

Abstract
Objective
The interleukin (IL)23 pathway contributes to IBD pathogenesis and is being actively studied as a therapeutic target in patients with IBD. Unexpected outcomes in these therapeutic trials have highlighted the importance of understanding the cell types and mechanisms through which IL23 regulates immune outcomes. How IL23 regulates macrophage outcomes and the consequences of the IL23R R381Q IBD-protective variant on macrophages are not well defined; macrophages are key players in IBD pathogenesis and inflammation.
Design
We analysed protein and RNA expression, signalling and localisation in human monocyte-derived macrophages (MDMs) through western blot, ELISA, real-time PCR, flow cytometry, immunoprecipitation and microscopy.
Results
IL23R was critical for optimal levels of pattern-recognition receptor (PRR)-induced signalling and cytokines in human MDMs. In contrast to the coreceptor IL12Rβ1, IL23 induced dynamic IL23R cell surface regulation and this required clathrin and dynamin-mediated endocytosis and endocytic recycling-dependent pathways; these pathways were essential for IL23R-mediated outcomes. The IBD-protective IL23R R381Q variant showed distinct outcomes. Relative to IL23R R381, HeLa cells expressing IL23R Q381 showed decreased IL23R recycling and reduced assembly of IL23R Q381 with Janus kinase/signal transducer and activator of transcription pathway members. In MDMs from IL23R Q381 carriers, IL23R accumulated in late endosomes and lysosomes on IL23 treatment and cells demonstrated decreased IL23R- and PRR-induced signalling and cytokines relative to IL23R R381 MDMs.
Conclusion
Macrophage-mediated inflammatory pathways are key contributors to IBD pathogenesis, and we identify an autocrine/paracrine IL23 requirement in PRR-initiated human macrophage outcomes and in human intestinal myeloid cells, establish that IL23R undergoes ligand-induced recycling, define mechanisms regulating IL23R-induced signalling and determine how the IBD-protective IL23R R381Q variant modulates these processes.
INTRODUCTION
The interleukin (IL)23/Th17 pathway is a major driver of intestinal inflammation1,2 and loss-of-function genetic variants in IL23R confer protection from IBD.3 As such, the IL23/Th17 pathway is being actively investigated as a therapeutic target in patients with IBD. Blocking antibodies to the shared IL23/ IL12p40 subunit demonstrated efficacy in phase III studies4 and have been approved for treatment in Crohn’s disease, with studies ongoing for UC. Phase III studies selectively blocking IL23p19 are ongoing following positive phase II outcomes.5,6 However, blocking IL17 directly was ineffective in Crohn’s disease,7 thereby highlighting that despite the contribution of IL23 to Th17 cells, IL17 produced from these cells does not account for the beneficial effects of blocking IL12p40 and/or IL23. Therefore, an active area of interest has been to more clearly define the cell types and mechanisms through which IL23 contributes to inflammation.
IL23R is expressed in multiple cell types, including myeloid cells.1 Studies examining responses to IL23 through IL23R have focused on IL17-producing cells such as Th17 cells, innate lymphoid cells and natural killer cells.1,2,8 Human T cell studies,9,10 including by us,11 found that the IBD12 and immune-disease13,14 protective loss-of-function rs11209026 IL23R polymorphism resulting in an Arg381Gln amino acid change in the cytoplasmic tail leads to a decrease in IL23R-dependent STAT3 and STAT4 activation and cell proliferation, and in circulating Th17 and Tc17 cells. Despite the critical role for myeloid cells in immune-mediated diseases,15 contributions of and mechanisms through which IL23R and the IL23R R381Q variant contribute to myeloid cell functions are unclear. Two studies have observed IL23-induced cytokine secretion from mouse myeloid cells,16,17 with no reports to our knowledge examining IL23R outcomes in human macrophages. Given the importance of macrophages in contributing to inflammation in IBD, and the ability of IBD risk polymorphisms to increase macrophage-mediated inflammation (eg, IRF5, TNFSF15, MAP3K8, PTPN2),18–21 we addressed critical questions regarding IL23R contributions in macrophages. Does IL23R regulate human macrophage outcomes on microbial product exposure? What signalling pathways and mechanisms does IL23R activate in macrophages? How is surface IL23R regulated and what are the mechanisms mediating this regulation? How does the IL23R R381Q protective variant regulate human macrophage outcomes?
In this study, we define key roles for IL23R in human macrophages, identify previously undefined mechanisms mediating these IL23R effects and elucidate how the IBD-protective IL23R R381Q variant modulates these outcomes.
RESULTS
Autocrine IL23 is required for optimal levels of NOD2-induced cytokine secretion in primary human MDMs and human intestinal myeloid cells
Properly regulating pattern-recognition receptor (PRR)-induced signalling and cytokines on encounter with microbial products in macrophages is critical for intestinal immune homeostasis.15 Autocrine/paracrine cytokines can dramatically amplify these outcomes.22,23 To address if autocrine IL23 contributes to PRR-induced outcomes, we examined NOD2, a PRR associated with Crohn’s disease.24 We first established that treating MDMs with the NOD2 ligand MDP induced IL23p19 secretion (figure 1A). We then reduced IL23R expression by siRNA as assessed using two independent methods, western blot (figure 1B) and flow cytometry (figure 1C). IL23R knockdown decreased NOD2-induced secretion of proinflammatory and anti-inflammatory cytokines (figure 1D). Consistently, IL23 treatment of MDMs was sufficient to induce cytokines (figure 1E). Moreover, autocrine/paracrine IL23 was required for optimal levels of cytokine secretion on stimulating multiple PRRs (figure 1F). MDMs were functional following IL23R knockdown as IL10 secretion was intact on dectin receptor stimulation (online supplementary figure 1A), which activates distinct pathways,25 and cell viability was intact (online supplementary figure 1B). Importantly, pathogenic bacteria can induce cytokine secretion from intestinal myeloid cells,26 and we found that autocrine/paracrine IL23 was crucial for this outcome on coculture with the enteric pathogen Salmonella Typhimurium and with AIEC (figure 1G), bacteria which are enriched in the ilea of patients with Crohn’s disease.27 A similar IL23-dependent role was observed for these live bacteria in MDMs (figure 1G). Therefore, autocrine/paracrine IL23 is required for optimal levels of cytokine secretion on stimulation of multiple PRRs in MDMs and in bacteria-exposed intestinal myeloid cells.
Figure 1.

Autocrine/paracrine IL23 is required for optimal levels of pattern-recognition receptor (PRR)-induced cytokines in monocyte-derived macrophages (MDMs). (A) Human MDMs (n=4 donors) were treated with 100 µg/mL MDP for 24 hours. Mean IL23 secretion+SEM. (B–D, F) MDMs were transfected with scrambled or IL23R siRNA. (B) Western blot. GAPDH is shown as a loading control. (C) Representative flow cytometry for one of six donors with mean fluorescence intensity (MFI) shown. Transfected cells were treated with: (D) 100 µg/mL MDP (n=4), or (F) 10 µg/mL Pam3Cys (TLR2), 100 µg/mL polyI:C (TLR3), 0.1 µg/mL lipid A (TLR4), 5 ng/mL flagellin (TLR5), 1 µg/mL CL097 (TLR7), or 10 µg/mL CpG DNA (TLR9) for 24 hours (n=4). (E) MDMs were treated with 10 ng/mL IL23 (n=4; similar results for an additional n=4). (G) Human intestinal myeloid cells (n=6) or MDMs (peripheral) (n=6) were preincubated with 4 µg/mL anti-IL23p19 neutralising antibody for 1 hour, and then cocultured with Salmonella Typhimurium or AIEC at multiplicity of infection (MOI) 10:1 for 24 hours. Similar results were observed for S. Typhimurium in an additional n=6. Mean cytokine secretion+SEM for (D–G). **P<0.01; ***P<0.001; †P<1×10−4; ††P<1×10−5. AIEC, adherent invasive Escherichia coli; GAPDH, glyceraldehyde 3-phosphate dehydrogenase; IL, interleukin; MDP, muramyl dipeptide; scr, scrambled; TNF, tumour necrosis factor; Tx, treatment.
IL23R signalling activates Janus kinase/signal transducer and activator of transcription pathway members which amplify PRR-induced cytokines in MDMs
IL23R activates specific Janus kinase/signal transducer and activator of transcription (JAK/STAT) pathway members in select cell types.9–11,28,29 However, cytokine-induced signalling pathways can vary in different cell types,30 and IL23-induced signalling has not been examined in macrophages. We established that optimal levels of JAK pathway activation occurred 1 hour following IL23 treatment (online supplementary figure 2A). IL23 most prominently induced JAK2 and TYK2 activation, with less JAK1 activation, whereas JAK3 was not activated in MDMs (figure 2A). Importantly, optimal levels of NOD2-induced JAK2 and TYK2 activation required autocrine IL23 (online supplementary figure 2B). To determine which JAKs are required for IL23-induced cytokines, we successfully knocked down each JAK member in MDMs (online supplementary figure 2C). Only JAK2 and TYK2 were required for IL23-induced cytokines (figure 2B). JAK1 and JAK3 knockdown reduced interferon gamma- and IL2-induced cytokines, respectively, thereby ensuring efficacy of these JAK member knockdowns (online supplementary figure 2D).
Figure 2.

IL23R-induced Janus kinase/signal transducer and activator of transcription (JAK/STAT) pathways are required for IL23-induced cytokines in monocyte-derived macrophages (MDMs). (A, C) MDMs (n=6 for each) were treated with 10 ng/mL IL23 for 1 hour. Representative flow cytometry with mean fluorescence intensity (MFI) values and summary graphs with fold increase MFI+SEM. Isotype control shown for treated cells. (B, D) MDMs (n=6 for each) were transfected with scrambled or the indicated (B) JAK member or (D) STAT member siRNA. Cells were then treated with 10 ng/ mL IL23 for 24 hours. Mean cytokine secretion+SEM. Significance is compared with scrambled siRNA-transfected, IL23-treated cells. Similar results were seen in an independent n=6 for (C) and (D), and n=4 for (B). *P<0.05; **P<0.01; ***P<0.001; †P<1×10−4; ††P<1×10−5. IL, interleukin; NS, not significant; NT, no treatment; scr, scrambled; TNF, tumour necrosis factor; Tx, treatment.
We next assessed STAT signalling and established that IL23 activated STAT3 most strongly with peak activation at 1 hour (figure 2C, online supplementary figure 2E), but also activated additional STAT members in MDMs (figure 2C). Through effective knockdown (online supplementary figure 2F), we found that each STAT protein was required for optimal levels of IL23-induced cytokines to varying degrees, with STAT3 requirements being most pronounced (figure 2D). Importantly, autocrine/ paracrine IL23 was required for optimal levels of NOD2-induced STAT3 activation (online supplementary figure 2G). With combined JAK2/TYK2 (online supplementary figure 2H) or STAT member (online supplementary figure 2I) knockdown, a residual level of IL23-induced cytokines persisted, indicating that additional signalling pathways may operate in human MDMs. We found that IL23 (online supplementary figure 3A,B) and NOD2-induced autocrine IL23 (online supplementary figure 3C,D) activated mitogen-activated protein kinase (MAPK) and NFκB pathways in MDMs. Further, through effective knockdown (online supplementary figure 3E), we found that these pathways were required for optimal levels of IL23-induced cytokines (online supplementary figure 3F). Therefore, IL23 is required for activation of the JAK2/TYK2, STAT, MAPK and NFκB pathways, and each pathway contributes to optimal levels of IL23-induced cytokines in MDMs.
IL23 mediates dynamic regulation of cell surface IL23R in MDMs
Whether IL23 regulates cell surface IL23R expression and how this affects outcomes has not been examined. On IL23 treatment of MDMs, IL23R surface protein expression increased within 15 min of IL23 treatment, decreased by 30 min, increased at 1 hour and then gradually declined (figure 3A,B). Total cellular IL23R protein levels remained unchanged during this time period as assessed by intracellular flow cytometry (figure 3C) and western blot (figure 3D). The dynamic pattern of cell surface protein regulation was also observed by microscopy (online supplementary figure 4A) and in freshly isolated monocytes (online supplementary figure 4B). Of note is that IL23 did not interfere with staining by the IL23R antibody, as when we transfected FLAG-IL23R into HeLa cells and treated with IL23, the same dynamic IL23R regulation was observed when using antibodies to IL23R or to FLAG (online supplementary figure 4C). These studies also demonstrate that the dynamic cell surface protein regulation of IL23R in response to IL23 treatment occurs in epithelial cells. Interestingly, NOD2 stimulation induced a similar IL23R regulation pattern in MDMs (online supplementary figure 4D), which depended on autocrine IL23 (online supplementary figure 4E). TLR4 stimulation and live bacteria (S. Typhimurium, AIEC) also induced this IL23R cell surface protein regulation pattern (online supplementary figure 4F,G).
Figure 3.
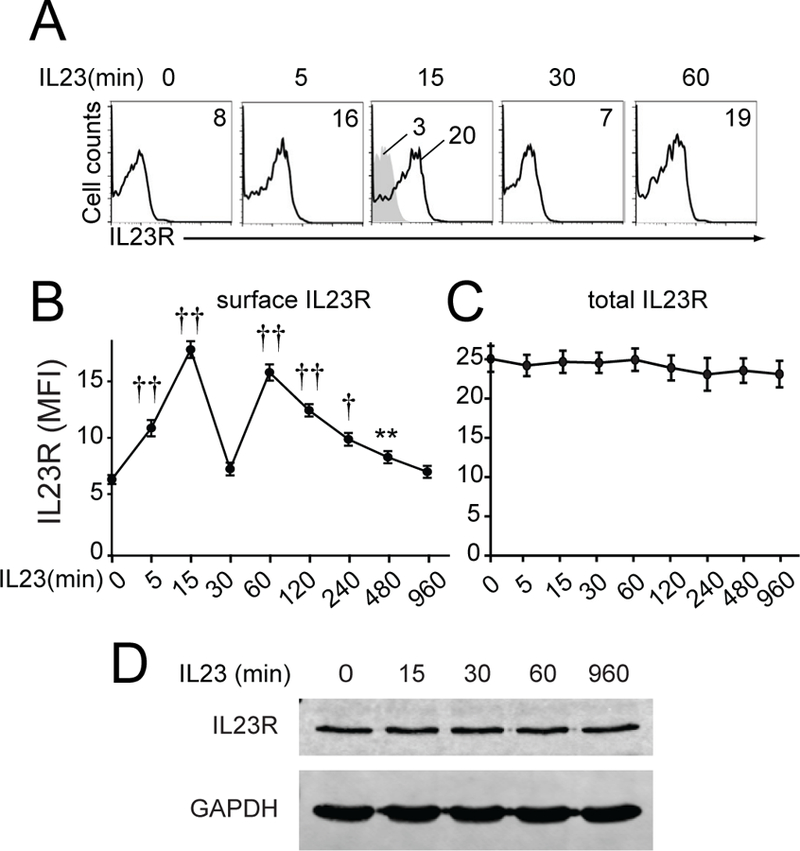
IL23 treatment induces dynamic cell surface regulation of IL23R in monocyte-derived macrophages (MDMs). MDMs were treated with 10 ng/mL IL23. (A) Representative surface flow cytometry for a subset of time points with MFI values. Isotype control for treated cells (shaded histogram). (B) Summary graph with surface MFI±SEM (n=12 pooled from two independent cohorts). (C) Summary graph of intracellular flow cytometry for cellular IL23R MFI±SEM (n=6). (D) Western blot with GAPDH as a loading control. **P<0.01; †P<1×10−4; ††P<1×10−5. GAPDH, glyceraldehyde 3-phosphate dehydrogenase; IL, interleukin; MFI, mean fluorescence intensity.
The increase in cell surface IL23R at 15 min and 1 hour did not require de novo transcription (per actinomycin) or translation (per cycloheximide) (online supplementary figure 5A). The inhibitors did not reduce cell viability (online supplementary figure 5B), but were effective as they decreased IL23-induced cytokines (data not shown). Importantly, the IL12Rβ1 coreceptor was expressed on MDMs but did not recycle with IL23 treatment (online supplementary figure 5C). Moreover, IL12 treatment induced IL12Rβ1 (online supplementary figure 5D) and IL12Rβ2 (online supplementary figure 5E) internalisation, but not recycling, demonstrating selectivity in receptor recycling. Notably, during the 1 hour period of dramatic changes in IL23R surface regulation after IL23 treatment, NOD2-induced cytokines depended on autocrine/paracrine IL23; this dependency decreased by 4 hours and particularly 8 hours after NOD2 stimulation (online supplementary figure 5F). Therefore, IL23 treatment induces dynamic regulation of IL23R surface expression which occurs within a window when autocrine IL23 is most operational.
IL23R surface regulation is dependent on endocytosis and recycling endosomes
We next asked if IL23R surface upregulation requires recycling endosomes. Effective Arf6, Rab11 (endocytic recycling31) and Rab22A (slow recycling endosomes31) knockdown (online supplementary figure 6A; supplementary table 1) demonstrated a requirement for recycling endosomes for cell surface IL23R upregulation at 15 and 60 min (figure 4A). Consistently, IL23R colocalised with Rab11 on IL23 treatment (online supplementary figure 6B,C). We next addressed mechanisms mediating IL23R internalisation, and found that with dynamin and AP2 knockdown (online supplementary figure 6D), molecules which mediate clathrin-dependent endocytosis,31,32 surface IL23R failed to decrease, and in fact increased (figure 4B). Cell viability was intact with these knockdowns (online supplementary figure 6E). We confirmed each of the above mechanisms through an independent approach using inhibitors: brefeldin A (recycling endosomes),33 bafilomycin (acidified endosomes),34 chlorpromazine (clathrin-mediated endocytosis) and dynasore (dynamin-mediated endocytosis) (online supplementary figure 7A–C). The inhibitors did not reduce cell viability (online supplementary figure 7D). Therefore, IL23R surface levels on IL23 treatment likely reflect the balance of both internalisation and recycling.
Figure 4.

Dynamic cell surface regulation of IL23R is dependent on recycling endosomes, and dynamin, clathrin-mediated endocytosis. Monocyte-derived macrophages (MDMs) were transfected with scrambled or the indicated siRNA. (A, B) Cells (n=6) were treated with 10 ng/mL IL23 for the indicated times. Summary graph with MFI of IL23R surface protein expression±SEM. Significance is compared with scrambled siRNA-transfected, IL23-treated cells at the corresponding time point. (C) Cells (n=4) were treated with 10 ng/mL IL23 for 24 hours. Mean cytokine secretion+SEM. **P<0.01; ***P<0.001; †P<1×10−4; ††P<1×10−5 (for Arf6, dynamin and (C)); ♯P<0.001; ‡P<1×10−5 (for Rab11 and AP2); £P<0.001; ǁP<1×10−4; ¶P<1×10−5 (for Rab22A). IL, interleukin; MFI, mean fluorescence intensity; scr, scrambled; TNF, tumour necrosis factor; Tx, treatment.
We next assessed if the identified pathways were required for IL23R-dependent signalling and cytokines. We selected Rab11 to represent endosomal recycling, and dynamin and AP2 to represent dynamin- and clathrin-dependent endocytosis. Each molecule was required for optimal levels of IL23-induced JAK/STAT (online supplementary figure 8A,B), MAPK (online supplementary figure 8C) and NFκB (online supplementary figure 8D) pathway activation and cytokine secretion (figure 4C). Therefore, IL23R cell surface regulation and IL23R-induced signalling and cytokines require recycling endosomes and dynamin- and clathrin-mediated endocytosis.
IL23-induced IL23R recycling corresponds to the dynamic assembly of IL23R signalling intermediates
We next assessed if the observed IL23R cell surface regulation corresponds to a dynamic assembly of signalling intermediates with IL23R. We treated MDMs with IL23, immunoprecipitated IL23R and found that the recruitment of the IL23R coreceptor IL12Rβ1, and of JAK2, TYK2 and STAT3 paralleled the increase and decrease in IL23R surface expression (figure 3A,B) at the respective time points examined (figure 5). Interestingly, we observed IL23 in the complex as well, with a similar dynamic association pattern (figure 5). In contrast, JAK3, which was not activated with IL23 treatment (figure 2A), was not recruited to IL23R (figure 5). Moreover, NOD2 stimulation induced a similar dynamic recruitment pattern of JAK2 to IL23R, and this was dependent on autocrine/paracrine IL23 (online supplementary figure 9), thereby establishing a clear role for IL23 in the recruitment of signalling molecules to IL23R. Therefore, cell surface IL23R recycling is paralleled by the dynamic recruitment of the IL23R signalling complex.
Figure 5.

The recruitment of the IL12Rβ1 coreceptor and signalling intermediates to IL23R corresponds to the kinetics of IL23R surface regulation. Monocyte-derived macrophages (MDMs) were treated with 10 ng/mL IL23 for the indicated times. IL23R was immunoprecipitated and the recruitment of IL12Rβ1 (representative 1 of 5), JAK2 (representative 1 of 6), TYK2 (representative 1 of 6), STAT3 (representative 1 of 5), IL23A (representative 1 of 3) and JAK3 (representative 1 of 2) was assessed by western blot (immunoblotting, IB). Equivalent expression for the respective proteins, along with IL23R and GAPDH (loading controls) in whole cell lysates (WCL). GAPDH, glyceraldehyde 3-phosphate dehydrogenase; IL, interleukin; IP, immunoprecipitation; Tx, treatment.
The IL23R Q381 variant decreases IL23-induced signalling and cytokines
We next assessed how the IBD-protective IL23R R381Q variant regulates IL23-induced outcomes in MDMs. We transfected FLAG-IL23R R381 or Q381 into MDMs from IL23R R381 carriers, so as to be able to distinguish the transfected FLAG-IL23R from endogenous IL23R in pertinent studies. Of note is that we observed similar IL23R cell surface regulation on IL23 treatment of transfected HeLa cells whether the FLAG tag was on the N-terminus or C-terminus of IL23R (online supplementary figure 10A). On IL23 treatment, IL23R R381-transfected MDMs showed increased JAK2, TYK2 (figure 6A), STAT3 (figure 6B), MAPK (online supplementary figure 10B) and NFκB (online supplementary figure 10C) pathway activation, and increased cytokine secretion (figure 6C) relative to empty vector-transfected MDMs. Importantly, relative to IL23R R381-transfected cells, IL23R Q381-transfected MDMs showed a decrease in each of these outcomes (figure 6A–C, online supplementary figure 10B,C). Interestingly, IL23R Q381-transfected MDMs also showed decreased IL23R-mediated outcomes relative to empty vector-transfected IL23R R381 MDMs (figure 6A–C, online supplementary figure 10B,C), suggesting that the IL23R Q381 variant may act as a dominant negative. Moreover, we observed similar differences in IL23R R381 versus IL23R Q381 regulation of signalling and cytokines with both N-terminus and C-terminus FLAG-labelled constructs (online supplementary figure 10D,E). Similar regulation was observed on MDP treatment (online supplementary figure 10F). Therefore, the IL23R Q381 IBD-protective variant results in decreased IL23R-induced signalling and cytokines in MDMs, and in turn, decreased PRR-induced cytokines.
Figure 6.

Compared with IL23R R381, IL23R Q381 leads to reduced cell surface recycling, altered cellular localisation, reduced assembly of the signalling complex and reduced signalling and cytokine secretion on IL23 treatment. (A–D) MDMs were transfected with empty vector (EV), FLAG-IL23R R381 or FLAG-IL23R Q381 vectors. (A, B) Cells (n=6) were treated with 10 ng/mL IL23 for 1 hour. Summary graphs with fold increase mean fluorescence intensity (MFI) of the indicated phosphoproteins+SEM. (C) Cells (n=6, similar results in an additional n=6) were treated with 10 ng/ mL IL23 for 24 hours. Mean cytokine secretion+SEM. (D) Cells (n=6, similar results in an additional n=10) were treated with 10 ng/mL IL23 for the indicated times. Summary graph of cell surface IL23R MFI (as detected by anti-FLAG antibody to detect the transfected IL23R variants)±SEM. (E, F) HeLa cells were transfected with IL12Rβ1, and either FLAG-IL23R R381 or FLAG-IL23R Q381 vectors. (E) Cells were treated with 10 ng/mL IL23 for the indicated times. IL23R was immunoprecipitated with anti-IL23R. IL12Rβ1, JAK2, TYK2 or STAT3 recruitment was assessed by western blot (IB). Equivalent expression for the respective proteins, GAPDH and IL23R (loading controls) in whole cell lysates (WCL). Left: Representative western blots. Right: Summary graphs for densitometry quantification of IL12Rβ1 (10 replicates), JAK2 (10 replicates), TYK2 (14 replicates), STAT3 (10 replicates) and IL23R (7 replicates)±SEM. (F) Cells were treated with 10 ng/mL IL23 for the indicated times and immunostained for IL23R, and Rab11, Rab7, or LysoTracker. Summary graphs+SEM (quantified for 50 cells/condition). Data represent one of two independent experiments. Significance is compared with: (A–C) EV-transfected cells or (D–F) the same condition in IL23R R381-transfected cells. *P<0.05; **P<0.01; ***P<0.001; †P<1×10−4; ††P<1×10−5. GAPDH, glyceraldehyde 3-phosphate dehydrogenase; IB, immunoblotting; IL, interleukin; IP, immunoprecipitation; MDMs, monocyte-derived macrophages; TNF, tumour necrosis factor; Tx, treatment.
IL23R Q381 reduces surface IL23R expression and decreases IL23-induced signalling complex recruitment relative to IL23R R381
We next addressed mechanisms for the reduced IL23R Q381-mediated signalling and cytokine secretion. Transfecting FLAG-IL23R R381 and FLAG-IL23R Q381-expressing vectors into MDMs led to equivalent IL23R baseline expression (figure 6D). However, IL23R Q381 surface protein expression at 15 min after IL23 (figure 6D) or MDP (online supplementary figure 10G) treatment was reduced relative to IL23R R381, and this reduction was particularly pronounced at 1 hour after treatment (figure 6D, online supplementary figure 10G). We therefore sought to assess if the altered IL23R Q381 cell surface regulation corresponds to altered recruitment to IL23R of the signalling intermediates identified in figure 5. To address this, we transfected IL23R into HeLa cells together with the coreceptor IL12Rβ1, which is also not constitutively expressed in HeLa cells. The two IL23R variants underwent IL23-dependent surface regulation similar to that observed in MDMs (online supplementary figure 10H), and the total cellular IL23R was not significantly decreased within the 1 hour period after IL23 treatment (figure 6E). Moreover, IL23-dependent kinetics in recruiting IL12Rβ1 and signalling intermediates to IL23R was similar when transfecting IL23R R381 into HeLa cells (figure 6E) to that observed in MDMs (figure 5). In contrast, IL23R Q381-transfected cells demonstrated decreased recruitment of each molecule as early as 15 min, but particularly 1 hour following IL23 treatment (figure 6E), consistent with the more pronounced decreased surface IL23R Q381 expression at this time.
The decreased IL23R Q381 relative to IL23R R381 surface protein expression might reflect altered trafficking through intracellular compartments. The IL23R Q381 variant showed decreased colocalisation with recycling endosomes (Rab11) at both 30 min and 1 hour after IL23 treatment (figure 6F; online supplementary figure 11A), consistent with lower surface expression of this variant in transfected HeLa cells at these time points (online supplementary figure 10H). We therefore assessed where IL23R Q381 may be localising at these points. We examined both late endosomes (Rab7) and lysosomes (LysoTracker) and observed increased IL23R Q381 colocalisation with both compartments at 30 min and 1 hour relative to the IL23R R381 variant (figure 6F; online supplementary figure 11B,C). While relative to IL23R R381, cellular expression of the IL23R Q381 variant was not decreased at 1 hour (figure 6E), cellular IL23R Q381 did decrease over an 8-hour period after IL23 treatment, consistent with degradative properties of lysosomes (online supplementary figure 10I).
We next assessed if the altered IL23R Q381 expression regulation is also IL23 independent. IL23R Q381 demonstrated decreased maturation and protein stability relative to IL23R R381 by inhibitors in an HEK293 overexpression system.29 However, we did not observe differences in: (1) the degradation rate between the two variants with cycloheximide, which inhibits de novo protein synthesis (online supplementary figure 12A); (2) the synthesis rates between the two variants or in the perceived maturation of the de novo synthesised protein after pulsing the transfected cells with cycloheximide to dramatically diminish IL23R expression (online supplementary figure 12B); or (3) the levels of glycosylated receptor and response to Endo H (N-glycosylated mannans35) between the two variants (online supplementary figure 12C). Therefore, relative to IL23R R381, on IL23 treatment the IL23R Q381 variant shows reduced colocalisation with recycling endosomes with less recycling to the cell surface, which in turn corresponds to decreased recruitment of the IL23R signalling complex. This is accompanied by increased colocalisation with late endosomes/lysosomes by later time points. These differences were not associated with differences in protein synthesis or degradation of the receptor under the non-IL23-treated conditions examined.
MDMs from IBD-protective IL23R R381/Q381 carriers show decreased IL23R and NOD2-induced outcomes relative to IL23R R381/R381 carriers
We next asked if human MDMs from IL23R R381/Q381 (rs11209026 GA) disease-protective carriers showed differential outcomes relative to IL23R R381/R381 (rs11209026 GG) carrier cells. Given the low AA homozygote frequency (1.6/100 individuals per dbSNP), we used cells from GG (WT) homozygotes and GA heterozygotes. GA heterozygotes demonstrate functional modulation in T cell studies.9–11 We first examined how the rs11209026 polymorphism regulates IL23R expression in MDMs. The rs11209026 A allele induced exon 9 skipping resulting in increased production of a soluble IL23R isoform in 293 T cells.36 Expression of full-length IL23R and the three IL23R isoforms identified through National Center for Biotechnology Information peaked 4 hours following IL23 stimulation (online supplementary figure 13A), but this was not modulated by R381Q genotype (online supplementary figure 13B). We next assessed IL23R surface regulation on IL23 treatment in MDMs. Cell surface upregulation at 15 and 60 min following IL23 or MDP treatment was reduced in R381/Q381 heterozygotes relative to R381/R381 homozygotes (figure 7A). Consistent with the transfected cell studies (figure 6F), on IL23 treatment we observed decreased IL23R colocalisation with recycling endosomes (Rab11) and increased colocalisation with late endosomes (Rab7) and lysosomes (LysoTracker) at 30 and 60 min in R381/ Q381 carrier relative to R381/R381 carrier MDMs (figure 7B; online supplementary figure 14). Also consistent with transfected cells (figure 6A,B), we observed reduced IL23R-induced JAK/STAT (figure 7C,D), and MAPK and NFκB pathway signalling (figure 7E,F) in R381/Q381 heterozygotes, with similar outcomes on NOD2 stimulation (online supplementary figure 13C–F). Finally, IL23R R381/Q381 MDMs secreted reduced levels of TNF on stimulation of IL23R (figure 7G) and of multiple PRRs (figure 7H) over several ligand doses. Therefore, IL23R R381/Q381 MDMs show distinct IL23R surface regulation with decreased IL23-induced IL23R expression, and reduced IL23R-and NOD2-initiated signalling and cytokines relative to IL23R R381/R381 MDMs.
Figure 7.

Monocyte-derived macrophages (MDMs) from IL23R R381/Q381 heterozygotes show distinct IL23 and pattern-recognition receptor (PRR)-dependent IL23R cell surface regulation and cellular localisation, decreased signalling and reduced cytokines relative to R381/R381 MDMs. (A) MDMs from R381/R381 or R381/Q381 carriers (n=13/genotype) were left untreated or treated with 10 ng/mL IL23 for the indicated times. Summarised cell surface IL23R MFI±SEM. (B) MDMs from R381/R381 or R381/Q381 carriers (n=10/genotype) were treated with 10 ng/mL IL23 for the indicated times. Cells were immunostained for IL23R, and Rab11, Rab7, or LysoTracker. Summary graphs+SEM (quantified for 25 cells/condition). Significance is compared with IL23R R381/R381 MDMs for each respective condition. (C–F) MDMs (n=15/genotype) were left untreated or treated with 10 ng/ mL IL23 for (C, D) 1 hour or (E, F) 30 min. Summary graphs with fold MFI of the indicated phosphoproteins+SEM. (G, H) MDMs (n=15/genotype) were treated for 24 hours with the indicated doses of: (G) IL23 or (H) MDP, Pam3Cys, polyI:C, lipid A, flagellin, CL097 or CpG. TNF secretion+SEM. *P<0.05; **P<0.01; ***P<0.001; †P<1×10−4; ††P<1×10−5. IL, interleukin; MDP, muramyl dipeptide; MFI, mean fluorescence intensity; TNF, tumour necrosis factor; Tx, treatment.
DISCUSSION
In this study we elucidate a critical role for autocrine/paracrine IL23 in amplifying PRR-induced outcomes relevant for IBD, dissect mechanisms regulating these outcomes and define how the IL23R R381Q disease-protective variant affects IL23R regulation, signalling and cytokines in human macrophages (online supplementary figure 15). In contrast to multiple cytokine receptors, which may undergo endocytosis, but not recycling,37 we observe an IL23-dependent rapid increase in cell surface expression and induction in IL23R recycling. Relative to the common IL23R R381 variant, IL23R Q381 surface expression is decreased after IL23R and PRR stimulation consistent with its reduced co-colocalisation to recycling endosomes and its partial colocalisation with lysosomes at later times.
We identify that on IL23 treatment, IL23R associates in a complex which includes IL12Rβ1, STAT3, JAK2, TYK2 and IL23. This is consistent with a recent study which shows that IL23 binding to IL23R results in a conformational change of IL23R which then recruits IL12Rβ1 and downstream signalling molecules.38 Further, we now find a dynamic pattern for this complex recruitment which parallels the oscillating pattern of IL23R surface protein expression that occurs on IL23 treatment.
Whereas IL17 blockade was ineffective in Crohn’s disease trials,7 phase II studies with antibodies selectively blocking IL23,5–7 and phase III studies with antibodies blocking the shared IL12p40 subunit (ustekinumab)4 demonstrated efficacy, such that ustekinumab has been approved for Crohn’s disease treatment. These contrasting results may in part reflect distinct IL17- and IL23-dependent outcomes in different cell types. Given that we now find that autocrine IL23 is critical for macrophage-mediated proinflammatory responses, macrophages might constitute a beneficial IL23 pathway therapeutic target in IBD patients, in addition to the likely beneficial outcome of targeting IL23 on T cells. As receptor recycling can affect therapeutic outcomes,39,40 understanding IL23R surface regulation might ultimately contribute to strategic design of therapies targeting this pathway. We identify IL23R as critical in signalling and amplification of PRR-induced cytokines in human macrophages, thereby further elucidating how IL23R and the IBD-protective IL23R variant regulate innate immune outcomes directly relevant in intestinal immune homeostasis.
METHODS
Patient recruitment and genotyping
Informed consent was obtained per protocol approved by the Institutional Review Board at Yale University. Healthy individuals were genotyped by TaqMan (Life Technologies, Grand Island, NY).
Myeloid cell culture
Human peripheral blood mononuclear cells (PBMC) were isolated using Ficoll-Paque (GE Dharmacon, Lafayette, CO). Monocytes were purified from PBMCs by positive CD14 selection (Miltenyi Biotec, Auburn, CA) and/or adhesion, tested for purity (>98% by CD11c expression) and differentiated as in ref 41. Intestinal myeloid cells (CD11c purity >75%) were isolated as in ref 42 from resection specimens from uninvolved colon in non-IBD patients undergoing surgery for colon cancer.
Monocyte-derived macrophage stimulation
Monocyte-derived macrophages (MDMs) were treated with muramyl dipeptide (MDP; Bachem, Torrance, CA), lipid A (Peptides International, Louisville, KY), Pam3Cys (EMD Millipore, Billerica, MA), polyI:C, flagellin, CL097, CpG (InvivoGen, San Diego, CA), IL23 (PeproTech), 10:1 multiplicity of infection Salmonella enterica serovar Typhimurium or adherent invasive Escherichia coli (AIEC) strain LF82. In some cases cells were incubated with neutralising anti-IL23p19 antibody (eBioscience, San Diego, CA) for 1 hour before the treatments. Supernatants were assayed for tumour necrosis factor (TNF), IL6, IL8, IL10 (BD Biosciences, San Jose, CA), IL1β or IL23 (coating antibody to IL23p19, clone eBio473P19, detection antibody to IL12/ IL23p40, clone C8.6) (eBioscience) by ELISA.
Transfection of small interfering RNAs and vectors
100 nM scrambled or ON-TARGETplus SMARTpool small interfering RNA (siRNA) against the indicated genes (Dharmacon, Lafayette, CO) (four pooled siRNAs for each gene) was transfected into MDMs using nucleofector (Amaxa, San Diego, CA) for 48 hours. Three micrograms (for MDMs) or 4 µg (for HeLa cells) empty vector (pcDNA3.0), FLAG-IL23R/R381 or FLAG-IL23R/Q381 (generated from IL23R cDNA (OriGene, Rockville, MD)) through mutagenesis (QuikChange Lightning Kit; Agilent Technologies) ±IL12Rβ1 (GeneCopoeia, Rockville, MD) was transfected into MDMs by nucleofector or into HeLa cells by Lipofectamine 2000 (Invitrogen).
Phosphoprotein and total protein detection
Proteins were detected by flow cytometry with Alexa Fluor 488 or phycoerythrin-labelled antibodies to IL23R (FAB41001P, R&D Systems), phospho-JAK1, phospho-JAK2, phospho-JAK3, phospho-TYK2, phospho-STAT1, phospho-STAT2, phospho-STAT3, phospho-STAT5, phospho-STAT6, FLAG (Cell Signaling) or phospho-STAT4 (BD Biosciences).
IL23R was immunoprecipitated using anti-IL23R antibody (Santa Cruz Technology, Santa Cruz, CA) bound to Protein A or Protein G Sepharose (EMD Millipore, Billerica, MA). Associated proteins were blotted with antibodies to JAK2, STAT3 (Cell Signaling), TYK2 (Abcam, Cambridge, MA), IL12Rβ1 (EMD Millipore), JAK3 (Santa Cruz Biotechnology) or IL23A (Proteintech). Control proteins were examined with glyceraldehyde 3-phosphate dehydrogenase or IL23R antibodies (EMD Millipore) as per ref 41.
Microscopy
Cells were fixed in 4% paraformaldehyde, permeabilised and incubated with allophycocyanin-labelled anti-IL23R (FAB14001P, R&D Systems), Rab11, Rab7 (Cell Signaling), or DAPI (Thermo Fisher) and Cy2-labelled secondary antibodies. For LysoTracker (Cell Signaling) cells were stained prior to fixation. Fluorescence microscopy used the Zeiss Axio Observer microscope (Carl Zeiss Microscopy, Thornwood, NY). Colocalisation was quantified using ImageJ.
Statistics
Significance was assessed using two-tailed t-test. To keep cytokines on same axis, a multiplier was applied for IL8 as shown in figure keys. Lines over bars indicate same significance values for these bars.
Supplementary Material
Significance of this study.
What is already known on this subject?
The interleukin (IL)23R R381Q variant is one of the most significant IBD-protective genetic variants.
Therapeutic targeting of the IL23 pathway is currently being examined in IBD trials.
IL23R signalling has been examined primarily in T cells, innate lymphoid cells and natural killer cells, where it contributes to inflammatory outcomes, but its role in macrophages is not well understood.
Significance of this study.
What are the new findings?
Autocrine/paracrine IL23 dramatically amplifies pattern-recognition receptor (PRR)-induced signalling and cytokine secretion in primary human macrophages; macrophages are key contributors to IBD pathogenesis.
IL23 is critical for bacterial pathogen-induced inflammatory cytokine secretion in human intestinal myeloid cells.
IL23 treatment results in recycling of cell surface IL23R; in contrast, the IL12Rβ1 coreceptor is not recycled.
Intact endosomal recycling and dynamin-dependent, clathrin-mediated endocytosis pathways are required for the dynamic IL23R recycling and for optimal levels of IL23R-induced signalling and cytokines.
In primary monocyte-derived macrophages (MDMs), IL23 treatment results in recruitment of IL12Rβ1 and the signalling intermediates JAK2, TYK2 and STAT3 to IL23R in an oscillating manner that parallels the pattern of IL23R recycling.
Relative to IL23R R381, overexpression of IL23R Q381 in HeLa cells results in reduced IL23-induced IL23R cell surface expression and assembly of signalling intermediates.
Relative to IL23R R381 MDMs, on IL23 treatment MDMs from disease-protective IL23R Q381 carriers show decreased IL23R cell surface expression, decreased colocalisation with recycling endosomes and increased colocalisation with late endosomes and lysosomes, as well as decreased IL23R- and PRR-initiated signalling and cytokine secretion
How might it impact on clinical practice in the foreseeable future?
This study identifies a previously undefined critical role for and mechanisms through which IL23 contributes to inflammatory outcomes in macrophages, which provides potential insight into unexpected outcomes in recent clinical trials in patients with IBD and might ultimately improve the therapeutic targeting for this actively investigated pathway.
Acknowledgements
We thank the blood donors for their participation.
Funding The study was funded by the National Institutes of Health; National Institute of Diabetes and Digestive and Kidney Diseases (grant numbers DK062422, DK099097, DK106593 and DKP30-34989).
Footnotes
Competing interests None declared.
Patient consent for publication Not required.
Ethics approval Institutional Review Board at Yale University
Provenance and peer review Not commissioned; externally peer reviewed.
References
- 1.Abraham C, Cho JH. IL-23 and autoimmunity: new insights into the pathogenesis of inflammatory bowel disease. Annu Rev Med 2009;60:97–110. [DOI] [PubMed] [Google Scholar]
- 2.Gaffen SL, Jain R, Garg AV, et al. The IL-23-IL-17 immune axis: from mechanisms to therapeutic testing. Nat Rev Immunol 2014;14:585–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jostins L, Ripke S, Weersma RK, et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature 2012;491:119–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Feagan BG, Sandborn WJ, Gasink C, et al. Ustekinumab as Induction and Maintenance Therapy for Crohn’s Disease. N Engl J Med 2016;375:1946–60. [DOI] [PubMed] [Google Scholar]
- 5.Sands BE, Chen J, Feagan BG, et al. Efficacy and Safety of MEDI2070, an Antibody Against Interleukin 23, in Patients With Moderate to Severe Crohn’s Disease: A Phase 2a Study. Gastroenterology 2017;153:77–86. [DOI] [PubMed] [Google Scholar]
- 6.Feagan BG, Sandborn WJ, D’Haens G, et al. Induction therapy with the selective interleukin-23 inhibitor risankizumab in patients with moderate-to-severe Crohn’s disease: a randomised, double-blind, placebo-controlled phase 2 study. Lancet 2017;389:1699–709. [DOI] [PubMed] [Google Scholar]
- 7.Abraham C, Dulai PS, Vermeire S, et al. Lessons Learned From Trials Targeting Cytokine Pathways in Patients With Inflammatory Bowel Diseases. Gastroenterology 2017;152:374–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Longman RS, Diehl GE, Victorio DA, et al. CX₃CR1⁺ mononuclear phagocytes support colitis-associated innate lymphoid cell production of IL-22. J Exp Med 2014;211:1571–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pidasheva S, Trifari S, Phillips A, et al. Functional studies on the IBD susceptibility gene IL23R implicate reduced receptor function in the protective genetic variant R381Q. PLoS One 2011;6:e25038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Di Meglio P, Di Cesare A, Laggner U, et al. The IL23R R381Q gene variant protects against immune-mediated diseases by impairing IL-23-induced Th17 effector response in humans. PLoS One 2011;6:e17160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sarin R, Wu X, Abraham C. Inflammatory disease protective R381Q IL23 receptor polymorphism results in decreased primary CD4+ and CD8+ human T-cell functional responses. Proc Natl Acad Sci U S A 2011;108:9560–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Duerr RH, Taylor KD, Brant SR, et al. A genome-wide association study identifies IL23R as an inflammatory bowel disease gene. Science 2006;314:1461–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Capon F, Di Meglio P, Szaub J, et al. Sequence variants in the genes for the interleukin-23 receptor (IL23R) and its ligand (IL12B) confer protection against psoriasis. Hum Genet 2007;122:201–6. [DOI] [PubMed] [Google Scholar]
- 14.Rueda B, Orozco G, Raya E, et al. The IL23R Arg381Gln non-synonymous polymorphism confers susceptibility to ankylosing spondylitis. Ann Rheum Dis 2008;67:1451–4. [DOI] [PubMed] [Google Scholar]
- 15.Abraham C, Medzhitov R. Interactions between the host innate immune system and microbes in inflammatory bowel disease. Gastroenterology 2011;140:1729–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Belladonna ML, Renauld JC, Bianchi R, et al. IL-23 and IL-12 have overlapping, but distinct, effects on murine dendritic cells. J Immunol 2002;168:5448–54. [DOI] [PubMed] [Google Scholar]
- 17.Cua DJ, Sherlock J, Chen Y, et al. Interleukin-23 rather than interleukin-12 is the critical cytokine for autoimmune inflammation of the brain. Nature 2003;421:744–8. [DOI] [PubMed] [Google Scholar]
- 18.Hedl M, Abraham C. IRF5 risk polymorphisms contribute to interindividual variance in pattern recognition receptor-mediated cytokine secretion in human monocyte-derived cells. J Immunol 2012;188:5348–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hedl M, Abraham C. A TNFSF15 disease-risk polymorphism increases pattern-recognition receptor-induced signaling through caspase-8-induced IL-1. Proc Natl Acad Sci U S A 2014;111:13451–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Spalinger MR, Manzini R, Hering L, et al. PTPN2 Regulates Inflammasome Activation and Controls Onset of Intestinal Inflammation and Colon Cancer. Cell Rep 2018;22:1835–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hedl M, Abraham C. A TPL2 (MAP3K8) disease-risk polymorphism increases TPL2 expression thereby leading to increased pattern recognition receptor-initiated caspase-1 and caspase-8 activation, signalling and cytokine secretion. Gut 2016;65:1799–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yarilina A, Park-Min KH, Antoniv T, et al. TNF activates an IRF1-dependent autocrine loop leading to sustained expression of chemokines and STAT1-dependent type I interferon-response genes. Nat Immunol 2008;9:378–87. [DOI] [PubMed] [Google Scholar]
- 23.Hedl M, Zheng S, Abraham C. The IL18RAP region disease polymorphism decreases IL-18RAP/IL-18R1/IL-1R1 expression and signaling through innate receptor-initiated pathways. J Immunol 2014;192:5924–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Abraham C, Cho JH, disease Ibowel N Engl J Med Overseas Ed 2009;361:2066–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brown GD, Gordon S. Immune recognition of fungal beta-glucans. Cell Microbiol 2005;7:471–9. [DOI] [PubMed] [Google Scholar]
- 26.Franchi L, Kamada N, Nakamura Y, et al. NLRC4-driven production of IL-1β discriminates between pathogenic and commensal bacteria and promotes host intestinal defense. Nat Immunol 2012;13:449–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Darfeuille-Michaud A, Boudeau J, Bulois P, et al. High prevalence of adherent-invasive Escherichia coli associated with ileal mucosa in Crohn’s disease. Gastroenterology 2004;127:412–21. [DOI] [PubMed] [Google Scholar]
- 28.Parham C, Chirica M, Timans J, et al. A receptor for the heterodimeric cytokine IL-23 is composed of IL-12Rbeta1 and a novel cytokine receptor subunit, IL-23R. J Immunol 2002;168:5699–708. [DOI] [PubMed] [Google Scholar]
- 29.Sivanesan D, Beauchamp C, Quinou C, et al. IL23R (Interleukin 23 Receptor) Variants Protective against Inflammatory Bowel Diseases (IBD) Display Loss of Function due to Impaired Protein Stability and Intracellular Trafficking. J Biol Chem 2016;291:8673–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.van Boxel-Dezaire AH, Rani MR, Stark GR. Complex modulation of cell type-specific signaling in response to type I interferons. Immunity 2006;25:361–72. [DOI] [PubMed] [Google Scholar]
- 31.Grant BD, Donaldson JG. Pathways and mechanisms of endocytic recycling. Nat Rev Mol Cell Biol 2009;10:597–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Le Roy C, Wrana JL. Clathrin- and non-clathrin-mediated endocytic regulation of cell signalling. Nat Rev Mol Cell Biol 2005;6:112–26. [DOI] [PubMed] [Google Scholar]
- 33.Lippincott-Schwartz J, Yuan L, Tipper C, et al. Brefeldin A’s effects on endosomes, lysosomes, and the TGN suggest a general mechanism for regulating organelle structure and membrane traffic. Cell 1991;67:601–16. [DOI] [PubMed] [Google Scholar]
- 34.Yoshimori T, Yamamoto A, Moriyama Y, et al. Bafilomycin A1, a specific inhibitor of vacuolar-type H(+)-ATPase, inhibits acidification and protein degradation in lysosomes of cultured cells. J Biol Chem 1991;266:17707–12. [PubMed] [Google Scholar]
- 35.Hebert DN, Garman SC, Molinari M. The glycan code of the endoplasmic reticulum: asparagine-linked carbohydrates as protein maturation and quality-control tags. Trends Cell Biol 2005;15:364–70. [DOI] [PubMed] [Google Scholar]
- 36.Yu RY, Brazaitis J, Gallagher G. The human IL-23 receptor rs11209026 A allele promotes the expression of a soluble IL-23R-encoding mRNA species. J Immunol 2015;194:1062–8. [DOI] [PubMed] [Google Scholar]
- 37.Cendrowski J, Mamińska A, Miaczynska M. Endocytic regulation of cytokine receptor signaling. Cytokine Growth Factor Rev 2016;32:63–73. [DOI] [PubMed] [Google Scholar]
- 38.Bloch Y, Bouchareychas L, Merceron R, et al. Structural activation of pro-inflammatory Human Cytokine IL-23 by Cognate IL-23 Receptor enables recruitment of the shared receptor IL-12Rβ1. Immunity 2018;48:45–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Umanah GKE, Pignatelli M, Yin X, et al. Thorase variants are associated with defects in glutamatergic neurotransmission that can be rescued by Perampanel. Sci Transl Med 2017;9:eaah4985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ahrens-Nicklas RC, Umanah GK, Sondheimer N, et al. Precision therapy for a new disorder of AMPA receptor recycling due to mutations in ATAD1. Neurol Genet 2017;3:e130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hedl M, Yan J, Abraham C. IRF5 and IRF5 Disease-risk variants increase glycolysis and human m1 macrophage polarization by regulating proximal signaling and Akt2 activation. Cell Rep 2016;16:2442–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hedl M, Lahiri A, Ning K, et al. Pattern recognition receptor signaling in human dendritic cells is enhanced by ICOS ligand and modulated by the Crohn’s disease ICOSLG risk allele. Immunity 2014;40:734–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.