

Abstract
Monitoring the aggregation of proteins within the cellular environment is key to investigating the molecular mechanisms underlying the formation of off-pathway protein assemblies associated with the development of disease and testing therapeutic strategies to prevent the accumulation of non-native conformations. It remains challenging, however, to couple protein aggregation events underlying the cellular pathogenesis of a disease to genetic circuits and monitor their progression in a quantitative fashion using synthetic biology tools. To link the aggregation propensity of a target protein to the expression of an easily detectable reporter, we investigated the use of a transcriptional AND gate system based on complementation of a split transcription factor. We first identified two-fragment tetracycline repressor (TetR) variants that can be regulated via ligand-dependent induction and demonstrated that split TetR variants can function as transcriptional AND gates in both bacteria and mammalian cells. We then adapted split TetR for use as an aggregation sensor. Protein aggregation was detected by monitoring complementation between a larger TetR fragment that serves as a “detector” and a smaller TetR fragment expressed as a fusion to an aggregation-prone protein that serves as a “sensor” of the target protein aggregation status. This split TetR represents a novel genetic component that can be used for a wide range of applications in bacterial as well as mammalian synthetic biology and a much needed cell-based sensor for monitoring a protein’s conformational status in complex cellular environments.
Keywords: split protein, split transcription factor, protein complementation, protein aggregation, solubility sensor, AND gate
Graphical Abstract
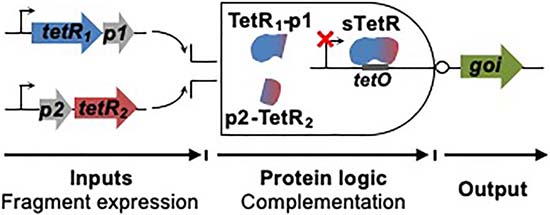
INTRODUCTION
The establishment of rapid and reliable techniques for assessing protein solubility is of vital importance to studying fundamental mechanisms of protein homeostasis, discovering therapeutics for protein misfolding diseases, and obtaining desired yields of high-value recombinant proteins. Protein aggregation arising from misfolding is associated with the cellular pathogenesis of numerous human diseases, such as Parkinson’s, Huntington’s, and Alzheimer’s diseases.1–3 While a number of misfolding and aggregation-prone proteins have been characterized,4,5 we lack reliable high-throughput methods to quantify how diverse genetic and chemical treatments affect the formation of these off-pathway aggregates in situ. Similarly, it remains challenging to study how protein aggregation events that are intimately associated with the regulation of cellular homeostasis are influenced by genetic background and cellular environment, such as protein aggregates that play a role in cell signaling and regulation.6–8 Furthermore, the accumulation of off-pathway protein aggregates often plagues the production of recombinant proteins.9 This aggregation can limit the yields of high value protein therapeutics for biotechnology and pharmaceutical applications, such as monoclonal antibodies and vaccines,10,11 and require significant optimization to identify conditions that maximize yields.
A wide range of tools has been developed for monitoring protein aggregation in situ, but existing approaches remain limited in their sensitivity, reliability, and modularity. For instance, antibodies can be used to monitor protein aggregation in living cells,12,13 provided that they are sufficiently specific against the desired epitope.14,15 While antibodies specific to different conformational states of disease-associated proteins have been developed, immunohistochemistry only provides a static snapshot of aggregation.16 Immunohistochemistry approaches are also low throughput, limiting their applications in screens for therapeutics. Genetic strategies for monitoring aggregation overcome some of these limitations. By expressing aggregation-prone proteins as fusions to fluorescent reporters, such as the green fluorescent protein (GFP), reporter folding and fluorescence can be used as a dynamic proxy for a protein’s solubility in living cells.17,18 However, fusion of large, stable reporter proteins can perturb the folding and solubility of the protein of interest. In addition, this approach is not effective at reporting when the kinetics of aggregation is slower than GFP chromophore formation. To overcome these limitations, a split GFP complementation assay was leveraged to monitor aggregation.19,20 With this protein fragment complementation assay, a small GFP fragment that functions as a solubility sensor is fused to the protein of interest to minimize the effects of fusion on folding and solubility of the protein of interest, and the second GFP fragment that functions as a detector is constitutively expressed. Protein aggregation in this expression system controls the soluble concentration of the smaller GFP fragment available to associate with the larger GFP fragment. This split protein approach has been successfully used to monitor the solubility of various disease associated proteins in situ, such as tau21,22 and α-synuclein.23 However, the assay’s sensitivity is limited by the concentration of the protein of interest that is present in soluble form, which controls the concentration of the sensor GFP fragment available to associate with the detector fragment, and the dynamic range of the fluorescent signal.
In synthetic biology, cells can be programed to generate robust and tunable reporter outputs (e.g., GFP signals) by creating genetic circuits that sense specific environmental conditions. With some design goals, natural proteins can be used to construct these genetic circuits. Many design goals, however, require the development of new transcriptional regulators that are engineered to display novel DNA-binding and sensing capabilities. Ligand and DNA-binding specificity has been diversified through a wide range of rational design and laboratory evolution approaches.24–28 In addition, new sensing capabilities have been developed by subjecting transcription factors to protein fragmentation. For example, the T7 RNA polymerase has been fragmented to create bipartite proteins that require fragment association to generate a transcriptional output.29,30 Similar to the split GFP used to monitor protein aggregation,23 the signal generated by two-fragment transcription factors depends upon the concentration of each fragment produced, suggesting that the activity of these split proteins could be regulated through fusion to aggregation-prone proteins, similar to that achieved with synthetic transcription factors in yeast.31
In this study, we describe the development of a ligand-dependent split transcriptional regulator whose DNA repression is inversely correlated with protein aggregation in living cells (Figure 1). We first used transposon mutagenesis to create combinatorial libraries expressing split tetracycline repressor (sTetR) proteins as fusions to two different pairs of interacting peptides in E. coli, and we employed a bacterial genetic circuit to screen for active sTetR. We then identified sTetR that can be used as genetic AND gates in E. coli and mammalian cells, and we characterized the response of these proteins induction with anhydro-tetracycline (aTc) and tetracycline (Tc). Among the sTetR presenting strong repression, we adapted the variant arising from peptide backbone fission proximal to the C-terminus as a solubility sensor. We show that when the smaller fragment of this sTetR is fused to proteins that are prone to aggregation, this transcriptional regulator yields a genetic output that is proportional to protein aggregation.
Figure 1.

Scheme illustrating the design of a solubility sensor. A protein of interest (POI) is fused to the C-terminus of the smaller fragment of the split transcription factor (TF2). When the protein of interest is soluble, the two fragments of the split transcription factor (TF1 and TF2) can associate into a functional transcription factor and repress the expression of the reporter output. Aggregation of the fusion protein precludes accessibility of the smaller fragment to the larger fragment, thus preventing fragment complementation and resulting in activation of the reporter output expression.
RESULTS
Isolation of active two-fragment TetR.
To identify split TetR (sTetR) whose fragments require association to generate a functional transcriptional regulator, we constructed a library of randomly fragmented TetR variants and expressed these as fusions to a pair of interacting peptides (IAAL-E3 and IAAL-K3) designed to associate into a parallel coiled coil with high affinity (KD = 70 nM).32,33 This “TetR-EK library” was created using a previously described transposon mutagenesis method,34 which creates sequence diversity by randomly inserting a transposon at different positions within the target gene sequence. The vector library was designed to express the TetR fragments independently under the control of the tac and T7 promoters, respectively (Figure 2A). In these vectors, the gene encoding the IAAL-E3 peptide is placed upstream of the fission site, resulting in an ORF that encodes a fusion of the IAAL-E3 peptide to the C-terminus of the first TetR fragment, while the gene encoding the IAAL-K3 peptide is downstream of the fission site, resulting in an ORF that encodes a fusion of the IAAL-K3 peptide to the N-terminus of the second TetR fragment.33 Each synthetic peptide was fused using a 12-residue glycine-rich linker.33
Figure 2.

Functional sTetR variants identified in the TetR EK library. (A) Schematic representation of the sTetR expression system where the transcription of the first and second fragments of sTetR is controlled by the IPTG-inducible tac and T7 promoters, respectively. Both fragments are repressed by the lac repressor. Fragment complementation results in formation of a functional repressor that binds to the tetracycline operator (TO) within the PLtet promoter and represses the expression of YFP. Addition of anhydro-tetracycline (aTc) results in displacement of sTetR from TO and activation of YFP expression. (B) Fission sites of active sTetR variants mapped onto TetR structure (PDB 4AC0). The first and second fragments of sTetR are shown in blue and red, respectively. The linker that is disordered in the TetR structure is shown as a dashed line. (C) Fluorescence of E. coli CS50-YFP cells (−) and E. coli CS50-YFP cells expressing TetR or sTetR variants identified in the TetR-EK library grown in the presence and absence of IPTG (10 μM) and aTc (200 ng/mL). Relative fluorescence values were calculated by normalizing the YFP fluorescence intensity to that of cells lacking TetR (−). (D) Fluorescence of E. coli CS50-YFP cells (−) and E. coli CS50-YFP cells expressing TetR and the sTetR variants reported in (C) but with the first TetR fragment lacking the IAAL-E3 peptide. Relative fluorescence was calculated as described in (C). All data are presented as mean ± s.d. (n=12, p<0.001, two tailed t-test).
To isolate sTetR variants that retain the ability to function as repressors in cells, we built a screening system (Figure S1A) that uses the tetracycline-inducible promoter PLtet to control the expression of the yellow fluorescent protein (YFP). YFP expression from this vector (pLtet-YFP) is repressed by TetR, which binds to the tetracycline operator (TO) within the PLtet promoter,35 and is induced upon addition of aTc addition, which results in displacement of TetR from TO.36 The screening system was tested by co-transforming E. coli CS50 cells with pLtet-YFP and a vector for expression of TetR under the control of the tac promoter (pTAC-TetR). YFP fluorescence of the resulting strain was compared to that of cells transformed with pLtet-YFP and an empty vector (pTAC). The YFP fluorescence of cells expressing TetR was ~20% of that of cells lacking TetR (Figure S1B, p<0.001). Addition of aTc significantly increased YFP fluorescence in cells transformed with pTAC-TetR (Figure S1B, p<0.001), while it did not affect the fluorescence of cells harboring the control plasmid. YFP fluorescence was not significantly affected by addition of isopropyl β-D-1-thiogalactopyranoside (IPTG), which induces TetR expression. This finding suggests that the tac promoter controlling TetR expression is leaky as previously observed.37 The coefficient of variance (CV) of the YFP fluorescence signal of cells harboring pLtet-YFP in this plasmid-based expression system was found to be high (~12%) and suboptimal for library screening.
To minimize the variability of the YFP signal, the PLtet-YFP fusion was chromosomally integrated to generate the strain E. coli CS50-YFP. This strain presented YFP fluorescence that was significantly higher than the unmodified parental strain, and a CV (~8%) that was 50% lower than that measured using the plasmid-encoded reporter (Figure S1C). When this strain was transformed with pTAC-TetR, the YFP signal decreased to ~10% of that of cells harboring an empty vector (Figure S1C, p<0.001). As observed with our plasmid-based screen system, addition of IPTG did not further enhance YFP repression. Moreover, addition aTc increased the YFP fluorescence of cells transformed with pTAC-TetR (Figure S1C, p<0.001), but it did not affect the fluorescence of cells containing the vector control (pTAC). Dual treatment with IPTG and aTc decreased the whole cell YFP fluorescence of cells containing pTAC-TetR, confirming that YFP expression depends on TetR levels.
To identify sTetR variants that retain activity, we screened the TetR-EK library for variants that decreased the YFP fluorescence of CS50-YFP cells. Single colonies (n = 720) of CS50-YFP cells transformed with the library were inoculated into LB liquid cultures containing ampicillin. Upon reaching stationary phase, cultures were diluted 1:10 into LB containing 10 μM IPTG, and cells were grown for 4 hours. YFP fluorescence and cell density were monitored using a plate reader, and cell fluorescence was normalized by cell density to account for variability in the cellular growth rate. IPTG was added to ensure detection of sTetR variants that exhibit a weaker DNA binding affinity than the parental TetR. Through this screening approach, a large number of clones (14%) presenting a YFP signal ≥5σ lower than that of cells lacking TetR were identified (Figure S2). Sequencing 97 clones revealed 3 unique variants that arose from peptide backbone fission after the codons encoding residues 180 (180-EK), 184 (184-EK) and 193 (193-EK). These three fission sites were all found to be within the regulatory core domain, and, more specifically, within the turn that connects helices α9 and α10 (180-EK), and within helix α10 (184-EK and 193-EK) (Figure 2B).38
Characterizing sTetR repression.
To better understand the activity of the sTetR variants identified by screening the TetR-EK library, we evaluated the effects of IPTG and aTc on the activity of each variant in CS50-YFP cells. In the absence of both inducers, the expression of all three sTetR variants decreased YFP fluorescence to ~10% of the signal displayed in cells without TetR (Figure 2C, p<0.001). This level of repression is comparable to that of cells expressing the parental TetR. IPTG-mediated induction of sTetR expression did not further decrease the YFP fluorescence. Addition of aTc increased YFP florescence 5.7 to 7.5-fold, indicating that the sTetR variants function as aTc-dependent repressors (Figure 2C, p<0.001). With variants 184-EK and 193-EK, the increase in YFP signal upon addition of aTc was significantly higher than that observed with cells expressing TetR (Figure 2C, p<0.001). In the presence of IPTG, aTc led to derepression of all three sTetR variants, albeit to a lesser extent than what observed with cells treated only with aTc, suggesting that the different aTc-bound sTetR exhibit non-specific binding to TO, as previously reported with full-length TetR.39,40 Moreover, all three sTetR presented aTc-induced derepression (+IPTG) that was significantly higher than cells expressing the parental TetR (Figure 2C, p<0.001). These results may be due to a reduced level of non-specific binding of sTetR to TO compared to TetR under the experimental conditions of this study.
To test the role that the IAAL-E3/IAAL-K3 peptides play in mediating TetR-fragment complementation, we generated vectors that express only one of the sTetR fragments as a fusion to a peptide. Specifically, the first TetR fragment was expressed alone, while the second TetR fragment was expressed as fusion to the IAAL-K3 peptide. This peptide was retained at the N-terminus of the second TetR fragment to avoid changes in the genetic context and strength of the RBS used to initiate translation.41 In the absence of inducers, CS50-YFP cells transformed with vectors expressing these split proteins (180-K, 184-K, and 193-K) presented YFP fluorescence that was 7 to 11-fold higher than that observed with cells expressing parental TetR (Figure 2D, p<0.001). With cells expressing 184-K and 193-K, induction with IPTG had no significant effect on the YFP fluorescence. However, cells expressing 180-K presented a decrease in YFP fluorescence corresponding to 38% of the signal obtained from cells transformed with the empty vector (Figure 2D, p<0.001). Addition of aTc and IPTG to cells expressing 180-K led to a considerably higher increase in YFP fluorescence compared to that observed upon addition of only IPTG (Figure 2D, p<0.001), indicating that this sTetR retains activity upon aTc induction. These data, taken together, indicate that TetR fragment complementation is enhanced by the EK peptides, albeit to varying levels that depend upon the site of protein fission.
We hypothesized that our initial library expressing TetR fragments as fusions to peptides that associate to form a parallel coiled coil might have resulted in isolation of a limited number of active sTetR variants. Fusion of the TetR fragments to the IAAL-E3/IAAL-K3 peptides results in a ~30 Å separation between the termini that become fused to the TetR fragments,42 which could be disruptive to the function of many two-fragment TetR variants. To investigate whether additional sTetR could be discovered, we created a second library in which the fragments of each sTetR were expressed as fusions to SYNZIP17 and SYNZIP18 peptides, which associate more strongly (KD<10 nM) to form an antiparallel coiled coil.43,44 Fusion of the TetR fragments to these peptides results in smaller separation between the peptide-fused termini (<10 Å).43,45 Screening clones from the TetR-SYNZIP library (n = 640) yielded 80 colonies with YFP fluorescence significantly (≥5σ) lower than that of cells lacking TetR (Figure S3). Sequencing revealed 6 unique sTetR including three variants having the same peptide backbone fission sites as the variants identified from the screening of the TetR-EK library (180-SZ17/18, 184-SZ17/18 and 193-SZ17/18) and three additional variants (74-SZ17/18, 166-SZ17/18 and 167-SZ17/18). The additional sTetR variants arise from peptide backbone fission within the loop that connects the DNA-binding domain and the regulatory domain (74-SZ17/18), and within the linker that connects helices α8 and α9 (166-SZ17/18 and 167-SZ17/18) (Figure 3A).38
Figure 3.

Functional sTetR variants identified in the TetR-SYNZIP library. (A) Fission sites of sTetR variants mapped onto TetR structure (PDB 4AC0). The first and second fragments of sTetR are shown in blue and red, respectively. The linker that is disordered in the TetR structure is shown as a dashed line. (B) Fluorescence of E. coli CS50-YFP cells (−) and E. coli CS50-YFP cells expressing TetR or functional sTetR variants found in the TetR SYNZIP library grown ±IPTG (10μM) and ±aTc (200 ng/mL). (C) Fluorescence of E. coli CS50-YFP cells (−) and E. coli CS50-YFP cells expressing TetR or the sTetR variants lacking the SYNZIP17 peptide. Relative fluorescence values were calculated by normalizing the YFP fluorescence intensity of each sample to that of cells lacking TetR (−). All data are presented as mean ± s.d. (n=3, p<0.001, two tailed t-test).
To better understand the relative activities of each sTetR variant isolated from the TetR-SYNZIP library, we analyzed their effects on the YFP fluorescence of CS50-YFP cells. In all cases, cells expressing sTetR presented a YFP signal that was significantly lower than cells lacking TetR (Figure 3B, p<0.001), and this repression was not enhanced by inducing expression of the TetR fragments using IPTG. Moreover, we observed an increase YFP fluorescence upon treatment of cells expressing each sTetR variant with aTc (Figure 3B, p<0.001), indicating that each sTetR displays aTc-dependent activity. Similar to what was observed in cells expressing TetR, the level of derepression was decreased upon induction of sTetR protein fragments using IPTG.
To investigate whether the SYNZIP17/SYNZIP18 peptides are required for TetR-fragment complementation, we generated vectors that express each sTetR as a fusion to only the SYNZIP18 peptide, which is fused to the C-terminal TetR fragment in each variant; the N-terminal fragments lacked a peptide. All of these sTetR variants displayed decreased repression activity in CS50-YFP compared to cognate split proteins fused to both SYNZIP17 and SYNZIP18 (Figure 3C). Many of the variants, however, displayed significant repression compared to cells transformed with the empty vector (Figure 3C, p<0.001). The repression achieved with three of these variants (167-SZ18, 180-SZ18, and 184-SZ18) was enhanced upon induction with IPTG (Figure 3C, p<0.001), an effect that was found to remain aTc-dependent.
sTetR functions as a genetic AND gate.
The variation in sTetR repression observed using sTetR ±SYNZIP17 suggested that many of these variants require both fragments for activity. To test this hypothesis, we further characterized the sTetR screened from the SYNZIP library. The fragments encoding each sTetR variant were cloned into separate vectors under the control of an IPTG-inducible promoter (PT5) and an arabinose-inducible promoter (PBAD), respectively. To evaluate whether both fragments are required for sTetR activity, CS50-YFP cells were transformed with these plasmids, and YFP fluorescence was analyzed upon addition of high levels of each inducer, IPTG and arabinose (±aTc). As controls, we monitored the fluorescence of cells lacking sTetR or expressing the parental TetR. The YFP fluorescence of cells expressing the individual fragments of 74-SZ17/18, 166-SZ17/18, 167-SZ17/18, 180-SZ17/18 and 184-SZ17/18 was comparable to that of cells lacking TetR, and was not affected by addition of IPTG and arabinose (Figure 4). These findings demonstrate that neither the N- or C-terminal fragment of these sTetR variants is sufficient alone to repress YFP expression. In contrast, the YFP fluorescence of cells expressing the first fragment of 193-SZ17/18 in the presence of IPTG and arabinose decreased to a level corresponding to 26% of the fluorescence of cells lacking TetR (Figure 4, p<0.001). This finding suggests that this sTetR variant, when expressed at high level, requires only the first fragment to function as a repressor.
Figure 4.

Two-input transcriptional regulation of sTetR in bacterial cells. E. coli CS50-YFP cells were transformed with pairs of vectors expressing the different fragments of the sTetR SYNZIP variants using IPTG- and arabinose-inducible promoters, respectively, or expressing TetR alone. Cells were grown in the presence of IPTG (1 mM) and arabinose (1 mM), and the signal was measured upon induction with aTc (2 μg/mL). Relative fluorescence values were calculated by normalizing the YFP fluorescence intensity of each sample to that of cells containing a pair of empty vectors (-,-). Data are presented as mean ± s.d. (n=6, p<0.001, two tailed t-test).
Co-expression of both fragments of the sTetR variants 166-SZ17/18, 167-SZ17/18, 180-SZ17/18 and 184-SZ17/18 resulted in a 93–99% decrease in YFP fluorescence (Figure 4, p<0.001). Moreover, addition of aTc to cells coexpressing both fragments caused an increase in YFP fluorescence similar to that observed upon expression of sTetR from the expression vectors used for library screening (Figure 4, p<0.001). The YFP fluorescence of cells expressing both fragments of 74-SZ17/18 was comparable to that of cells not expressing TetR, but was found to decrease in the presence of IPTG and arabinose (Figure 4, p<0.001). These results indicate that all the sTetR variants screened with the exception of 193-SZ17/18 exhibit an AND gate genetic logic, which can be regulated through addition of aTc.
To investigate whether sTetR can function as a transcriptional regulator in mammalian cells, we evaluated the ability of 184-EK and 184-SZ17/18 to repress GFP expression in cell cultures. These variants were chosen because they display TetR-like repression in the absence of aTc, and they were generated from fission proximal to the C-terminus of TetR. The fragments of each sTetR variant were cloned into separate vectors and placed under the control of a constitutive CMV promoter (Figure 5A). In each vector, the gene encoding the TetR fragments was followed by an IRES46 sequence and the gene encoding eqFP65047 or iRFP,48 respectively. These fluorescent proteins served as transfection controls. To evaluate the activities of each sTetR, HEK 293T and HeLa cells were transfected with a reporter plasmid (pTO_GFP)40 encoding GFP under the control of the Tc-inducible CMV/TO promoter and either one or both plasmids encoding the first and second fragments of 184-EK or of 184-SZ17/18. When HEK 293T (Figure 5B) or HeLa (Figure 5C) cells were transfected for the expression of only one of the sTetR fragments, the GFP fluorescence was comparable to that of cells lacking TetR (±Tc), suggesting that the individual fragments of each sTetR are not sufficient to repress GFP expression. The GFP fluorescence of HeLa cells transfected to express both fragments of the 184-SZ17/18 variant decreased to a level corresponding to 30% of the value observed in control cells lacking TetR (Figure 5B, p<0.001). Similar results were obtained when experiments were conducted using HEK 293T cells (Figure 5C). In contrast, expression of both fragments of 184-EK was found to decrease the GFP fluorescence of HeLa cells to a level corresponding to 75% of the value observed in control cells lacking TetR (Figure 5B, p<0.001), and did not to alter the GFP fluorescence of HEK 293T cells. These findings suggest that the efficiency of transcriptional repression achieved with the sTetR may be cell-type specific. These results also support the notion that the difference in transcriptional repression observed upon expression of sTetR variants based on the EK or SYNZIP interacting pairs is likely due to the higher binding affinity of the SYNZIP17/SYNZIP18 peptides compared to the IAAL-E3/IAAL-K3 peptides.
Figure 5.

sTetR functions as an AND gate in mammalian cells. (A) Schematic representation of the sTetR expression system in mammalian cells. TetR fragment complementation results in formation of a functional repressor that binds to the tetracycline operator (TO) within the CMV/TO promoter and represses the expression of GFP. Addition of tetracycline (Tc) displaces TetR from TO and activates GFP expression. (BC) Comparison of 184-EK and 184-SZ17/18 AND gate logic in (B) HeLa and (C) HEK 293T cells. Fluorescence of HeLa cells transfected for the expression of GFP under the control of CMV/TO and a plasmid encoding TetR, or the two plasmids encoding the sTetR fragments (F1 and F2), or a control plasmid lacking the gene encoding for either sTetR fragment and cultured ±Tc (2 μg/mL). Relative GFP fluorescence values were calculated by normalizing the GFP fluorescence intensity of transfected cells (iRFP-positive) of each sample to the iRFP fluorescence intensity, and dividing the resulting GFP signals to the normalized signal of cells not expressing TetR (−). All data are presented as mean ± s.d. (n=3, p<0.001, two tailed t-test).
To test whether 184-SZ17/18 retains ligand-dependent activity in mammalian cells, we evaluated the effect of Tc concentration on the GFP fluorescence of HEK 293T cells transfected for the expression of this sTetR variant. GFP fluorescence increased as a function of Tc dosage (Figure S4), ranging from 9 to 80% of the signal obtained with cells lacking TetR. The Tc-dependent signal increase in HEK 293T cells expressing 184-SZ17/18 was comparable to that of cells expressing the parental TetR, which presented a signal ranging from 1% to 70% of that observed in cells lacking TetR over the same range of Tc concentration. This finding demonstrates that peptide backbone fission does not affect TetR binding to TO or Tc when the fragments are fused to SYNZIP17 and SYNZIP18 peptides,49 and demonstrates that the circuit output (GFP fluorescence) can be tuned by varying the Tc dosage (Figure S4).
Using sTetR as a protein solubility sensor.
To evaluate whether the sTetR that functions in mammalian cells can be used to build a cell-based sensor that detects and quantifies protein aggregation in mammalian cells (Figure 6A), we developed a protein solubility sensor based on the expression of GFP under the control of the CMV/TO promoter. To couple GFP fluorescence to the aggregation of a protein of interest, the smaller fragment of 184-SZ17/18 was fused to an aggregation-prone protein and co-expressed with the larger fragment of 184-SZ17/18. We hypothesized that complementation of the TetR fragments, and thus repression of GFP, would be contingent upon the solubility of the protein of interest as aggregation of the protein of interest would result in sequestration of the smaller TetR fragment into aggregates, preventing TetR fragment complementation.
Figure 6.

Monitoring protein aggregation using the sTetR-based protein solubility sensor. (A) Design of the sTetR-based protein solubility sensor. By linking protein aggregation to the GFP output, sTetR serves as a protein solubility sensor that generates a positive correlation between protein aggregation and GFP output signal. A protein of interest (POI) is fused to the C-terminus of the small sTetR fragment (TetR2). When the POI is soluble, sTetR fragments (TetR1 and TetR2) can associate and repress GFP expression. Aggregation of the fusion protein prevents the smaller fragment from binding the larger fragment, thus preventing complementation of the sTetR fragments and activating GFP expression. (B) Fluorescence of HEK 293T cells transfected for the expression of GFP under the control of CMV/TO and the two plasmids encoding the large sTetR fragment (F1) and the small sTetR fragment fused to different POI (F2-Q23, F2-Q73, F2-Q103, or a control plasmid lacking a POI). The GFP fluorescence intensity of transfected cells (iRFP-positive) of each sample was divided by iRFP fluorescence intensity, and normalized to that of cells not expressing TetR (−). Data are presented as mean ± s.d. (n=3, *p<0.05, **p<0.01, two tailed t-test). (C) Total protein aggregation of HEK 293T cells transfected as described in (B) evaluated by quantifying binding of the ProteoStat dye of transfected cells and calculating the APF of cells expressing the protein of interest as fusion to the small sTetR fragment relative to cells expressing the small sTetR fragment without fusion to the target protein as described in Materials and Methods. Data are presented as mean ± s.d. (n=3, *p<0.05, two tailed t-test). (D) Quantification of aggregation of HTT-Q103 and α-synuclein using the sTetR sensor. HEK 293T cells were transfected as described in (B) with TetR2 fused to HTT-Q103 or α-synuclein. Data are presented as mean ± s.d. (n=3, p<0.001, two tailed t-test). n.s., not significant.
To first calibrate how the output signal of our solubility sensor relates to the relative amount of the sTetR and GFP, HEK 293T cells were transfected with different ratios (1:3, 1:6 and 1:12) of reporter plasmid (pTO_GFP) and a plasmid that expresses full-length TetR or 184-SZ17/18 (Figure S5). Experiments based on the expression of full-length TetR yielded similar GFP signals with the three plasmid ratios, and a similar GFP increase upon addition of Tc. Similar results were obtained upon expression of 184-SZ17/18. Because these results indicate that the dynamic range of the solubility sensor is not affected by the relative expression level of GFP and TetR under the condition tested, we evaluated the utility of the sTetR-based expression system as a solubility sensor using a 1:6 ratio of pTO_GFP and the plasmids encoding the sTetR.
To evaluate 184-SZ17/18 sTetR as a genetic tool to track protein aggregation, three variants of the N-terminal fragment of human Huntingtin (HTT) exon 1 containing 23, 73 and 103 glutamine repeats (Q23, Q73, and Q103, respectively) were targeted for analysis. Because previous studies have demonstrated a correlation between HTT aggregation and the number of glutamine repeats in the protein sequence,50,51 we expected to observe an increase in GFP fluorescence as a function of the number of glutamine repeats in the HTT variants. To perform these experiments, we compared the fluorescence of HEK 293T cells transfected with the reporter plasmid (pTO_GFP) and vectors for the expression of sTetR with its smaller fragment produced as a fusion to the protein of interest (pF1-SZ17, and pSZ18-F2-POI) to that of cells transfected with reporter plasmid and plasmids expressing the sTetR without protein fusion (pF1-SZ17, and pSZ18-F2). The plasmids encoding the sTetR were designed to link the expression of the small and large sTetR fragments to that of eqFP and iRFP, respectively, through the use of internal ribosome entry site (IRES) sequences. In all experiments, GFP fluorescence was normalized to that of iRFP to account for variations in the protein expression levels. The GFP fluorescence of cells transfected for the expression of the three different HTT variants was higher than that of control cells (Figure 6B). Specifically, cells expressing the solubility sensor fused to HTT containing Q23, Q73, and Q103 were observed to display 2.3-, 3.8- and 4.4-fold increases in GFP fluorescence compared to cells expressing the solubility sensor lacking HTT (Figure 6B, *p<0.05, **p<0.01).
To confirm that fusion of the HTT variants to the TetR fragment does not affect HTT aggregation propensity, we monitored the extent of total protein aggregation in cells expressing the HTT variant fusion constructs (pF1-SZ17, and pSZ18-F2-POI). Specifically, we quantified the aggregation propensity factor (APF) of HEK 293T cells transfected identically as in the experiments conducted to evaluate whole cell fluorescence. APF measurements revealed an increase in total protein aggregation in cells expressing HTT variants compared to control cells expressing the TetR fragments without fusion to the HTT variants and this increase correlated with the number of glutamine repeats in the HTT variants (Figure 6C, *p<0.05). These results confirm that fusion of the HTT variants to the smaller fragment of sTetR does not abolish HTT aggregation. More importantly, the changes in total protein aggregation upon expression of the HTT variants correlate with the changes in GFP fluorescence observed in the context of the solubility sensor, demonstrating a correlation between the GFP signal from the sTetR-based aggregation sensor and the aggregation status of the fusion protein. The limit of detection of the solubility sensor could also be easily tuned using tetracycline (Figure S6). This property provides a simple mechanism to to tune the sensitivity of sTetR-based solubility sensor for quantifying changes in the soluble levels of proteins with particularly low aggregation propensity.
To investigate the generality of the sTetR-based solubility sensor for monitoring aggregation of different cellular proteins, we also evaluated the GFP signal of this solubility sensor upon expression of α-synuclein (α-syn), an unstructured and misfolding-prone protein associated with the development of Parkinson’s disease.52 When the smaller TetR fragment was expressed as a fusion to α-syn, whole cell GFP fluorescence increased ~3-fold compared to cells expressing the same TetR fragment without fusion to an aggregation-prone protein (Figure 6D, p<0.001). The GFP signal of cells expressing the solubility sensor fused to HTT-Q103 is ~40% higher than that of cells expressing α-syn, which is consistent with results from APF measurements (Figure S7). These results suggest that the sTetR-based solubility sensor is sensitive to small changes in aggregation of two unrelated proteins of similar size and different aggregation propensities, indicating that the expression of the output signal does not depend on the size of the target protein. Taken together, these results demonstrate that the sTetR-based solubility sensor generated in this study can be used in a genetic assay to monitor the aggregation status of different proteins in mammalian cells and the effects of mutations on the proteins’ aggregation propensity.
DISCUSSION
Monitoring protein aggregation in mammalian cells is critically important to establishing the molecular and cellular mechanisms underlying the formation of off-pathway intermediates, which are often associated with the development of a large range of human diseases.1–3,53 This study aimed to develop a genetic tool for monitoring protein aggregation in cells that can serve as an alternative to existing approaches based on purified proteins,54,55 antibody-based solubility screens,56,57 split GFP complementation assays,19,20,58,59 fusion to fluorescent reporters,17,18 and to synthetic genetic regulators.31 To link the target protein aggregation status to a reporter output with minimal perturbation of the target’s aggregation propensity, we explored fission of the ligand-dependent transcription factor TetR, identified active split TetR variants, and demonstrated their function as logic AND gates in mammalian cells. Specifically, this study generated a set of split TetR variants that regulate gene expression and respond to induction with aTc and Tc in bacteria and mammalian cells, respectively. The aggregation sensor was built by coexpressing the fragments of a sTetR consisting of a large “detector” fragment and a small “sensor” fragment fused to the target protein. We demonstrated that fusion to the small “sensor” fragment does not affect the aggregation propensity of the target proteins tested in this study, namely variants of HTT exon 1 with glutamine repeats and α-syn, as indicated by the correlation between the output signal of the solubility sensor and a previously established tool to monitor protein aggregation (APF). Aggregation of the target protein results in sequestration of the “sensor” fragment, which prevents complementation and results in increase in expression of the fluorescent reporter.
Unlike existing approaches to monitor protein aggregation, this sTetR-based sensor allows linking aggregation of a target protein within cells to a readily detectable output and responds to control through a small molecule ligand. Control of sTetR through a small molecule inducer is likely to provide a means to finely tune the sensor for monitoring the status of target proteins with different aggregation propensities with superior sensitivity.40 In addition to enabling quantification of protein aggregation with high sensitivity, this method is also likely not to be plagued by the high rate of false positive signal of split GFP complementation assays that arise from formation of a stable fluorophore preceding aggregation of the target.58,59
Our library screening identified five sTetR that could be used as AND gates in synthetic circuits for a range of applications. Previous studies have described the development of transcriptional activators through protein fission, such as bipartite and tripartite T7 RNA polymerases,25,29,60 which can be used to control resource allocation in cells. However, their transcriptional regulation cannot be tuned directly through fast ligand binding. In contrast, all of sTetR variants identified as part of the present study display aTc-dependent repression in E. coli. Similar protein design approaches could be applied to other members of the TetR family of regulators to diversity the array of ligand-dependent split regulators available for synthetic biology applications.61 While transcriptional AND gates have previously been discovered in bacteria29,30 and yeast,62 this study also provides the first report of a split transcription factor that can be used to construct transcriptional logic gates in mammalian cells. The sTetR variants generated as part of the present study thus provide a novel and much needed part for the mammalian synthetic biology toolbox that enables the creation of transcriptional AND gates. We anticipate that sTetR could be further optimized by engineering the interacting peptides assisting complementation,33 and the affinity and specificity of DNA binding.63
In summary, we reported a series of sTetR variants for building AND logic gates within complex genetic circuit and the use of sTetR for generating a cell-based solubility sensor to quantify protein aggregation, which could be ultimately used to study the cellular pathogenesis of protein misfolding diseases and investigate approaches to interfere with the accumulation of proteinaceus aggregates in cells for therapeutic applications.
MATERIALS AND METHODS
Strain constructions.
The promoter-RBS-YFP gene fusion from pLtet-YFP (a generous gift from Dr Matthew Bennett) was integrated into E. coli CS5044 using Clonetegration64 at the 186 primary integration site. The YFP gene fusion was amplified from pLtet-YFP and cloned into the pOSIP-KO (Addgene #45985) using BamHI-HF and PstI (New England Biolabs). The resulting vector was transformed into E. coli CS50, cells were plated on LB-agar containing kanamycin (25 μg/mL), and plates were incubated at 30°C for 24 hours. Colony PCR was used to identify a strain with the desired chromosomal insert. These cells were made competent using the Mix & Go Competent Cells kit (Zymo Research), and the resulting cells were transformed with the pE-FLP (Addgene #45978) plasmid using heat shock, which encodes the flipase enzyme to remove the integration module from pOSIP-KO. Cells were plated on LB-agar plates containing ampicillin (50 μg/mL) and incubated at 30°C for 24 hours to excise the integration module. To screen for cells where the integration module was excised, individual colonies were streaked on LB-agar plates containing or lacking kanamycin. A colony that grew only in the absence of kanamycin but retained the integrated YFP module was identified and designated E. coli CS50-YFP. This strain was used to screen sTetR libraries for functional protein variants.
Library construction.
A library of randomly fragmented TetR variants was constructed using pTAC_TetR, a pGEX-2TK derivative that contains the TetR gene under the control of the tac promoter. First, a Mu library of TetR variants was created by randomly inserting a minitransposon into the TetR gene as previously described.65 Briefly, a NotI-flanked minitransposon encoding the kanamycin resistance cassette (M1-KanR, Thermo Scientific) was inserted into pTAC_TetR using the HyperMu MuA transposase (Epicentre Biotechnologies) by incubating 20 μl reactions containing 300 ng of pTAC_TetR, 100 ng of M1-KanR, and 1 Unit of HyperMu MuA transposase in HyperMu buffer at 37°C for 16 h. The products of the transposase reactions were purified using the Zymo DNA Clean & Concentrator kit (Zymo Research) and transformed into ElectroMAX DH10B cells (Invitrogen). Transformed cells were plated onto LB-agar plates containing 25 μg/ml kanamycin and incubated at 37°C for 16 h. Colonies were harvested and pooled, and the plasmid DNA was purified using a Qiagen Miniprep Kit to obtain the TetR-Mu library. The TetR-Mu library (200 ng) was digested with restriction enzymes BamHI-HF and KpnI-HF (New England Biolabs) that cut at sites flanking the tetR gene. DNA fragments were separated using agarose gel electrophoresis, and the tetR-transposon hybrids (1.7 kb) were purified from the rest of the fragments, which included the 0.6 kb parental tetR fragment, the 4.3 kb backbone fragment, and 5.4 kb vector backbone containing a 1.1 kb transposon fragment. The ensemble of tetR-transposon hybrid fragments was subcloned into the pTAC expression vector to create a size-selected library (SS library).65 The minitransposon in the tetR gene variants was replaced by subcloning the DNA cassettes ek-kanR or synzip-kanR into the NotI sites, thus creating two libraries of vectors that express different fragmented TetR, including: (i) the TetR-EK library which expresses N-terminal and C-terminal TetR fragments fused to the IAAL-E3 and IAAL-K3 peptides at their C- and N-terminal ends, respectively and (ii) the TetR-SYNZIP library which expresses TetR fragments fused to the SYNZIP17 and SYNZIP18 peptides at their C- and N-terminal ends, respectively. The theoretical size of each sTetR library that can be generated through random gene fission (1246 variants) was determined by multiplying the number of possible split sites in the TetR gene (623) by the number of orientations (2) of the synthetic DNA insert encoding the interacting peptides (ek-kanR or synzip-kanR).33 We estimated the size of the library obtained upon transformation of E. coli CS50-YFP cells to be more than 10,000 colonies, which is about 10-fold higher than the maximum theoretical number of sTetR variants. Randomly chosen clones were sequenced with a primer that hybridizes within the ek-kanR and synzip-kanR insert.65 Both sTetR variants with EK or SYNZIP peptide fusion are referred to as “sTetR variants” hereafter unless otherwise specified.
Screening.
E. coli CS50-YFP cells were transformed with pTAC, pTAC-TetR, or with a TetR library and grown on LB-agar plates supplemented with 100 μg/mL ampicillin overnight at 37°C. Single colonies from these plates were used to inoculate 1mL LB cultures containing 50 μg/mL ampicillin in 96-well deep well plates, and plates were incubated for 16 h at 37°C while shaking at 250 rpm. Aliquots (20 μL) of the stationary phase cultures were diluted 1:10 into LB containing 50 μg/mL ampicillin and 10 μM IPTG in clear polystyrene 96-well flat bottom plates (Costar). These cultures were grown for 4 h at 37°C, and whole cell fluorescence (λex = 510 nm; λem = 527 nm) was measured using a TECAN infinite M1000 PRO plate reader and normalized to the cell absorbance (600 nm). Samples yielding normalized fluorescence at least 5σ lower than that of the sample transformed with pTAC were selected, purified vectors were sequenced, and the sTetR variants were named after the last residue of the N-terminal TetR fragment followed by the letters designating the coiled-coil fused to the termini at the split site, where EK signifies IAAL-E3 and IAAL-K3 and SZ17/18 signifies SYNZIP17 and SYNZIP18.
Bacterial vector construction.
The TetR gene and pGEX vector backbone were PCR amplified from pcDNA6/TR (Invitrogen) and pGEX2TK-IFP1.4,33 respectively, and these were assembled to create a plasmid that expresses TetR using tac promoter (pTAC-TetR) via Gibson Assembly66 using the Gibson Assembly® Master Mix (New England Biolabs). pTAC-TetR was digested with BamHI-HF and KpnI-HF (New England Biolabs), treated with Mung Bean Nuclease (New England Biolabs) to create blunt ends, and self-ligated to generate pTAC, the control vector lacking the TetR gene. To generate pair of vectors for independent expression of the TetR fragments encoding each SYNZIP variant, the individual fragments were PCR amplified from the vector identified in our library screen, the ORFs encoding the first TetR fragments fused to SYNZIP17 was cloned into pQE80 (QIAGEN), and the ORFs encoding SYNZIP18 fused to the second TetR fragments were cloned into pTara.67 An IPTG-inducible T5 promoter controls expression in the plasmid pQE80, while pTara uses an arabinose-inducible araBAD promoter. Experiments using these vectors were performed in the presence of both 1 mM IPTG and 1 mM arabinose.
Bacterial fluorescence measurements.
E. coli CS50-YFP were transformed with vectors encoding the sTetR variants and plated onto LB-agar plates supplemented with 100 μg/mL ampicillin. Plates were incubated overnight at 37°C, and individual colonies were inoculated into 96-well deep well plates containing 1mL LB and 50 μg/mL ampicillin. Liquid cultures were incubated for 16 h at 37°C while shaking at 250 rpm. Aliquots (20 μL) of the stationary phase cultures were then diluted 1:10 into LB containing 50 μg/mL ampicillin, 10 μM IPTG, and 200 ng/mL aTc in 96-well flat bottom plates. Whole cell fluorescence (λex = 510 nm; λem = 527 nm) and absorbance (600 nm) were acquired after 4 h of incubation at 37°C using a TECAN infinite M1000 PRO plate reader. Fluorescence measurements were normalized to absorbance measurements of each well, and expressed as fluorescence relative to that of cells lacking tetR.
Mammalian vector construction.
PCR amplifications of small DNA fragments (shorter than 2000 bp) were performed using Vent® DNA polymerase (New England Biolabs), while PCR amplifications of large fragments (longer than 2000 bp) were performed using KAPA HiFi™ HotStart DNA polymerase (KAPA Biosystems), according to the manufacturer’s protocol. All mammalian expression vectors were constructed via Gibson Assembly66 using the Gibson Assembly® Master Mix (New England Biolabs) according to the manufacturer’s protocol, and verified by DNA sequencing.
pTO_GFP, which encodes emGFP under the control of the CMV/TO promoter, was generated as previously described.40 The 184-EK and 184-SZ TetR variants were amplified via PCR and cloned into ptTA as follows. The larger TetR fragments were cloned into ptTA40 by replacing the tTA cassette with a fragment consisting of the larger TetR variant F1-E3 or F1-SZ17, an IRES (internal ribosome entry site) sequence amplified from pMSCV PIG (Addgene plasmid#21654), and the gene encoding eqFP650 amplified from ptTA, thereby generating pF1-E3 and pF1-SZ17, respectively. The smaller TetR fragments were cloned into ptTA by replacing the tTA cassette with a fragment consisting of the smaller TetR variant K3-F2 or SZ18-F2, the IRES sequence, and the gene encoding iRFP amplified from piRFP (Addgene plasmid#31857), thereby generating pK3-F2 and pSZ18-F2, respectively.
The plasmids used for protein solubility assays were constructed by cloning genes encoding aggregation-prone proteins, HTT (human huntingtin) exon 1 Q23, Q53, Q73, Q103 and α-synuclein, at the C-terminal of the gene encoding the smaller fragment of TetR in the pSZ18-F2 plasmid, thereby generating plasmids pSZ18-F2-Q23, pSZ18-F2-Q53, pSZ18-F2-Q73, pSZ18-F2-Q103 and pSZ18-F2-α-syn, respectively. The cDNAs of HTT exon 1 Q23 and Q7351 were amplified from pEGFP-Q23 (Addgene plasmid#40261) and pEGFP-Q74 (Addgene plasmid#40262), respectively. The sequence encoding HTT exon 1 Q73 and Q103 were purchased (GenScript). The cDNA of α-synuclein was amplified from pcDNA6.2+ α-syn-emGFP plasmid.23
Cell cultures and flow cytometry studies.
HEK 293T cells (ATCC) were cultured in high glucose DMEM (Invitrogen) supplemented with 10% fetal bovine serum (FBS, Sigma) and 1% Penicillin-Streptomycin-Glutamine (PSQ, Hyclone) and incubated at 37 °C in humidified 5% CO2 atmosphere. HeLa cells (ATCC) were cultured in DMEM (Lonza) supplemented with 10% FBS and 1% PSQ and incubated at 37 °C and 5% CO2. Cells were added to 12-well plates and transfected using JetPrime (Polyplus transfection) according to the manufacturer’s protocol with 500 ng total DNA unless otherwise stated. Cells were transfected using pTO_GFP and the plasmids expressing the first and second fragments of the sTetR variants in a 1:6:6 ratio. The culture medium of transfected cells was replaced with fresh medium containing 2 μg/mL tetracycline (Invitrogen) 24 h post-transfection. Cells were washed with PBS (Lonza) and harvested using TrypLE (GIBCO® Invitrogen) 48 h post-transfection for analysis.
Flow cytometry analyses were conducted using a FACSCanto II flow cytometer (BD, San Jose, CA) to measure the fluorescence intensity of GFP (488 nm laser, 530/30 nm emission filter), eqFP650 (633 nm laser, 675/25 nm emission filter), and iRFP (633 nm laser, 780/60 nm emission filter). At least 10,000 cells were recorded for analysis in each sample. For transient transfection experiments, GFP fluorescence intensity was measured within iRFP-positive cells to monitor changes within transfected cells. The reported output signal was calculated by normalizing the intensity of GFP by iRFP to eliminate differences arising from transfection efficiencies.
Protein aggregation analyses.
Protein aggregation was measured using the ProteoStat Aggregation detection kit (Enzo Life Sciences) as previously described.68 HEK 293T cells were added to 6-well plates and transfected with 1 μg total DNA using JetPrime. Cells were transfected using a 1:6:6 ratio of pTO_GFP, pF1-SZ17 and pSZ18-F2 or pSZ18-F2-POI and the aggregation propensity factor (APF) was calculated using the following formula: APF = 100 × (MFItreated – MFIcontrol)/MFItreated where MFI is the mean fluorescence intensity of the ProteoStat® dye and cells transfected with pTO_GFP, pF1-SZ17 and pSZ18-F2 were used as control. Fluorescence intensity was measured by flow cytometry (488nm laser, 585/40nm emission filter).
Statistical analyses.
All data are presented as mean ± s.d (n≥3 biological replicates). Statistical significance was calculated using a two-tailed t-test in all experiments.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Dr. Matthew R. Bennett for providing pLtet-YFP. This work was funded by the National Science Foundation (MCB-1615562 and 1150138) and the Welch Foundation (C-1824). AMJ was supported by the National Science Foundation Graduate Research Fellowship Program under grant number (R3E821), and EET was partially funded by a training fellowship from the Keck Center of the Gulf Coast Consortia, on the Houston Area Molecular Biophysics Program, National Institute of General Medical Sciences (NIGMS) T32GM008280.
Footnotes
SUPPORTING INFORMATION
The Supporting Information is available free of charge on the ACS Publications website at DOI:
Data showing the high-throughput screen, library screening results, Tc-dependent activity in mammalian cells, and effect of plasmid transfection ratios on circuit signal (PDF)
REFERENCES
- (1).Thomas PJ, Qu B-H, and Pedersen PL (1995) Defective protein folding as a basis of human disease. Trends Biochem. Sci 20, 456–459. [DOI] [PubMed] [Google Scholar]
- (2).Dobson CM (1999) Protein Misfolding, Evolution and Disease. Trends Biochem. Sci 24, 329–332. [DOI] [PubMed] [Google Scholar]
- (3).Douglas PM, and Dillin A (2010) Protein homeostasis and aging in neurodegeneration. J. Cell Biol 190, 719–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Kopito RR (2000) Aggresomes, inclusion bodies and protein aggregation. Trends Cell Biol. 10, 524–530. [DOI] [PubMed] [Google Scholar]
- (5).Dobson CM (2003) Protein folding and misfolding. Nature 426, 884–90. [DOI] [PubMed] [Google Scholar]
- (6).Majumdar A, Cesario WC, White-Grindley E, Jiang H, Ren F, Khan MR, Li L, Choi EML, Kannan K, Guo F, Unruh J, Slaughter B, and Si K (2012) Critical role of amyloid-like oligomers of Drosophila Orb2 in the persistence of memory. Cell 148, 515–529. [DOI] [PubMed] [Google Scholar]
- (7).Cai X, Chen J, Xu H, Liu S, Jiang QX, Halfmann R, and Chen ZJ (2014) Prion-like polymerization underlies signal transduction in antiviral immune defense and inflammasome activation. Cell 156, 1207–1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Protter DSW, and Parker R (2016) Principles and Properties of Stress Granules. Trends Cell Biol. 26, 668–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Mahler H-C, Friess W, Grauschopf U, and Kiese S (2009) Protein Aggregation: Pathways, Induction Factors and Analysis. J. Pharm. Sci 98, 2909–2934. [DOI] [PubMed] [Google Scholar]
- (10).Harris RJ, Shire RJ, and Winter C (2004) Commercial manufacturing scale formulation and analytical characterization of therapeutic recombinant antibodies. Drug Dev. Res 61, 137–154. [Google Scholar]
- (11).Roberts CJ (2014) Therapeutic protein aggregation: Mechanisms, design, and control. Trends Biotechnol. 32, 372–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Taschenberger G, Garrido M, Tereshchenko Y, Bähr M, Zweckstetter M, and Kügler S (2011) Aggregation of αsynuclein promotes progressive in vivo neurotoxicity in adult rat dopaminergic neurons. Acta Neuropathol. 123, 671–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Johnston JA, Ward CL, and Kopito RR (1998) Aggresomes: A Cellular Response to Misfolded Proteins Aggresomes : Cell 143, 1883–1898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Rideout HJ, Larsen KE, Sulzer D, and Stefanis L (2001) Proteasomal inhibition leads to formation of ubiquitin/α-synuclein-immunoreactive inclusions in PC12 cells. J. Neurochem 78, 899–908. [DOI] [PubMed] [Google Scholar]
- (15).Webb JL, Ravikumar B, Atkins J, Skepper JN, and Rubinsztein DC (2003) α-synuclein Is Degraded by Both Autophagy and the Proteasome. J. Biol. Chem 278, 25009–25013. [DOI] [PubMed] [Google Scholar]
- (16).Paxinou E, Chen Q, Weisse M, Giasson BI, Norris EH, Rueter SM, Trojanowski JQ, Lee VM, and Ischiropoulos H (2001) Induction of alpha-synuclein aggregation by intracellular nitrative insult. J. Neurosci 21, 8053–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Lecerf JM, Shirley TL, Zhu Q, Kazantsev A, Amersdorfer P, Housman DE, Messer A, and Huston JS (2001) Human single-chain Fv intrabodies counteract in situ huntingtin aggregation in cellular models of Huntington’s disease. Proc. Natl. Acad. Sci. U. S. A 98, 4764–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).McLean PJ, Kawamata H, and Hyman BT (2001) α-Synuclein-enhanced green fluorescent protein fusion proteins form proteasome sensitive inclusions in primary neurons. Neuroscience 104, 901–912. [DOI] [PubMed] [Google Scholar]
- (19).Cabantous S, Terwilliger TC, and Waldo GS (2005) Protein tagging and detection with engineered self-assembling fragments of green fluorescent protein. Nat. Biotechnol 23, 102–7. [DOI] [PubMed] [Google Scholar]
- (20).Cabantous S, and Waldo GS (2006) In vivo and in vitro protein solubility assays using split GFP. Nat. Methods 3, 845–854. [DOI] [PubMed] [Google Scholar]
- (21).Chun W, Waldo GS, and Johnson GVW (2007) Split GFP complementation assay: a novel approach to quantitatively measure aggregation of tau in situ: effects of GSK3β activation and caspase 3 cleavage. J. Neurochem 103, 2529–2539. [DOI] [PubMed] [Google Scholar]
- (22).Chun W, Waldo GS, and Johnson GVW (2011) Split GFP Complementation Assay for Quantitative Measurement of Tau Aggregation In Situ. Methods Mol. Biol 670, 169–89. [DOI] [PubMed] [Google Scholar]
- (23).Kothawala A, Kilpatrick K, Novoa JA, and Segatori L (2012) Quantitative analysis of α-synuclein solubility in living cells using split GFP complementation. PLoS One 7, e43505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Lohmueller JJ, Armel TZ, and Silver P. a. (2012) A tunable zinc finger-based framework for Boolean logic computation in mammalian cells. Nucleic Acids Res. 40, 5180–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Boch J, Scholze H, Schornack S, Landgraf A, Hahn S, Kay S, Lahaye T, Nickstadt A, and Bonas U (2009) Breaking the code of DNA binding specificity of TAL-type III effectors. Science (80-.). 326, 1509–12. [DOI] [PubMed] [Google Scholar]
- (26).Qi LS, Larson MH, Gilbert L. a, Doudna J. a, Weissman JS, Arkin AP, and Lim W. a. (2013) Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell 152, 1173–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Guido NJ, Wang X, Adalsteinsson D, McMillen D, Hasty J, Cantor CR, Elston TC, and Collins JJ (2006) A bottom-up approach to gene regulation. Nature 439, 856–860. [DOI] [PubMed] [Google Scholar]
- (28).Shis DL, Hussain F, Meinhardt S, Swint-Kruse L, and Bennett MR (2014) Modular, Multi-Input Transcriptional Logic Gating with Orthogonal LacI/GalR Family Chimeras. ACS Synth. Biol 3, 645–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Shis DL, and Bennett MR (2013) Library of synthetic transcriptional AND gates built with split T7 RNA polymerase mutants. Proc. Natl. Acad. Sci. U. S. A 110, 5028–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Schaerli Y, Gili M, and Isalan M (2014) A split intein T7 RNA polymerase for transcriptional AND-logic. Nucleic Acids Res. 42, 12322–12328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Newby GA, Kiriakov S, Hallacli E, Kayatekin C, Tsvetkov P, Mancuso CP, Bonner JM, Hesse WR, Chakrabortee S, Manogaran AL, Liebman SW, Lindquist S, and Khalil AS (2017) A Genetic Tool to Track Protein Aggregates and Control Prion Inheritance. Cell 171, 966–979.e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Nguyen PQ, Liu S, Thompson JC, and Silberg JJ (2008) Thermostability promotes the cooperative function of split adenylate kinases. Protein Eng. Des. Sel 21, 303–310. [DOI] [PubMed] [Google Scholar]
- (33).Pandey N, Nobles CL, Zechiedrich L, Maresso AW, and Silberg JJ (2015) Combining Random Gene Fission and Rational Gene Fusion to Discover Near-Infrared Fluorescent Protein Fragments That Report on Protein-Protein Interactions. ACS Synth. Biol 4, 615–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Mehta MM, Liu S, and Silberg JJ (2012) A transposase strategy for creating libraries of circularly permuted proteins. Nucleic Acids Res. 40, e71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Chrast-Balz J, and Hooft van Huijsduijnen R (1996) Bi-directional gene switching with the tetracycline repressor and a novel tetracycline antagonist. Nucleic Acids Res. 24, 2900–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Lutz R, and Bujard H (1997) Independent and tight regulation of transcriptional units in Escherichia coli via the LacR/O, the TetR/O and AraC/I1–I2 regulatory elements. Nucleic Acids Res. 25, 1203–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Khlebnikov a, Risa O, Skaug T, Carrier T. a, and Keasling JD (2000) Regulatable arabinose-inducible gene expression system with consistent control in all cells of a culture. J. Bacteriol 182, 7029–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Ramos JL, Martínez-bueno M, Antonio J, Terán W, Watanabe K, Gallegos MT, Brennan R, Martı M, Molina-henares AJ, and Tera W (2005) The TetR Family of Transcriptional Repressors. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Kim HJ, Gatz C, Hillen W, and Jones TR (1995) Tetracycline repressor-regulated gene repression in recombinant human cytomegalovirus. J. Virol 69, 2565–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Zhao W, Bonem M, McWhite C, Silberg JJ, and Segatori L (2014) Sensitive detection of proteasomal activation using the Deg-On mammalian synthetic gene circuit. Nat. Commun 5, 1–12. [DOI] [PubMed] [Google Scholar]
- (41).Salis HM, Mirsky EA, and Voigt CA (2009) Automated design of synthetic ribosome binding sites to control protein expression. Nat. Biotechnol 27, 946–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Lindhout DA, Litowski JR, Mercier P, Hodges RS, and Sykes BD (2004) NMR solution structure of a highly stable de novo heterodimeric coiled-coil. Biopolymers 75, 367–375. [DOI] [PubMed] [Google Scholar]
- (43).Reinke AW, Grant RA, and Keating AE (2010) A Synthetic Coiled-Coil Interactome Provides Heterospecific Modules for Molecular Engineering. J. Am. Chem. Soc 6025–6031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Thomas EE, Pandey N, Knudsen S, Ball ZT, and Silberg JJ (2017) Programming Post-Translational Control over the Metabolic Labeling of Cellular Proteins with a Noncanonical Amino Acid. ACS Synth. Biol 6, 1572–1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Thompson KE, Bashor CJ, Lim W. a., and Keating AE (2012) Synzip protein interaction toolbox: In vitro and in vivo specifications of heterospecific coiled-coil interaction domains. ACS Synth. Biol 1, 118–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Jang SK, Kräusslich HG, Nicklin MJ, Duke GM, Palmenberg AC, and Wimmer E (1988) A segment of the 5’ nontranslated region of encephalomyocarditis virus RNA directs internal entry of ribosomes during in vitro translation. J. Virol 62, 2636–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Shcherbo D, Shemiakina II, Ryabova AV, Luker KE, Schmidt BT, Souslova E. a, Gorodnicheva TV, Strukova L, Shidlovskiy KM, Britanova OV, Zaraisky AG, Lukyanov K. a, Loschenov VB, Luker GD, and Chudakov DM (2010) Near-infrared fluorescent proteins. Nat. Methods 7, 827–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Filonov GS, Piatkevich KD, Ting LM, Zhang J, Kim K, and Verkhusha VV (2011) Bright and stable near-infrared fluorescent protein for in vivo imaging. Nat Biotechnol 29, 757–761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Gossen M, and Bujard H (1992) Tight control of gene expression in mammalian cells by tetracycline-responsive promoters. Proc. Natl. Acad. Sci. U. S. A 89, 5547–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Li SH, and Li XJ (1998) Aggregation of N-terminal huntingtin is dependent on the length of its glutamine repeats. Hum. Mol. Genet 7, 777–782. [DOI] [PubMed] [Google Scholar]
- (51).Narain Y, Wyttenbach a, Rankin J, Furlong R. a, and Rubinsztein DC (1999) A molecular investigation of true dominance in Huntington’s disease. J. Med. Genet 36, 739–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Shults CW (2006) Lewy bodies. Proc. Natl. Acad. Sci. U. S. A 103, 1661–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Gestwicki JE, and Garza D (2012) Protein quality control in neurodegenerative disease. Prog. Mol. Biol. Transl. Sci. 1st ed Elsevier Inc. [DOI] [PubMed] [Google Scholar]
- (54).Wang W, Perovic I, Chittuluru J, Kaganovich A, Nguyen LTT, Liao J, Auclair JR, Johnson D, Landeru A, Simorellis AK, Ju S, Cookson MR, Asturias FJ, Agar JN, Webb BN, Kang C, Ringe D, Petsko G. a, Pochapsky TC, and Hoang QQ (2011) A soluble α-synuclein construct forms a dynamic tetramer. Proc. Natl. Acad. Sci. U. S. A 108, 17797–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Etzkorn M, Böckmann A, and Baldus M (2011) Kinetic analysis of protein aggregation monitored by real-time 2D solid-state NMR spectroscopy. J. Biomol. NMR 49, 121–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Knaust RK, and Nordlund P (2001) Screening for soluble expression of recombinant proteins in a 96-well format. Anal. Biochem 297, 79–85. [DOI] [PubMed] [Google Scholar]
- (57).Vincentelli R, Canaan S, Offant J, Cambillau C, and Bignon C (2005) Automated expression and solubility screening of His-tagged proteins in 96-well format. Anal. Biochem 346, 77–84. [DOI] [PubMed] [Google Scholar]
- (58).Kazantsev A, Preisinger E, Dranovsky A, Goldgaber D, and Housman D (1999) Insoluble detergent-resistant aggregates form between pathological and nonpathological lengths of polyglutamine in mammalian cells. Proc. Natl. Acad. Sci. U. S. A 96, 11404–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Wigley WC, Stidham RD, Smith NM, Hunt JF, and Thomas PJ (2001) Protein solubility and folding monitored in vivo by structural complementation of a genetic marker protein. Nat. Biotechnol 19, 131–136. [DOI] [PubMed] [Google Scholar]
- (60).Segall-Shapiro TH, Meyer AJ, Ellington AD, Sontag ED, and Voigt CA (2014) A “resource allocator” for transcription based on a highly fragmented T7 RNA polymerase. Mol. Syst. Biol 10, 742–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Routh MD, Su CC, Zhang Q, and Yu EW (2009) Structures of AcrR and CmeR: Insight into the mechanisms of transcriptional repression and multi-drug recognition in the TetR family of regulators. Biochim. Biophys. Acta - Proteins Proteomics 1794, 844–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Fields S, and Sternglanz R (1994) The two-hybrid system: an assay for protein-protein interactions. Trends Genet. 10, 286–292. [DOI] [PubMed] [Google Scholar]
- (63).Helbl V, and Hillen W (1998) Stepwise selection of TetR variants recognizing tet operator 4C with high affinity and specificity 1 1Edited by R. Ebright. J. Mol. Biol 276, 313–318. [DOI] [PubMed] [Google Scholar]
- (64).St-Pierre F, Cui L, Priest DG, Endy D, Dodd IB, and Shearwin KE (2013) One-step cloning and chromosomal integration of DNA. ACS Synth. Biol 2, 537–541. [DOI] [PubMed] [Google Scholar]
- (65).Segall-Shapiro TH, Nguyen PQ, Dos Santos ED, Subedi S, Judd J, Suh J, and Silberg JJ (2011) Mesophilic and hyperthermophilic adenylate kinases differ in their tolerance to random fragmentation. J. Mol. Biol 406, 135–48. [DOI] [PubMed] [Google Scholar]
- (66).Gibson DG, Young L, Chuang R, Venter JC, H. CA Iii, Smith HO, and America, N. (2009) Enzymatic assembly of DNA molecules up to several hundred kilobases 6, 12–16. [DOI] [PubMed] [Google Scholar]
- (67).Wycuff DR, and Matthews KS (2000) Generation of an AraC-araBAD Promoter-Regulated T7 Expression System. Anal. Biochem 277, 67–73. [DOI] [PubMed] [Google Scholar]
- (68).Kilpatrick K, Zeng Y, Hancock T, and Segatori L (2015) Genetic and Chemical Activation of TFEB Mediates Clearance of Aggregated α-Synuclein. PLoS One 10, e0120819. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.