

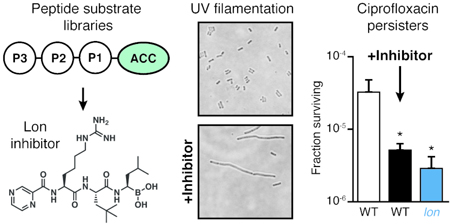
Abstract
Lon is a widely-conserved housekeeping protease found in all domains of life. Bacterial Lon is involved in the recovery from various types of stress, including tolerance to fluoroquinolone antibiotics, and is linked to pathogenesis in a number of organisms. However, detailed functional studies of Lon have been limited by the lack of selective, cell-permeant inhibitors. Here we describe the use of positional scanning libraries of hybrid peptide substrates to profile the primary sequence specificity of bacterial Lon. In addition to identifying optimal natural amino acid binding preferences, we identified several non-natural residues that were leveraged to develop optimal peptide substrates as well as a potent peptidic boronic acid inhibitor of Lon. Treatment of Escherichia coli with this inhibitor promotes UV-induced filamentation and reduces tolerance to ciprofloxacin, phenocopying established lon-deletion phenotypes. It is also non-toxic to mammalian cells due to its selectivity for Lon over the proteasome. Our results provide new insight into the primary substrate specificity of Lon and identify substrates and an inhibitor that will serve as useful tools for dissecting the diverse cellular functions of Lon.
Graphical Abstract
INTRODUCTION
Lon is a widely-conserved housekeeping protease, found in bacteria, archaea, and eukaryotic mitochondria and chloroplasts1. All Lon orthologs feature a AAA+ ATPase domain that unfolds protein substrates and a proteolytic domain that catalyzes the hydrolysis of those substrates2. The importance of bacterial Lon has been determined mostly through studies using Escherichia coli lon mutants and via biochemical analyses of recombinant enzyme. Lon has myriad regulatory functions related to stress-response3,4, including roles in the SOS response to DNA damage5, defense against reactive oxygen species6, heat shock7, amino acid starvation8, and phage integration9. Phenotypic consequences of lon deletion include the inability to recover normally from UV-induced DNA damage and the reduced persistence of lon mutants following fluoroquinolone treatment10,11. In the context of pathogenesis, lon mutants of many bacteria are defective for infection. These include Pseudomonas aeruginosa in lung infection models of mice and rats12, Salmonella enterica in macrophages and systemic infection of mice13, and Brucella abortis in macrophages and spleen infections of mice14.
Due to its roles in stress-response, Lon is an interesting target for small-molecule inhibition. A selective inhibitor would enable dynamic studies of Lon proteolysis in a variety of physiological contexts and, based on the links between Lon and pathogenesis, has the potential to be useful as a therapeutic agent15. Furthermore, specific inhibition of Lon protease activity would allow separation of its proteolytic functions from those involving chaperone activity or binding of DNA and polyphosphate. For example, controlled inhibition of Lon would be useful for clarifying the role the protease plays in persistence. While a defect in fluoroquinolone tolerance in lon mutants has been established for many years16, there has been substantial debate about the mechanism by which Lon contributes to this phenomenon17,18. A recently-proposed model for the role of Lon in persistence which involves the degradation of toxin-antitoxin modules has since been disproven19–21. The current model involves regulation through degradation of the cell-division inhibitor SulA, the same mechanism by which Lon directs recovery from other sources of DNA damage. According to this model, Lon proteolytic activity would be important primarily when SulA is overexpressed as part of the SOS response. The ability to precisely control Lon inactivation (i.e., by addition of a small-molecule inhibitor) would be critical to test this hypothesis.
While a number of small-molecule Lon inhibitors have been identified, to our knowledge, none have been used to test the consequences of Lon inhibition within live bacterial cells. Lon features a serine-lysine dyad in its active site, notably different from the canonical serine-histidine-aspartic acid found in many serine proteases22,23. A likely consequence of its noncanonical active site is that many broad-spectrum serine protease inhibitors have poor activity against the enzyme. Early studies noted that E. coli Lon could be inhibited by the serine protease inhibitors diisopropyl fluorophosphate24 and dansyl fluoride,25 but only at mM concentrations. Inhibitors with slightly greater potency include 3,4-dichloroisocoumarin, other coumarin derivatives26, and oleanane triterpenoids27. Another class of Lon inhibitors comprises peptidic compounds that couple an amino acid recognition moiety with an electrophilic ‘warhead’ that covalently reacts with the active-site serine to inactivate the enzyme. Examples of such inhibitors with activity for Lon include Z-Gly-Leu-Phe-chloromethylketone28, as well as the human proteasome inhibitors MG13229,30 (featuring an aldehyde warhead), and MG262 and bortezomib (BZ)31,32 (featuring boronic acid warheads). In addition, a larger, hexapeptide boronic acid inhibitor of Lon was generated from the amino acid sequence of the natural λN Lon substrate33. These peptidic inhibitors take advantage of amino acid sequences that are tolerated by Lon, but none have been optimized for the enzyme, nor counter-screened for potential off-target binding or inhibition.
One of the most significant issues for current Lon inhibitors is their high level of cross reactivity with the proteasome. This leads to significant toxicity, making them ineffective as tools to study Lon function in cells. It is striking that many proteasome inhibitors have cross-reactivity with Lon, considering the differences in the active-sites: hydrolysis by the proteasome is catalyzed by an N-terminal threonine. Crystal structures of Meiothermus taiwanensis Lon revealed that the boronic acid warheads of MG262 and BZ bind covalently to the active-site serine, like their covalent modification of the threonine hydroxyl in the proteasome32. This strong covalent reactivity of boronates towards active site hydroxyls explains the dual potency for Lon and the proteasome.
We set out to develop selective inhibitors that could be used to specifically block Lon protease activity in cells. We hypothesized that the identification of highly selective peptide substrates could be leveraged to generate an optimized inhibitor using established electrophilic warheads. This strategy builds on a body of work from our groups and others describing the conversion of peptide substrates to inhibitors and activity-based probes for diverse protease targets34, including caspases35, cathepsins36, human neutrophil elastase37, human neutrophil serine protease 438, both the human39 and Plasmodium40,41 proteasome, and proteases important in Mycobacterium tuberculosis pathogensis42 and Zika virus infection43. By screening a large combinatorial library of peptide substrates, we identified a sequence of amino acids optimized for Lon. Based on this screening data, we designed a peptidic boronic acid inhibitor with potent activity for Lon and reduced potency for the human 20S proteasome. This compound was non-toxic to mammalian macrophages and is able to phenocopy classic lon deletion phenotypes in E. coli. We expect this compound to serve as a tool for studying the role of Lon-mediated proteolysis during stress response and pathogenesis.
RESULTS AND DISCUSSION
The primary sequence specificity of Lon has been examined using individual fluorogenic peptide substrates28 as well as by identifying the cleavage sites for a number of endogenous Lon substrates44–46. However, there has not yet been a comprehensive and unbiased profiling of its amino acid preferences. We therefore performed a screen for fluorogenic peptide substrates using a hybrid combinatorial substrate library (HyCoSuL) that has been successfully applied to other protease targets37, 39, 47. Lon was purified after recombinant expression in E. coli (Figure S1). We chose to use libraries of tetrapeptides in which the P1 residue directly adjacent to the site of hydrolysis was fixed as a phenylalanine in order to ensure recognition by Lon. These libraries are made up of a set of sub-libraries in which 121 natural and non-natural amino acids are scanned through each of the P2 and P3 positions on the substrate (Figure 1a). Cleavage by Lon of each sub-library containing a fixed P2 or P3 residue is used to determine the overall specificity patterns at those positions. Initial analysis of the natural amino acid libraries provides some insight into the potential cleavage sites of native protein substrates. We found that at both the P2 and P3 positions, multiple residues are accepted, suggesting an overall broad specificity of the protease (Figure 1b). At each position, bulky or hydrophobic residues yielded the best substrates. This result is consistent with previous reports of favored peptide substrates that feature Ala, Leu, and Phe residues, and various analyses of the cleavage sites within protein substrates. In addition, these results support the model in which Lon is involved in degrading unfolded hydrophobic domains of endogenous substrates. It should be noted that, while information about peptide substrate preferences may be useful for determining preferred cleavage sites and cleavage rates of natural substrates, data from our peptide library screens cannot be used to determine preferences for protein substrates that are dictated by interactions with other domains of the enzyme (e.g., the substrate recognition domain).
Figure 1. HyCoSuL screening of Lon substrates.
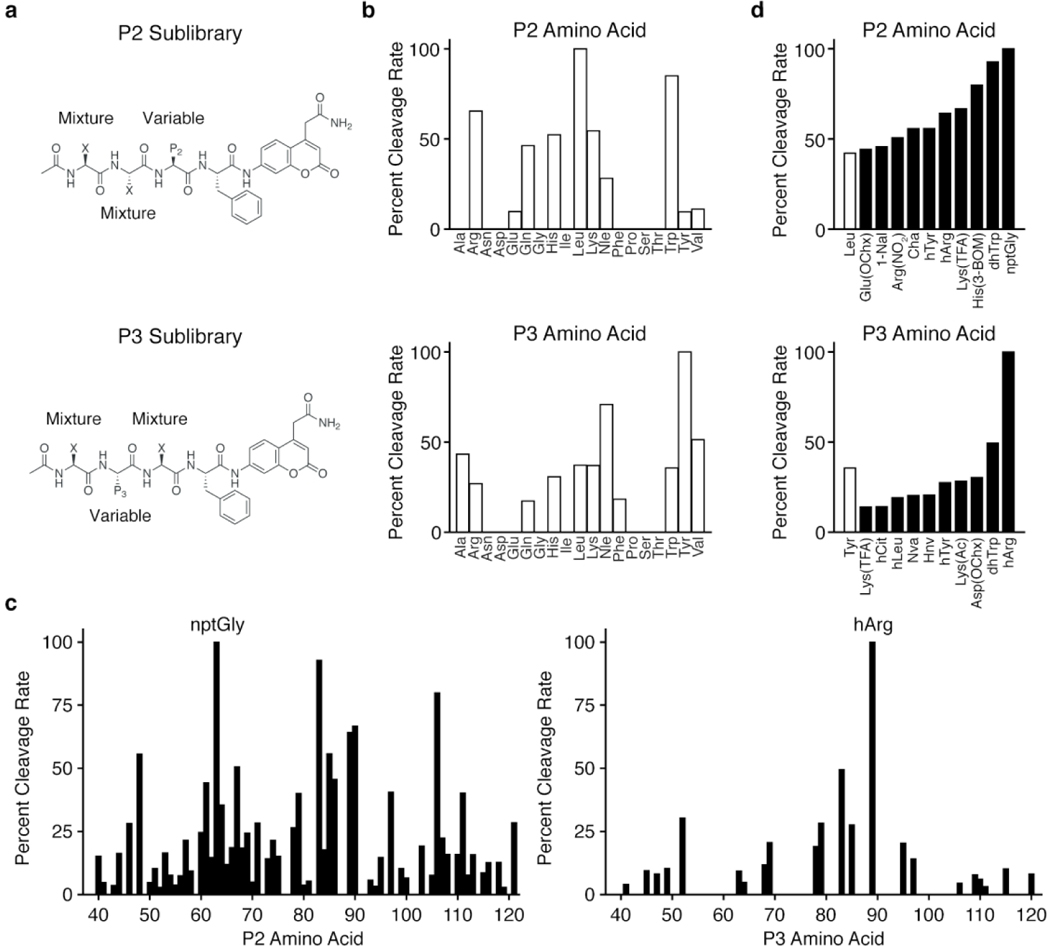
Structures of (a) P2 and P3 fluorogenic HyCoSuL libraries. Positions comprising equimolar mixtures of 18 natural amino acids and norleucine (Nle) as a substitute for Met and Cys (not included in the library) are designated by an X. The remaining variable position is held constant as the indicated natural or non-natural amino acid for each sub-library. Plot of the relative cleavage rates for (b) natural amino acids in the P2 (top) or P3 (bottom) positions and (c) non-natural amino acids in the P2 (left) or P3 (right) positions. (d) Plots of cleavage rates of the best non-natural amino acids for the P2 (top) or P3 (bottom) positions. The best natural amino acid (white bar) at each position is included for comparison. Results for each sub-library were normalized to the amino acid with the fastest cleavage rate, indicated by an arrow, and data represent the mean of two independent screens. Amino acid type is indicated by white (natural) or black (non-natural) bars. D-amino acids (Nos. 20-36) exhibited no cleavage and were excluded from the plot in (c). Amino acid abbreviations and structures are as reported in Kasperkiewicz, et al37. The list of amino acids and normalized cleavage rates can be found in Table S1.
We next performed substrate cleavage analysis using the libraries containing non-natural amino acids to get a broader perspective on the substrate specificity of Lon (Figure 1c–d; Table S1). Interestingly, Lon accepted a diverse array of non-natural amino acids at the P2 position, with more than 50% of the library exhibiting measurable cleavage. In contrast, it was more stringent at the P3 position and showed a strong preference for a single non-natural amino acid, L-homoarginine (hArg). For both positions, the most-preferred amino acids contained bulky side chains. To verify the results of the combinatorial library screening, we generated a set of fluorogenic tri-peptide substrates that contained the newly identified P3 hArg as well as the fixed P1 Phe and a morpholine acetate N-terminal cap. We then varied the P2 position using amino acids selected from the best substrates identified in the substrate screen (Figure 2a). For comparison, we used Mo-Leu-Leu-Phe-ACC (1), a peptide substrate containing only natural amino acids. This substrate yielded kinetic parameters similar to those previously reported for the Lon substrate, Glt-Ala-Ala-Phe-MNA28,48. In contrast, all of the substrates containing the P3 hArg greatly outperformed 1, with specificity constants (kcat/KM) as much as 12-fold higher for the best substrate, 5, which contains a neopentylglycine (nptGly) at the P2 position (Figure 2b, Figure S2a).
Figure 2. Design of selective Lon substrates.
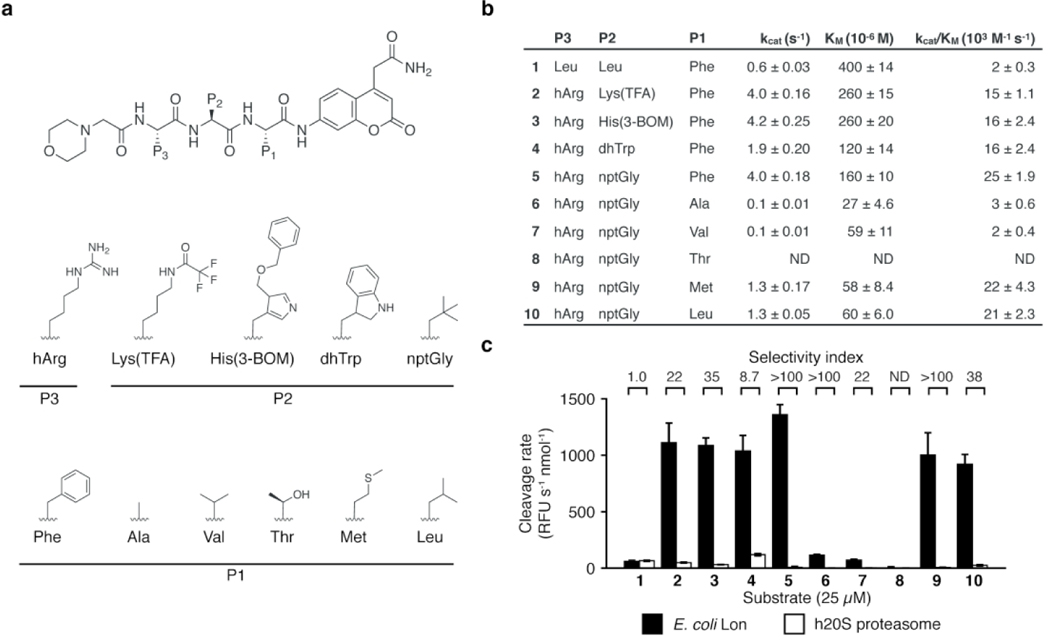
(a) General structure of fluorogenic Lon substrates. Amino acids used in the P3, P2, and P1 positions are shown. (b) Kinetic parameters for cleavage by Lon for each substrate (mean ± standard deviation, n=3). ND, not determined. (c) Cleavage rates for 25 μM of each substrate by Lon (black) and h20S (white) (mean ± standard deviation, n=3). Rates were normalized to total enzyme amount. Selectivity values were calculated as the ratio of cleavage rates. Raw kinetic data are shown in Figure S2.
For many proteases, the P1 position adjacent to the scissile bond is critical for recognition of substrates. To evaluate the importance of this position in combination with the optimized hArg and nptGly residues, we generated a set of substrates featuring P1 amino acids found in endogenous Lon substrates: Ala, Val, Thr, Met, Leu44–46 (6–10, Figure 2a). Substrates with Ala (6), Val (7), and Thr (8) at the P1 position exhibited low cleavage rates while substrates with the bulkier amino acids Met (9) and Leu (10) had catalytic efficiencies similar to 5, with nearly 3-fold lower KM values (Figure 2b, Figure S2a). The decrease in activity observed for some P1 variants highlights the importance of this position for the design of efficient Lon substrates.
Having determined optimal substrates for Lon, we set out to use these scaffolds to build a potent, covalent inhibitor of Lon. The fact that several classes of covalent inhibitors have been reported suggests that the choice of electrophile is important for the optimal inhibitor design. We therefore screened our existing focused library of electrophilic protease inhibitors49 to identify an appropriate electrophile. This set of compounds includes diverse, reactive moieties that form permanent covalent bonds with active-site serine, threonine, or cysteine residues, including diphenyl phosphonates, vinyl sulfones, epoxy ketones, chloroisocoumarins, vinyl ketones, and triazole ureas. To screen this set of ~1,200 compounds we established an in vitro enzyme assay using our optimized fluorogenic peptide substrate 5. Our initial screen at a high concentration (10 μM) of the compounds identified a small number of hits that abolished Lon activity (Figure S3a–b). While we identified hits within all warhead classes, even the most potent compounds from the screen had IC50 values well above that of the human proteasome inhibitor BZ, which has previously been reported as an inhibitor of Lon (Figure S3c). We therefore decided to focus on using the reversible covalent boronic acid electrophile in BZ to make an optimized Lon inhibitor.
We suspected that converting any one of the Lon substrates to a boronic acid would yield a potent inhibitor. Because peptide boronic acids have been shown to be highly effective inhibitors of the human proteasome, counter screening for proteasome inhibition is essential to avoid high toxicity due to this cross-reactivity. To identify peptide scaffolds that would likely yield a selective Lon inhibitor, we evaluated cleavage of the substrates by both Lon and the human 20S proteasome (h20S). These results showed that the non-optimized substrate 1 containing the Leu-Leu-Phe sequence was cleaved equally effectively by both Lon and the proteasome while substrates containing the optimized P3 hArg were primarily cleaved by Lon and not the human proteasome. In fact, cleavage of 2–5 by the proteasome was so weak that it did not saturate and as a result, we were unable to determine kinetic parameters for those substrates (Figure S2b–c). In lieu of kinetic constants, we compared normalized cleavage rates for a fixed substrate concentration (Figure 2c). These results confirmed that substrates 2–10 were selective for Lon, with 5 being the most selective. This substrate showed essentially no detectable cleavage by the h20S. This result is consistent with a HyCoSuL screen of h20S that showed that peptides featuring hArg in the P3 position are poor substrates for the β1 and β5 subunits39.
To leverage the identified substrate specificity of Lon into the design of a selective inhibitor, we synthesized a hybrid compound containing the P1, P2, and P3 positions from substrate 10 combined with the boronic acid warhead and N-terminal pyrazinamide cap of BZ to generate Pyz-hArg-nptGly-Leu-B(OH)2 (11, Figure 3a). Though substrates 5 and 9 exhibited higher selectivity indices, we chose to use Leu at the P1 position (i) for the low KM observed for 10 which indicates tight binding to the Lon active site, (ii) for ease of comparison with BZ which also features a Leu in the P1 position, and (iii) for synthetic simplicity (i.e., Fmoc-Leu-boronate is commercially available). Both 11 and BZ exhibited potent, time-dependent inhibition of recombinant Lon (Figure S4a–b), with IC50 values after 60 min of inhibitor pre-incubation approaching the active-site concentration used in the assay (Figure S4c), suggesting covalent inhibition. Kinetic analyses (Figure 3b–c, Figure S4d–e) showed 11 to be a more potent Lon inhibitor than BZ with a two-fold higher kinact/KI driven primarily by improved potency (i.e., a lower KI value) (Figure 3d). To test for activity toward the human proteasome, we pre-treated purified h20S with each compound and then labeled subunit active sites with the fluorescent, activity-based probe MV151 (Figure 3e)50. Competition for active-site labeling of β1 and β5 subunits of h20S required a 10-fold higher concentration of 11 than BZ (50 vs 5 μM). Inhibition assays using fluorogenic peptides specific for each subunit similarly showed an increase in IC50 values for 11 compared to BZ for the β1 and β5 subunits (Table 1, Figure S4f). Surprisingly, we also saw some inhibition of the β2 subunit by 11, despite the strong preference of this “trypsin-like” subunit for Arg at the P1 position. Together these results confirm that the slight increase in Lon potency of 11 compared to BZ was accompanied by a substantial reduction in binding to the β1 and β5 subunits of the proteasome. More importantly, the Lon inhibitor 11 was not cytotoxic to murine macrophages at doses as high as 10 μM. This is in stark contrast to BZ which kills the same cells with an EC50 of 160 nM (Figure 3f). Thus, the drop in potency of 11 towards the proteasome is sufficient to eliminate the toxicity in mammalian cells and suggests that it should be a valuable new compound for use in cell biological studies of Lon function.
Figure 3. Design of a selective Lon inhibitor.
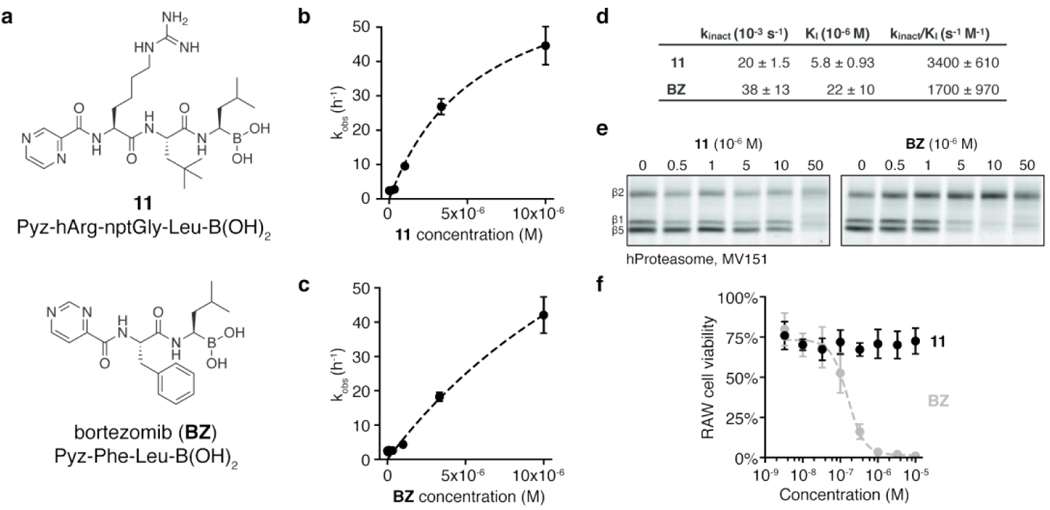
(a) Structures of the designed Lon inhibitor 6 and BZ. Kinetic analysis of time-dependent inhibition of Lon in vitro by (b) 11 and (c) BZ (mean ± standard deviation, n=3). Dashed line indicates the fit used to calculate kinetic parameters. Raw kinetic data are shown in Figure S4. (d) Kinetic parameters of time-dependent inhibition of Lon (mean ± standard deviation, n=3). (e) h20S treated with 11 or BZ, then labeled with MV151. Proteins were separated by SDS-PAGE and scanned for MV151 fluorescence. Images are representative of two independent experiments. (f) Viability of murine RAW macrophages following 24 h treatment with 11 (black) or BZ (gray). Dashed line indicates the fit used to obtain the IC50 for BZ. Viability was quantified by normalizing CellTiter-Blue fluorescence to that of untreated cells (mean ± standard deviation, n=3).
Table 1.
IC50 values and relative activities for inhibitors against bacterial Lon and the human proteasome
| Enzyme (subunit) |
Inhibitor | IC50 (10−9 M)a | Relative activityb |
|---|---|---|---|
| Lon | 11 | 430±140 | 1.2 |
| BZ | 500±230 | ||
| h20S (β1) | 11 | 1900±140 | 0.2 |
| BZ | 380±20 | ||
| h20S (β2) | 11 | 430±40 | ND |
| BZ | ND | ||
| h20S (β5) | 11 | 290±6.4 | 0.1 |
| BZ | 37±7.1 | ||
Measured after 60 min of enzyme-inhibitor pre-incubation (mean ± standard deviation, n=3).
Ratio of BZ IC50 to 11 IC50
Although Lon plays important roles in stress response and pathogenesis, lon is a nonessential gene and deletion mutants grow normally in the absence of exogenous stress. We generated a clean-deletion of lon (Figure S5a–b) and found that neither genetic disruption of lon nor treatment with 100 μM 11 or BZ had an effect on exponential growth rates (Figure S5c). One of the first observed consequences of lon mutation in E. coli was the filamentation of cells after UV-induced DNA damage51. DNA damage causes upregulation of the cell-division inhibitor SulA as part of the SOS response. Lon-mediated degradation of SulA allows cells to resume division after recovery from stress. In the absence of Lon, SulA concentrations remain high and cells grow but cannot divide, resulting in extended filaments. We hypothesized that if 11 was a selective inhibitor of Lon then treatment of E. coli should phenocopy the eponymous “long” filamentation phenotype found in lon cells. As expected, outgrowth following UV-stress resulted in long filaments in the lon deletion strain but not in wild-type or sulA mutant cells (Figure 4a). Treatment with 11 during outgrowth following UV-stress led to a dose-dependent increase in filamentation (Figure 4b, Figure S6a). In a sulA mutant strain, 11 had no effect on filamentation, similar to observations of lon sulA double mutants52. Quantification of cell area for more than 500 cells per condition showed increases in the maximum cell area and in the percent of cells that were filamented (i.e., with area greater than 4 μm2, Figure 4c).
Figure 4. Lon inhibition induces E. coli filamentation following UV stress.
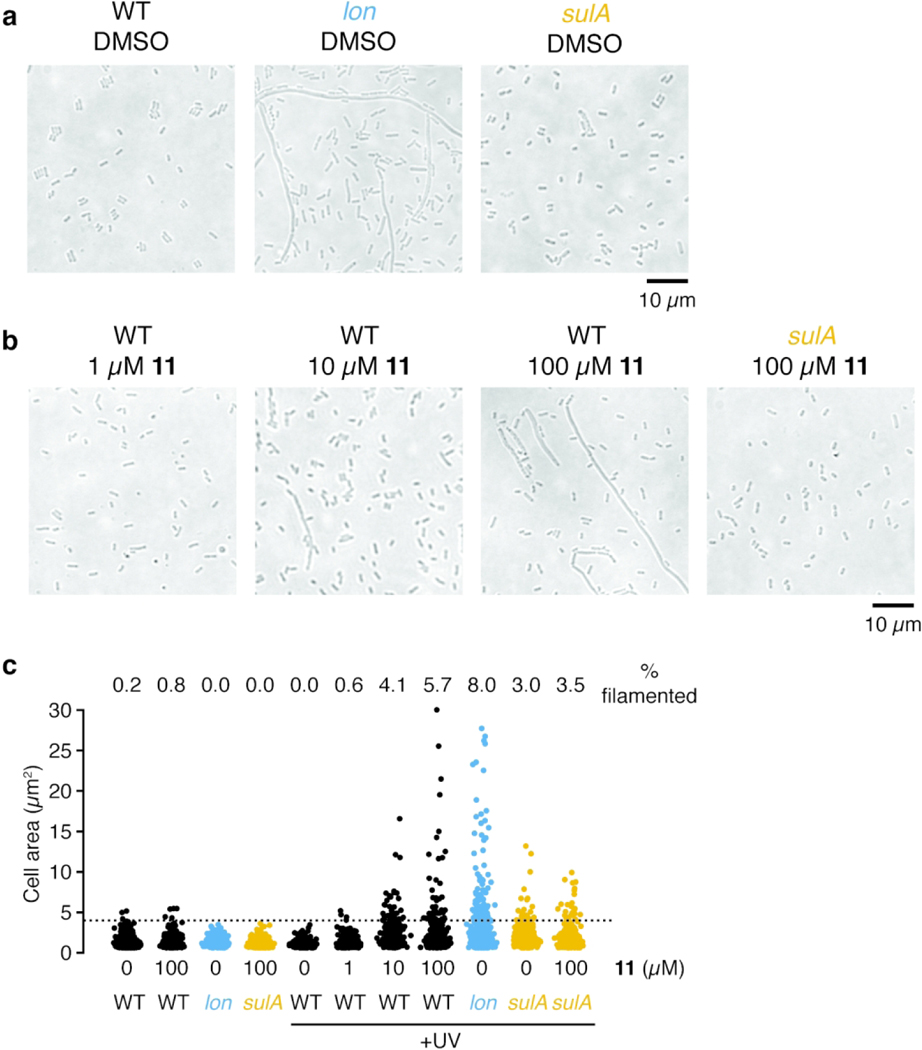
(a-b) Representative phase contrast images of E. coli strains exposed to UV light and then diluted into LB containing (a) DMSO or (b) various concentrations of 11. Cultures were grown for 6 h after UV-exposure and then imaged. Images are representative of two independent experiments. (c) Quantification of cell area for cells without UV-treatment or cells treated as in (a-b). The percent of cells with area greater than 4 μm2 (dashed line) is indicated (n<500 for each condition). Additional images are presented in Figure S6a.
Lon is also implicated in recovery from DNA damage caused by fluoroquinolone antibiotics16, 53. Most cells treated with such antibiotics die, but a small subpopulation (typically 0.01% of the initial population) tolerate antibiotic exposure and can replicate after removal of the antibiotic. So-called persister cells54 are reduced in a lon knockout strain. Like UV-induced filamentation, Lon’s role in persistence depends on the presence of SulA, with lon sulA double mutant strains producing a similar number of persisters as wild-type cells10, 17, 55. We therefore predicted that co-treatment of cells with 11 and ciprofloxacin would reduce persister cell formation. Neither lon deletion nor treatment of wild-type cells with 100 μm 11 altered the overall MIC of ciprofloxacin (0.0125 μg/ml). However, compared to wild type, we consistently observed a statistically-significant reduction in the fraction of cells that tolerated ciprofloxacin for both the lon mutant strain and wild-type cells treated with 11 in both rich (LB, Figure 5a) and minimal media (M9, Figure S6b). Importantly, this effect was abrogated in the sulA mutant strain, suggesting it results from inhibition of Lon. Furthermore, the effect was time-dependent, with both lon and 11-treated cells exhibiting faster death than wild type (Figure 5b). The effect was also concentration-dependent, with the extent of effect from 11 treatment matching that of the lon knockout strain at high concentrations (Figure 5c).
Figure 5. Lon inhibition decreases ciprofloxacin persister cells.
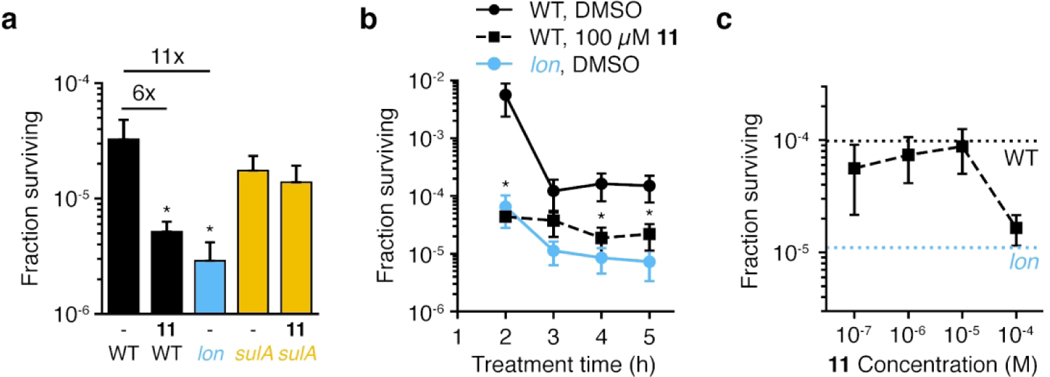
All panels show the fraction of E. coli cells surviving treatment with 10 μg/mL ciprofloxacin in LB. (a) Cells were co-treated with ciprofloxacin and either DMSO (-) or 100 μM 11 for 4 h (mean ± standard deviation, n=3; unpaired t-tests comparing each sample to wild-type cells treated with DMSO: *, p<0.05). Fold reduction compared to wild-type treated with DMSO is indicated above. (b) Time course of killing for cells co-treated with ciprofloxacin and DMSO or 100 μM 11 (mean ± standard deviation, n=3; unpaired t-tests comparing each sample to wild-type cells treated with DMSO at the same time point: *, p<0.05; lon was significantly different at all time points). (c) Dose-dependent effect of 11 in reducing cell survival. Cells were co-treated with ciprofloxacin and the indicated dose of 11 for 4 h (mean ± standard deviation, n=3; unpaired t-tests comparing each sample to wild-type cells treated with DMSO: p=0.054 for 100 μM 11). Dashed lines indicate the fraction surviving of wildtype or lon cells treated with DMSO. For all experiments, cultures were centrifuged, washed, and cell numbers were determined by spot dilution plating on LB agar. Panel (a) is representative of four independent experiments.
The SulA-dependent effects of 11 on UV-induced filamentation and ciprofloxacin persister formation strongly suggest that the compound inhibits Lon in cells. Incomplete phenocopying and the requirement for a high dose (e.g., 100 μM for cellular effects) are likely due to some combination of active efflux of the compound and permeability barriers. Both of these issues are common challenges for treating gram-negative bacteria with small molecules56. Encouragingly, there is evidence that other boronic acid inhibitors can enter E. coli cells57, 58, so we expect that modifications to increase the permeability of 11 will lead to further improved potency against live cells.
We leveraged amino acid preferences of Lon to develop both an improved fluorogenic substrate and a boronic acid inhibitor of Lon with increased selectivity over the proteasome. Our substrate screening results build on previous observations that Lon prefers to cleave peptides with bulky, hydrophobic residues, consistent with its role in degrading denatured proteins during stress responses. In our initial screen for inhibitors, Lon was poorly inhibited by electrophiles such as diphenyl phosphonates and chloroisocoumarins, which are potent inhibitors of many proteases with canonical serine-histidine-aspartic acid catalytic triads. This observation, along with the potency of proteasome inhibitors toward Lon, highlight the unusual nature of the serine-lysine dyad in its active site. Structural analyses of Lon inhibition by 11 would confirm the hypothesized covalent interaction with the active-site serine and would help to explain the structural basis for Lon’s preferences for bulky amino acids and the role that hArg plays in enhancing substrate and inhibitor binding. In the future, novel Lon inhibitors may be identified by exploring alternative warheads such as β-lactams59, or nitriles60 which have activity toward serine-lysine dyads in signal peptidases and the UmuD family of proteases.
We expect 11 to be a useful compound for studying the roles that Lon plays in stress-response and pathogenesis. The use of a small molecule inhibitor rather than genetic disruptions (e.g., lon deletion or active-site mutants) introduces a level of dynamic flexibility to studies of Lon. Additionally, it provides a means to disentangle Lon’s proteolytic activity from other functions of the multidomain complex, such as ATPase activity and its ability to bind and respond to DNA. In our cellular experiments, we observed 11-mediated effects on cellular physiology both when the compound was added during recovery from (outgrowth after UV exposure) or concurrent with (co-treatment with ciprofloxacin) stress. These observations suggest that Lon inhibition during or after stress has similar effects, at least for the SulA-mediated models of stress response tested here. Our data also show that 11 is not toxic to macrophages, meaning it can be used to test inhibition of bacterial Lon in cell culture models of infection and pathogenesis. Finally, because it is a covalent inhibitor, it can be converted to a fluorescent or otherwise affinity-labeled probe in order to visualize Lon activity within living cells. This compound should therefore greatly expand the scope of future studies of Lon function.
METHODS
HyCoSuL screens
HyCoSuL screens were performed in Corning opaque 96-well plates. Each well contained 99 μl of Lon in assay buffer (250 mM Tris, pH 8.0, 1 M KCl, 100 mM MgCl2, and 1 mM ATP). Lon was added to a final hexamer concentration of 190 nM (P2 library) or 570 nM (P3 library). HyCoSuL substrates were added to a final concentration of 100 μM and kinetic fluorescence measurements (ex. 355 nm, em. 460 nm) were taken at 37 °C for at least 30 min starting immediately after substrate addition (Spectramax Gemini XPS, Molecular Devices). The substrate hydrolysis rate (RFU s−1) was calculated from the linear portion of each progress curve. The amino acid with the highest cleavage rate was set to 100%, and remaining amino acids were adjusted accordingly. Each library was screened twice and results are presented as mean values.
Kinetic analysis of substrates
Lon and h20S substrate cleavage assays were performed in black 96- or 384-well plates. For Lon experiments, each well contained 25 μl 2X Lon assay buffer, 0.5 μl of 100 mM ATP (1 mM final concentration), and 40 nM final concentration of Lon hexamer. For ATP regeneration, 0.75 μl of 5 mg ml−1 creatine kinase (75 μg ml−1 final concentration) and 4 μl of 50 mM creatine phosphate (4 mM final concentration) were included. Water was added to a final volume of 40 μl. For h20S experiments, each well contained 25 μl 2X h20S buffer (100 mM Tris, pH 7.5, 200 mM NaCl), 1 mM DTT, 2 nM final concentration of h20S (BostonBiochem), 24 nM final concentration of PA28 (BostonBiochem), and water to a final volume of 40 μl. To begin the reaction, 10 μl of each substrate was added from a 5X stock, and fluorescence (ex. 360 nm, em. 460 nm) was measured every minute for 1 h at 37 °C in a microplate reader (BioTek Cytation 3).
Kinetic analysis of inhibitors
Inhibition assays were performed under the same conditions as for substrate kinetics. Compounds were added from a 100X stock in DMSO (0.5 μl). For pre-incubation, compounds were added to the enzyme mixture in each well and plates were incubated at 37 °C for the indicated time. For experiments without pre-incubation, compounds were added to the working stock of substrate. Substrates (10 μl of 250 μM working stock) were added to the enzyme mixture and fluorescence (ex. 360 nm, em. 460 nm) was measured every minute for 1 h at 37 °C in a microplate reader (BioTek Cytation 3). For Lon, 5 was the substrate. For h20S, Z-LLE-AMC, Boc-LRR-AMC, and Suc-LLVY-AMC were substrates specific for the β1, β2, and β5 subunits, respectively. Inhibition data for the proteasome was determined following 60 min pre-incubation with the enzyme. Proteasome substrates were purchased from BostonBiochem.
Proteasome labeling
For each compound, 1 μl of 20X stock in DMSO was added to a sample of h20S (10 nM) in 19 μl labeling buffer (50 mM Tris, pH 7.5, 5 mM MgCl2, 1 mM DTT) and incubated for 1 h at 37 °C. To label proteasome subunits, 0.5 μl of 80 μM MV151 (final concentration 2 μM) was added and incubated for an additional 2 h at 37 °C. Labeling was quenched by addition of 4X Laemmli sample buffer, samples were incubated for 5 min at 95 °C, and samples were separated by SDS-PAGE. MV151 fluorescence was imaged using a Typhoon 9410 Imager on the Cy3 channel (Amersham Biosciences).
RAW cell viability
RAW 264.7 murine macrophages were cultured in DMEM with 4.5 g l−1 glucose, 4 mM L- glutamine, and 10% v/v FBS (Invitrogen) at 37 °C with 5% CO2. Cells were split and seeded into a 96 well-plate to 5×103 cells per well with 50 μl of medium. To each well was added 49 μl of medium with 1 μl of 100X compound in DMSO (1% v/v final DMSO concentration). Cells were incubated with compound for 24 h then treated with 20 μl CellTiter-Blue (Promega) for 4 h. Cell viability was quantified by measuring fluorescence in a microplate reader (BioTek Cytation 3). Fluorescence values were normalized to untreated cells. Incubation with 1% v/v DMSO reduced cell viability compared to untreated cells, but the effect was independent of compound or dose.
Bacterial strains and growth conditions
Bacteria were cultured with shaking in LB (Fisher), 2xYT (Teknova), or M9 at 37°C, unless otherwise indicated. M9 contained 6 g l−1 Na2HPO4, 3 g/l KH2PO4, 1 g l−1 NH4Cl, 0.5 g l−1 NaCl, 0.5% w/v glucose, 1 mM MgSO4, 0.1 mM CaCl2, and 0.34 mg l−1 thiamine HCl. The lon mutant strain was generated by clean deletion of the coding region of lon using homologous recombination with CRISPR-Cas9 selection61. The sulA mutant strain (sulA773(del)::kan) was obtained from the Keio Collection62. Growth rates were determined by measuring OD600 of 100 μl cultures grown at 37°C in a 96-well plate in a microplate reader overnight.
UV treatment and microscopy
Overnight cultures of wild-type, lon, or sulA strains were grown in LB. Cultures were diluted to OD600 0.1 in LB and grown for 1 h. Cells were pelleted by centrifugation (8,000 rcf for 5 min), resuspended in 0.1 volume of 10 mM MgSO4 and transferred to glass tubes. Cells were irradiated with 900 J cm−2 254 nm light (Stratagene Stratalinker 2400). Control cultures were resuspended in MgSO4 as above, but were not irradiated. Cells were diluted 1:25 into LB with compound added from 100X stock in DMSO (1% v/v final DMSO concentration) and grown for 6 h at 37 °C with shaking in the dark. For imaging, 4 μl of each culture was applied to 2% w/v agarose pads63. Phase contrast microscopy was performed on a Zeiss LSM700 confocal microscope with a Plan- Apochromat 63x/1.4 objective. Twenty-five images were captured via tile scan for each condition. Quantification of cell area was performed with the MicrobeJ64 plugin for ImageJ. Regions of interest containing at least 500 cells were analyzed using default settings for bacterial detection. A minimum cell area of 0.9 μm2 was used to exclude non-cellular debris.
Ciprofloxacin treatment and persister cell quantification
For MIC measurements, overnight cultures of wild-type or lon strains were grown in LB, then diluted 1:50 into Mueller Hinton Broth 2 (Sigma). Diluted cultures (50 μl) were aliquoted into wells in a 96-well plate, each containing 50 μl medium and 2X the final concentration of ciprofloxacin. For persister experiments, overnight cultures of wild-type, lon, or sulA strains were grown in LB or M9. Cultures were diluted to OD600 0.01 in the same medium and incubated for 2 h. Cultures were treated with 10 μg ml−1 ciprofloxacin (Sigma) from a 100X stock in water and compound from a 100X stock in DMSO (1% v/v final DMSO concentration). Aliquots (100 μl) of each culture were removed at the indicated time, pelleted by centrifugation (8,000 rcf for 5 min), washed once with PBS, and resuspended in 100 μl PBS. Cells were serially diluted in PBS, 10 μL spots were spotted onto LB agar plates, and plates were incubated for 16–24 h at 37 °C. Colonies were counted to determine CFU.
Software
Statistical analysis, fitting, and plotting were performed with Python v. 3.6.0, Scipy v. 1.1.0, Numpy v. 1.13.3, Matplotlib v. 3.0.3, and Seaborn v. 0.9.0. Microscopy data were analyzed in ImageJ. DNA sequence analysis was performed in SnapGene 4.3.10. Figures were assembled in Adobe Illustrator CS6.
Supplementary Material
ACKNOWLEDGEMENTS
The authors appreciate critical feedback of the manuscript from M. Lakemeyer.
FUNDING SOURCES
B.M.B. was supported by the Stanford Dean’s Fellowship and the A. P. Giannini Foundation. The work was funded in part by grants from the National Institutes of Health (R01EB026332, R01EB026285 to M.B. and T32AI07328 to B.M.B.). P.K. is the beneficiary of a L’Oreal Poland and the Polish Ministry of Science and Higher Education and START Foundation for Polish Science scholarships. The Drag laboratory is supported by the “TEAM/ 2017–4/32” project, which is carried out within the TEAM program of the Foundation for Polish Science co-financed by the European Union under the European Regional Development Fund. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Supporting Information contains Supplemental Methods, Supplemental Synthesis Methods, Compound Characterization, Supplemental References, Supplemental Figures, and Supplemental Tables. This material is available free of charge via the internet at http://pubs.acs.org.
REFERENCES
- 1.Gur E (2013) The Lon AAA+ protease, In Regulated Proteolysis in Microorganisms. (Dougan D, Ed.), pp 35–51, Springer, Dordrecht. [Google Scholar]
- 2.Olivares AO, Baker TA, and Sauer RT (2016) Mechanistic insights into bacterial AAA+ proteases and protein-remodelling machines, Nat. Rev. Microbiol. 14, 33–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gottesman S (2003) Proteolysis in bacterial regulatory circuits, Annu. Rev. Cell Dev. Biol. 19, 565–587. [DOI] [PubMed] [Google Scholar]
- 4.Tsilibaris V, Maenhaut-Michel G, and Van Melderen L (2006) Biological roles of the Lon ATP-dependent protease, Res. Microbiol. 157, 701–713. [DOI] [PubMed] [Google Scholar]
- 5.Mizusawa S, and Gottesman S (1983) Protein degradation in Escherichia coli: The Ion gene controls the stability of sulA protein, Proc. Natl. Acad. Sci. U. S. A. 80, 358–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Griffith KL, Shah IM, and E. Wolf R (2004) Proteolytic degradation of Escherichia coli transcription activators SoxS and MarA as the mechanism for reversing the induction of the superoxide (SoxRS) and multiple antibiotic resistance (Mar) regulons, Mol. Microbiol. 51, 1801–1816. [DOI] [PubMed] [Google Scholar]
- 7.Bissonnette SA, Rivera-Rivera I, Sauer RT, and Baker TA (2010) The IbpA and IbpB small heat-shock proteins are substrates of the AAA+ Lon protease, Mol. Microbiol. 75, 1539–1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Christensen SK, Mikkelsen M, Pedersen K, and Gerdes K (2001) RelE, a global inhibitor of translation, is activated during nutritional stress, Proc. Natl. Acad. Sci. U. S. A. 98, 14328–14333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gottesman S, Gottesman M, Shaw JE, and Pearson ML (1981) Protein Degradation in E. coli: The lon Mutation and Bacteriophage Lambda N and cll Protein Stability, Cell 24, 225–233. [DOI] [PubMed] [Google Scholar]
- 10.Theodore A, Lewis K, and Vulic M (2013) Tolerance of Escherichia coli to fluoroquinolone antibiotics depends on specific components of the SOS response pathway, Genetics 195, 1265–1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Helaine S, Cheverton AM, Watson KG, Faure LM, Matthews SA, and Holden DW (2014) Internalization of Salmonella by macrophages induces formation of nonreplicating persisters, Science 343, 204–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Breidenstein EB, Janot L, Strehmel J, Fernandez L, Taylor PK, Kukavica-Ibrulj I, Gellatly SL, Levesque RC, Overhage J, and Hancock RE (2012) The Lon protease is essential for full virulence in Pseudomonas aeruginosa, PLoS One 7, e49123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Takaya A, Suzuki M, Matsui H, Tomoyasu T, Sashinami H, Nakane A, and Yamamoto T (2003) Lon, a stress-induced ATP-dependent protease, is critically important for systemic Salmonella enterica serovar typhimurium infection of mice, Infect. Immun. 71, 690–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Robertson GT, Kovach ME, Allen CA, Ficht TA, and Roop II RM (2000) The Brucella abortus Lon functions as a generalized stress response protease and is required for wild-type virulence in BALB/c mice, Mol. Microbiol. 35, 577–588. [DOI] [PubMed] [Google Scholar]
- 15.Raju RM, Goldberg AL, and Rubin EJ (2012) Bacterial proteolytic complexes as therapeutic targets, Nat. Rev. Drug Discovery 11, 777–789. [DOI] [PubMed] [Google Scholar]
- 16.Piddock LJ, and Walters RN (1992) Bactericidal activities of five quinolones for Escherichia coli strains with mutations in genes encoding the SOS response or cell division, Antimicrob. Agents Chemother. 36, 819–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shan Y, Brown Gandt A, Rowe SE, Deisinger JP, Conlon BP, and Lewis K (2017) ATP-dependent persister formation in Escherichia coli, mBio 8, e02267–02216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wood TK, Song S, and Yamasaki R (2019) Ribosome dependence of persister cell formation and resuscitation, J. Microbiol. (Seoul-Repub. Korea) 57, 213–219. [DOI] [PubMed] [Google Scholar]
- 19.Maisonneuve E, Castro-Camargo M, and Gerdes K (2018) Retraction Notice to: (p)ppGpp Controls Bacterial Persistence by Stochastic Induction of Toxin-Antitoxin Activity, Cell 172, 1135. [DOI] [PubMed] [Google Scholar]
- 20.(2018) Retraction for Maisonneuve et al. , Bacterial persistence by RNA endonucleases, Proc. Natl. Acad. Sci. U. S. A. 115, E2901–E2901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Harms A, Fino C, Sorensen MA, Semsey S, and Gerdes K (2017) Prophages and Growth Dynamics Confound Experimental Results with Antibiotic-Tolerant Persister Cells, mBio 8, e01964–01917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ekici OD, Paetzel M, and Dalbey RE (2008) Unconventional serine proteases: variations on the catalytic Ser/His/Asp triad configuration, Protein Sci. 17, 2023–2037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rotanova TV, Mel’nikov EE, and Tsirulnikov KB (2003) A catalytic Ser-Lys dyad in the active site of the ATP-dependent lon protease from Escherichia coli, Russ. J. Bioorg. Chem. 29, 85–87. [DOI] [PubMed] [Google Scholar]
- 24.Swamy KHS, and Goldberg AL (1981) E. coli contains eight soluble proteolytic activities, one being ATP dependent, Nature 292, 652–654. [DOI] [PubMed] [Google Scholar]
- 25.Waxman L, and Goldberg AL (1982) Protease La from Escherichia coli hydrolyzes ATP and proteins in a linked fashion, Proc. Natl. Acad. Sci. U. S. A. 79, 4883–4887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bayot A, Basse N, Lee I, Gareil M, Pirotte B, Bulteau AL, Friguet B, and Reboud-Ravaux M (2008) Towards the control of intracellular protein turnover: mitochondrial Lon protease inhibitors versus proteasome inhibitors, Biochimie 90, 260–269. [DOI] [PubMed] [Google Scholar]
- 27.Bernstein SH, Venkatesh S, Li M, Lee J, Lu B, Hilchey SP, Morse KM, Metcalfe HM, Skalska J, Andreeff M, Brookes PS, and Suzuki CK (2012) The mitochondrial ATP-dependent Lon protease: a novel target in lymphoma death mediated by the synthetic triterpenoid CDDO and its derivatives, Blood 119, 3321–3329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Waxman L, and Goldberg AL (1985) Protease La, the lon gene product, cleaves specific fluorogenic peptides in an ATP-dependent reaction, J. Biol. Chem. 260, 12022–12028. [PubMed] [Google Scholar]
- 29.Granot Z, Geiss-Friedlander R, Melamed-Book N, Eimerl S, Timberg R, Weiss AM, Hales KH, Hales DB, Stocco DM, and Orly J (2003) Proteolysis of normal and mutated steroidogenic acute regulatory proteins in the mitochondria: the fate of unwanted proteins, Mol. Endocrinol. 17, 2461–2476. [DOI] [PubMed] [Google Scholar]
- 30.Bezawork-Geleta A, Brodie EJ, Dougan DA, and Truscott KN (2015) LON is the master protease that protects against protein aggregation in human mitochondria through direct degradation of misfolded proteins, Sci. Rep. 5, 17397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Frase H, Hudak J, and Lee I (2006) Identification of the proteasome inhibitor MG262 as a potent ATP-dependent inhibitor of the Salmonella enterica serovar Typhimurium Lon protease, Biochemistry 45, 8264–8274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liao JH, Ihara K, Kuo CI, Huang KF, Wakatsuki S, Wu SH, and Chang CI (2013) Structures of an ATP-independent Lon-like protease and its complexes with covalent inhibitors, Acta Crystallogr., Sect. D: Biol. Crystallogr. 69, 1395–1402. [DOI] [PubMed] [Google Scholar]
- 33.Frase H, and Lee I (2007) Peptidyl boronates inhibit Salmonella enterica serovar Typhimurium Lon protease by a competitive ATP-dependent mechanism, Biochemistry 46, 6647–6657. [DOI] [PubMed] [Google Scholar]
- 34.Kasperkiewicz P, Poreba M, Groborz K, and Drag M (2017) Emerging challenges in the design of selective substrates, inhibitors and activity-based probes for indistinguishable proteases, FEBS J. 284, 1518–1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Poreba M, Kasperkiewicz P, Snipas SJ, Fasci D, Salvesen GS, and Drag M (2014) Unnatural amino acids increase sensitivity and provide for the design of highly selective caspase substrates, Cell Death Differ. 21, 1482–1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Poreba M, Rut W, Vizovisek M, Groborz K, Kasperkiewicz P, Finlay D, Vuori K, Turk D, Turk B, Salvesen GS, and Drag M (2018) Selective imaging of cathepsin L in breast cancer by fluorescent activity-based probes, Chem. Sci. 9, 2113–2129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kasperkiewicz P, Poreba M, Snipas SJ, Parker H, Winterbourn CC, Salvesen GS, and Drag M (2014) Design of ultrasensitive probes for human neutrophil elastase through hybrid combinatorial substrate library profiling, Proc. Natl. Acad. Sci. U. S. A. 111, 2518–2523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kasperkiewicz P, Poreba M, Snipas SJ, Lin SJ, Kirchhofer D, Salvesen GS, and Drag M (2015) Design of a Selective Substrate and Activity Based Probe for Human Neutrophil Serine Protease 4, PLoS One 10, e0132818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rut W, Poreba M, Kasperkiewicz P, Snipas SJ, and Drag M (2018) Selective Substrates and Activity-Based Probes for Imaging of the Human Constitutive 20S Proteasome in Cells and Blood Samples, J. Med. Chem. 61, 5222–5234. [DOI] [PubMed] [Google Scholar]
- 40.Li H, O’Donoghue AJ, van der Linden WA, Xie SC, Yoo E, Foe IT, Tilley L, Craik CS, da Fonseca PCA, and Bogyo M (2016) Structure-and function-based design of Plasmodium-selective proteasome inhibitors, Nature 530, 233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yoo E, Stokes BH, de Jong H, Vanaerschot M, Kumar TRS, Lawrence N, Njoroge M, Garcia A, Van der Westhuyzen R, Momper JD, Ng CL, Fidock DA, and Bogyo M (2018) Defining the Determinants of Specificity of Plasmodium Proteasome Inhibitors, J. Am. Chem. Soc. 140, 11424–11437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lentz CS, Ordonez AA, Kasperkiewicz P, La Greca F, O’Donoghue AJ, Schulze CJ, Powers JC, Craik CS, Drag M, Jain SK, and Bogyo M (2016) Design of Selective Substrates and Activity-Based Probes for Hydrolase Important for Pathogenesis 1 (HIP1) from Mycobacterium tuberculosis, ACS Infect. Dis. 2, 807–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rut W, Zhang L, Kasperkiewicz P, Poreba M, Hilgenfeld R, and Drag M (2017) Extended substrate specificity and first potent irreversible inhibitor/activity-based probe design for Zika virus NS2B-NS3 protease, Antiviral Res. 139, 88–94. [DOI] [PubMed] [Google Scholar]
- 44.Nishii W, Maruyama T, Matsuoka R, Muramatsu T, and Takahashi K (2002) The unique sites in SulA protein preferentially cleaved by ATP-dependent Lon protease from Escherichia coli, Eur. J. Biochem. 269, 451–457. [DOI] [PubMed] [Google Scholar]
- 45.Maurizi MR (1987) Degradation in vitro of bacteriophage lambda N protein by Lon protease from Escherichia coli, J. Biol. Chem. 262, 2696–2703. [PubMed] [Google Scholar]
- 46.Ondrovicova G, Liu T, Singh K, Tian B, Li H, Gakh O, Perecko D, Janata J, Granot Z, Orly J, Kutejova E, and Suzuki CK (2005) Cleavage site selection within a folded substrate by the ATP-dependent lon protease, J. Biol. Chem. 280, 25103–25110. [DOI] [PubMed] [Google Scholar]
- 47.Kasperkiewicz P, Kolt S, Janiszewski T, Groborz K, Porçba M, Snipas SJ, Salvesen GS, and Drąg M (2018) Determination of extended substrate specificity of the MALT1 as a strategy for the design of potent substrates and activity-based probes, Sci. Rep. 8, 15998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lee I, and Berdis AJ (2001) Adenosine triphosphate-dependent degradation of a fluorescent lambda N substrate mimic by Lon protease, Anal. Biochem. 291, 74–83. [DOI] [PubMed] [Google Scholar]
- 49.Arastu-Kapur S, Ponder EL, Fonovic UP, Yeoh S, Yuan F, Fonovic M, Grainger M, Phillips CI, Powers JC, and Bogyo M (2008) Identification of proteases that regulate erythrocyte rupture by the malaria parasite Plasmodium falciparum, Nat. Chem. Biol. 4, 203–213. [DOI] [PubMed] [Google Scholar]
- 50.Verdoes M, Florea BI, Menendez-Benito V, Maynard CJ, Witte MD, van der Linden WA, van den Nieuwendijk AM, Hofmann T, Berkers CR, van Leeuwen FW, Groothuis TA, Leeuwenburgh MA, Ovaa H, Neefjes JJ, Filippov DV, van der Marel GA, Dantuma NP, and Overkleeft HS (2006) A fluorescent broad-spectrum proteasome inhibitor for labeling proteasomes in vitro and in vivo, Chem. Biol. (Oxford, U. K.) 13, 1217–1226. [DOI] [PubMed] [Google Scholar]
- 51.Howard-Flanders P, Simson E, and Theriot L (1964) A locus that controls filament formation and sensitivity to radiation in Escherichia coli K-12, Genetics 49, 237–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gottesman S, Halpern E, and Trisler P (1981) Role of sulA and sulB in Filamentation by Lon Mutants of Escherichia coli K-12, J. Bacteriol. 148, 265–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yamaguchi Y, Tomoyasu T, Takaya A, Morioka M, and Yamamoto T (2003) Effects of disruption of heat shock genes on susceptibility of Escherichia coli to fluoroquinolones, BMC Microbiol. 3, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Balaban NQ, Helaine S, Lewis K, Ackermann M, Aldridge B, Andersson DI, Brynildsen MP, Bumann D, Camilli A, Collins JJ, Dehio C, Fortune S, Ghigo JM, Hardt WD, Harms A, Heinemann M, Hung DT, Jenal U, Levin BR, Michiels J, Storz G, Tan MW, Tenson T, Van Melderen L, and Zinkernagel A (2019) Definitions and guidelines for research on antibiotic persistence, Nat. Rev. Microbiol. 17, 441–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ghosh A, Baltekin O, Waneskog M, Elkhalifa D, Hammarlof DL, Elf J, and Koskiniemi S (2018) Contact-dependent growth inhibition induces high levels of antibiotic-tolerant persister cells in clonal bacterial populations, EMBO J. 37, e98026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Masi M, Refregiers M, Pos KM, and Pages JM (2017) Mechanisms of envelope permeability and antibiotic influx and efflux in Gram-negative bacteria, Nat. Microbiol. 2, 17001. [DOI] [PubMed] [Google Scholar]
- 57.Dzhekieva L, Kumar I, and Pratt RF (2012) Inhibition of bacterial DD-peptidases (penicillin-binding proteins) in membranes and in vivo by peptidoglycan-mimetic boronic acids, Biochemistry 51, 2804–2811. [DOI] [PubMed] [Google Scholar]
- 58.Venturelli A, Tondi D, Cancian L, Morandi F, Cannazza G, Segatore B, Prati F, Amicosante G, Shoichet BK, and Costi MP (2007) Optimizing cell permeation of an antibiotic resistance inhibitor for improved efficacy, J. Med. Chem. 50, 5644–5654. [DOI] [PubMed] [Google Scholar]
- 59.Paetzel M, Dalbey RE, and Strynadka NCJ (1998) Crystal structure of a bacterial signal peptidase in complex with a p-lactam inhibitor, Nature 396, 186–190. [DOI] [PubMed] [Google Scholar]
- 60.Smith PA, Koehler MFT, Girgis HS, Yan D, Chen Y, Chen Y, Crawford JJ, Durk MR, Higuchi RI, Kang J, Murray J, Paraselli P, Park S, Phung W, Quinn JG, Roberts TC, Rouge L, Schwarz JB, Skippington E, Wai J, Xu M, Yu Z, Zhang H, Tan MW, and Heise CE (2018) Optimized arylomycins are a new class of Gram-negative antibiotics, Nature 561, 189–194. [DOI] [PubMed] [Google Scholar]
- 61.Jiang Y, Chen B, Duan C, Sun B, Yang J, and Yang S (2015) Multigene editing in the Escherichia coli genome via the CRISPR-Cas9 system, Appl. Environ. Microbiol. 81, 2506–2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Baba T, Ara T, Hasegawa M, Takai Y, Okumura Y, Baba M, Datsenko KA, Tomita M, Wanner BL, and Mori H (2006) Construction of Escherichia coli K-12 inframe, single-gene knockout mutants: the Keio collection, Mol. Syst. Biol. 2, 2006.0008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Levin PA (2002) Light microscopy techniques for bacterial cell biology, Methods Microbiol. 115–132. [Google Scholar]
- 64.Ducret A, Quardokus EM, and Brun YV (2016) Microbe J, a tool for high throughput bacterial cell detection and quantitative analysis, Nat. Microbiol. 1, 16077. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.