

Abstract
Objective
The objective of this study is to examine the risk of serious infections (SIs) associated with biological disease‐modifying antirheumatic drugs (bDMARDs) compared with conventional synthetic disease‐modifying antirheumatic drugs (csDMARDs) in patients with rheumatoid arthritis (RA).
Methods
We studied patients with RA who initiated bDMARDs or csDMARDs from 2001 to 2016 in FORWARD–The National Databank for Rheumatic Diseases. Disease‐modifying antirheumatic drugs (DMARDs) were categorized into three groups: (1) csDMARDs (bDMARD‐naïve; reference), (2) tumor necrosis factor α inhibitors (TNFis), and (3) non‐TNFi biologics (abatacept, rituximab, tocilizumab, and anakinra). SIs were defined as those requiring intravenous antibiotics or hospitalization or those resulting in death. We calculated the propensity score (PS), which reflected the probability of receiving a specific DMARD group, and estimated the hazard ratio (HR) (with the 95% confidence interval [CI]) for SI from multivariable Cox models, adjusting for PS and time‐varying confounders.
Results
A total of 694 (5.9%) first SIs were identified in 11 623 patients with RA during 27 552 patient‐years of follow‐up. The SI incidence rate per 1000 patient‐years was 22.4 (95% CI 19.2‐26.1) for csDMARDs, 26.9 (95% CI 24.5‐29.6) for TNFis, and 23.3 (95% CI 19.0‐28.5) for non‐TNFi bDMARDs. Adjusted HRs for SIs were 1.33 (95% CI 1.05‐1.68) for TNFis and 1.48 (95% CI 1.02‐2.16) for non‐TNFi bDMARDs, compared with csDMARDs. The SI risk with non‐TNFi bDMARDs versus TNFis was not different. Other risk factors for SI were older age, higher comorbidity burden (particularly pulmonary disease), higher weighted cumulative prednisone dose, disability and disease activity, and number of prior csDMARD failures.
Conclusion
TNFis and non‐TNFi bDMARDs were associated with an increased SI risk compared with csDMARDs in RA, even after accounting for risk‐associated patient characteristics.
Introduction
Rheumatoid arthritis (RA) is associated with increased risk of serious infections (SIs), which significantly contribute to increased overall mortality 1, 2, 3. This increased infection risk in RA has been attributed to complex interactions of disease‐associated immune dysregulation, accompanying comorbidities, and use of immunosuppressive medications 4. With the advent of biologic disease‐modifying antirheumatic drugs (bDMARDs), better inflammation control and, consequently, reduction in associated morbidity and mortality have been achieved in RA 5. However, because of their mechanism of action, targeting key cytokines and cells of host immune response, concerns about infection risk have been raised. Several randomized clinical trials, observational studies, and meta‐analyses have evaluated this issue, and some, but not all, have showed an increased risk of infection with bDMARDs compared with conventional synthetic disease‐modifying antirheumatic drugs (csDMARDs) with different risk magnitudes 2, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16. Studies comparing the risk of infections across different bDMARDs, both in classes and as individual drugs, have also reported discordant results, although most have suggested a similar infection risk 6, 17, 18, 19, 20, 21.
Because the risk of infection in patients with RA is determined by both drug‐specific parameters (agent, dose, and duration) 13, 14 and patient‐specific characteristics (age, sex, extra‐articular manifestations, comorbidities, and comedications, particularly glucocorticoids [GCs]) 14, 22, it is methodologically challenging to estimate bDMARD‐associated infection risk without assessing all of these factors. Considering the significant variations of previous studies in study design and population (biologic registries and administrative or pharmacy data), comparator drug cohorts (csDMARDs or methotrexate only or one of the bDMARDs), follow‐up duration, and assessment of confounders, further characterization of the risk of SI attributable to bDMARDs and other patient factors in RA is needed.
In this study, we assessed the risk of SIs associated with bDMARDs compared with csDMARDs in a US‐wide observational RA cohort using propensity score (PS) analysis.
Patients and methods
Patients were participants in FORWARD–The National Databank for Rheumatic Diseases, a longitudinal prospective observational study 23. The study included patients with RA who completed at least two semiannual questionnaires and initiated a new course (incident users) of either bDMARDs (tumor necrosis factor α inhibitors [TNFis] or non‐TNFi bDMARDs) or csDMARDs during the period of January 2001 through December 2016. Patients with RA with a diagnosis of cancer during the follow‐up before the diagnosis of SI were excluded.
Disease‐modifying antirheumatic drug exposure
Treatment exposure was measured at enrollment and every 6 months using comprehensive questionnaires 23. For disease‐modifying antirheumatic drug (DMARD) use, we defined three mutually exclusive categories based on the initiated treatment: 1) csDMARDs (reference) (bDMARD‐naïve); 2) TNFis (infliximab, etanercept, adalimumab, certolizumab, and golimumab), independent of other csDMARDs; and 3) non‐TNFi bDMARDs (abatacept, rituximab, tocilizumab, and anakinra), independent of other csDMARDs and prior TNFi exposure. Each patient contributed to one specific DMARD group reached last during the follow‐up. According to this design, as an example, a patient whose last DMARD was a non‐TNFi bDMARD initiated during the follow‐up period would only contribute to the non‐TNFi biologic group regardless of prior csDMARD‐only or TNFi exposure periods. A diagram showing DMARD‐exposure assignment with sample patients is provided in the Supplementary Material. Treatment categorization was based on the last DMARD because it reflected the trajectory of the disease process and was a way of fixing the treatment regimen for each patient.
Outcome and follow‐up
The primary outcome was incident SI, which was defined as an infection requiring intravenous antibiotics or hospitalization or resulting in death. Only SIs that were confirmed by medical review or death records were included (Supplementary Material). SIs were attributed to the corresponding DMARD group when the treatment was ongoing or discontinued 3 months or fewer before SI. This risk window was extended to 12 or fewer months for rituximab, considering its long‐term effects on B cells 24.
Follow‐up started at the time of treatment initiation with any of the DMARD groups mentioned previously and ended at the first SI, at treatment discontinuation, at death, at loss to follow‐up, or on December 31, 2016, the study end. The risk window after discontinuation of a DMARD was included in the follow‐up period for patients who discontinued therapy for a reason other than SIs.
Statistical analysis
Baseline characteristics of the patients with RA at the time of DMARD initiation, by future SIs and by DMARD group, were compared using either χ2, analysis of variance (ANOVA), or nonparametric tests (Kruskal‐Wallis test), as applicable. Crude incidence rates for all patients with RA, and by DMARD group, were calculated by dividing the number of events per 1000 patient‐years of follow‐up and are presented with 95% confidence intervals (CIs).
To estimate risk differences between DMARD groups, a survival analysis was performed using a Cox proportional hazards model. The multiple‐PS method was applied to minimize the channeling bias related to baseline clinical characteristics of patients 25. PSs reflecting the probability of receiving csDMARDs, TNFis, or non‐TNFi bDMARDs were calculated by using multinomial logistic regression models (csDMARDs vs TNFis and non‐TNFi DMARDs). The PS was estimated based on the following characteristics at the time of treatment initiation: age, sex, ethnicity (white vs other), insurance status (Medicare vs others), annual income, RA disease duration, smoking status, Rheumatic Diseases Comorbidity Index (RDCI) score, Health Assessment Questionnaire (HAQ) score, pain and patient global assessment scores by visual analogue scales (0‐10), GC use (yes/no), total number of csDMARDs and bDMARDs used prior to the initiated DMARD in the cohort, prior SI history, and calendar year of treatment initiation.
Because of differences in timing of bDMARD availability for use, restricted cubic spline function of the calendar year was employed in the PS model to deal with nonlinearity. Two‐way ANOVA for continuous variables and logistic regression models for binary variables were used to assess if the PSs were balanced. In these models, the dependent variable was each covariate, and the other factors were the exposure variable (DMARD group), the PSs, and the interaction terms between the two. The PSs were both used as continuous variables and as split‐into quintiles. The nonstatistical significance of the main effects of exposure or of the interaction of exposure with the PS or its quintiles indicated a balanced score for that covariate. The distribution of the multiple PSs within each treatment group was also assessed, confirming a substantial overlap of the PS values. All patient characteristics were balanced between DMARD groups after correction with the multiple‐PS method.
To examine the SI risk associated with DMARDs, we constructed several multivariable models. First, the PS was added in the Cox models as a continuous variable instead of potential confounders. Second, considering the potential influence of increasing age, disease duration, new comorbidities, added csDMARDs, disability, disease severity measures, and cumulative GC dose over time, time‐varying age, disease duration, comorbidities, the HAQ score, pain and patient global assessment scores, weighted cumulative exposure (WCE) of GCs, and number of prior csDMARDs and bDMARDs were added to the Cox model along with the PS. As a disease activity measure, the patient activity scale (PAS), which is a composite index that uses the HAQ score and patient global assessment and pain scores, was also assessed in a different model. A PAS score of 3.7 or lower was defined as remission or low disease activity and a score greater than 3.7 was defined as moderate or high disease activity 26.
The WCE model for GCs (WCE‐prednisone), which combines information about duration, intensity, and timing of exposure into a summary measure, was assessed as the weighted sum of past oral doses (prednisolone equivalent). The weights assigned to past doses were estimated using a flexible cubic spline‐based method 27, 28. The time window (past GC exposure affects the current risk of SI) was determined based on the methodology applied by Dixon et al 28. Details of the WCE model are described in the Supplementary Material.
In sensitivity analyses, the PS was added to the Cox model as a categorical variable in quintiles because stratifying on the PS quintiles of a continuous variable can alleviate the bias due to the measured confounders when estimating a treatment effect. Before using the quintiles in the models, we confirmed that all propensity quintiles had sufficient numbers in each group. Because patients in the csDMARDs group were required to have no prior bDMARD exposure, a PS model without the number of prior bDMARDs was also assessed. Potential interactions of different DMARDs with the comorbidity burden were also tested. For this analysis, regardless of the comorbidity type, the RDCI (which includes lung disease, heart attack, other cardiovascular diseases, stroke, hypertension, fracture, depression, diabetes, cancer, and gastrointestinal problems) 29 score of patients was categorized as 0 (no comorbidity), 1 or 2, or 3 or more. Use of csDMARDs without any comorbidity was set as the reference. Additionally, SI risk in patients who initiated TNFis was compared with that of non‐TNFi bDMARD initiators. Lastly, we assessed the SI risk with bDMARDs at different durations compared with the same duration of use of csDMARDs by restricting the time at risk to the first 3, 6, and 12 months and after 12 months of treatment.
Missing covariate data were replaced by using multiple imputation by chained equations to create multiple imputed data sets for analyses (annual income had 4% missing; all other variables had less than 1% missing) 30. The proportional hazards assumption was assessed by testing the Schoenfeld residuals. No violation of the proportional hazard assumption was observed. All tests were two‐sided and were considered statistically significant at P < 0.05. All statistical analyses were performed using Stata version 14.0 (StataCorp).
Results
The study included 11 623 patients with RA, of whom 2 737 were csDMARD incident users, 7 210 were TNFi incident users, and 1 676 were non‐TNFi bDMARDs incident users. The baseline characteristics of the patients by DMARD group and future SI are presented in Table 1 and Supplementary Table 1. Patients who initiated TNFis and non‐TNFi bDMARDs had significantly higher disease duration, disease activity, disability, and comorbidity scores than those who initiated csDMARDs at the time of treatment initiation (Table 1). During the 27 552 patient‐years of follow‐up, 694 (5.9%) SIs were identified, yielding a crude incidence rate of 25.2 (95% CI 23.4‐27.2) per 1000 patient‐years. The incidence rate for the TNFi‐exposed group was slightly higher than that of other DMARD‐exposure groups (Figure 1). The cumulative incidences of SIs in different DMARD groups during the first 2 years of treatment are also shown in Figure 1.
Table 1.
Characteristics of patients by DMARD group at the time of treatment initiation
| Variables | csDMARDs (n = 2737) | TNFis, (n = 7210) | Non‐TNFi bDMARDs (n = 1676) | P |
|---|---|---|---|---|
| Age, mean (SD), y | 60.3 (13.4) | 59.9 (14.1) | 58.9 (12.9) | 0.001 |
| Female sex, % | 79.4 | 79.6 | 86.4 | <0.001 |
| White, % | 93.6 | 94.8 | 93.4 | 0.061 |
| Education level, mean (SD), y | 13.3 (2.3) | 13.5 (2.3) | 14.1 (2.2) | <0.001 |
| Annual income (by $1000), mean (SD) | 46.3 (30.7) | 49.8 (32.5) | 55.9 (33.8) | <0.001 |
| Rural residence, % | 30.9 | 26.4 | 25.3 | <0.001 |
| Medicare, % | 51.4 | 54.6 | 49.9 | <0.001 |
| Disease duration, mean (SD), y | 15.2 (13.6) | 15.9 (12.9) | 17.3 (12.6) | 0.002 |
| Ever used GCs, % | 49.6 | 61.2 | 77.9 | <0.001 |
| Current GC use, with doses (as prednisone equivalents), % | <0.001 | |||
| None | 64.4 | 64.2 | 47.3 | … |
| <5 mg/d | 7.3 | 7.3 | 8.5 | … |
| 5‐10 mg/d | 17 | 17.8 | 25.6 | … |
| ≥10 mg/d | 11.3 | 10.7 | 18.7 | … |
| Rheumatic Disease Comorbidity Index (0‐9), mean (SD) | 1.9 (1.6) | 1.8 (1.6) | 2.1 (1.6) | <0.001 |
| Ever smoked, % | 42.7 | 42.1 | 44.9 | 0.738 |
| Diabetes, % | 13.7 | 13.4 | 15.5 | 0.445 |
| Pulmonary disease, %a | 15.8 | 15.9 | 23.7 | <0.001 |
| Prior serious infection history, % | 4.2 | 4.6 | 5.6 | 0.373 |
| HAQ disability score (0‐3), mean (SD) | 1.06 (0.74) | 1.13 (0.73) | 1.27 (0.71) | <0.001 |
| Pain score (0‐10), mean (SD) | 4.0 (2.9) | 4.2 (2.8) | 5.0 (2.7) | <0.001 |
| Patient global assessment score (0‐10), mean (SD) | 3.6 (2.5) | 3.8 (2.5) | 4.4 (2.5) | <0.001 |
| PAS (0‐10), mean (SD) | 3.7 (2.3) | 3.9 (2.2) | 4.5 (2.1) | <0.001 |
| No. of prior bDMARDs, mean (SD) | 0.00 | 1.0 (0.9) | 2.6 (1.2) | <0.001 |
| No. of prior csDMARDs, mean (SD) | 2.0 (1.2) | 2.2 (1.6) | 2.9 (1.8) | <0.001 |
Abbreviation: bDMARD, biologic disease‐modifying antirheumatic drug; csDMARD, conventional synthetic disease‐modifying antirheumatic drug; DMARD, disease‐modifying antirheumatic drug; GC, glucocorticoid; HAQ, Health Assessment Questionnaire; PAS, patient activity scale; TNFi, tumor necrosis factor α inhibitor.
Pulmonary disease included chronic obstructive lung disease, asthma, and interstitial lung disease.
Figure 1.
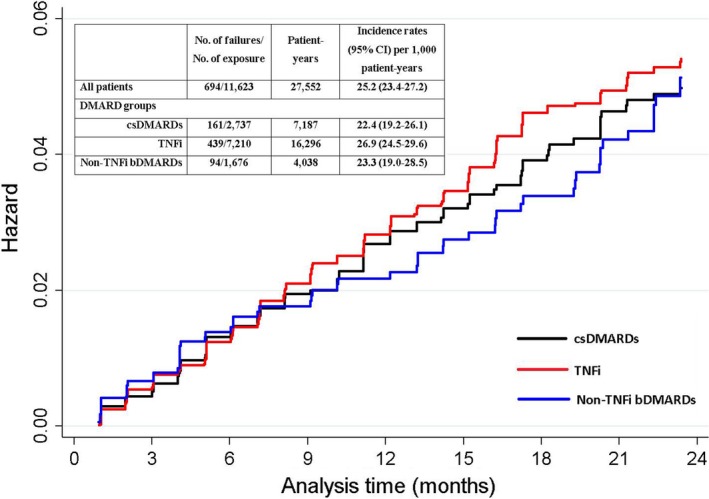
Cumulative incidence of serious infections by disease‐modifying antirheumatic drug (DMARD) groups during the first 2 years of treatment. The y‐axis shows the hazard of serious infection. bDMARD, biological disease‐modifying antirheumatic drug; CI, confidence interval; csDMARD, conventional synthetic disease‐modifying antirheumatic drug; TNFi, tumor necrosis factor α inhibitor.
In PS‐only–adjusted models, TNFis (hazard ratio [HR] 1.11; 95% CI 0.91‐1.34) and non‐TNFi bDMARDs (HR 1.26; 95% CI 0.95‐1.68) were associated with a nonsignificant SI‐risk increase, whereas further adjustment for time‐varying confounders revealed significantly increased SI risk with both TNFis (HR 1.33; 95% CI 1.05‐1.68) and non‐TNFi bDMARDs (HR 1.48; 95% CI 1.02‐2.16) compared with csDMARDs (Table 2). In the assessment of SI risk at different treatment durations, incidence rates of SI were higher in the first year of the treatment compared with those in the later years of the treatment in each DMARD group (Supplementary Table 2). Although the number of patients and events was lower when the time at risk was restricted according to the treatment duration, we observed a trend of increased SI risk early in the treatment with bDMARDs versus csDMARDs compared with later years of the treatment (first 3 months > 6 months > 12 months > after 12 months) (Supplementary Table 2).
Table 2.
Risk of serious infections in RA by treatment; results from the Cox proportional hazard models with PSs and time‐varying confounders
| No. of Events/No. of Exposures | Incidence Rate per 1000 Patient‐Years, (95% CI) | Adjusted for Age and Sex Only, HR (95% CI) | Adjusted for PS Only, HR (95% CI) | Adjusted for PS and Time‐Varying Confounders, HR (95% CI)a | |
|---|---|---|---|---|---|
| csDMARDs | 161/2737 | 22.4 (19.2‐26.1) | Reference | Reference | Reference |
| TNFis | 439/7210 | 26.9 (24.5‐29.6) | 1.30 (1.00‐1.44) | 1.11 (0.91‐1.34) | 1.33 (1.05‐1.68) |
| Non‐TNFi bDMARDs | 94/1676 | 23.3 (19.0‐28.5) | 1.07 (0.83‐1.56) | 1.26 (0.95‐1.68) | 1.48 (1.02‐2.16) |
Abbreviation: bDMARD, biologic disease‐modifying antirheumatic drug; CI, confidence interval; csDMARD, conventional synthetic disease‐modifying antirheumatic drug; HR, hazard ratio; PS, propensity score; RA, rheumatoid arthritis; TNFi, tumor necrosis factor α inhibitor; WCE, weighted cumulative exposure.
Adjusted for age, RA disease duration, annual income, Rheumatic Diseases Comorbidity Index, selected comorbidities (including diabetes and pulmonary diseases), Health Assessment Questionnaire score, pain and patient global assessment scores, cumulative GC exposure as WCE‐prednisone, and the number of prior csDMARDs and bDMARDs.
Other factors associated with the risk of SI were older age, lower annual income, higher comorbidity scores, pulmonary disease, higher disability and patient global assessment scores, being exposed to several csDMARDs previously, and higher weighted cumulative prednisone doses (Table 3). Also, higher disease activity was associated with increased SI risk, with an HR of 1.18 (95% CI 1.14‐1.23) for each unit increase in PAS and an HR of 1.82 (95% CI 1.52‐2.17) for moderate or high disease activity compared with remission or low disease activity.
Table 3.
Factors associated with serious infection risk in rheumatoid arthritis
| Adjusted HR (95% CI)a | P | |
|---|---|---|
| Age, y | ||
| <50 | Reference | … |
| 50‐64 | 1.39 (1.03‐1.89) | 0.034 |
| ≥65 | 2.31 (1.70‐3.12) | <0.001 |
| Annual income > $45 000 | 0.70 (0.57‐0.84) | 0.001 |
| Rheumatic Disease Comorbidity Index | 1.20 (1.13‐1.26) | <0.001 |
| Selected comorbidities | ||
| Diabetes | 1.15 (0.94‐1.41) | 0.174 |
| Pulmonary disease | 1.46 (1.21‐1.77) | <0.001 |
| Disease duration, y | 1.00 (0.99‐1.01) | 0.852 |
| HAQ disability | 1.27 (1.10‐1.47) | 0.001 |
| Pain score | 1.04 (1.00‐1.08) | 0.050 |
| Patient global assessment score | 1.06 (1.02‐1.11) | 0.008 |
| WCE‐prednisone | 1.33 (1.22‐1.45) | <0.001 |
| No. of prior csDMARDs | 1.11 (1.05‐1.17) | <0.001 |
| No. of prior bDMARDs | 0.90 (0.80‐1.02) | 0.095 |
Abbreviation: bDMARD, biological disease‐modifying antirheumatic drug; CI, confidence interval; csDMARD, conventional synthetic disease‐modifying antirheumatic drug; HAQ, Health Assessment Questionnaire; HR, hazard ratio; WCE, weighted cumulative exposure.
Adjusted for propensity scores and DMARD exposure.
In the sensitivity analyses using PS in quintiles and PS calculated without the number of prior bDMARDs, the risk of SIs associated with bDMARDs compared with csDMARDs was similar to that in primary analyses, although SI risk with TNFis did not reach statistical significance when the PS without the number of prior bDMARDs was used (Table 4). Additionally, the risk of SIs in non‐TNFi bDMARD users was not significantly different compared with that in TNFi users (reference) in both PS‐adjusted and PS‐ and time‐varying confounders–adjusted models (HR 1.14 [95% CI 0.88‐1.46]; HR 1.13 [95% CI 0.85‐1.50], respectively) (Table 4).
Table 4.
Sensitivity analyses: risk of serious infections by using different PSs
| csDMARDs | TNFis | Non‐TNFi bDMARDs | |
|---|---|---|---|
| PS in quintiles | |||
| Adjusted HR (95% CI) for PS only | Reference | 1.06 (0.87‐1.28) | 1.22 (0.92‐1.62) |
| Adjusted HR (95% CI) for PS and time‐varying confounders | Reference | 1.33 (1.06‐1.70) | 1.53 (1.05‐2.23) |
| PS continuous: Non‐TNFi bDMARDs vs TNFis | |||
| Adjusted HR (95% CI) for PS only | … | Reference | 1.14 (0.88‐1.46) |
| Adjusted HR (95% CI) for PS and time‐varying confounders | … | Reference | 1.13 (0.85‐1.50) |
| PS without the No. of prior bDMARDs | |||
| Adjusted HR (95% CI) for PS only | Reference | 1.11 (0.92‐1.35) | 1.21 (0.92‐1.59) |
| Adjusted HR (95% CI) for PS and time‐varying confounders | Reference | 1.39 (1.10‐1.75) | 1.55 (1.07‐2.25) |
Abbreviation: bDMARD, biological disease‐modifying antirheumatic drug; CI, confidence interval; csDMARD, conventional synthetic disease‐modifying antirheumatic drug; HR, hazard ratio; PS, propensity score; TNFi, tumor necrosis factor α inhibitor; WCE, weighted cumulative exposure.
When the interaction of DMARD type with comorbidity burden was examined, csDMARD users were also found to have a significant SI‐risk increase in the presence of RDCI scores of 3 or higher compared with csDMARD users with no comorbidities (HR 3.68; 95% CI 1.95‐5.94). Although TNFi and non‐TNFi biologic users without any comorbidity still had an increased SI risk compared with csDMARD users with no comorbidities (RDCI = 0 and TNFi use: HR 2.07 [95% CI 1.05‐4.06]; RDCI = 0 and non‐TNFi use: HR 2.95 [95% CI 1.20‐7.28]), the risk was markedly higher in patients who had a higher comorbidity burden (RDCI score of 3 or more and TNFi use: HR 4.49 [95% CI 2.38‐8.45]; RDCI score of 3 or more and non‐TNFi use: HR 5.15 [95% CI 2.51‐10.54]) (Figure 2).
Figure 2.
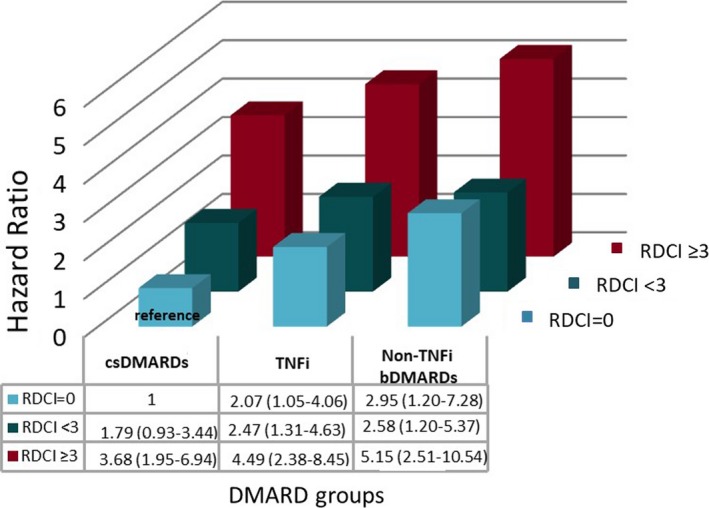
Hazard ratios of serious infections for the interaction of treatment with different disease‐modifying antirheumatic drugs (DMARDs) and comorbidity burden. The reference is treatment with conventional synthetic disease‐modifying antirheumatic drugs (csDMARDs) and having a Rheumatic Diseases Comorbidity Index (RDCI) (0‐9) score of 0. bDMARD, biological disease‐modifying antirheumatic drug; TNFi, tumor necrosis factor α inhibitor.
Discussion
In this large US‐wide observational cohort study, we found that TNFis and non‐TNFi bDMARDs were associated with an increased risk of SI compared with csDMARDs. However, SI risk associated with non‐TNFi bDMARDs was not significantly different from t SI risk associated with TNFis. Comorbidity burden significantly increased the SI risk observed across all treatment groups, but SI risk was markedly increased in patients with RA with high comorbidity burden who were receiving TNFis and non‐TNFi bDMARDs. We also found that older age, pulmonary disease, higher disability, higher disease activity, and cumulative GC exposure were predictive of SI.
Although previous studies showed an increased incidence of SI in patients with RA compared with the non‐RA population 1, there are discrepancies in the literature regarding the risk of SI associated with bDMARDs. Randomized controlled trials of bDMARDs have not reported a consistent pattern of SI risk with different bDMARDs compared with csDMARD 7, 31, 32, 33, 34, 35, probably because of power restraints and relatively shorter follow‐up durations. A recent meta‐analysis of 106 randomized controlled trials (wide range of study selection from 1992 to 2014; included 42 330 patients with RA; also examining doses of bDMARD) showed a 31% increase in SI risk with standard‐dose bDMARDs in patients with RA compared with csDMARDs, particularly in studies that only had 6‐12 months of follow‐up 13. Although randomized controlled trials typically include patients with higher disease activity and less comorbidity 36, our findings of an increased SI risk with bDMARDs were consistent with those found in this meta‐analysis.
The results from several observational studies were again inconsistent. The observed SI risk ratio with TNFis compared with csDMARDs ranged between 2.4‐ to 1.1‐fold in the studies that showed an increased SI risk 2, 4, 9, 14, 37, 38, 39. However, others reported either a risk increase only during early treatment or with certain TNFis such as infliximab or no increase at all with TNFis 8, 10, 40, 41. For the newer non‐TNFi bDMARDs, there are limited observational studies examining the risk of SI. Similar to TNFis, some, but not all, studies reported increased SI risk compared with csDMARDs (HR ranged from 1.1 to 6.9) 6, 16, 19. Our findings of a 33% and 48% SI‐risk increase with TNFis and non‐TNFi bDMARDs compared with csDMARDs, respectively, are in keeping with those of previous studies that reported increased risk 2, 6, 9, 12, 14, 38, 39. Regarding the comparison of SI risk across bDMARDs, the risk we observed with non‐TNFi bDMARDs compared with TNFis was also consistent with previous studies that showed no clear risk‐increase pattern with bDMARD type despite the selection of different comparator bDMARDs in each study 6, 17, 18, 19, 20, 21.
Notably, similar findings were obtained despite substantial heterogeneity in study design, such as study population (biologic registries or administrative/pharmacy databases) 10, 14, 21, treatment episodes (incident/prevalent use) 9, 14, 19, 21, study outcomes (only bacterial or hospitalized infections not including death) 10, 17, 39, comparator groups (methotrexate only/all csDMARDs/different bDMARDs) 9, 14, 21, 22, examined bDMARDs, included covariates (lack of disease severity measures in administrative databases), and methods for addressing confounding by indication 8, 9, 14, 21, 22, 28.
The crude incidence rates and SI risk with bDMARDs compared with csDMARDs tended to be higher during the first months of treatment than during long‐term use, although our sample size was small to show any significant association. Few observational studies using bDMARD registries also showed a similar trend for TNFis 2, 8, 40. Although bDMARDs can lead to a reduction in GC doses and improvement in function, which, in turn, may decrease SI risk, this finding should be interpreted cautiously because one of the reasons for SI‐risk decline over time can be bDMARD termination or loss to follow‐up in patients at increased risk 14.
Because this was an observational study, treatment selection was informed. Thus, factors that determine treatment choice may also influence the outcome. We minimized this potential bias by incorporating PS into multivariable models. PSs calculated using robust registry data, including demographics, comorbidities, RA disease severity, prior SI and csDMARD/bDMARD/GC exposure, and calendar year at the time of treatment initiation, were predictive of both treatment choice and risk of SI. Because the PS was calculated using single time‐point measurements, we also included time‐varying variables in the models to account for the changes in clinical factors that can alter the SI risk. Only a few studies used PS methodology, including three from biologic/clinical registries that reported an increased SI risk with TNFis compared with csDMARDs 9, 12, 14 and two from administrative databases that showed no risk increase 10, 41. Besides the differences in covariables, study populations, and risk windows, unlike ours, all of these prior studies allowed patients to contribute to more than one treatment group, which may introduce bias. Furthermore, to account for the variation in timing, duration, and intensity of the GC exposure, we calculated WCE of GCs, which has been shown to be a superior method of GC‐exposure assessment than conventional approaches (current or previous use) in examining SI risk 28. Our study is one of the largest observational studies to assess SI risk by using PS and WCE of GCs.
Our results also provide key information about the contribution of patient factors, disease characteristics, and GC use on SI risk in RA. As shown previously, we found that older age, a higher comorbidity burden (particularly the presence of pulmonary disease), worse physical disability, higher patient global assessment scores, a greater number of prior csDMARD failures, and cumulative GC exposure were strong predictors of SI in patients with RA 14, 28, 39, 42, 43. Despite the lack of tender/swollen joint counts or acute‐phase reactants, we observed about a doubled SI risk in patients who were in a moderate or high disease activity state compared with those in remission or a low disease activity state by PAS. The limited evidence on the influence of disease activity suggested that higher disease activity, measured by the 28‐joint Disease Activity Score, was associated with increased SI risk in RA 44. However, our study examined this association along with a comprehensive DMARD‐ and GC‐exposure assessment. Given the close relation between disease activity and GC use, it has also been shown that GC use is strongly associated with increased SI risk in a dose‐ and duration‐dependent manner 28, 39, 42, 44. Our results were also consistent with the previous studies when GC exposure was assessed as both WCE and time‐varying categorical (data not shown) daily doses.
Notably, although chronic pulmonary disease was the most important comorbidity influencing SI risk in RA, we also observed that the SI risk increased substantially as the number of comorbidities increased, regardless of the type of comorbidities. Higher comorbidity burden, assessed with the RDCI, a validated comorbidity index 45, was a better predictor of SI than individual comorbidities included in the RDCI, other than pulmonary disease. This association between RDCI and SI was also observed when the patients without chronic pulmonary disease were analyzed separately (data not shown). Earlier studies indicated increased SI risk with several individual comorbidities, such as pulmonary disease, diabetes, and kidney disease 14, 39, 42, 43; however, so far, only one study assessed comorbidity burden using the number of comorbidities and reported no association with SI risk 39. Evaluating comorbidity burden in RA is critical to achieving optimal long‐term outcomes because of an increasing prevalence of multiple chronic conditions and an aging population.
Our study has some limitations. First, we intentionally limited our analysis to DMARD‐initiators and did not allow patients to contribute to multiple treatment groups. Although this method provided a less biased approach, it decreased our sample size and prohibited us from examining individual bDMARDs and site‐specific infections. Second, we evaluated only the first SI for each patient. However, this approach limited the bias resulting from bDMARD discontinuation caused by an SI development. Third, as an observational study, there is the potential that channeling bias and unmeasured confounding occurred. However, using PS, time‐varying disease severity measures, and patient characteristics–adjusted multivariable models should have decreased this bias. Lastly, we may not have captured all SIs because patients who are possibly in better health may be more likely to participate in FORWARD–The National Databank for Rheumatic Diseases than those who are frail and at higher risk of infection. This participation bias can also explain our relatively lower SI incidence rates. However, the design of the analysis and the inclusion of comparison arms helped mitigate complications with data capture.
In conclusion, we observed that TNFis and non‐TNFi bDMARDs were associated with an increased SI risk in RA compared with csDMARDs, with no difference in SI risk between TNFis and non‐TNFi bDMARDs. SI risk was also associated with older age, comorbidity burden, pulmonary disease, higher disability, higher disease activity, and cumulative GC exposure. Our study both strengthens the evidence of increased SI risk with bDMARDs and indicates that patient characteristics also determine SI risk. Therefore, despite the efficacy of bDMARDs in reducing disease activity, disability, and, potentially, GC exposure, the risk/benefit ratio should be weighed carefully when treating patients with RA, particularly older individuals with multiple comorbidities and disability, with bDMARDs. Patients should be monitored closely for symptoms and signs of infection, and assessment and possible modification of risk factors should be continued throughout the bDMARD treatment.
Author contributions
All authors were involved in drafting the article or revising it critically for important intellectual content, and all authors approved the final version to be published. Dr. Ozen, Ms. Pedro, and Drs. England, Wolfe, and Michaud had full access to all of the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis.
Study concept and design
Ozen, Pedro, England, Mehta, Wolfe, Michaud.
Analysis of data
Ozen, Pedro.
Analysis and interpretation of data
Ozen, Pedro, England, Mehta, Wolfe, Michaud.
Supporting information
Acknowledgments
We thank all of the patients who participated in FORWARD that made this important study possible.
Dr. England has received grant support from the University of Nebraska Medical Center Internal Medicine Scientist Development Award, the University of Nebraska Medical Center Physician‐Scientist Training Program, and the University of Nebraska Medical Center Mentored Scholars Program. Dr. Michaud has received grant support from the Rheumatology Research Foundation and Pfizer. No other disclosures relevant to this article were reported.
References
- 1. Doran MF, Crowson CS, Pond GR, O'Fallon WM, Gabriel SE. Frequency of infection in patients with rheumatoid arthritis compared with controls: a population‐based study. Arthritis Rheum 2002;46:2287–93. [DOI] [PubMed] [Google Scholar]
- 2. Galloway JB, Hyrich KL, Mercer LK, Dixon WG, Fu B, Ustianowski AP, et al. Anti‐TNF therapy is associated with an increased risk of serious infections in patients with rheumatoid arthritis especially in the first 6 months of treatment: updated results from the British Society for Rheumatology Biologics Register with special emphasis on risks in the elderly. Rheumatology (Oxford) 2011;50:124–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ogdie A, Maliha S, Shin D, Love TJ, Baker J, Jiang Y, et al. Cause‐specific mortality in patients with psoriatic arthritis and rheumatoid arthritis. Rheumatology (Oxford) 2017;56:907–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Listing J, Gerhold K, Zink A. The risk of infections associated with rheumatoid arthritis, with its comorbidity and treatment. Rheumatology (Oxford) 2013;52:53–61. [DOI] [PubMed] [Google Scholar]
- 5. Lacaille D, Avina‐Zubieta JA, Sayre EC, Abrahamowicz M. Improvement in 5‐year mortality in incident rheumatoid arthritis compared with the general population: closing the mortality gap. Ann Rheum Dis 2017;76:1057–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Aaltonen KJ, Joensuu JT, Virkki L, Sokka T, Aronen P, Relas H, et al. Rates of serious infections and malignancies among patients with rheumatoid arthritis receiving either tumor necrosis factor inhibitor or rituximab therapy. J Rheumatol 2015;42:372–8. [DOI] [PubMed] [Google Scholar]
- 7. Bongartz T, Sutton AJ, Sweeting MJ, Buchan I, Matteson EL, Montori V. Anti‐TNF antibody therapy in rheumatoid arthritis and the risk of serious infections and malignancies: systematic review and meta‐analysis of rare harmful effects in randomized controlled trials [published erratum appears in JAMA 2006;295:2482]. JAMA 2006;295:2275–85. [DOI] [PubMed] [Google Scholar]
- 8. Dixon WG, Symmons DP, Lunt M, Watson KD, Hyrich KL, British Society for Rheumatology Biologics Register Control Centre C , et al. Serious infection following anti‐tumor necrosis factor α therapy in patients with rheumatoid arthritis: lessons from interpreting data from observational studies. Arthritis Rheum 2007;56:2896–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Greenberg JD, Reed G, Kremer JM, Tindall E, Kavanaugh A, Zheng C, et al. Association of methotrexate and tumour necrosis factor antagonists with risk of infectious outcomes including opportunistic infections in the CORRONA registry. Ann Rheum Dis 2010;69:380–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Grijalva CG, Chen L, Delzell E, Baddley JW, Beukelman T, Winthrop KL, et al. Initiation of tumor necrosis factor‐α antagonists and the risk of hospitalization for infection in patients with autoimmune diseases. JAMA 2011;306:2331–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Leombruno JP, Einarson TR, Keystone EC. The safety of anti‐tumour necrosis factor treatments in rheumatoid arthritis: meta and exposure‐adjusted pooled analyses of serious adverse events. Ann Rheum Dis 2009;68:1136–45. [DOI] [PubMed] [Google Scholar]
- 12. Listing J, Strangfeld A, Kary S, Rau R, von Hinueber U, Stoyanova‐Scholz M, et al. Infections in patients with rheumatoid arthritis treated with biologic agents. Arthritis Rheum 2005;52:3403–12. [DOI] [PubMed] [Google Scholar]
- 13. Singh JA, Cameron C, Noorbaloochi S, Cullis T, Tucker M, Christensen R, et al. Risk of serious infection in biological treatment of patients with rheumatoid arthritis: a systematic review and meta‐analysis. Lancet 2015;386:258–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Strangfeld A, Eveslage M, Schneider M, Bergerhausen HJ, Klopsch T, Zink A, et al. Treatment benefit or survival of the fittest: what drives the time‐dependent decrease in serious infection rates under TNF inhibition and what does this imply for the individual patient? Ann Rheum Dis 2011;70:1914–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Thompson AE, Rieder SW, Pope JE. Tumor necrosis factor therapy and the risk of serious infection and malignancy in patients with early rheumatoid arthritis: a meta‐analysis of randomized controlled trials. Arthritis Rheum 2011;63:1479–85. [DOI] [PubMed] [Google Scholar]
- 16. Cobo‐Ibanez T, Descalzo MA, Loza‐Santamaria E, Carmona L, Munoz‐Fernandez S. Serious infections in patients with rheumatoid arthritis and other immune‐mediated connective tissue diseases exposed to anti‐TNF or rituximab: data from the Spanish registry BIOBADASER 2.0. Rheumatol Int 2014;34:953–61. [DOI] [PubMed] [Google Scholar]
- 17. Curtis JR, Yang S, Patkar NM, Chen L, Singh JA, Cannon GW, et al. Risk of hospitalized bacterial infections associated with biologic treatment among US veterans with rheumatoid arthritis. Arthritis Care Res (Hoboken) 2014;66:990–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Johnston SS, Turpcu A, Shi N, Fowler R, Chu BC, Alexander K. Risk of infections in rheumatoid arthritis patients switching from anti‐TNF agents to rituximab, abatacept, or another anti‐TNF agent, a retrospective administrative claims analysis. Semin Arthritis Rheum 2013;43:39–47. [DOI] [PubMed] [Google Scholar]
- 19. Lampropoulos CE, Orfanos P, Bournia VK, Karatsourakis T, Mavragani C, Pikazis D, et al. Adverse events and infections in patients with rheumatoid arthritis treated with conventional drugs or biologic agents: a real world study. Clin Exp Rheumatol 2015;33:216–24. [PubMed] [Google Scholar]
- 20. Sakai R, Cho SK, Nanki T, Watanabe K, Yamazaki H, Tanaka M, et al. Head‐to‐head comparison of the safety of tocilizumab and tumor necrosis factor inhibitors in rheumatoid arthritis patients (RA) in clinical practice: results from the registry of Japanese RA patients on biologics for long‐term safety (REAL) registry. Arthritis Res Ther 2015;17:74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yun H, Xie F, Delzell E, Levitan EB, Chen L, Lewis JD, et al. Comparative risk of hospitalized infection associated with biologic agents in rheumatoid arthritis patients enrolled in medicare. Arthritis Rheumatol 2016;68:56–66. [DOI] [PubMed] [Google Scholar]
- 22. Curtis JR, Xie F, Chen L, Baddley JW, Beukelman T, Saag KG, et al. The comparative risk of serious infections among rheumatoid arthritis patients starting or switching biological agents. Ann Rheum Dis 2011;70:1401–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Michaud K. The National Data Bank for Rheumatic Diseases (NDB). Clin Exp Rheumatol 2016;34 Suppl 101:S100–1. [PubMed] [Google Scholar]
- 24. Leandro MJ, Cooper N, Cambridge G, Ehrenstein MR, Edwards JC. Bone marrow B‐lineage cells in patients with rheumatoid arthritis following rituximab therapy. Rheumatology (Oxford) 2007;46:29–36. [DOI] [PubMed] [Google Scholar]
- 25. Spreeuwenberg MD, Bartak A, Croon MA, Hagenaars JA, Busschbach JJ, Andrea H, et al. The multiple propensity score as control for bias in the comparison of more than two treatment arms: an introduction from a case study in mental health. Med Care 2010;48:166–74. [DOI] [PubMed] [Google Scholar]
- 26. Wolfe F, Michaud K, Pincus T. A composite disease activity scale for clinical practice, observational studies, and clinical trials: the patient activity scale (PAS/PAS‐II). J Rheumatol 2005;32:2410–5. [PubMed] [Google Scholar]
- 27. Sylvestre MP, Abrahamowicz M. Flexible modeling of the cumulative effects of time‐dependent exposures on the hazard. Stat Med 2009;28:3437–53. [DOI] [PubMed] [Google Scholar]
- 28. Dixon WG, Abrahamowicz M, Beauchamp ME, Ray DW, Bernatsky S, Suissa S, et al. Immediate and delayed impact of oral glucocorticoid therapy on risk of serious infection in older patients with rheumatoid arthritis: a nested case‐control analysis. Ann Rheum Dis 2012;71:1128–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Michaud K, Wolfe F. Comorbidities in rheumatoid arthritis. Best Pract Res Clin Rheumatol 2007;21:885–906. [DOI] [PubMed] [Google Scholar]
- 30. Rubin DB. Multiple imputation for nonresponse in surveys. New York: John Wiley & Sons; 1987. [Google Scholar]
- 31. Cohen SB, Emery P, Greenwald MW, Dougados M, Furie RA, Genovese MC, et al. Rituximab for rheumatoid arthritis refractory to anti‐tumor necrosis factor therapy: results of a multicenter, randomized, double‐blind, placebo‐controlled, phase III trial evaluating primary efficacy and safety at twenty‐four weeks. Arthritis Rheum 2006;54:2793–806. [DOI] [PubMed] [Google Scholar]
- 32. Genovese MC, Becker JC, Schiff M, Luggen M, Sherrer Y, Kremer J, et al. Abatacept for rheumatoid arthritis refractory to tumor necrosis factor α inhibition [published erratum appears in N Engl J Med 2005;353:2311]. N Engl J Med 2005;353:1114–23. [DOI] [PubMed] [Google Scholar]
- 33. Klareskog L, van der Heijde D, de Jager JP, Gough A, Kalden J, Malaise M, et al. Therapeutic effect of the combination of etanercept and methotrexate compared with each treatment alone in patients with rheumatoid arthritis: double‐blind randomised controlled trial. Lancet 2004;363:675–81. [DOI] [PubMed] [Google Scholar]
- 34. Lipsky PE, van der Heijde DM, St Clair EW, Furst DE, Breedveld FC, Kalden JR, et al, and the Anti‐Tumor Necrosis Factor Trial in Rheumatoid Arthritis with Concomitant Therapy Study Group . Infliximab and methotrexate in the treatment of rheumatoid arthritis. N Engl J Med 2000;343:1594–602. [DOI] [PubMed] [Google Scholar]
- 35. Smolen JS, Beaulieu A, Rubbert‐Roth A, Ramos‐Remus C, Rovensky J, Alecock E, et al. Effect of interleukin‐6 receptor inhibition with tocilizumab in patients with rheumatoid arthritis (OPTION study): a double‐blind, placebo‐controlled, randomised trial. Lancet 2008;371:987–97. [DOI] [PubMed] [Google Scholar]
- 36. Vashisht P, Sayles H, Cannella AC, Mikuls TR, Michaud K. Generalizability of patients with rheumatoid arthritis in biologic agent clinical trials. Arthritis Care Res (Hoboken) 2016;68:1478–88. [DOI] [PubMed] [Google Scholar]
- 37. Komano Y, Tanaka M, Nanki T, Koike R, Sakai R, Kameda H, et al. Incidence and risk factors for serious infection in patients with rheumatoid arthritis treated with tumor necrosis factor inhibitors: a report from the Registry of Japanese Rheumatoid Arthritis Patients for Longterm Safety. J Rheumatol 2011;38:1258–64. [DOI] [PubMed] [Google Scholar]
- 38. Lane MA, McDonald JR, Zeringue AL, Caplan L, Curtis JR, Ranganathan P, et al. TNF‐α antagonist use and risk of hospitalization for infection in a national cohort of veterans with rheumatoid arthritis. Medicine (Baltimore) 2011;90:139–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Curtis JR, Patkar N, Xie A, Martin C, Allison JJ, Saag M, et al. Risk of serious bacterial infections among rheumatoid arthritis patients exposed to tumor necrosis factor α antagonists. Arthritis Rheum 2007;56:1125–33. [DOI] [PubMed] [Google Scholar]
- 40. Askling J, Fored CM, Brandt L, Baecklund E, Bertilsson L, Feltelius N, et al. Time‐dependent increase in risk of hospitalisation with infection among Swedish RA patients treated with TNF antagonists. Ann Rheum Dis 2007;66:1339–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Grijalva CG, Kaltenbach L, Arbogast PG, Mitchel EF Jr, Griffin MR. Initiation of rheumatoid arthritis treatments and the risk of serious infections. Rheumatology (Oxford) 2010;49:82–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Van Dartel SA, Fransen J, Kievit W, Dutmer EA, Brus HL, Houtman NM, et al. Predictors for the 5‐year risk of serious infections in patients with rheumatoid arthritis treated with anti‐tumour necrosis factor therapy: a cohort study in the Dutch Rheumatoid Arthritis Monitoring (DREAM) registry. Rheumatology (Oxford) 2013;52:1052–7. [DOI] [PubMed] [Google Scholar]
- 43. Doran MF, Crowson CS, Pond GR, O'Fallon WM, Gabriel SE. Predictors of infection in rheumatoid arthritis. Arthritis Rheum 2002;46:2294–300. [DOI] [PubMed] [Google Scholar]
- 44. Au K, Reed G, Curtis JR, Kremer JM, Greenberg JD, Strand V, et al. High disease activity is associated with an increased risk of infection in patients with rheumatoid arthritis. Ann Rheum Dis 2011;70:785–91. [DOI] [PubMed] [Google Scholar]
- 45. England BR, Sayles H, Mikuls TR, Johnson DS, Michaud K. Validation of the rheumatic disease comorbidity index. Arthritis Care Res (Hoboken) 2015;67:865–72. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials