order to cluster different pathologists’ assessments of the same patient and different patient assessments by the same pathologist. All logistic regression models are summarized in terms of odds ratios and 95% confidence intervals (CIs). Classification accuracy of pathologist assessments was evaluated in terms of C‐statistics. A C‐statistic can be interpreted as the probability that a randomly selected event/case is given a higher score by the model than a randomly selected nonevent/control. A C‐statistic close to 0.5 indicates that the model performs no better than random noise in discriminating between cases and controls, whereas a C‐statistic close to 1.0 indicates that the model provides perfect discrimination 16. Linear mixed effects models were summarized in terms of mean differences and 95% CI. Youden's J‐maximizing cutoffs for predicting PH were determined for all CP measures. P values less than 0.05 were considered statistically significant. All analyses were performed using SAS v.9.4 (SAS Institute Inc.).
Results
Thirty‐one SSc‐PF patients were studied with lung pathology obtained at autopsy (n = 9) or at lung transplantation (n = 22) and were classified either with (n = 22) or without (n = 9) PH based on the cardiac catheterization data. There were a total of 360 stained (324 H&E and TRI‐EVG; 36 CD31) histopathology slides (226 with PH; 98 without PH) reviewed by two pathologists. Six to 18 slides were analyzed for each patient (median 11 slides per patient). Subjects with PH had increased FVC% predicted (49 ± 16 versus 36 ± 13; P = 0.04) and forced expiratory volume in a second percentage predicted (53 ± 18 versus 32 ± 9; P = 0.003) compared with those without PH. The PH group had an mPAP of 48 ± 8 versus 18 ± 3 mmHg. Other baseline variables between the subgroups, including demographics, pulmonary and systemic hemodynamics, and pulmonary function, are reported in Table 1.
Pulmonary microcirculation changes associated with pulmonary hypertension
The interpathologist reliability was significant for the presence of CP (P < 0.001) with κ‐statistic (95% CI) of 0.69 (0.41, 0.97). Similarly, the interpathologist reliability by Spearman correlation for maximum CP and CP% involvement was 0.74 and 0.75, respectively (Supplement E1A and B). As a consensus binary measure, CP was associated with PH (P = 0.01; C‐statistic 0.75). All measures of CP, including maximum CP and CP % involvement, were significantly associated with PH with C‐statistic values that were 0.74 or greater (Table 2). Maximum CP median (the median of the two pathologists’ maximum assessments of CP) was the CP measure most strongly associated with PH (P = 0.01; C‐statistic 0.87). A maximum CP maximum of at least two layers and CP% involvement maximum of at least 10% were optimal thresholds for predicting PH, both with a sensitivity of 56% and a specificity of 91% (Supplemental Table E6). The maximum CP (mean ± SD layers) for the PH group, compared with the group without PH, was 3.1 (1.9) and 1.4 (0.8) (P = 0.001) and, similarly, CP% involvement (based on decile) was 3.2 (3.0) and 0.7 (1.8) (P = 0.006), respectively (Table 2). The associations between all measures of CP and PH remained significant when adjusted for FVC, supporting the concept that the degree of lung fibrosis did not significantly contribute to these findings (Supplement Table E7). Figure 1 shows the histologic features of CP typically seen in the SSc‐PF‐PH subgroup. H&E images (Figure 1A and B) show lung parenchyma with no interstitial fibrosis but with areas of CP that demonstrate multiple congested lumens, which are haphazardly oriented, with few areas of adjacent normal alveolar capillary architecture. The CD31 stain (Figure 1C) demonstrates alveolar wall capillary dilatation and proliferation.
Table 2.
C‐statistics for models predicting pulmonary hypertension (PH) in terms of pathologist consensus measures and measures of capillary proliferation from a systemic sclerosis–related pulmonary fibrosis cohort stratified by the presence or absence of PH (based on the analysis of 324 pathology slides by two pathologists)
| Effect | C‐Statistic | P value |
|---|---|---|
| Consensus CP vs. consensus non‐CP | 0.753 | 0.011 |
| Maximum CP median | 0.869 | 0.013 |
| Maximum CP maximum | 0.821 | 0.011 |
| CP % involvement median | 0.838 | 0.027 |
| CP % involvement maximum | 0.737 | 0.029 |
| Consensus NSIP (NSIP alone or NSIP + UIP) vs. consensus non‐NSIP (only UIP) | 0.558 | 0.651 |
| Consensus small‐vessel vasculopathy vs. consensus non–small‐vessel vasculopathy | 0.720 | 0.029 |
| PH | No PH | P Value | |
|---|---|---|---|
| (n = 452) | (n = 196) | ||
| Maximum CP | 0.001 | ||
| Mean (SD) | 3.12 (1.92) | 1.38 (0.84) | |
| Median (Q1, Q3) | 3 (1, 4) | 1 (1, 1) | |
| Min, Max | 1, 10 | 1, 5 | |
| Missing | 128 | 50 | |
| CP% Involvement | |||
| Mean (SD) | 3.21 (3.01) | 0.66 (1.76) | 0.006 |
| Median (Q1, Q3) | 3 (0, 5) | 0 (0, 0) | |
| Min, Max | 0, 11 | 0, 9 | |
| Missing | 127 | 50 |
Abbreviation: CP, capillary proliferation; NSIP, nonspecific interstitial pneumonia; UIP, usual interstitial pneumonia.
Figure 1.
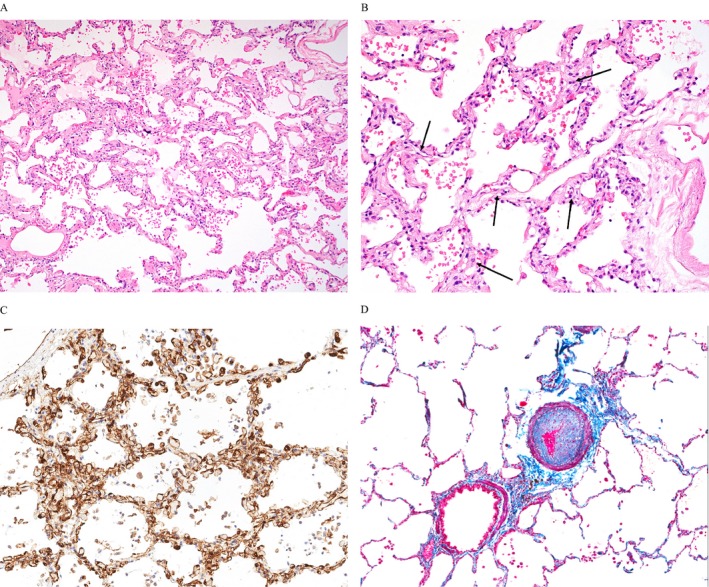
Typical capillary proliferation (CP) and small‐vessel vasculopathy in lungs with advanced systemic sclerosis–related pulmonary fibrosis. (A) Diffuse distribution of CP (hematoxylin and eosin [H&E] stain; original magnification ×100); (B) Higher power example of CP; the alveolar walls have irregularly dilated capillaries with more than two layers (arrows) (H&E stain; original magnification ×200); (C) CP is demonstrated by highlighting endothelial cells, (CD31 immunohistochemistry stain; original magnification ×200); (D) Bronchovascular bundle demonstrating prominent pulmonary arterial intimal fibrosis (trichrome/elastin stain; original magnification ×100).
As a consensus binary measure, small‐vessel vasculopathy (presence of abnormal arteries and/or veins) was associated with PH (P = 0.03; C‐statistic 0.72), although with a lower C‐statistic than any CP parameter (Table 2). Finally, the presence of hemosiderin‐laden macrophages trended to an association with PH (P = 0.06).
Relationship of CP to the remaining pulmonary microcirculation
As a consensus binary measure, small‐vessel vasculopathy was associated with CP as a consensus binary measure (P = 0.04), maximum CP median (P < 0.05), and CP% involvement maximum (P = 0.04) (Table 3).
Table 3.
Mixed effects ordinal logistic regression models of small‐vessel vasculopathy in terms of capillary proliferation measures
| Effect | Small‐Vessel Vasculopathy OR (95% CI) | P value |
|---|---|---|
| Maximum CP median | 2.03 (1.00, 4.12) | 0.049 |
| Maximum CP maximum | 1.44 (0.98, 2.12) | 0.061 |
| CP% involvement median | 1.37 (0.95, 1.97) | 0.094 |
| CP% involvement maximum | 1.32 (1.01, 1.72) | 0.043 |
| CP (yes/no) | 7.47 (1.15, 48.37) | 0.036 |
Abbreviation: CI, confidence interval; CP, capillary proliferation; OR, Odds Ratio.
Relationship of CP to the pattern of PF
More CP was seen in the UIP pattern of background PF. Compared with NSIP and combined UIP/NSIP, three of the four CP measures were significantly associated with a background of isolated UIP (maximum CP median, CP% involvement maximum, and CP% involvement median) (Supplement Table E8).
SEM and sequential lung tissue analysis
Supplement Table E9 displays the pulmonary hemodynamics and pulmonary function testing for two additional SSc‐PF subjects, one SSc‐PF‐PH with SEM (autopsy) images (Supplement Table E9A), and another with sequential lung histopathology (Supplement Table E9B; initial surgical biopsy [PH absent] and subsequent explant with the interval development of PH). Standard light microscopy and SEM performed on the autopsy SSc‐PF lung with PH showed CP in areas of lung with preserved architecture (Figure 2A) and abnormal capillary bed morphology and ultrastructure by SEM (Figure 2B compared with normal SEM in Figure 2C), respectively. In addition, standard light microscopy on lung tissue obtained sequentially from another SSc‐PF subject initially at surgical lung biopsy (SSc‐PF without PH; Figure 3A and B) and subsequently at lung transplantation (SSc‐PF with PH; Figure 3C and D) exhibit the interval development of CP associated with clinical PH.
Figure 2.
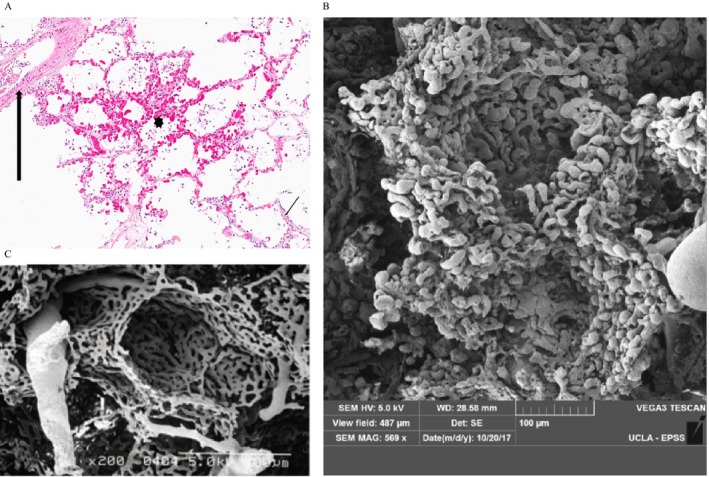
Light microscopy evaluation of alveolar tissue from the systemic sclerosis–related pulmonary fibrosis–pulmonary hypertension (SSc‐PF‐PH) autopsy that demonstrates irregular alveolar wall capillary proliferation in areas without interstitial fibrosis: (A) capillary proliferation (asterisk) adjacent to normal artery (thick arrow) and normal alveolar capillaries (thin arrow) (hematoxylin and eosin [H&E] stain; original magnification ×200); (B) scanning electron microscopy (SEM) image of a vascular cast from the same SSc‐PF‐PH autopsy specimen highlighting extensive and crowded capillary duplication in the walls of two alveolar sacs. Note the abnormal pulmonary angiogenesis in the microvasculature with bulbous budding alternating with pinched areas in the alveolar capillary bed (SEM; original magnification ×569); (C) SEM of normal lung microvasculature and capillary bed. Note the much thinner capillary bed with more orderly arrangement of capillary architecture with few capillaries showing budding or abrupt termination of blind pouches (photos courtesy of Dr. Kazufumi Nakamura), (SEM, original magnification ×200.
Figure 3.
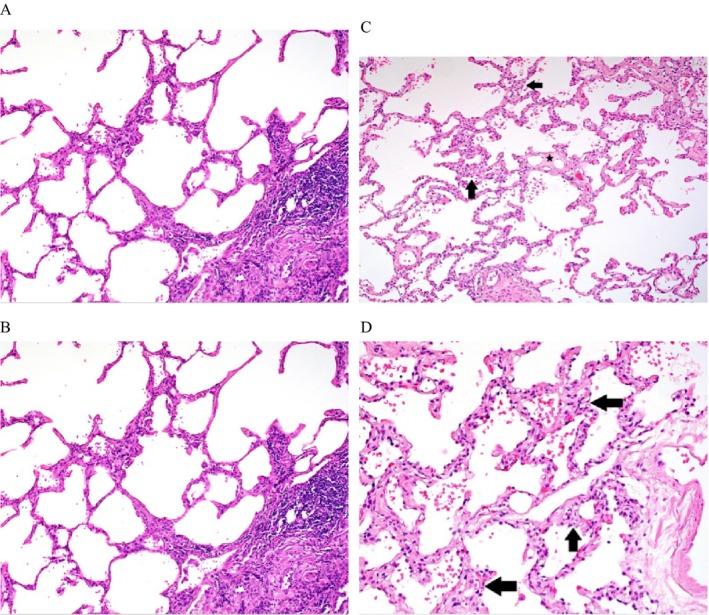
(A) Alveolar tissue at surgical lung biopsy of a systemic sclerosis (SSc)–pulmonary fibrosis (PF) subject without pulmonary hypertension (PH) with normal and inconspicuous capillary architecture adjacent to area of interstitial pneumonia (hematoxylin and eosin stain [H&E]; original magnification ×100); (B) Higher‐power view of alveolar tissue with normal architecture and inconspicuous capillaries (H&E stain; original magnification ×200); (C) Alveolar tissue from the same SSc‐PF subject (hemodynamic data in Supplemental Table E9B) after the interval development of PH from explanted lung tissue in areas without interstitial pneumonia but with alveolar capillary prominence and proliferation (arrows) and venular fibrosis (star) (H&E stain; original magnification ×100); (D) Higher‐power view of alveolar tissue from explant specimen showing alveolar capillary proliferation (arrows) (H&E stain; original magnification ×100).
Discussion
This study systematically evaluated the pulmonary microcirculation, with a focus on abnormal capillary structure, in a well‐characterized SSc‐PF cohort with and without PH, as demonstrated by RHC. We specifically sought to determine if CP could distinguish SSc‐PF with and without PH. Importantly, we focused on the pathology of the pulmonary microcirculation only in areas with preserved lung architecture without significant interstitial fibrosis because fibrosis may obscure microcirculatory changes and/or affect morphology 9. Our main finding was that, compared with small‐vessel vasculopathy (abnormal small arteries and veins), CP was the histopathologic element of the pulmonary microcirculation most capable of distinguishing SSc‐PF‐PH from SSc‐PF without PH.
The data were presented as consensus measures of two pathologists with good interpathologist reliability with regard to measures of CP. Although all CP parameters were significant, the maximum CP had the strongest association with PH (C‐statistic 0.87). The capillary changes were usually multifocal and were associated with the UIP pattern of background PF (as opposed to the NSIP pattern). Compared with the SSc‐PF group without PH, the SSc‐PF‐PH group had, on average, three layers of capillaries (versus less than 1.4 layers) involving 20%‐30% (versus less than 10%) of the examined architecturally preserved area. A maximum CP of at least two layers and CP% involvement of at least 10% were optimal thresholds for predicting PH, both with a sensitivity of 56% and a specificity of 91%. Small‐vessel vasculopathy (abnormal small arteries and veins) had a significant but weaker association with PH when compared with CP.
In all cases, the areas of CP were readily identifiable with H&E staining alone after the pathologists were trained on the diagnostic features of CP, and areas with architectural remodeling or artifact were excluded. The CD31 stain helped to highlight the CP by staining endothelial cells in areas of interest (Figure 1C), but the distinctive features of CP demonstrated by H&E, along with the nonspecific CD31 staining (including macrophages and larger vessels), limited the use of CD31 staining to evaluate CP in all cases. By delineating the intima between the endothelium and the elastic lamina, the TRI‐EVG stain highlighted abnormal small artery and vein intimal fibrosis indicative of small vessel vasculopathy (Figure 1D). However, despite distinct anatomic location differences 14 and a focus in areas of nonfibrotic lung, the pathologic distinction between abnormal small artery and vein was not always clear and suffered from poor interpathologist reliability. Consequently, we elected to report abnormal small arteries and veins collectively as a single category that we called “small‐vessel vasculopathy.” Although small‐vessel vasculopathy was found to be significantly associated with PH, this association was weaker when compared with CP. Ultimately, pathologic review of the alveolar capillary bed by routine H&E staining alone was reliable in distinguishing PH from non‐PH clinical phenotypes. In our analysis, when PH complicates SSc‐PF, the entire pulmonary microcirculation is significantly abnormal, specifically in areas of nonfibrotic lung; however, the SSc‐PF‐PH phenotype is best distinguished from SSc‐PF without PH by the increased presence and extent of CP.
The concept of vascular remodeling in PF was first suggested by Turner‐Warwick 17 in the form of systemic‐to‐pulmonary circulation neovascularization. Subsequently, human IPF lung homogenates have shown a proangiogenesis signature 18, and in this context, alveolar capillaries have also demonstrated distinctly abnormal morphology based on SEM analysis in rats with bleomycin‐induced PF 19. In human IPF, Ebina and coworkers 8 established an inverse relationship between the CD34+ alveolar capillary “vascular density” and the degree of pathologic fibrosis, with similar findings described in SSc‐PF human lung specimens 20. In a study of IPF, Colombat and coworkers studied the pulmonary microcirculation within areas of preserved lung architecture and reported significant panvascular pathology, including CP. Interestingly, they found no relationship between the presence of venopathy in architecturally preserved areas of IPF lung and RHC‐determined PH; however, arteriopathy and CP were not evaluated in this context.
Abnormal capillary pathology has been reported in SSc and other connective tissue diseases (CTD) with PH without clinically significant fibrosis 5, 15, 16. Odronic and colleagues 6 reported four cases of pulmonary capillary hemangiomatosis (PCH)‐like lesions in CTD‐associated PAH, similar to the CP that we observed. Other reports describe a pattern of small‐vessel vasculopathy and a lack of plexiform lesions as being unique to SSc‐PAH (without PF), as compared with idiopathic PAH 15. A PVOD‐like pattern and secondary capillary congestion has also been described in CTD‐associated PAH 15, 16. This capillary congestion is readily discernable from the CP that we report, although CP has been observed by SEM in clinical PVOD 26. It is also important to distinguish the CP that we report from the capillary congestion reported in long‐standing pulmonary venous congestion 12. In this setting, capillary congestion causes an expansion of the lymphatics with dilated interseptal spaces, characteristics that we did not generally find or avoided by focusing on alveolar rather than lobular septal capillaries. In addition, none of our patients had the clinical picture of left heart failure.
The finding of CP and its association with PH also serves to increase the heterogeneity of abnormal pulmonary microcirculation and, as a result, complicates the ability to predict the clinical PH phenotype. For instance, it is generally accepted that in the context of clinical PVOD, as a consequence of downstream venous obstruction, capillaries may undergo angiogenesis, similar to the CP we report, and additional secondary changes can manifest, including mild PF 21. Alternatively, our data suggest that PF may similarly function as a primary trigger for CP (in a background of small‐vessel vasculopathy) and associated PH, perhaps by providing the appropriate microenvironment favoring angiogenesis 18, 22. In consideration of this hypothesis, we demonstrated the interval development of CP (in a background of small‐vessel vasculopathy) involving areas of architecturally preserved lung for the isolated SSc‐PF subject in our cohort with accessible lung tissue, obtained before and after the development of RHC‐proven PH. Our findings support the concept that within this panvasculopathy, CP had the best association with clinical PH in SSc‐PF, whereas the presence of small‐vessel vasculopathy (abnormal small arteries and veins) had a limited (inferior C‐statistic), albeit significant, association with PH. Finally, the trend toward an increased presence of hemosiderin‐laden macrophages in our SSc‐PF‐PH subgroup, a well‐documented finding in pulmonary vascular disease 21 as well as in PF with PH 21, 22, further supports an abnormal capillary bed.
CP has also been shown in PH associated with congenital heart disease 23 and chronic thromboembolic disease (WHO Group 4: Chronic Thromboembolic Pulmonary Hypertension) 5. A focus on the capillary bed in the evaluation of pulmonary microcirculation pathology has recently gained attention and is timely, given the finding suggesting that only ~30% of the pulmonary vascular resistance in PAH may be explained by the extent of arteriopathy 24. Although this concept raises the question of arteriolar rarefaction as a contributing factor to elevated pulmonary vascular resistance in PAH, abnormal capillary and venular pathology may also play a role, particularly in the setting of PH outside of WHO Group 1: Pulmonary Arterial Hypertension.
The cast vasculature seen in our SEM images of the PH lungs with what we have termed CP confirmed several haphazardly arranged layers in the alveolar walls. The capillaries had heterogeneous shapes, including irregular luminal diameter and budding, which suggests abnormal capillary neogenesis. These misshapen capillaries would not provide a clear pathway through the vascular bed and may contribute to increased vascular resistance. The lack of normal capillary structure would also affect both pulmonary hemodynamics and gas exchange. These abnormal capillaries have been reported in several conditions with a pathologic picture reminiscent of the PCH‐PVOD spectrum of disease and are often associated with clinically progressive PH and hypoxemia 25, 26, 27, 28.
CP has been reported to be a feature of architecturally preserved lung in the background of idiopathic UIP 9, 29. Our study builds on this concept by reporting concordant findings in an SSc‐PF cohort inherently at risk for UIP and/or NSIP 30. CP was significantly associated with a background of isolated UIP pathology, as opposed to either NSIP or combined NSIP/UIP. Although the UIP fibrosis phenotype was not itself associated with PH in our study, we hypothesize that capillary rarefaction is likely greater in areas of UIP fibrosis compared with NSIP. If correct, the UIP fibrosis phenotype may be uniquely predisposed to angiogenesis and aberrant CP in areas of architecturally preserved lung and, in some cases, this progression may give rise to clinically significant PH.
There are several limitations to the study, including its retrospective study design and small sample size from a single institution. However, it is the largest SSc pathology cohort studied to date. It would have been ideal to include SSc‐PAH without PF in our study; however, such tissue was simply unavailable as all of our SSc explants had significant PF. Surgical lung biopsies are uncommon in SSc lung disease because they rarely alter clinical management; consequently, all pathology was obtained in the setting of advanced PF and results may not be generalizable to milder cases. Our pathologic assessments are semiquantitative, but this limitation was mitigated by restricting pathologic evaluation to experienced pulmonary pathologists practicing at a large lung transplant center. Furthermore, the pathologists met and agreed upon standards before any evaluations, and we used consensus statistical measures. Finally, there are limiting factors based on the pathology itself. Distinguishing small arteries and veins by light microscopy proved to be difficult in terms of interpathologist correlation despite the use of strict topographic criteria 31 when labeling vessels as arteries and veins, a focus in parenchymal areas not obscured by fibrosis, and the use of larger lung specimens to maximize the availability of such areas. Nevertheless, our analysis of the data combining abnormal small arteries and/or veins (small‐vessel vasculopathy) provides more of a real‐world pathology approach. The fibrosis and CP are a heterogeneous process with great individual variation in the severity of the lesions and extent in different lungs. Even extensive sampling may not give accurate representation of the full pathology, so no quantitative conclusions can be made. Finally, because the two SSc‐PF subgroups were broadly separated with regard to pulmonary hemodynamics and had minimal within‐group variation, we chose not to define the correlation between pulmonary pressure (as a continuous variable) and CP.
We have demonstrated that CP in the architecturally preserved areas of patients with advanced SSc‐PF is associated with PH, as determined by RHC. In the context of a panvasculopathy, CP appears to be the element of the microcirculation most closely associated with PH. These data add to our understanding of the pathophysiology of PH in SSc‐PF and suggest that aberrant capillaries and angiogenesis may be a major causative factor and future target for therapy in this population.
Author Contributions
Study Design
AS, RS, RS, DK, JS, MF, DW, HX
Data Collection
MD, KS, RS, JS, DC, DK, SS, DM, LS, FS
Pathology assessment
GF, HX, DW, AS, MF, ML, DS
Data Analysis
ZA, RS, SV
Manuscript preparation
RS, ZA, AS, GF, SV, DR, JP, JL, AD, JB, AS, EB, LG, SW
Guarantor of the paper
Rajan Saggar
Supporting information
Acknowledgements
All authors thank Dr. Kazufumi Nakamura for providing the scanning electron microscopy image from figure 2.
Drs. Seki, Anklesaria, Wallace, and Rajan Saggar contributed equally to this work.
No authors have any stated conflicts of interest or financial/nonfinancial disclosures.
References
- 1. Condliffe R, Kiely DG, Peacock AJ, Corris PA, Gibbs JS, Vrapi F, et al. Connective tissue disease‐associated pulmonary arterial hypertension in the modern treatment era. Am J Respir Crit Care Med 2009;179:151–7. [DOI] [PubMed] [Google Scholar]
- 2. Trad S, Amoura Z, Beigelman C, Haroche J, Costedoat N, Boutin le TH, et al. Pulmonary arterial hypertension is a major mortality factor in diffuse systemic sclerosis, independent of interstitial lung disease. Arthritis Rheumatol 2006;54:184–91. [DOI] [PubMed] [Google Scholar]
- 3. Volkmann ER, Saggar R, Khanna D, Torres B, Flora A, Yoder L, et al. Improved transplant‐free survival in patients with systemic sclerosis‐associated pulmonary hypertension and interstitial lung disease. Arthritis Rheumatol 2014;66:1900–8. [DOI] [PubMed] [Google Scholar]
- 4. Le Pavec J, Girgis RE, Lechtzin N, Mathai SC, Launay D, Hummers LK, et al. Systemic sclerosis‐related pulmonary hypertension associated with interstitial lung disease: impact of pulmonary arterial hypertension therapies. Arthritis Rheumatol 2011;63:2456–64. [DOI] [PubMed] [Google Scholar]
- 5. Dorfmuller P, Humbert M, Perros F, Sanchez O, Simonneau G, Müller KM, et al. Fibrous remodeling of the pulmonary venous system in pulmonary arterial hypertension associated with connective tissue diseases. Hum Pathol 2007;38:893–902. [DOI] [PubMed] [Google Scholar]
- 6. Odronic SI, Narula T, Budev M, Farver C. Pulmonary capillary hemangiomatosis associated with connective tissue disease: a report of 4 cases and review of the literature. Ann Diagn Pathol 2015;19:149–53. [DOI] [PubMed] [Google Scholar]
- 7. Overbeek MJ, Vonk MC, Boonstra A, Voskuyl AE, Vonk‐Noordegraaf A, Smit EF, et al. Pulmonary arterial hypertension in limited cutaneous systemic sclerosis: a distinctive vasculopathy. Eur Respir J 2009;34:371–9. [DOI] [PubMed] [Google Scholar]
- 8. Ebina M, Shimizukawa M, Shibata N, Kimura Y, Suzuki Y, Endo M, et al. Heterogeneous increase in CD34‐positive alveolar capillaries in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2004;169:1203–8. [DOI] [PubMed] [Google Scholar]
- 9. Colombat M, Mal H, Groussard O, Capron F, Thabut G, Jebrak G, et al. Pulmonary vascular lesions in end‐stage idiopathic pulmonary fibrosis: histopathologic study on lung explant specimens and correlations with pulmonary hemodynamics. Hum Pathol 2007;38:60–5. [DOI] [PubMed] [Google Scholar]
- 10. Wagenvoort CA, Beetstra A, Spijker J. Capillary haemangiomatosis of the lungs. Histopathology 1978;2:401–6. [DOI] [PubMed] [Google Scholar]
- 11. Nathan SD, Shlobin OA, Ahmad S, Koch J, Barnett SD, Ad N, et al. Serial development of pulmonary hypertension in patients with idiopathic pulmonary fibrosis. Respiration 2008;76:288–94. [DOI] [PubMed] [Google Scholar]
- 12. Matsukuma S, Sato K. Pulmonary capillary haemangiomatosis‐like lesions in severely congested lungs. Histopathology 2011;59:876–81. [DOI] [PubMed] [Google Scholar]
- 13. Travis WD, Costabel U, Hansell DM, King TE Jr, Lynch DA, Nicholson AG, et al. An official American Thoracic Society/European Respiratory Society statement: update of the international multidisciplinary classification of the idiopathic interstitial pneumonias. Am J Respir Crit Care Med 2013;188:733–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Tuder RM, Archer SL, Dorfmüller P, Erzurum SC, Guignabert C, Michelakis E, et al. Relevant issues in the pathology and pathobiology of pulmonary hypertension. J Am Coll Cardiol 2013;62:D4–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Miura A, Nakamura K, Kusano KF, Matsubara H, Ogawa A, Akagi S, et al. Three‐dimensional structure of pulmonary capillary vessels in patients with pulmonary hypertension. Circulation 2010;121:2151–3. [DOI] [PubMed] [Google Scholar]
- 16. Pencina MJ, D'Agostino RB, Sr . Evaluating discrimination of risk prediction models: the C statistic. JAMA 2015;314:1063–4. [DOI] [PubMed] [Google Scholar]
- 17. Turner‐Warwick M. Precapillary systemic‐pulmonary anastomoses. Thorax 1963;18:225–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Keane MP, Arenberg DA, Lynch JP, Whyte RI, Iannettoni MD, Burdick MD, et al. The CXC chemokines, IL‐8 and IP‐10, regulate angiogenic activity in idiopathic pulmonary fibrosis. J Immunol 1997;159:1437–43. [PubMed] [Google Scholar]
- 19. Schraufnagel DE, Mehta D, Harshbarger R, Treviranus K, Wang NS. Capillary remodeling in bleomycin‐induced pulmonary fibrosis. Am J Pathol 1986;125:97–106. [PMC free article] [PubMed] [Google Scholar]
- 20. Renzoni EA, Walsh DA, Salmon M, Wells AU, Sestini P, Nicholson AG, et al. Interstitial vascularity in fibrosing alveolitis. Am J Respir Crit Care Med 2003;167:438–43. [DOI] [PubMed] [Google Scholar]
- 21. Lantuéjoul S, Sheppard MN, Corrin B, Burke MM, Nicholson AG. Pulmonary veno‐occlusive disease and pulmonary capillary hemangiomatosis: a clinicopathologic study of 35 cases. Am J Surg Pathol 2006;30:850–7. [DOI] [PubMed] [Google Scholar]
- 22. Sakashita N, Motooka Y, Suganuma M, Ohnishi K, Fujiwara Y, Nakagawa T, et al. A case of pulmonary capillary hemangiomatosis with pulmonary fibrosis associated with MMP‐9 related pulmonary remodeling. Pathol Int 2011;61:306–12. [DOI] [PubMed] [Google Scholar]
- 23. Aiello VD, Thomaz AM, Pozzan G, Lopes AA. Capillary hemangiomatosis like‐lesions in lung biopsies from children with congenital heart defects. Pediatr Pulmonol 2014;49:E82–5. [DOI] [PubMed] [Google Scholar]
- 24. Rol N, Timmer EM, Faes TJ, Vonk Noordegraaf A, Grünberg K, Bogaard HJ, et al. Vascular narrowing in pulmonary arterial hypertension is heterogeneous: rethinking resistance. Physiol Rep 2017;5:e13159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. DuBrock HM, Kradin RL, Rodriguez‐Lopez JM, Channick RN. Pulmonary capillary hemangiomatosis: the role of invasive cardiopulmonary exercise testing. Pulm Circ 2015;5:580–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lee C, Suh RD, Krishnam MS, Lai CK, Fishbein MC, Wallave WD, et al. Recurrent pulmonary capillary hemangiomatosis after bilateral lung transplantation. J Thorac Imaging 2010;25:W89–92. [DOI] [PubMed] [Google Scholar]
- 27. Güttinger E, Vrugt B, Speich R, Ulrich S, Schwitz F, Arrigo M, et al. Reactive pulmonary capillary hemangiomatosis and pulmonary veno‐occlusive disease in a patient with repaired scimitar syndrome. Case Rep Cardiol 2016;2016:9384126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Varnholt H, Kradin R. Pulmonary capillary hemangiomatosis arising in hereditary hemorrhagic telangiectasia. Hum Pathol 2004;35:266–8. [DOI] [PubMed] [Google Scholar]
- 29. Kim KH, Maldonado F, Ryu JH, Eiken PW, Hartman TE, Bartholmai BJ, et al. Iron deposition and increased alveolar septal capillary density in nonfibrotic lung tissue are associated with pulmonary hypertension in idiopathic pulmonary fibrosis. Respir Res 2010;11:37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Park JH, Kim DS, Park IN, Jang SJ, Kitaichi M, Nicholson AG, et al. Prognosis of fibrotic interstitial pneumonia: idiopathic versus collagen vascular disease‐related subtypes. Am J Respir Crit Care Med 2007;175:705–11. [DOI] [PubMed] [Google Scholar]
- 31. Tuder RM, Abman SH, Braun T, Capron F, Stevens T, Thistlethwaite PA, et al. Development and pathology of pulmonary hypertension. J Am Coll Cardiol 2009;54:S3–9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials