

Graphical Abstract
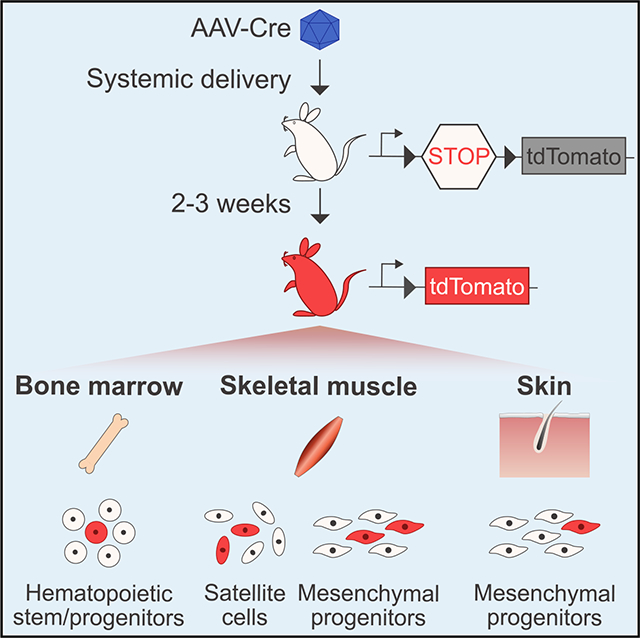
In Brief
Goldstein et al. demonstrate in vivo transduction of endogenous tissue stem cells in the muscle, blood, and skin by systemic or local administration of adeno-associated viruses (AAVs) encoding genome-modifying enzymes. They report that AAV-transduced and genome-modified stem and progenitor cells maintain their capacity to differentiate and engraft following transplantation.
SUMMARY
In vivo delivery of genome-modifying enzymes holds significant promise for therapeutic applications and functional genetic screening. Delivery to endogenous tissue stem cells, which provide an enduring source of cell replacement during homeostasis and regeneration, is of particular interest. Here, we use a sensitive Cre/lox fluorescent reporter system to test the efficiency of genome modification following in vivo transduction by adeno-associated viruses (AAVs) in tissue stem and progenitor cells. We combine immunophenotypic analyses with in vitro and in vivo assays of stem cell function to reveal effective targeting of skeletal muscle satellite cells, mesenchymal progenitors, hematopoietic stem cells, and dermal cell subsets using multiple AAV serotypes. Genome modification rates achieved through this system reached >60%, and modified cells retained key functional properties. This study establishes a powerful platform to genetically alter tissue progenitors within their physiological niche while preserving their native stem cell properties and regulatory interactions.
INTRODUCTION
Effective organ function requires both homeostatic maintenance of appropriate cell numbers and injury-induced repair responses that can replace damaged cells, both processes that rely on tissue stem cells. Studies spanning multiple decades have sought to define key molecular regulators of tissue stem cell function; however, the pace at which investigators have been able to interrogate and define such mediators has been constrained by the technological limitations of generating genetically engineered mice and of stem cell transplantation models typically used for such studies. In particular, transgenic and gene knockout-based approaches require the generation and breeding of multiple distinct genetically engineered deletion and/or floxed alleles to disrupt genes of interest in a ubiquitous or tissue-specific manner, and this challenge is exacerbated when the combinatorial effects of perturbing several genes are of interest. Likewise, ex vivo genomic manipulation of stem cells requires the isolation and transplantation of these cells, which disturbs key regulatory interactions present in endogenous stem cell niches and can profoundly modify normal stem cell properties (Wagers, 2012). Thus, the field would benefit tremendously from the availability of a programmable, in vivo platform to manipulate gene expression in endogenous stem cells without the need to isolate them or to generate complex, multiallelic transgenic animals.
Previous work from our laboratory used a fluorescent reporter system to monitor in vivo delivery of DNA encoding Cre recombinase, a sequence-targeted genome-modifying enzyme, to skeletal muscle stem cells (also known as muscle satellite cells) using adeno-associated viruses (AAVs) (Tabebordbar et al., 2016). In that study, systemic Cre delivery to neonatal mice harboring a Rosa26-tdTomato Ai9 reporter allele using AAV serotype 9 (AAV9-Cre) resulted in modification of ~10% of the total satellite cell pool (as indicated by tdTomato fluorescence). Analogous administration of AAV-CRISPR, a system composed of two AAV vectors (one encoding Staphylococcus aureus Cas9 [saCas9] and a second encoding dual U6-driven guide RNAs targeting sequences flanking the STOP cassette upstream of the Ai9 reporter allele), induced tdTomato fluorescence in 2%–4% of endogenous satellite cells (Tabebordbar et al., 2016). These results suggest that endogenous muscle satellite cells in neonatal animals are accessible to systemically administered AAVs and can be modified following transduction by these vectors carrying genome-targeting enzymes.
Encouraged by these initial results, we have applied this same tdTomato reporter system in this study to investigate whether systemic AAV administration can also transduce satellite cells in adult animals and whether this approach might be extended to additional AAV serotypes and distinct tissue stem cells and progenitor populations. We report efficient transduction of adult mouse satellite cells following in vivo systemic delivery of AAVCre, reaching >60% of the total satellite cell pool and representing a 6-fold increase over our previous study in neonatal mice (Tabebordbar et al., 2016). We further reveal that this transduction capacity is not limited to AAV9 but extends to additional AAV serotypes, including AAV8 and Anc80L65 (hereafter designated Anc80). Finally, we report the transduction and genome modification of multiple non-myogenic stem and progenitor cells, including mesenchymal progenitors in the skeletal muscle and dermis, as well as hematopoietic stem and progenitor cells in the bone marrow. Subsequent isolation, differentiation, and transplantation studies confirm that the targeted tissue stem cells retain their regenerative functions following in situ AAV transduction and genome modification. Collectively, these studies document efficient in vivo genome modification of distinct lineages of stem and progenitor cells across multiple anatomical niches using AAV delivery in adult mammals. This system presents exciting opportunities to pursue in vivo gene activation, disruption, and editing strategies in tissue-resident stem cells for therapeutic purposes, as well as approaches to induce or inactivate transgenic or endogenous alleles to uncover novel molecular regulators of stem and progenitor cells within their native niches.
RESULTS
We previously demonstrated the feasibility of in vivo gene modification in satellite cells by AAV9-mediated delivery of genome-modifying enzymes (Tabebordbar et al., 2016). As noted earlier, we observed transduction of satellite cells after intraperitoneal delivery of AAV9-Cre in neonatal mice (~10%) and following intramuscular delivery of AAV9-Cre in adult mice (~35%). However, we did not evaluate adult satellite cell transduction rates following intravenous injection (Tabebordbar et al., 2016). Thus, in the current study, we asked whether systemic AAV administration to adult animals might target satellite cells and whether this systemic delivery approach in adults may likewise target other stem cell and progenitor populations in distinct anatomical locations.
To perform this proof-of-concept study, we produced AAV particles encoding Cre recombinase (AAV-Cre) downstream of a cytomegalovirus (CMV) promoter and a chimeric intron packaged with AAV8, AAV9, or Anc80 (Zinn et al., 2015) and injected them intravenously into young adult (6 weeks old) Ai9 mice (on an mdx background: mdx;Ai9 mice) (Figure 1A). The Ai9 allele encodes a Rosa26-LoxP-STOP-LoxP-tdTomato reporter, which enables irreversible labeling of targeted cells with tdTomato fluorescence, detectable with single-cell resolution, after Cre-mediated excision of the STOP cassette (Madisen et al., 2010). AAVCre particles were injected intravenously by either the tail vein or the retro-orbital sinus at a low dose (5.5 × 1011 to 8 × 1011 viralgenomes [vg] per mouse) or a high dose (2.2 × 1012 vg per mouse). Tissues were then analyzed 2–3 weeks after injection for the presence of tdTomato fluorescence in three well-defined regenerative adult precursor cell populations—skeletal muscle satellite cells, muscle-resident mesenchymal progenitor cells, and bone marrow-localized hematopoietic stem and progenitor cells—all of which are easily identifiable by extensively validated cell surface marker profiles (Cerletti et al., 2008; Kiel et al., 2005; Maesner et al., 2016; Sherwood et al., 2004).
Figure 1. Systemic AAV-Cre Administration Transduces Muscle Satellite Cells.

(A) Experimental design. AAVs encoding Cre recombinase were injected into mdx;Ai9 mice, which carry a Rosa26-LSL-tdTomato reporter. i.v., intravenous; LSL, LoxP-STOP-LoxP.
(B) Representative flow cytometric analysis of skeletal muscle satellite cells isolated from mdx;Ai9 mice injected intravenously with AAV-Cre packaged in various serotypes at a low dose (~5.5 × 1011 to 8 × 1011 vg) or a high dose (~2.2 × 1012 vg). MuSCs, muscle satellite cells.
(C) Quantification of the frequency of AAV-transduced and Cre-recombined tdTomato+ muscle satellite cells.
Data points from individual mice are overlaid with mean ± SD. n = 4 mice injected with each AAV serotype per group, n = 3 vehicle-injected mice per group. vg, viral genomes; Anc80, Anc80L65.
See also Figures S1, S2, S5, and S6 and Table S1.
We detected tdTomato+ muscle satellite cells, defined as CD45− CD11b (Mac-1)− Ter119− Sca-1– β1-integrin+ CXCR4+ cells (Figure S1C) (Cerletti et al., 2008; Maesner et al., 2016; Sherwood et al., 2004), at frequencies ranging from 8% to 62% in all animals injected with AAV-Cre (n = 24 mice) irrespective of the anatomical location of intravenous injection (retro-orbital or tail vein), indicating robust and reproducible Cre-dependent recombination following in vivo transduction of these cells by AAV (Figures 1B and 1C). No tdTomato+ cells were detected in vehicle-injected controls (n = 6) (Figures 1B and 1C). Comparing mice injected with the same serotype of AAV, the percentage of tdTomato+ satellite cells roughly correlated with the dose of AAV injected, and on average, more than half of the total pool of satellite cells was transduced and genetically modified with the higher doses of either AAV8-Cre or Anc80-Cre (Figure 1C). We validated these findings by immunofluorescence analysis in a subset (n = 3) of high-dose AAV8-Cre-injected animals, confirming the presence of tdTomato+ muscle satellite cells (marked by Pax7 expression and by their localization beneath the basal lamina) (Seale et al., 2000) in 29 of 30 of sections analyzed from three AAV-Cre-injected mice (Figure S1A). No tdTomato+ cells were detected in sections from uninjected controls. Quantification of these microscopy data across all three animals indicated an average ~34% of Pax7+ satellite cells that were also tdTomato+ (Figure S1B), similar to results obtained by flow cytometry (Figure 1C). We observed substantial variability in the frequency of tdTomato+ Pax7+ muscle satellite cells present in different sections prepared from the same muscle (Figure S1B), suggesting that AAV transduction, and subsequent genome modification events, may not be distributed evenly throughout the muscle tissue.
We next investigated whether in vivo AAV-transduced and genome-modified satellite cells maintained their myogenic capacity (Figure 1A). Satellite cells sorted from the muscles of AAV-Cre-injected mice retained their ability to proliferate, differentiate, and fuse to form tdTomato+ myosin heavy chain (MHC)-expressing myotubes when cultured ex vivo (Figure 2A). The relative intensity of the tdTomato signal in fused myotubes appeared to reflect the transduction efficiencies of the sorted populations (as determined by fluorescence-activated cell sorting [FACS]), with more highly transduced populations generating visibly brighter tdTomato+ myotubes (Figure 2A). Consistent with the unperturbed proliferation and differentiation properties of AAV-Cre-transduced tdTomato+ satellite cells, relative quantification of myogenic gene expression indicated equivalent levels of Pax7, Myf5, and Myog expression, with a slight (~25%) reduction in Myod1 in tdTomato+ satellite cells compared to tdTomato– satellite cells (Figure S1D). Finally, we confirmed that AAV-transduced muscle satellite cells retain their myogenic stem cell properties using stringent in vivo transplantation assays. tdTomato+ satellite cells preserved their capacity to engraft, generate tdTomato+ muscle fibers (Figure 2C), and replenish the pool of sublaminar Pax7+ satellite cells (Figure 2B) following isolation and transplantation into preinjured recipient muscle. Collectively, these results confirm retention of key myogenic features and satellite cell functions after in vivo AAV transduction of muscle satellite cells in adult animals.
Figure 2. AAV-Transduced Muscle Satellite Cells Retain Differentiation and Engraftment Potential.

(A) Representative immunofluorescence images of myosin heavy chain (MHC)-stained myotubes differentiated from muscle satellite cells isolated from mice injected with different AAV serotypes at low or high doses. Green, MHC; red, tdTomato; blue, Hoechst. Scale bar, 100 μm. Anc80, Anc80L65.
(B) Representative immunofluorescence images analyzing satellite cell engraftment in tibialis anterior (TA) muscles from mdx mice that either were not transplanted (top row, control) or were transplanted with 50,000 FACS-purified tdTomato+ muscle satellite cells from high-dose AAV8-Cre-injected Ai9 mice (bottom row). TA muscles from 2 transplanted legs and 2 non-transplanted contralateral legs were analyzed 3 weeks post-transplantation. White arrowheads denote Pax7+ tdTomat− muscle satellite cells. White arrows denote Pax7+ tdTomato+ muscle satellite cells. tdTomato+ myofibers are also apparent. Blue, DAPI; white, laminin; green, Pax7; red, tdTomato. Scale bar, 20 μm.
(C) Representative images analyzing muscle fiber engraftment in TA muscles from mdx mice 3 weeks after injection of vehicle only (top row, control) or of muscle satellite cells harvested from mdx;Ai9 mice injected previously with high-dose AAV8-Cre (bottom row). Red, tdTomato; green, wheat germ agglutinin (WGA); blue, DAPI. Scale bar, 100 μm.
See also Figures S1, S2, S5, and S6 and Table S1.
In addition to the satellite cell compartment, we detected tdTomato+ cells in a subset (ranging from 8% to 46%) of muscle-resident Sca-1+ mesenchymal progenitors, which can differentiate to form adipocytes (Figures S1C, S1F, and S1G) (Hettmer et al., 2011; Schulz et al., 2011; Sherwood et al., 2004; Tan et al., 2011), and a subset (ranging from 4% to 33%) of muscle-infiltrating hematopoietic cells (marked by CD45, Mac-1, and/or Ter119) within the muscles of animals injected with different serotypes of AAVCre (Figures S1C and S1E). A separate experiment evaluating systemic administration of AAV6, AAV8, or AAV9 encoding Cre recombinase under the control of the CMV promoter similarly showed transduction of muscle satellite cells (ranging from 6% to 13%) and muscle-localized Sca-1+ progenitors (ranging from 5% to 6%), emphasizing the reproducibility of this in vivo delivery system (Table S1).
Finally, we assessed local delivery methods to target muscle progenitors using AAV-Cre. For these studies, we used mdx;Ai9 animals carrying a Pax7-ZsGreen allele to transgenically mark muscle satellite cells (Figure S2A) (Arpke et al., 2013; Bosnakovski et al., 2008; Maesner et al., 2016). We delivered 6 3 × 1011 vg/mouse of AAV-Cre using serotypes 1, 8, or 9 via direct intramuscular injection, and similar to our results with systemic delivery, analysis of skeletal muscles 3 weeks after local injection of AAV-Cre revealed tdTomato expression in a substantial fraction of hematopoietic lineage cells (ranging from 33% to 62%) (Figure S2B), Sca-1+ mesenchymal progenitors (ranging from 62% to 80%) (Figure S2C), and satellite cells (ranging from 10% to 23%) (Figure S2D). Although this genetic approach to mark muscle satellite cells using a Pax7 reporter allele in mdx muscle has been applied previously by our group and others (Filareto et al., 2015; Tabebordbar et al., 2016), we cannot rule out that the Pax7-ZsGreen allele may also mark some myoblast cells in the mdx mouse. Nonetheless, together with the results obtained following systemic AAV delivery, these data demonstrate efficient, dose-dependent, and reproducible transduction and delivery of a genome-modifying enzyme by multiple AAV serotypes into skeletal muscle satellite cells and mesenchymal progenitors within their native niche.
To determine whether additional stem cell populations in other anatomical niches might be similarly targeted by systemic AAV administration, we analyzed bone marrow cells harvested from mice injected intravenously with AAV8, AAV9, or Anc80 encoding Cre recombinase (Figure 3A). We observed detectable tdTomato expression within immunophenotypically identified hematopoietic stem cells (HSCs; defined as Lin−Sca-1+c-kit+CD48−CD150+) (ranging from 3% to 38%) (Figures 3B and 3C). These results were reproducible using intravenous delivery through either the tail vein or the retro-orbital sinus (Figure 3C) and in a separate experiment in which we injected CMV-driven Cre using AAV6, AAV8, or AAV9-Cre (Table S1). Similar to our results with skeletal muscle satellite cells, bone marrow HSCs were transduced and modified by multiple AAV serotypes with efficiencies that roughly correlated with viral titer (Figure 3C).
Figure 3. Systemic Injection of AAV-Cre Transduces Functional Hematopoietic Stem Cells.

(A) Experimental design. AAVs encoding Cre recombinase were injected into mdx;Ai9 mice carrying the Rosa26-LSL-tdTomato reporter. i.v., intravenous; LSL, LoxP-STOP-LoxP; HSPCs, hematopoietic stem and progenitor cells.
(B) HSC gating strategy (Lin−Sca-1+c-Kit+CD48−CD150+) from bone marrow (BM) and representative flow cytometric analysis of tdTomato expression within HSCs. LSK: Lin−Sca-1+c-Kit+.
(C) Frequency of HSCs expressing tdTomato. Data points from individual mice are overlaid with mean ± SD. n = 4 mice injected with each AAV serotype per group, n = 3 vehicle-injected mice per group.
(D) Percentage of donor chimerism among live peripheral blood cells of primary recipients at monthly intervals post-transplantation. Data points represent mean ± SD. n = 8 primary recipients per group.
(E) Frequency of primary recipients with tdTomato+ multilineage engraftment within the indicated peripheral blood lineages at 16 weeks post-transplant. For each lineage, tdTomato+ engraftment was scored based on satisfaction of two criteria: >1% CD45.2+ cells and >1% tdTomato+ among CD45.2+ cells.
(F–I) Frequency of tdTomato+ cells among live CD45.2+ peripheral blood (F) T cells, (G) B cells, (H) monocytes, and (I) neutrophils in primary recipients at 16 weeks post-transplantation. For each lineage, only recipients exhibiting >1% CD45.2+ cells within that lineage are shown. Individual data points are shown overlaid with mean ± SD. n = 6–8 primary recipients per group.
(J) Percentage of tdTomato+ HSCs among CD45.2+ HSCs in primary recipients at 6 months post-transplantation Individual data points are shown overlaid with mean ± SD. n = 6–8 primary recipients per group.
vg, viral genomes; Anc80, Anc80L65.
See also Figures S3–S6 and Tables S1 and S2.
In addition to HSCs, we investigated the targeting of more committed hematopoietic precursors in the bone marrow by systemically delivered AAV-Cre. tdTomato fluorescence was detected in multiple subsets of bone marrow progenitors, including common myeloid progenitors (CMPs) (Lin−Sca-1−c-Kit+CD34+FcγRlow, ranging from 4% to 26%), granulocyte monocyte progenitors (GMPs) (Lin−Sca-1−c-Kit+CD34+FcγR+, ranging from 5% to 17%), and megakaryocyte erythroid progenitors (MEPs) (Lin−Sca-1−c-Kit+CD34−FcγR−, ranging from 2.5% to 12%) (Figures S3B and S3F–S3H). tdTomato+ cells were also detected among lineage-committed erythroid precursors, including EryA cells (basophilic erythroblasts) (Ter119+CD71+FSChigh, ranging from 23% to 74%), EryB cells (late basophilic and polychromatic erythroblasts) (Ter119+CD71+FSClow, ranging from 9% to 20%), and a small fraction of EryC cells (orthochromatic erythroblasts and reticulocytes) (Ter119+CD71−FSClow, ranging from 0.8% to 2.5%) (Figures S3A and S3C–S3E). Detection of tdTomato+ lineage-restricted hematopoietic progenitors suggests that these cells were either directly transduced by AAV-Cre or that they arose from a more upstream precursor cell within the 14 days after AAV injection.
To determine whether the in vivo transduced tdTomato+ HSCs detected by immunophenotyping retained stem cell engraftment, self-renewal, and differentiation potential, we applied rigorous primary and secondary transplantation assays to evaluate the long-term reconstituting function of HSCs transduced by AAV-Cre. We isolated Linlow tdTomato+ bone marrow cells by FACS from AAV-Cre-injected Ai9 mice expressing the CD45.2 allotype and transplanted these cells into lethally irradiated congenic CD45.1+ recipients (Figure 3A). Monthly assessments of peripheral blood chimerism revealed high levels of donor (CD45.2+) cell engraftment among all primary transplant recipients (Figure 3D). In addition, all recipients exhibited enduring contributions of tdTomato+ CD45.2+ donor-derived peripheral blood cells in multiple hematopoietic lineages at 16 weeks post-transplant (Figures 3E–3I). Analysis of tdTomato expression in the bone marrow further revealed the presence of donor-derived CD45.2+ tdTomato+ HSCs in 45 of 46 recipient mice analyzed (Figure 3J). The detection of tdTomato− CD45.2+ donor-derived peripheral blood cells and HSCs in the transplanted recipients likely reflects our decision to perform single, rather than double, sorts to maximize cell yield over cell purity (Figures 3F–3J).
We next performed secondary transplantation of bone marrow cells from a subset of previously engrafted primary recipient mice (Figure 3A). Analysis of donor chimerism levels within secondary recipients revealed a range of approximately 20%–80% among peripheral blood cells (Figure 4A). Of these secondary recipients, a majority showed multilineage engraftment by tdTomato+ donor cells at 16 weeks post-transplant (Figures 4B–4F). Moreover, tdTomato+ donor-derived HSCs were detected in the bone marrow of secondary recipients 4–5 months post-transplant (Figure 4G), providing functional evidence that AAV-Cre transduced long-term reconstituting HSCs within the initial injected animals, rather than long-lived oligolineage progenitors (Busch et al., 2015; Sawai et al., 2016; Sun et al., 2014). We also evaluated transduction of HSCs following local injection of AAV serotypes 1, 2, 5, 6, 8, 9, 10, and Anc80, with immunophenotypically (Figures S4A and S4B) and functionally (Figures S4C–S4F) defined tdTomato+ HSCs and tdTomato+ hematopoietic progenitors (Table S2) detected in the bone marrow in multiple AAV-injected animals. Altogether, these data provide strong evidence that local or systemic injection of any of several AAV serotypes encoding a sequence-specific DNA-modifying enzyme can allow for transduction of multiple subsets of endogenous bone marrow stem and progenitor cells, including the most primitive, long-term reconstituting HSCs. This strategy enables irreversible genomic modification, sustained through multiple rounds of hematopoietic regeneration as assayed in transplantation assays, of long-term reconstituting HSCs residing within their native niche.
Figure 4. AAV-Transduced Hematopoietic Stem Cells Exhibit Long-Term Reconstitution Potential following Secondary Transplantation.

(A) Percentage of donor chimerism among live peripheral blood cells in secondary recipients at monthly intervals post-transplantation. Data points represent mean ± SD. n = 6–9 secondary recipients per group.
(B) Frequency of secondary recipients with tdTomato+ multilineage engraftment within indicated peripheral blood lineages at 16 weeks post-transplant. tdTomato+ engraftment was determined using the same criteria as in Figure 3E.
(C–F) Frequency of tdTomato+ cells among live CD45.2+ peripheral blood (C) T cells, (D) B cells, (E) monocytes, and (F) neutrophils in secondary recipients at 16 weeks post-transplantation. For each lineage, only recipients exhibiting >1% CD45.2+ cells within that lineage are shown. Individual data points are shown overlaid with mean ± SD. n = 6–9 mice per group.
(G) Percentage of tdTomato+ HSCs among CD45.2+ HSCs in secondary recipients at 4–5 months post-transplantation. Individual data points are shown overlaid with mean ± SD. n = 6–9 secondary recipients per group.
vg, viral genomes; Anc80, Anc80L65.
See also Figures S5 and S6 and Table S1.
Lastly, to extend our analysis beyond skeletal muscle and hematopoietic tissues, we collected skin tissue from a subset of animals injected with high-dose AAV8-Cre and evaluated whether systemic delivery of AAV might also transduce discrete precursor cell populations within this well-defined anatomical compartment. We observed tdTomato fluorescence within hematopoietic cells localized in the skin (CD45+, ranging from 7% to 22%) (Figures S5A, S5B, and S5H), as expected from our bone marrow analyses, and in dermal fibroblasts (Figure S5C) marked by CD140a expression (also known as Pdgfra) (Collins et al., 2011). Using flow cytometry, we evaluated tdTomato expression within four populations of CD45−CD140a+ dermal fibroblasts, distinguishable by combinatorial expression of CD24 and Sca-1 (Figure S5A) (Zhang et al., 2016). tdTomato fluorescence was detected in each of these dermal subsets, including adipocyte precursors (CD24+Sca-1+, ranging from 8% to 26%) (Figures S5E and S5H), dermal papilla cells (CD24−Sca-1−, ranging from 13% to 20%) (Figures S5F and S5H), and two populations of dermal fibroblasts (CD24+Sca-1−, ranging from 2% to 4%, and CD24−Sca-1+, ranging from 16% to 27%) (Figures S5D, S5G, and S5H). Thus, systemically administered AAVs target multiple immunophenotypically distinct cell populations in mammalian skin, including precursors of dermal adipose and connective tissue that support epidermal homeostasis and regeneration (reviewed in Driskell et al., 2014). In concert with the analyses of skeletal muscle and bone marrow cell populations presented earlier, these data demonstrate the utility of systemic AAV administration to accomplish direct genome modification in multiple adult cell lineages and within anatomically distinct tissue compartments.
DISCUSSION
In this study, we document efficient delivery of DNA-modifying enzymes by AAVs to enable permanent genome modifications within tissue stem cell populations in vivo. This highly programmable approach demonstrates effectiveness in three lineages of stem and progenitor cells (myogenic, mesenchymal, and hematopoietic), and in three distinct anatomical niches (skeletal muscle, bone marrow, and skin) and is highly likely to be generalizable to other DNA-modifying enzymes, including CRISPR/Cas-based systems, as well as additional regenerative cell types in other organ systems and species. A distinct advantage of our approach is that it allows for manipulation of stem cell genomes in situ without requiring cell isolation, culture, or subsequent transplantation, thereby preserving native regulatory interactions and extant stem cell properties. This system also mitigates challenges and toxicities associated with ex vivo stem cell modification and subsequent transplantation, such as the failure of in vitro conditions to maintain robust stem cell function, the necessary use of ablative conditioning, and the unavoidable risk of graft failure (Morgan et al., 2017). We anticipate that the opportunity to directly transduce endogenous tissue stem cells with DNA-modifying enzymes will be of direct and specific relevance for ongoing academic and commercial efforts aimed at therapeutic gene editing in stem cells, all of which have, to this point, considered it necessary to purify stem cells for ex vivo modification and re-infusion (Morgan et al., 2017).
A key finding of our work is that AAVs can efficiently transduce multiple adult tissue stem and progenitor cell types in vivo. These results contrast to some degree with previous reports concluding that AAVs do not effectively transduce skeletal muscle satellite cells in adult animals following local or systemic administration (Arnett et al., 2014; Chamberlain and Chamberlain, 2017; Chang et al., 2016). In one of these prior studies (Arnett et al., 2014), systemic administration of AAV6 encoding CMV-driven EGFP into 4-week-old mice did not result in detectable Pax7+/EGFP+ satellite cells 4 weeks after injection. Meanwhile, intramuscular administration of AAV6 or AAV9 encoding CMV-EGFP into 8-week-old mice resulted in no detectable Pax7+/EGFP+ satellite cells 2 weeks post-injection, while similar administration of AAV8-CMV-EGFP resulted in a minority (<5%) of the satellite cell compartment expressing EGFP (Arnett et al., 2014). One potential explanation for these discrepancies could be that our study used a more sensitive Cre/lox-driven system to robustly drive tdTomato fluorescence from the mouse genome in the AAV-transduced cells and their progeny, as opposed to monitoring expression of the introduced EGFP protein encoded by the AAV genome, which may exhibit transient expression. Ultimately, our results demonstrate that AAVs are capable of transducing satellite cells at efficiencies required for therapeutic gene editing. Our approach, in which cells are permanently modified following AAV transduction, also allowed us to detect delivery of AAV cargo to non-myogenic stem and progenitor cells, including bone marrow localized HSCs. While detection in recent years of naturally occurring clade F AAV variants in healthy human CD34+ hematopoietic cells (termed AAVHSC) (Smith et al., 2014) suggests the existence of AAVs that can transduce such cells, our report demonstrates the efficient in vivo transduction of these cells by recombinant AAV vectors and confirms the preservation of HSC function post-transduction using immunophenotyping and transplantation assays. Still, it will be important in future studies to investigate whether AAV transduction influences global gene expression levels within stem cell populations and whether AAVs preferentially transduce certain subsets of tissue stem cells over others.
In addition to targeting muscle satellite cells and HSCs, our finding that AAVs transduce mesenchymal progenitor cells in vivo has important potential applications for this cell population. Mesenchymal progenitors are well-established regulators of myogenesis (Joe et al., 2010), and they represent a major source of fibrosis and adipose deposition in aging and muscle disease (Joe et al., 2010; Lounev et al., 2009; Uezumi et al., 2010, 2014). Thus, these cells are potential targets for anti-fibrotic interventions or for manipulations that will promote their promyogenic activities. In addition, because little is known about the mechanisms that govern the actions of these cells, including regulators of their proliferation, differentiation, or role in trophic support for muscle, we anticipate that targeting this population of cells could be useful for revealing such mechanisms and for advancing our understanding of muscle maintenance and regeneration. This in turn could lead to therapeutic interventions that mitigate the detrimental impacts of these cells and promote their pro-myogenic activity. Given the large number of studies that have used Cre models to manipulate gene expression in these cells for such purposes (Heredia et al., 2013; Kopinke et al., 2017; Lees-Shepard et al., 2018; Roberts et al., 2013), we anticipate that our system provides an alternative and in some cases more facile system for gene manipulation, which will be of interest to researchers seeking to uncover the biology of this unique class of tissue progenitors.
Our current and prior efforts to target tissue stem and progenitor cells using AAV-mediated delivery of Cre or CRISPR complexes (Tabebordbar et al., 2016) establish a broadly accessible platform for in vivo delivery of genome-modifying enzymes. One key difference between using AAVs to deliver Cre recombinase and using CRISPR is whether a single- or dual-vector system is necessary to deliver the desired components. In this study, we delivered Cre-recombinase via a single AAV, such that any cell that received the AAV cargo would have sufficient genetic material to execute Cre-mediated genome modification. However, extending this approach to more complex CRISPR/Cas-based systems involving Cas9 and multiple guide RNAs may require co-transduction with multiple AAV vectors due to the limited ~ 4.7-kb packaging capacity of AAVs. In our previously published study (Tabebordbar et al., 2016), we co-administered two AAVs to perform in vivo CRISPR editing: one AAV encoding CMV-driven saCas9 and a second AAV encoding two U6-driven guide RNAs targeting the locus of interest (Tabebordbar et al., 2016). While we did observe CRISPR-mediated targeting using this dual-vector system, future efforts to harness minimal stem cell-specific promoters could reduce this approach to a single-vector system, which may improve editing efficiency.
An important consideration regarding the potential applicability of AAV-mediated transduction of tissue stem cells to humans is the impact of an induced immune response to repeated AAV dosing. This issue has been extensively reviewed elsewhere, and it has not precluded clinical testing of AAV-based therapeutics for various diseases in human populations (Colella et al., 2017; Mingozzi and High, 2011). An additional consideration for translating these findings to humans is the dose of AAV necessary to achieve sufficient transduction of tissue stem and progenitor cells, because a report suggested that high-dose systemic administration of AAV can induce liver toxicity (Hinderer et al., 2018). The AAV doses used in our studies reported here are comparable with those used in many therapeutically oriented preclinical studies in mice (Bengtsson et al., 2017; Chamberlain and Chamberlain, 2017; Long et al., 2016; Nelson et al., 2016; Tabebordbar et al., 2016; Yang et al., 2016) and canines (Amoasii et al., 2018), as well as those used in successful human trials for spinal muscular atrophy gene therapy (Mendell et al., 2017). While we did not observe overt signs of liver toxicity in the mice in our study (Figure S6), we acknowledge that this issue remains an important consideration for future development of AAV-based therapeutics.
The in vivo AAV system described here also overcomes critical technological and practical limitations associated with current experimental systems for interrogating stem cell function. In particular, commonly employed transgenic and gene knockout-based models frequently require complex breeding schemes to introduce multiple alleles, necessitating significant investment of both time and resources. Such approaches become even more challenging when assessing gene-targeting effects in aged animals, in non-standard genetic backgrounds, or in a combinatorial fashion. In contrast, AAV-mediated delivery of programmable DNA-modifying enzymes can be applied across a range of animal ages and strains and to various individual and multiplexed genetic loci. In addition, the AAV-Cre strategy used here provides an alternative and complementary approach to tamoxifen-inducible Cre- (CreER) dependent gene activation or inactivation strategies that additionally enables researchers to bypass the potentially confounding effects of hormone administration (Patel et al., 2017). This technology is therefore likely to have important applications in accelerating the pace at which gene functions and interactions can be interrogated in vivo and in tissue progenitors. Our results also indicate that multiple AAV serotypes exhibit tropism for tissue stem and progenitor cells, establishing a foundation for the development of more specific and selective systems, including identification of AAV capsid variants that transduce particular stem cell populations and of naturally occurring or synthetic gene regulatory elements that restrict expression of AAV-encoded genes to only these cell types. Ultimately, this system may be adapted to enable rapid and direct in vivo screening of candidate and unknown gene targets suspected to specifically influence stem cell phenotypes.
STAR★METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Amy Wagers (amy_wagers@harvard.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
Mice were housed at the animal facility at the Harvard University/Faculty of Arts and Sciences Biological Research Infrastructure, which is accredited by the Association for Assessment and Accreditation of Laboratory Animal Care (AALAC). All procedures were performed under protocols approved by the Institutional Animal Care and Use Committee (IACUC). Mice were housed in standard ventilated racks at a maximum density of 5 mice per cage. Room temperature was maintained at 22°C ± 1°C with 30%–70% humidity. Mice were kept on a 12-hour light/dark cycle and provided food and water ad libitum. Breeder mice were kept on irradiated PicoLab Mouse Diet 20 5058 (LabDiet, St. Louis, MO), and non-breeder mice were kept on irradiated LabDiet Prolab Isopro RMH 3000 5P75 (LabDiet, St. Louis, MO). Cages were filled with 1/4 inch Anderson’s Bed o Cob bedding (The Andersons, Inc., Maumee, OH) and each contained one nestlet (2 × 2” compressed cotton square, Ancare, Bellmore, NY) and one red mouse hut (certified poly-carbonate; 3 3/4” wide x 1 7/8” tall x 3” long, BioServ, Flemington, NJ). Cage changes were performed at least every 14 days, and more frequently if necessary. Animal health surveillance was performed quarterly by PCR testing of index animals and through swabs from rack plenums.
C57BL/10ScSn-Dmdmdx/J (mdx; C57BL/10ScSnJ background), B6;129S6-Gt(ROSA)26Sortm9(CAG-tdTomato)Hze/J (Ai9; B6/129S6 background), B6.SJL-Ptprca Pepcb/BoyJ (CD45.1; C57BL/6J background), B6.Cg-Tg(Pax7-ZsGreen)1Kyba/J (Pax7-ZsGreen; C57BL/6J background) and C57BL/6J mouse strains were used for this study. To generate mdx; Ai9 homozygous mice, mdx mice were bred to Ai9 homozygous mice. mdx; Ai9 mice were bred with Pax7-Zsgreen mice to generate mdx;Ai9;Pax7-Zsgreen animals. The Pax7-zsGreen allele has been extensively validated in multiple studies to mark skeletal muscle satellite cells by flow cytometry (Arpke et al., 2013; Bosnakovski et al., 2008; Maesner et al., 2016). Animals were randomly assigned to experimental and control groups. All mice used for experiments were immunocompetent.
For intrafemoral injection experiments, 6–9-month-old male and female Ai9 homozygous animals were injected with AAV-Cre or vehicle. 6 weeks post injection, mice were euthanized and hematopoietic cells were isolated for flow cytometry analysis and FACS. For systemic injection experiments, 6-week-old male mdx; Ai9 homozygous mice were injected with AAV-Cre or vehicle via tail vein or retroorbital injection. 2 weeks post injection, mice were harvested and liver, skeletal muscle and hematopoietic cells were collected for analysis. Histopathological assessment of hematoxylin & eosin stained sections of liver tissue from a subset of AAV-Cre and control-injected animals, performed in a blinded fashion by a skilled mouse pathologist (R. Bronson), did not reveal signs of tissue pathology (Figure S6B).
For hematopoietic cell transplantation experiments, bone marrow cells from homozygous CD45.1 mice were collected and depleted for Sca-1 to use for helper marrow. Homozygous CD45.1 mice were used as recipients for primary and secondary hematopoietic stem and progenitor cell (HSPC) transplants. Following HSPC transplantation, recipient animals were kept on antibiotic water containing 0.67mg/mL Sulfadiazine Trimethoprim for 4 weeks post-transplant.
METHOD DETAILS
AAV selection and production
Multiple AAV serotypes were used, with a particular emphasis on serotypes with broad biodistribution (Zincarelli et al., 2008). For the intrafemoral injection experiments, we evaluated a panel of 7 AAV serotypes (1, 2, 5, 6, 8, 9 and 10) and identified those that targeted HSCs with the greatest efficiency. A smaller panel of 3 AAV serotypes (1, 8, 9) was used for the intramuscular injection experiments. We did not move forward with AAV1 for the systemic injection experiments because it is known to induce a greater inflammatory response than the other AAV serotypes (Lu and Song, 2009), which may limit its usefulness in preclinical testing or in disease modeling applications. The selection of AAV serotypes used for the systemic injection experiments was based on our previous work showing that AAV9 effectively transduces muscle satellite cells in vivo (Tabebordbar et al., 2016). For our preliminary analyses (Table S1), we administered AAV9 via tail vein injection along with two additional serotypes: AAV6 and AAV8, both of which have been shown to target skeletal muscle following in vivo delivery (Blankinship et al., 2004; Wang et al., 2005). Building off of these results, we next performed systemic tail vein administration of AAV8 and AAV9, and also included Anc80L65, an ancestral AAV with robust tropism for skeletal muscle and other organs (Zinn et al., 2015), to express Cre recombinase downstream of CMV promoter and a chimeric intron. For systemic retro-orbital AAV administration, we focused particularly on Anc80 and AAV8, both of which exhibited higher targeting efficiency for blood and muscle stem cells in the tail vein injection experiments. Furthermore, we included two doses of AAV8 to assess if injecting a higher concentration of AAV resulted in a greater transduction efficiency.
For systemic injection experiments using AAV8, AAV9 and Anc80L65, AAV production was performed by the Gene Transfer Vector Core (GTVC) at the Grousbeck Gene Therapy Center at the Schepens Eye Research Institute and Massachusetts Eye and Ear as described previously (Lock et al., 2010; Zinn et al., 2015). For intrafemoral injection experiments using AAV serotypes 1, 2, 5, 6, 8, 9 and 10, AAVs were produced at the University of Pennsylvania Penn Vector Core. For the systemic injection experiment using AAV6, AAV8, and AAV9, AAV production was done by the UMASS medical school vector core.
Muscle stem cell isolation and in vitro differentiation
Muscle stem cell isolation from mdx;Ai9 mice systemically injected with AAV-Cre was performed as previously described (Cerletti et al., 2008; Sherwood et al., 2004). Briefly, tibialis anterior (TA), gastrocnemius, triceps, quadriceps and abdominal muscles were digested with collagenase and dispase and myofiber-associated cells were isolated by centrifugation. The cells were stained with an antibody mix (APC-Cy7 anti-CD45 (1:200), APC-Cy7 anti-CD11b (1:200), APC-Cy7 anti-TER119 (1:200), APC anti-Sca-1 (1:200), FITC anti-CD29 (β1-integrin) (1:100), Biotin anti-CD184 (CXCR4) (1:100)) for 30 min on ice, followed by a secondary staining with Streptavidin-PE-Cy7 (1:100) for 15 minutes on ice. Propidium iodide (PI) and calcein blue were used to discriminate dead and live cells, respectively. CD45− CD11b (Mac-1)− Ter119− Sca-1− β1-integrin+ CXCR4+ cells were isolated by FACS as muscle satellite cells;Sca-1+, CD45− CD11b− Ter119− cells were isolated as mesenchymal progenitors; Lineage (CD45, CD11b, Ter119)+ cells were gated as Lin+ blood cells for analysis. For in vitro expansion of satellite cells isolated from AAV-transduced muscles, satellite cells were seeded on collagen/laminin-coated plates in F10 containing 20% donor horse serum, 1% penicillin-streptomycin, and 1% glutamax. 5 ng/ml bFGF was added to the medium daily. Media was refreshed every other day. After 5 days, satellite cells were harvested, cell numbers were counted and cells were re-plated in multiple wells of a 96 well plate for differentiation. The next day, media was changed to DMEM containing 2% donor horse serum and 1% penicillin-streptomycin. Myotubes were fixed with 4% paraformalde-hyde after 60 or 72 hours in differentiation media.
Muscle stem cell isolation from AAV-Cre injected TA muscles of mdx;Pax7-ZsGreen+/−;Ai9 mice was performed as previously described (Tabebordbar et al., 2016). Briefly, individual TA muscles were separately minced using scissors followed by two digestion steps in collagenase and dispase. Muscles were triturated in between and after the digestion steps. The homogenate was centrifuged, re-suspended, and filtered before another round of centrifugation and resuspension. Cells were stained with an antibody mix of APC anti-Sca-1 (1:200), APC-Cy7 anti-CD45 (1:200), APC-Cy7 anti-CD11b (Mac-1) (1:200), and APC-Cy7 anti-Ter119 (1:200). Propidium iodide (PI) and calcein blue were used to discriminate dead and live cells, respectively. CD45− CD11b (Mac-1)−Ter119− Sca-1− Pax7-ZsGreen+ cells were isolated as muscle satellite cells. This transgenic line has been used previously in multiple studies for the identification and isolation of muscle satellite cells, with extensive validation studies performed by multiple groups (Arpke et al., 2013; Bosnakovski et al., 2008; Maesner et al., 2016), including a study from our lab which specifically addressed the issue of satellite cell identification by flow cytometry (Maesner et al., 2016).
Muscle Sca-1+ progenitor isolation and adipogenic differentiation
Sca-1+ progenitor (Sca-1+, CD45− CD11b (Mac-1)− Ter119−) isolation from mdx;Ai9 mice systemically injected with AAV-Cre was performed as previously described (Hettmer et al., 2011; Schulz et al., 2011; Tan et al., 2011). Briefly, freshly sorted Sca-1+ cells were plated in collagen/laminin-coated 96 well plates and expanded for 8 days in growth medium (F10 containing 20% donor horse serum, 1% penicillin-streptomycin, and 1% glutamax) and provided with 5 ng of bFGF daily with fresh media changes every other day. After 8 days in growth medium, cells were switched to Adipogenic Induction Media (DMEM containing 10% FBS, 1% penicillin/streptomycin, 1 mM Dexamethasone, 100 nM Insulin, 1 mM Rosiglitazone and 0.5 mM 3-isobutyl-1-methylxanthine) for 2 days and then switched to Adipogenic Differentiation Media (DMEM containing 10% FBS, 1% penicillin/streptomycin and 100 nM Insulin) for an additional 8 days. Media was changed every other day, and cells were stained with BODIPY™ 493/503 as per manufacturer’s instructions.
Muscle satellite cell transplantation
One day before transplantation, 25 μL of 10 μM Cardiotoxin gamma from Naja pallida was injected to the tibialis anterior (TA) muscles of anesthetized 7–8 week-old male mdx recipient mice. Up to 50,000 single-sorted tdTomato+ satellite cells or vehicle (staining media) alone was injected into the pre-injured tibialis anterior (TA) muscles. The injected TA muscles were harvested 3 weeks post-transplantation for cryosectioning and fluorescence detection.
Immunofluorescence
For myosin heavy chain (MHC) staining of in vitro differentiated myotubes, myotubes were permeabilized using 0.5% Triton X-100 for 15 minutes at room temperature, washed 2 × 5 minutes with DPBS, and blocked with 5% Normal Goat Serum, 2% BSA, 2% protein concentrate (from M.O.M.™ Basic Kit), and 0.1% Tween-20 for 1 hour at room temperature. Cells were washed 2 × 5 minutes with DPBS, incubated with anti-skeletal myosin type II (fast-twitch) (1:200) and anti-skeletal myosin type I (slow-twitch) (1:100) at 4°C overnight, washed 3 × 5 minutes with DPBS, and incubated with goat anti-mouse IgG Alexa 488 conjugate secondary antibody (1:250). After washing for 3 × 5 minutes with DPBS, cells were stained with 10 mg/ml Hoechst.
For staining of skeletal muscle tissue, tibialis anterior (TA) muscles were dissected and immediately fixed in 1% PFA for one hour at room temperature, washed with DPBS, and cryopreserved in 30% sucrose at 4°C overnight. Tissues were then embedded in O.C.T. compound, cryopreserved in super-cooled isopentane, and sectioned using a Leica CM1860 cryostat (Leica Biosystems). Sections were post-fixed in 2% PFA for 5 minutes, incubated in 0.1 M glycine buffer for 5 minutes, and permeabilized in 0.3% Triton X-100/DPBS for 20 minutes. Sections were blocked with 10% M.O.M. IgG blocking reagent, and further blocked with 1% M.O.M. IgG blocking reagent, 3% BSA, 8% M.O.M protein concentrate, and 5% Normal Goat Serum. Primary antibody staining was performed using anti-Pax7 (15 μg/mL) and Rabbit anti-laminin (1:200) at 4°C overnight. The following day, sections were washed and stained with goat anti-mouse IgG1 Alexa Fluor 488 (1:250) and goat anti-rabbit IgG H+L Alexa Fluor 647 (1:250). Slides were mounted with VectashieldHardSet Antifade Mounting Medium containing DAPI, and images were captured using a Zeiss LSM 880 inverted confocal microscope. For quantification of Pax7+ tdTomato+ sublaminar cells by immunofluorescence analysis, 60–80 Pax7+ sublaminar cells were quantified per animal among 10 0.181mm2 fields.
For staining of skin tissue, dorsal skin samples were fixed for 15 minutes using 4% PFA at room temperature, washed extensively with PBS, immersed in 30% sucrose overnight at 4°C and embedded in O.C.T. 30 μM sections were blocked for 1–2 hours (5% Donkey serum, 1% BSA, 2% Cold water fish gelatin in 0.3% Triton in PBS), incubated with anti-CD140a (1:200) and anti-RFP (1:1000) antibodies overnight at 4°C and incubated for 2–4 hours at room temperature with Donkey anti-goat IgG Alexa 488 (1:400) or Donkey anti-rabbit Cy3 (1:400) secondary antibodies. DAPI was used for counterstaining.
RNA isolation, cDNA synthesis and Real-time PCR
Satellite cells were sorted directly into Trizol LS and stored at −80°C. Total RNA was isolated according to manufacturer’s instructions. cDNA was synthesized using the SuperScript IV VILO Master Mix with ezDNase Enzyme kit. Quantitative PCR was performed on a QuantStudio 6 Flex Real-Time PCR system. The following TaqMan assays were used to evaluate relative gene expression: Pax7 (Mm01354484_m1), Myf5 (Mm00435125_m1), Myod1 (Mm00440387_m1), Myog (Mm00446194_m1) and Gapdh (Mm99999915_g1).
HSPC Isolation and Flow Cytometry Analysis/FACS
Bone marrow cells were flushed from all 4 long bones (2 femurs and 2 tibias) with a 21-gauge needle into staining media (HBSS containing 2%FBS), resuspended, and filtered through a 40 μm cell strainer. Cells were pelleted and subjected to ACK lysis for 5 minutes on ice (except when analyzing erythroid precursor cells), re-filtered through a 40 μm strainer, and washed with staining media.
To identify HSCs from AAV-injected mice, cells were stained with the following antibodies for 45 minutes on ice: CD3-ef450 or CD3-biotin (1:100), B220-ef450 or B220-biotin (1:200), Ter119-ef450 or Ter119-biotin (1:100), Gr-1-ef450 or Gr-1-biotin (1:400), CD11b-APC-Cy7 (1:200), c-Kit-APC (1:200), Sca-1-PE-Cy7 (1:200), CD48-FITC (1:200), and CD150-BV510 (1:50). When biotinylated antibodies were used, cells were washed and incubated with Streptavidin-ef450 (1:200) for 30 minutes on ice.
To identify HSCs from transplanted mice, cells were stained with the following antibodies for 45 minutes on ice: Lineage Cocktail-Pacific Blue (1:20), c-Kit-APC (1:200), Sca-1-PE-Cy7 (1:200), CD48-FITC (1:200), CD150-BV510 (1:50), and CD45.2-AF700 (1:100).
To identify myeloid progenitors from AAV-injected mice, cells were stained with the following antibodies for 60 minutes on ice: CD3-ef450 or CD3-biotin (1:100), B220-ef450 or B220-biotin (1:200), Ter119-ef450 or Ter119-biotin (1:100), Gr-1-ef450 or Gr-1-biotin (1:400), CD11b-APC-Cy7 (1:200), c-Kit-APC (1:200), Sca-1-PE-Cy7 (1:200), CD34-FITC (1:25) and CD16/CD32-AF700 (1:100). For biotinylated antibodies, cells were washed and incubated with Streptavidin-ef450 (1:200) for 30 minutes on ice.
To identify erythroid precursor cells from AAV-injected mice, cells were stained with the following antibodies for 30 minutes on ice: CD71-FITC (1:200) and Ter119-APC (1:200).
For all HSPC staining panels, cells were washed after antibody/streptavidin incubation and resuspended in staining media, and Sytox Blue (1:1,000) was added immediately prior to FACS to mark dead cells. Cells were analyzed on a BD LSR II flow cytometer and sorting was performed on a BD FACS Aria II. Data analysis was performed using BD FACS Diva and FlowJo software.
HSPC Transplantation
For each animal, Linlow tdTomato+ live bone marrow cells were singly sorted from bone marrow into staining media containing 10% FBS. As CD11b (Mac-1) expression has been reported to increase on HSCs in certain contexts of inflammation or stress (Randall and Weissman, 1997), we did not sort tdTomato+ bone marrow cells for transplantation on the basis of CD11b expression, in case this marker might be altered in its expression following AAV administration. Cells from each donor animal were combined with 6×105 Sca-1-depleted radioprotective CD45.1 helper bone marrow cells and injected intravenously into two lethally irradiated (950 rads, split dose) anesthetized CD45.1 recipient animals (3×105 helper marrow cells per recipient). Cells were injected with a 28-gauge insulin syringe into the retroorbital sinus.
Sca-1 depletion was performed by staining ACK-lysed CD45.1 bone marrow cells with Sca-1-APC (1:200) for 10 minutes on ice, washing and incubating with anti-APC microbeads for 20 minutes on ice. Cells were washed and depleted using the AutoMACS Separator (Depletes program). Depletion efficiency was confirmed by flow cytometry analysis. 7-AAD (1:20) was used as a viability dye and was added to cells immediately prior to analysis.
Peripheral blood collection and flow cytometry analysis
Donor chimerism and tdTomato analysis was performed from peripheral blood samples of primary and secondary transplant recipients at 4, 8, 12 and 16 weeks post-transplant. Peripheral blood was collected via the tail vein into a 1.5mL tube containing 100 μL of PBS/10mM EDTA and stored on ice. 1mL of 2%Dextran/PBS was added to the samples, mixed and incubated at 37°C for 30 minutes to sediment red blood cells. The remaining cells were washed and subjected to ACK lysis for 5 minutes on ice. Cells were washed, filtered through a 40 μm cell strainer, and resuspended in HBSS/2% FBS/10mM EDTA containing anti-CD16/CD32 (1:50) for 5 minutes on ice. Cells were stained with the following antibodies for 30 minutes on ice: CD3-PE-Cy7 (1:65), B220-FITC (1:100), CD11b-APC-Cy7 (1:200), Gr-1-Biotin (1:400), CD45.1-Pacific Blue (1:100) and CD45.2-APC (1:100). Cells were washed and stained with Streptavidin-Pacific Orange (1:500) for 30 minutes on ice. Cells were washed, resuspended in HBSS/2% FBS/10mM EDTA, and 7-AAD (1:20) was added immediately prior to analysis. Cells were analyzed on a BD LSR II flow cytometer, and data analysis was performed using BD FACS Diva and FlowJo software.
Liver histology
Liver tissue was harvested and fixed in 4% paraformaldehyde for approximately 24 hours, then transferred to 70% ethanol. Samples were submitted to the Rodent Histopathology Core at the Dana-Farber/Harvard Cancer Center, embedded in paraffin, sectioned and stained with hematoxylin and eosin. Slides were analyzed and scored for histopathology by a Rodent Histopathologist in a blinded manner.
Skin cell isolation and flow cytometry analysis
Mouse back skin was dissected. Dissected skin (dermis facing down) was incubated in 0.25% Collagenase in HBSS for 40–60 minutes at 37°C. The dermis was then scraped using a surgical scalpel. Collected cells were centrifuged for 8 minutes at 350xg at 4°C.Single cell suspensions were obtained by incubation with trypsin-EDTA at 37°C for 10 minutes and filtering through 70 μM and 40 μM filters. Single cell suspensions were then centrifuged for 8 minutes at 350xg at 4°C, re-suspended in 5% FBS in PBS and stained for 30 minutes. The following antibodies were used: CD45-ef450 (1:250), CD140a-biotin (1:250), Streptavidin-APC (1:500), CD24-FITC (1:200), and Sca-1-PerCP-Cy5.5 (1:1000). DAPI was used to exclude dead cells. Blood-lineage cells were gated as CD45+. Dermal fibroblasts were gated as CD45− CD140a+, then further divided based on CD24 and Sca-1 expression. Cells were analyzed on a BD FACS Aria II, and data analysis was performed using BD FACS Diva and FlowJo software.
QUANTIFICATION AND STATISTICAL ANALYSIS
GraphPad Prism software was used to perform statistical analyses. For the real-time PCR analysis, paired t test was used to calculate statistical significance between the two groups. Repeated-measures ANOVA was used for longitudinal analyses of donor chimerism. Results with a p value < 0.05 were considered statistically significant. Information on replicates is reported in the figure legends.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Armenian Hamster Anti-Mouse CD3e ePluor® 450 | Thermo Fisher Scientific | Cat #48-0031-80; RRID: AB_10733280 |
| Rat Anti-Human/Mouse CD45R (B220) eFluor® 450 | Thermo Fisher Scientific | Cat #48-0452-80; RRID: AB_1548763 |
| Rat Anti-Mouse TER-119 eFluor® 450 | Thermo Fisher Scientific | Cat #48-5921-80; RRID: AB_1518809 |
| Rat Anti-Mouse Ly-6G/Ly-6C (Gr-1) eFluor® 450 | Thermo Fisher Scientific | Cat #48-5931-82; RRID: AB_1548788 |
| Rat Anti-Mouse/Human CD11b APC-Cy7 | Biolegend | Cat#101226; RRID: AB_830642 |
| Rat Anti-Mouse Ly-6A/E (Sca-1) PE-Cy7 | Biolegend | Cat#108114; RRID: AB_493596 |
| Rat Anti-Mouse CD117 (c-Kit) APC | BD Biosciences | Cat #553356; RRID: AB_398536 |
| Armenian Hamster Anti-Mouse CD48 PITC | Biolegend | Cat#103404; RRID: AB_313019 |
| Rat Anti-Mouse CD150 (SLAM) Brilliant Violet 510 | Biolegend | Cat#115929; RRID: AB_2562189 |
| Rat Anti-Mouse CD16/CD32 (Mouse BD PC Block) | BD Biosciences | Cat #553142; RRID: AB_394657 |
| Rat Anti-Mouse CD3 antibody PE-Cy7 | Biolegend | Cat#100220; RRID: AB_1732057 |
| Rat Anti-Human/Mouse CD45R (B220) PITC | Thermo Fisher Scientific | Cat #11-0452-85; RRID: AB_465055 |
| Rat Anti-Mouse Ly-6G/Ly-6C (Gr-1)Biotin | Biolegend | Cat #108404; RRID: AB_313369 |
| Mouse Anti-Mouse CD45.1 Pacific Blue | Biolegend | Cat #110722; RRID: AB_492866 |
| Mouse Anti-Mouse CD45.2 APC | Biolegend | Cat#109814; RRID: AB_389211 |
| Rat Anti-Mouse Lineage Cocktail Pacific Blue | Biolegend | Cat#133310; RRID: AB_11150779 |
| Mouse Anti-Mouse CD45.2 Alexa Fluor® 700 | Biolegend | Cat #109822; RRID: AB_493731 |
| Rat Anti-Mouse TER-119 APC | Biolegend | Cat#116212; RRID: AB_313713 |
| Rat Anti-Mouse CD71 PITC | BD Biosciences | Cat #553266; RRID: AB_394743 |
| Armenian Hamster Anti-Mouse CD3e Biotin | Biolegend | Cat#100304; RRID: AB_312669 |
| Rat Anti-Human/Mouse CD45R (B220) Biotin | Thermo Fisher Scientific | Cat #13-0452-82; RRID: AB_466449 |
| Rat Anti-Mouse CD19 Biotin | Biolegend | Cat #115504; RRID: AB_313639 |
| Rat Anti-Mouse TER-119 Biotin | Biolegend | Cat#116204; RRID: AB_313705 |
| Rat Anti-Mouse Ly-6G/Ly-6C (Gr-1)Biotin | Biolegend | Cat #108404; RRID: AB_313369 |
| eBioscience Streptavidin ePluor450 | Thermo Fisher Scientific | Cat #48-4317-82; RRID: AB_10359737 |
| Rat Anti-Mouse CD34 PITC | Thermo Fisher Scientific | Cat #11-0341-85; RRID: AB_465022 |
| Rat Anti-Mouse CD16/CD32 Alexa Fluor® 700 | Thermo Fisher Scientific | Cat #56-0161-82; RRID: AB_493994 |
| Rat Anti-Mouse/Human CD11b APC | Biolegend | Cat#101212; RRID: AB_312795 |
| Rat Anti-Mouse Ly-6G Pacific Blue | Biolegend | Cat#127612; RRID: AB_2251161 |
| Rat Anti-Mouse Ly-6A/E (Sca-1) APC | Biolegend | Cat#108112; RRID: AB_313349 |
| Rat Anti-Mouse CD45 APC-Cy7 | BD Biosciences | Cat #557659; RRID: AB_396774 |
| Rat Anti-Mouse CD11b APC-Cy7 | BD Biosciences | Cat #557657; RRID: AB_396772 |
| Rat Anti-Mouse Ter-119 APC-Cy7 | Biolegend | Cat#116223; RRID: AB_2137788 |
| Rat Anti-Mouse Ly-6A/E (Sca-1) APC | Thermo Fisher Scientific | Cat #17-5981-82; RRID: AB_469487 |
| Hamster Anti-Mouse/Rat CD29 (β1-integrin) PITC | Biolegend | Cat#102206; RRID: AB_312883 |
| Rat Anti-Mouse CD184 (CXCR4) Biotin | BD Biosciences | Cat #551968; RRID: AB_394307 |
| Anti-Myosin (Skeletal, Past) antibody | Millipore Sigma | Cat #M4276; RRID: AB_477190 |
| Anti-Myosin (Skeletal, Slow) antibody | Millipore Sigma | Cat #M8421; RRID: AB_477248 |
| Mouse Anti-Pax7 antibody | Developmental Studies Hybridoma Bank | Cat #AB_528428; RRID: AB_528428 |
| Rabbit Anti-Laminin antibody | Millipore Sigma | Cat #AB2034; RRID: AB_91209 |
| Goat Anti-mouse IgG1, Alexa Fluor 488 | Thermo Fisher Scientific | Cat #A-21121; RRID: AB_2535764 |
| Goat Anti-rabbit IgG (H+L), Alexa Fluor 647 | Thermo Fisher Scientific | Cat #A-21244; RRID: AB_2535812 |
| Mouse on Mouse (M.O.M.™) Basic Kit | Vector Laboratories | Cat #BMK-2202; RRID: AB_2336833 |
| Streptavidin-PE-Cy7 | BD Biosciences | Cat #557598; RRID: AB_I0049577 |
| CD45-ePluor450 | Thermo Fisher Scientific | Cat #48-0451-82; RRID: AB_1518806 |
| CD140a (PDGPRa) -biotin | Thermo Fisher Scientific | Cat #13-1401-82; RRID: AB_466607 |
| CD24-PITC | Thermo Fisher Scientific | Cat #11-0242-82; RRID: AB_464988 |
| Sca-1-PerCP-Cy5.5 | Thermo Fisher Scientific | Cat #45-5981-82; RRID: AB_914372 |
| Goat anti-mouse CD140a (PDGPRa) | R&D systems | Cat #AP1062-SP; RRID: AB_2236897 |
| Rabbit anti-RPP | Rockland | Cat #600-401-379; RRID: AB_2209751 |
| Alexa Fluor 488 Affinipure Donkey anti-goat IgG | Jackson Immunoresearch | Cat #705-545-147; RRID: AB_2336933 |
| Cy ™ 3 Affinipure Donkey anti-rabbit IgG | Jackson Immunoresearch | Cat #711-165-152; RRID: AB_2307443 |
| 4’,6-diamino-2-phenyNndole (DAPI) | Thermo Fisher Scientific | Cat #D1306; RRID: AB_2629482 |
| SYTOX Blue Dead Cell Stain, for flow cytometry | Thermo Fisher Scientific | Cat #S34857 |
| 7-AAD | BD Biosciences | Cat #559925 |
| Streptavidin, Pacific Orange conjugate | Thermo Fisher Scientific | Cat #S32365 |
| Anti-APC Microbeads | Miltenyi Biotec | Cat#120-001-265 |
| Wheat Germ Agglutinin, Alexa Fluor 488 Conjugate | Thermo Fisher Scientific | Cat#W11261 |
| Propidium Iodide | Millipore Sigma | Cat#P4170-25MG |
| Calcein Blue, AM | Thermo Fisher Scientific | Cat#C1429 |
| eBioscience™ Streptavidin-APC | Thermo Fisher Scientific | Cat#17-4317-82 |
| Bacterial and Virus Strains | ||
| ElectroMAX™ Stbl4™ Competent Cells | Thermo Fisher Scientific | Cat#11635018 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| HBSS, calcium, magnesium, no phenol red | Thermo Fisher Scientific | Cat#14025-134 |
| PBS (for cell culture) | GE Healthcare (Hyclone) | Cat#SH30071.03 |
| PBS (for staining media) | Millipore Sigma | Cat#P6178-500mL |
| Dulbecco’s Phosphate-Buffered Saline (DPBS), no calcium, no magnesium | Thermo Fisher Scientific | Cat#14190-250 |
| Collagen I Rat Protein | Thermo Fisher Scientific | Cat#A1048301 |
| Natural Mouse Laminin, 1mg | Thermo Fisher Scientific | Cat#23017015 |
| PGP-Basic, recombinant | Millipore Sigma | Cat #P0291 |
| DMEM, high glucose | Thermo Fisher Scientific | Cat#11965118 |
| Ham’s P10 Nutrient Mix, 500ml bottle | Thermo Fisher Scientific | Cat#11550043 |
| Donor Horse Serum | Atlanta Biologicals | Cat#S12150 |
| Penicillin-Streptomycin | Thermo Fisher Scientific | Cat#15140122 |
| GlutaMAX Supplement | Thermo Fisher Scientific | Cat#35050061 |
| Dexamethasone | Millipore Sigma | Cat#D1756 |
| Insulin solution from bovine pancreas | Millipore Sigma | Cat#I0516 |
| Rosiglitazone | Cayman Chemical | Cat#71740 |
| 3-isobutyl-1-methylxanthine | Millipore Sigma | Cat #I5879 |
| Paraformaldehyde, 32% solution | VWR | Cat#100496-496 |
| Triton X-100 | Millipore Sigma | Cat #T9284 |
| Triton X-100 | Thermo Fisher Scientific | Cat#BP151 |
| Normal Goat Serum, Jackson Immuno | VWR | Cat#102643-594 |
| Donkey Serum | Millipore Sigma | Cat #D9663 |
| Bovine Serum Albumin (BSA) | Millipore Sigma | Cat #A9647 |
| Bovine Serum Albumin (BSA) | Millipore Sigma | Cat #A7030 |
| Gelatin from cold water fish skin | Millipore Sigma | Cat #G7765 |
| Tween 20 | Millipore Sigma | Cat #P1379 |
| BODIPY™ 493/503 | Thermo Fisher Scientific | Cat #D3922 |
| Hoechst 33342, Trihydrochloride, Trihydrate | Thermo Fisher Scientific | Cat #H1399 |
| Cardiotoxin gamma from Naja pallida | Latoxin | Cat #L8102 |
| EDTA (0.5M), pH 8.0 | Thermo Fisher Scientific | Cat #AM9261 |
| Dextran | Millipore Sigma | Cat #31392 |
| ACK Lysing Buffer | Thermo Fisher Scientific | Cat #A1049201 |
| Iodixanol | Axis-Shield PoC AS | Cat #AXS-1114542-5 |
| Trizol LS | Thermo Fisher Scientific | Cat #1029610 |
| Isopentane (2-Methylbutane) | Millipore Sigma | Cat #M32631 |
| Paraformaldehyde (PPA), 32% solution, EM grade | VWR | Cat #100496-496 |
| Vectashield HardSet Antifade Mounting Medium with DAPI | Vector Laboratories | Cat #H-1200 |
| Critical Commercial Assays | ||
| SuperScript IV VILO Master Mix with ezDNase Enzyme | Thermo Fisher Scientific | Cat #11766050 |
| Pax7 TaqMan assay | Thermo Fisher Scientific | Mm01354484_m1 |
| Myf5 TaqMan assay | Thermo Fisher Scientific | Mm00435125_m1 |
| Myod1 TaqMan assay | Thermo Fisher Scientific | Mm00440387_m1 |
| Myog TaqMan assay | Thermo Fisher Scientific | Mm00446194_m1 |
| Gapdh TaqMan assay | Thermo Fisher Scientific | Mm99999915_g1 |
| TaqMan Past Advanced Master Mix | Thermo Fisher Scientific | Cat #4444557 |
| Experimental Models: Cell Lines | ||
| HEK293 | ATCC | Cat #CRL-1573; RRID: CVCL_0045 |
| Experimental Models: Organisms/Strains | ||
| Mouse: C57B/10ScSn-Dmdmdx/J | The Jackson Laboratory | JAX: 001801; RRID: IMSR_JAX:001801 |
| Mouse: B6;129S6-Gt(ROSA)26Sortm9 (CAG-tdTomato)Hze/J | The Jackson Laboratory | JAX: 007905; RRID: IMSR_JAX:007905 |
| Mouse: B6.Cg-Tg(Pax7-ZsGreen)1Kyba/J | The Jackson Laboratory | JAX: 029549; RRID: IMSR_JAX:029549 |
| Mouse: B6.SJL-Ptprca Pepcb/BoyJ | The Jackson Laboratory | JAX: 002014; RRID: IMSR_JAX:002014 |
| Mouse: C57BL/6J | The Jackson Laboratory | JAX: 000664; RRID: IMSR_JAX:000664 |
| Oligonucleotides | ||
| Pax7_zsGreen_F; CTGCATGTACCACGAGTCCA | N/A | |
| Pax7_zsGreen_R; GTCAGGTGCCACTTCTGGTT | N/A | |
| tdTomato_WT_F; AAGGGAGCTGCAGTGGAGTA | N/A | |
| tdTomato_WT_R; CCGAAAATCTGTGGGAAGTC | N/A | |
| tdTomato_Knock-in_F; CTGTTCCTGTACGGCATGG | N/A | |
| tdTomato_Knock-in_R; GGCATTAAAGCAGCGTATCC | N/A | |
| Recombinant DNA | ||
| Package plasmid (pAAV2/Anc80) L65AAP) | Addgene | Cat #92307 |
| Transgene plasmid (pAAV Cre) | GTVC | In house production |
| pAAV-Cre control plasmid | Cell Biolabs | AAV-401 |
| Helper plasmid (ΔP6) | GTVC | In house production |
| Package plasmid (pKAAV2/8, pKAAV2/9) | GTVC | In house production |
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
| Software and Algorithms | ||
| BD PACSDiva™ Software v8.0.2 | BD Biosciences |
http://www.bdbiosciences.com/us/instruments/research/software/flow-cytometry-acquisition/bd-facsdiva-software/m/111112/overview
; RRID: SCR_001456 |
| FlowJo v10.1r5 | FlowJo |
https://www.flowjo.com/solutions/flowjo; RRID: SCR_008520 |
| Other | ||
| Covidien Monoject Softpack Insulin Syringe, 1/2mL, 28Gx 1/2” | Covidien | 1188528012 |
| Equisul-SDT® (Sulfadiazine Trimethoprim) | Aurora Pharmaceutical | Cat #28002 |
| Irradiated PicoLab Mouse Diet 20 5058 | LabDiet, St. Louis, MO | Cat #0007689 |
| Irradiated LabDiet Prolab Isopro RMH 3000 5P75 | LabDiet, St. Louis, MO | Cat #0006972 |
| Tissue-Tek® O.C.T. Compound, Sakura® Pinetek | VWR | 25608-930 |
Highlights.
Multiple AAV serotypes transduce tissue stem cells via systemic or local delivery
AAV-delivered Cre recombinase modifies stem cells in multiple anatomical niches
In vivo AAV transduction does not require stem cell isolation or transplantation
AAV-transduced stem cells retain differentiation and engraftment capacities
ACKNOWLEDGMENTS
This work was supported in part by awards and grants from Harvard University (Star Family Challenge Award, Dean’s Initiative Fund, and Harvard Stem Cell Institute Blood Program Pilot Award), the New York Stem Cell Foundation, and NIH grants R01 HL135287 and DP1 AG048917 (to A.J.W.); a Harvard Stem Cell Institute Junior Faculty Award, the Pew Charitable Trusts, a Smith Family Foundation Odyssey Award, the American Cancer Society, and NIH grant R01 AR070825 (to Y.-C.H.); Lonza Houston (to L.H.V.); Editas Medicine, Inc.; NIH postdoctoral fellowship F32AG050395 (to J.M.G.); a St. Baldrick’s Foundation Scholar Award and a Damon Runyon-Sohn Foundation Pediatric Cancer Research Fellowship (to L.D.W.); and a Helen Hay Whitney Foundation postdoctoral fellowship and an award of the Weizmann Institute of Science National Postdoc Award Program for Advancing Women in Science (to Y.S.). We thank J. LaVecchio and S. Ionescu for assistance with FACS, R. Bronson for liver histology analysis, and M. Fernandez-Alfara, C. Unzu, C. Rios, and A. Almada for technical assistance.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2019.03.105.
DECLARATION OF INTERESTS
C.A. is an employee of Editas Medicine, Inc., and J.K.W.C. and C.A. are stock owners of Editas Medicine, Inc. L.H.V. holds equity in and chairs the scientific advisory board of Akouos, a gene therapy company focused on hearing disorders. L.H.V. is also an inventor and has a royalty interest in various AAV technologies, including AAV9 and ancestral AAVs (AncAAVs), which have been licensed to several companies. In addition, L.H.V. is a consultant to Nightstar Therapeutics, Cobalt, Lonza Houston, Exonics, and Selecta Biosciences and has received travel reimbursement from AveXis (now Novartis). L.D.W. holds equity in and is a consultant to Magenta Therapeutics, M.T. is a consultant to Solid Biosciences, and A.J.W. is a consultant to Frequency Therapeutics. J.M.G., M.T., K.Z., L.D.W., M.G.-C., Y.-C.H., and A.J.W. are inventors on patent applications filed through Harvard University regarding in vivo transduction of stem cells by AAVs. L.H.V. receives research funding from Lonza Houston, Oxford Biomedica, Selecta Biosciences, and Solid Biosciences. A.J.W. is a co-founder of Elevian, a company that aims to develop medicines to restore regenerative capacity, in addition to being a recipient of sponsored research support, a member of the scientific advisory board, and holding private equity in the company.
REFERENCES
- Amoasii L, Hildyard JCW, Li H, Sanchez-Ortiz E, Mireault A, Caballero D, Harron R, Stathopoulou TR, Massey C, Shelton JM, et al. (2018). Gene editing restores dystrophin expression in a canine model of Duchenne muscular dystrophy. Science 362, 86–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnett AL, Konieczny P, Ramos JN, Hall J, Odom G, Yablonka-Reuveni Z, Chamberlain JR, and Chamberlain JS (2014). Adeno-associated viral (AAV) vectors do not efficiently target muscle satellite cells. Mol. Ther. Methods Clin. Dev 1, 14038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arpke RW, Darabi R, Mader TL, Zhang Y, Toyama A, Lonetree CL, Nash N, Lowe DA, Perlingeiro RC, and Kyba M (2013). A new immuno-, dystrophin-deficient model, the NSG-mdx(4Cv) mouse, provides evidence for functional improvement following allogeneic satellite cell transplantation. Stem Cells 31, 1611–1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bengtsson NE, Hall JK, Odom GL, Phelps MP, Andrus CR, Hawkins RD, Hauschka SD, Chamberlain JR, and Chamberlain JS (2017). Muscle-specific CRISPR/Cas9 dystrophin gene editing ameliorates patho-physiology in a mouse model for Duchenne muscular dystrophy. Nat. Commun 8, 14454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blankinship MJ, Gregorevic P, Allen JM, Harper SQ, Harper H, Halbert CL, Miller AD, and Chamberlain JS (2004). Efficient transduction of skeletal muscle using vectors based on adeno-associated virus serotype 6. Mol. Ther 10, 671–678. [DOI] [PubMed] [Google Scholar]
- Bosnakovski D, Xu Z, Li W, Thet S, Cleaver O, Perlingeiro RC, and Kyba M (2008). Prospective isolation of skeletal muscle stem cells with a Pax7 reporter. Stem Cells 26, 3194–3204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busch K, Klapproth K, Barile M, Flossdorf M, Holland-Letz T, Schlenner SM, Reth M, Höfer T, and Rodewald HR (2015). Fundamental properties of unperturbed haematopoiesis from stem cells in vivo. Nature 518, 542–546. [DOI] [PubMed] [Google Scholar]
- Cerletti M, Jurga S, Witczak CA, Hirshman MF, Shadrach JL, Goodyear LJ, and Wagers AJ (2008). Highly efficient, functional engraftment of skeletal muscle stem cells in dystrophic muscles. Cell 134, 37–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamberlain JR, and Chamberlain JS (2017). Progress toward Gene Therapy for Duchenne Muscular Dystrophy. Mol. Ther 25, 1125–1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang NC, Chevalier FP, and Rudnicki MA (2016). Satellite Cells in Muscular Dystrophy—Lost in Polarity. Trends Mol. Med 22, 479–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colella P, Ronzitti G, and Mingozzi F (2017). Emerging Issues in AAV-Mediated In Vivo Gene Therapy. Mol. Ther. Methods Clin. Dev 8, 87–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins CA, Kretzschmar K, and Watt FM (2011). Reprogramming adult dermis to a neonatal state through epidermal activation of b-catenin. Development 138, 5189–5199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Driskell RR, Jahoda CA, Chuong CM, Watt FM, and Horsley V (2014). Defining dermal adipose tissue. Exp. Dermatol 23, 629–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filareto A, Rinaldi F, Arpke RW, Darabi R, Belanto JJ, Toso EA, Miller AZ, Ervasti JM, McIvor RS, Kyba M, and Perlingeiro RC (2015). Pax3-induced expansion enables the genetic correction of dystrophic satellite cells. Skelet. Muscle 5, 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heredia JE, Mukundan L, Chen FM, Mueller AA, Deo RC, Locksley RM, Rando TA, and Chawla A (2013). Type 2 innate signals stimulate fibro/adipogenic progenitors to facilitate muscle regeneration. Cell 153, 376–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hettmer S, Liu J, Miller CM, Lindsay MC, Sparks CA, Guertin DA, Bronson RT, Langenau DM, and Wagers AJ (2011). Sarcomas induced in discrete subsets of prospectively isolated skeletal muscle cells. Proc. Natl. Acad. Sci. USA 108, 20002–20007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinderer C, Katz N, Buza EL, Dyer C, Goode T, Bell P, Richman LK, and Wilson JM (2018). Severe Toxicity in Nonhuman Primates and Piglets Following High-Dose Intravenous Administration of an Adeno-Associated Virus Vector Expressing Human SMN. Hum. Gene Ther 29, 285–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joe AW, Yi L, Natarajan A, Le Grand F, So L, Wang J, Rudnicki MA, and Rossi FM (2010). Muscle injury activates resident fibro/adipogenic progenitors that facilitate myogenesis. Nat. Cell Biol. 12, 153–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiel MJ, Yilmaz OH, Iwashita T, Yilmaz OH, Terhorst C, and Morrison SJ (2005). SLAM family receptors distinguish hematopoietic stem and progenitor cells and reveal endothelial niches for stem cells. Cell 121, 1109–1121. [DOI] [PubMed] [Google Scholar]
- Kopinke D, Roberson EC, and Reiter JF (2017). Ciliary Hedgehog Signaling Restricts Injury-Induced Adipogenesis. Cell 170, 340–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lees-Shepard JB, Yamamoto M, Biswas AA, Stoessel SJ, Nicholas SE, Cogswell CA, Devarakonda PM, Schneider MJ Jr., Cummins SM, Legendre NP, et al. (2018). Activin-dependent signaling in fibro/adipogenic progenitors causes fibrodysplasia ossificans progressiva. Nat. Commun 9, 471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lock M, Alvira M, Vandenberghe LH, Samanta A, Toelen J, Debyser Z, and Wilson JM (2010). Rapid, simple, and versatile manufacturing of recom binant adeno-associated viral vectors at scale. Hum. Gene Ther. 21, 1259–1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long C, Amoasii L, Mireault AA, McAnally JR, Li H, Sanchez-Ortiz E, Bhattacharyya S, Shelton JM, Bassel-Duby R, and Olson EN (2016). Postnatal genome editing partially restores dystrophin expression in a mouse model of muscular dystrophy. Science 351, 400–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lounev VY, Ramachandran R, Wosczyna MN, Yamamoto M, Maidment AD, Shore EM, Glaser DL, Goldhamer DJ, and Kaplan FS (2009). Identification of progenitor cells that contribute to heterotopic skeletogenesis.J. Bone Joint Surg. Am 91, 652–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y, and Song S (2009). Distinct immune responses to transgene products from rAAV1 and rAAV8 vectors. Proc. Natl. Acad. Sci. USA 106, 17158–17162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madisen L, Zwingman TA, Sunkin SM, Oh SW, Zariwala HA, Gu H, Ng LL, Palmiter RD, Hawrylycz MJ, Jones AR, et al. (2010). A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat. Neurosci 13, 133–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maesner CC, Almada AE, and Wagers AJ (2016). Established cell surface markers efficiently isolate highly overlapping populations of skeletal muscle satellite cells by fluorescence-activated cell sorting. Skelet. Muscle 6, 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendell JR, Al-Zaidy S, Shell R, Arnold WD, Rodino-Klapac LR, Prior TW, Lowes L, Alfano L, Berry K, Church K, et al. (2017). Single-Dose Gene-Replacement Therapy for Spinal Muscular Atrophy. N. Engl. J. Med 377, 1713–1722. [DOI] [PubMed] [Google Scholar]
- Mingozzi F, and High KA (2011). Therapeutic in vivo gene transfer for genetic disease using AAV: progress and challenges. Nat. Rev. Genet 12, 341–355. [DOI] [PubMed] [Google Scholar]
- Morgan RA, Gray D, Lomova A, and Kohn DB (2017). Hematopoietic Stem Cell Gene Therapy: Progress and Lessons Learned. Cell Stem Cell 21, 574–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson CE, Hakim CH, Ousterout DG, Thakore PI, Moreb EA, Castellanos Rivera RM, Madhavan S, Pan X, Ran FA, Yan WX, et al. (2016). In vivo genome editing improves muscle function in a mouse model of Duchenne muscular dystrophy. Science 351, 403–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel SH, O’Hara L, Atanassova N, Smith SE, Curley MK, Rebourcet D, Darbey AL, Gannon AL, Sharpe RM, and Smith LB (2017). Low-dose tamoxifen treatment in juvenile males has long-term adverse effects on the reproductive system: implications for inducible transgenics. Sci. Rep 7, 8991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Randall TD, and Weissman IL (1997). Phenotypic and functional changes induced at the clonal level in hematopoietic stem cells after 5-fluorouracil treatment. Blood 89, 3596–3606. [PubMed] [Google Scholar]
- Roberts EW, Deonarine A, Jones JO, Denton AE, Feig C, Lyons SK, Espeli M, Kraman M, McKenna B, Wells RJ, et al. (2013). Depletion of stromal cells expressing fibroblast activation protein-a from skeletal muscle and bone marrow results in cachexia and anemia. J. Exp. Med 210, 1137–1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawai CM, Babovic S, Upadhaya S, Knapp DJHF, Lavin Y, Lau CM, Goloborodko A, Feng J, Fujisaki J, Ding L, et al. (2016). Hematopoietic Stem Cells Are the Major Source of Multilineage Hematopoiesis in Adult Animals. Immunity 45, 597–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz TJ, Huang TL, Tran TT, Zhang H, Townsend KL, Shadrach JL, Cerletti M, McDougall LE, Giorgadze N, Tchkonia T, et al. (2011). Identification of inducible brown adipocyte progenitors residing in skeletal muscle and white fat. Proc. Natl. Acad. Sci. USA 108, 143–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seale P, Sabourin LA, Girgis-Gabardo A, Mansouri A, Gruss P, and Rudnicki MA (2000). Pax7 is required for the specification of myogenic satellite cells. Cell 102, 777–786. [DOI] [PubMed] [Google Scholar]
- Sherwood RI, Christensen JL, Conboy IM, Conboy MJ, Rando TA, Weissman IL, and Wagers AJ (2004). Isolation of adult mouse myogenic progenitors: functional heterogeneity of cells within and engrafting skeletal muscle. Cell 119, 543–554. [DOI] [PubMed] [Google Scholar]
- Smith LJ, Ul-Hasan T, Carvaines SK, Van Vliet K, Yang E, Wong KK Jr., Agbandje-McKenna M, and Chatterjee S (2014). Gene transfer properties and structural modeling of human stem cell-derived AAV. Mol. Ther 22, 1625–1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun J, Ramos A, Chapman B, Johnnidis JB, Le L, Ho YJ, Klein A, Hofmann O, and Camargo FD (2014). Clonal dynamics of native haematopoiesis. Nature 514, 322–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabebordbar M, Zhu K, Cheng JKW, Chew WL, Widrick JJ, Yan WX, Maesner C, Wu EY, Xiao R, Ran FA, et al. (2016). In vivo gene editing in dystrophic mouse muscle and muscle stem cells. Science 351, 407–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan KY, Eminli S, Hettmer S, Hochedlinger K, and Wagers AJ (2011). Efficient generation of iPS cells from skeletal muscle stem cells. PLoS ONE 6, e26406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uezumi A, Fukada S, Yamamoto N, Takeda S, and Tsuchida K (2010). Mesenchymal progenitors distinct from satellite cells contribute to ectopic fat cell formation in skeletal muscle. Nat. Cell Biol 12, 143–152. [DOI] [PubMed] [Google Scholar]
- Uezumi A, Fukada S, Yamamoto N, Ikemoto-Uezumi M, Nakatani M, Morita M, Yamaguchi A, Yamada H, Nishino I, Hamada Y, and Tsuchida K (2014). Identification and characterization of PDGFRa+ mesenchymal progenitors in human skeletal muscle. Cell Death Dis. 5, e1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagers AJ (2012). The stem cell niche in regenerative medicine. Cell Stem Cell 10, 362–369. [DOI] [PubMed] [Google Scholar]
- Wang Z, Zhu T, Qiao C, Zhou L, Wang B, Zhang J, Chen C, Li J, and Xiao X (2005). Adeno-associated virus serotype 8 efficiently delivers genes to muscle and heart. Nat. Biotechnol 23, 321–328. [DOI] [PubMed] [Google Scholar]
- Yang Y, Wang L, Bell P, McMenamin D, He Z, White J, Yu H, Xu C, Morizono H, Musunuru K, et al. (2016). A dual AAV system enables the Cas9-mediated correction of a metabolic liver disease in newborn mice. Nat. Biotechnol 34, 334–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Tsai PC, Gonzalez-Celeiro M, Chung O, Boumard B, Perdi-goto CN, Ezhkova E, and Hsu YC (2016). Hair follicles’ transit-amplifying cells govern concurrent dermal adipocyte production through Sonic Hedgehog. Genes Dev. 30, 2325–2338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zincarelli C, Soltys S, Rengo G, and Rabinowitz JE (2008). Analysis of AAV serotypes 1–9 mediated gene expression and tropism in mice after systemic injection. Mol. Ther 16, 1073–1080. [DOI] [PubMed] [Google Scholar]
- Zinn E, Pacouret S, Khaychuk V, Turunen HT, Carvalho LS, Andres-Mateos E, Shah S, Shelke R, Maurer AC, Plovie E, et al. (2015). In Silico Reconstruction of the Viral Evolutionary Lineage Yields a Potent Gene Therapy Vector. Cell Rep. 12, 1056–1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.