

Abstract
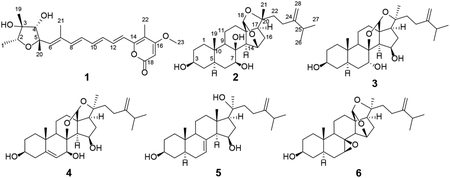
Four new metabolites, 4-epi-citreoviridin (1), auransterol (3), and two analogues (2 and 4) of paxisterol (6), together with two known metabolites (15R*,20S*)-dihydroxyepisterol (5) and (6), were isolated from cultures of the fungal associate, Penicillium aurantiacobrunneum of the lichen, Niebla homalea, endemic to California and Baja California. The structures of all compounds were determined by comprehensive spectroscopic and spectrometric methods, including single-crystal X-ray diffraction for the determination of the absolute configuration of 3. Compound 1 showed selective cytotoxicity towards MCF-7 breast and A2780 ovarian cells with IC50 values of 4.2 and 5.7 μM respectively.
GRAPHICAL ABSTRACT
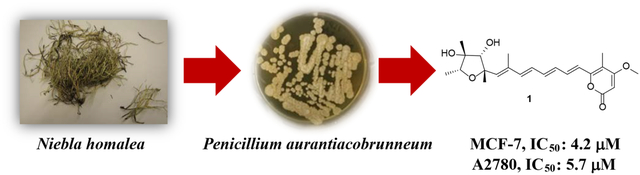
Lichens are composite organisms consisting of photobionts (cyanobacteria and/or microalgae) and mycobionts (fungi) living in symbiosis and have been harnessed for their medicinal properties. For instance, Irish moss, Cetraria islandica (L.) Acharius s.l., has been used for demulcent, appetite stimulation, and topical wound treatment,1 and numerous species from the Usnea genus for antibacterial2 and analgesic purposes.3 Recent investigations of lichens have revealed various examples of biologically active secondary metabolites such as the antimicrobial usnic acid,4 lobaric acid, an arachidonate 5-lipoxygenase inhibitor from Stereocaulon alpinum,5 buellin, a cytotoxic agent against B16 murine melanoma cells and HaCaT human keratinocytes,6 and tumidulin from Patagonian species of Vermilacinia that reduced the stemness potential of colorectal cancer cells.7 The production of these secondary metabolites has been attributed mainly to the mycobionts, present either in the symbiotic or aposymbiotic state, and these mycobionts are capable of producing different profiles of metabolites when stressed.8
Bioactive endemic lichens that inhabit coastal regions of the United States and elsewhere are the consequences of interactions between microorganisms in both marine and terrestrial ecological habitats. We hypothesized that the symbiotic relationship between the exclusive sets of photobionts and mycobionts can produce novel molecular scaffolds, hence providing new pharmacophores for drug targets. In the present pursuit for novel antiproliferative compounds from these niche sources, the fungal associates have been investigated for an unexplored species of a fruticose lichen endemic to the coastal fog regions of western United States and Mexico, Niebla homalea (Ach.) Runder & Bowler, in the family Ramalinaceae. From this species, 18 different fungal strains have been isolated with axenic culture. Of these, Penicillium aurantiacobrunneum, was strategically selected for investigation of its metabolic profile as its extract was shown to be cytotoxic towards two cancer cell lines, MCF-7 (human breast adenocarcinoma) and A2780 (human ovarian carcinoma). After fungal extracts were prepared by fermentations on brown rice, potato dextrose agar (PDA) and potato dextrose broth (PDB) of P. aurantiacobrunneum, four new (1-4) and two known compounds (5 and 6) were isolated. A configurational uncertainty was resolved in the structure of the previously known compound 5 and the structure of compound 6 was confirmed using its previously reported spectroscopic data as paxisterol, which was first isolated from P. paxilli.9
RESULTS AND DISCUSSION
Structure Elucidation of Compounds 1-5.
Compound 1 was isolated as a yellow oil. Its molecular formula, C23H30O6, was supported by the presence of a sodiated molecular ion peak in the high-resolution ESIMS (HRESIMS) and from its 13C NMR spectrum. The 1H NMR spectrum of compound 1 exhibited signals for three singlet methyl groups at δH 1.19, δH 1.34, and δH 3.92 (s, 3H each), a secondary methyl group at δH 1.17 (d, J = 6.4 Hz, 3H), with its respective methine at δH 3.76 (q, J = 6.4 Hz, 1H), two singlet olefinic methyl groups at δH 1.92 (brs, 3H) and δH 2.02 (s, 3H), a singlet corresponding to an oxygen-bearing methine at δH 3.92 (1H), and eight olefinic protons at δH 5.63 (s, 1H), δH 5.96 (brs, 1H), δH 6.39 (dd, J = 15.0, 9.5 Hz, 1H), δH 6.43 (d, J = 15.0 Hz, 1H), δH 6.51 (dd, J = 15.0, 11.0 Hz, 1H), δH 6.55 (d, J = 15.0 Hz, 1H), δH 6.64 (ddd, J = 15.0, 9.5, 1.4 Hz, 1H), δH 7.16 (ddd, J = 15.0, 11.0, 1.4 Hz, 1H). The UV spectrum of 1 showed multiple maxima, particularly at a longer wavelength of 385 nm, suggesting the presence of a highly conjugated system. A broad band at 3398 cm−1 and a sharp band at 1691 cm−1 in the IR spectrum indicated the presence of hydroxy and unsaturated carbonyl functionalities, respectively. The planar structure was determined by 2D NMR spectroscopy using the HSQC and HMBC spectra and 1D selective NMR experiments (Figure 1) to be the same as that reported for the closely-related mycotoxin, (−)-citreoviridin (7), which was first identified from a species from the same genus, P. citreoviride.10
Figure 1.

Key COSY ( ), TOCSY (
), TOCSY ( ) and HMBC (
) and HMBC ( ) correlations of 1
) correlations of 1
Upon comparison of the 13C NMR spectroscopic data of both compounds (Table 1), the resonances of 1 were generally observed to be slightly more upfield (ca. –1.4 ppm) than those of 7. However, noticeable differences in two 13C NMR chemical shifts between 1 and 7 were observed at C-7 (δC 132.2 for 1 and δC 137.4 for 7) and C-12 (δC 136.0 for 1 and δC 133.6 for 7), suggesting that the two compounds have at least one configuration difference in either the tetrahydrofuran ring or the geometry of the double bonds. To account for these differences, one-dimensional selective nuclear Overhauser effect Spectroscopy (1D NOESY) experiments were conducted to determine the relative configuration of the tetrahydrofuran moiety in 1. When the signal at δH 3.76 (H-2) was irradiated, the resonances at δH 1.19 (CH3-19) and δH 1.34 (CH3-20) were enhanced. In addition, irradiation of the signal at δH 3.92 (H-4) affected the proton signal at δH 1.19 (CH3-19). From these observations, the relative configuration of the tetrahydrofuran was assigned as in Figure 2. Next, the all-trans configuration in the polyene moiety was deduced based on the coupling constants of the olefinic protons as delineated from a series of 1D correlation spectroscopy (1D COSY) spectroscopic data and comparison of previously reported data for 7 (Table 1).
Table 1.
1H and 13C NMR Data for a Compound 1 and the Known Compound (−)-citreoviridin (7)11
| 1a | 7a | |||
|---|---|---|---|---|
| position | δC | δH, m, (J in Hz) | δC | δH, m, (J in Hz) |
| 1 | 11.6 | 1.17, d, (6.4) | 13.0 | 1.13, d, (6.3) |
| 2 | 77.7 | 3.76, q, (6.4) | 79.1 | 3.73, q, (6.3) |
| 3 | 80.0 | - | 81.4 | - |
| 4 | 85.4 | 3.92, s | 86.8 | 3.90, s |
| 5 | 84.5 | - | 85.9 | - |
| 6 | 143.2 | 5.96, brs | 144.7 | 5.94, s |
| 7 | 132.2 | - | 137.4 | - |
| 8 | 141.3 | 6.43, d, (15.0) | 142.7 | 6.41, d, (15.0) |
| 9 | 126.3 | 6.39, dd, (15.0, 9.5) | 127.7 | 6.34, dd, (15.3, 9.0) |
| 10 | 138.9 | 6.64, ddd, (15.0, 9.5, 1.4) | 140.3 | 6.62, dd, (14.8, 9.0) |
| 11 | 130.6 | 6.51, dd, (15.0, 11.0) | 132.0 | 6.51, dd, (15.0, 11.1) |
| 12 | 136.0 | 7.16, ddd, (15.0, 11.0, 1.4) | 133.6 | 7.15, dd, (15.0, 11.2) |
| 13 | 118.2 | 6.55, d, (15.0) | 119.6 | 6.49, d, (15.0) |
| 14 | 154.7 | - | 156.1 | - |
| 15 | 108.1 | - | 109.5 | - |
| 16 | 171.7 | - | 173.1 | - |
| 17 | 87.5 | 5.63, s | 88.9 | 5.61, s |
| 18 | 165.1 | - | 166.5 | - |
| 19 | 18.0 | 1.19, s | 19.4 | 1.16, s |
| 20 | 20.5 | 1.34, s | 22.0 | 1.31, s |
| 21 | 12.3 | 1.92, brs | 13.7 | 1.90, d, (1.2) |
| 22 | 7.4 | 2.02, s | 8.9 | 1.99, s |
| 23 | 55.8 | 3.92, s | 57.3 | 3.89, s |
Measured in MeOD
Figure 2.

Key NOESY ( ) correlations of 1 to determine the relative configuration of the highly oxygenated tetrahydrofuran moiety
) correlations of 1 to determine the relative configuration of the highly oxygenated tetrahydrofuran moiety
After the discovery of 7 from P. citeoviride, several other analogues of citreoviridin have been isolated from multiple species of the genus Penicillium. It is noteworthy that both enantiomers of citreoviridin (7a and 7b) have been reported, yet they displayed the same sign of specific rotation.10–14 Two other analogues of citreoviridin, namely, neocitreoviridin (8) and isocitreoviridin (9), were consequently characterized based on the two different illustrations of citreoviridin (Figure 3). Compound 1 was named 4-epi-citreoviridin using the convention set by Steyn et al. for 7a.12
Figure 3.

Two derivatives of citreoviridin, isocitreoviridin (8) and neocitreoviridin (9) that were defined based on different analogues of the same compound, (−)-citreoviridin (7a/7b)
Compound 2 was isolated as a white amorphous solid. The molecular formula was established as C28H44O5 using HRESIMS from the sodium adduct ion peak and from its 13C NMR data. Its 1H NMR spectrum exhibited signals of two singlet methyl groups at δH 0.98 and δH 1.37 (3H each), an isopropyl group at δH 1.06 (d, J = 6.8 Hz, 6H) and at δH 2.28 (sept, J = 6.8 Hz, 1H), three oxygen-bearing methines at δH 3.58 (ddd, J = 15.8, 10.9, 4.7 Hz, 1H), δH 4.34 (brs, 1H), δH 3.74 (t, J = 2.6 Hz, 1H), one acetal proton at δH 5.54 (s, 1H), and one exocyclic methylene group at δH 4.73 and δH 4.79 (brs, 1H each). All 28 carbon signals of compound 2, which were detected using 13C and HSQC experiments, were similar to those of the known compound, paxisterol (6).9
Compound 2 was postulated to be derived from 6 by hydration, as evident in their respective molecular formulas (C28H44O5 in 2 and C28H42O4 in 6). Comparison of the 13C NMR data of 2 and 6 (Table 3) pinpointed significant differences in the resonances of C-7 (δC 71.4 in 2 and δC 56.6 in 6) and C-8 (δC 73.9 in 2 and δC 60.7 in 6), which implied hydration across the C-7−C-8 epoxide of 6. Selective irradiation of the oxygen-bearing methine signal at δH 3.58 (H-3) of 1 in a 1D TOCSY experiment was used to locate a hydroxy group at C-7 conclusively as it showed correlations with H-7 [H-1 (δH 1.02), H-2 (δH 1.75), H-4 (δH 1.48), H-5 (δH 1.68) and H-6 (δH 1.26)]. The remaining planar structure of the molecule was established subsequently by HSQC and HMBC correlations (Figure 4).
Table 3.
13C NMR Data for 2−6 in MeOD
| position | 2a | 3a | 4b | 5b | 6a |
|---|---|---|---|---|---|
| 1 | 37.5 | 37.4 | 36.7 | 38.2 | 37.6 |
| 2 | 30.2 | 30.0 | 30.1 | 32.2 | 30.0 |
| 3 | 70.6 | 70.3 | 70.3 | 71.5 | 70.1 |
| 4 | 36.7 | 36.9 | 41.5 | 38.7 | 36.8 |
| 5 | 36.2 | 35.85 | 143.7 | 41.4 | 39.3 |
| 6 | 32.1 | 33.3 | 121.4 | 42.9 | 28.3 |
| 7 | 71.4 | 67.9 | 65.6 | 120.7 | 56.6 |
| 8 | 73.9 | 93.9 | 93.4 | 137.1 | 60.7 |
| 9 | 48.1c | 49.3c | 46.1 | 51.2 | 48.2c |
| 10 | 35.4 | 35.90 | 37.5 | 35.4 | 33.5 |
| 11 | 18.4 | 20.4 | 20.1 | 22.3 | 21.2 |
| 12 | 28.2 | 31.9 | 32.0 | 31.0 | 27.8 |
| 13 | 57.2 | 63.9 | 63.1 | 44.3 | 58.6 |
| 14 | 55.0 | 61.8 | 62.2 | 60.5 | 54.0 |
| 15 | 74.3 | 73.5 | 74.1 | 71.5 | 73.1 |
| 16 | 34.8 | 37.8 | 37.6 | 37.5 | 34.7 |
| 17 | 49.1c | 51.9 | 51.9 | 59.8 | 48.7 |
| 18 | 107.3 | 108.6 | 108.8 | 17.4 | 106.5 |
| 19 | 11.5 | 11.0 | 18.6 | 13.3 | 11.8 |
| 20 | 85.0 | 88.5 | 88.5 | 75.7 | 85.5 |
| 21 | 26.2 | 23.8 | 23.7 | 25.7 | 26.2 |
| 22 | 39.6 | 36.8 | 36.8 | 43.4 | 39.6 |
| 23 | 29.1 | 27.9 | 27.9 | 29.8 | 29.0 |
| 24 | 155.7 | 155.5 | 155.5 | 157.7 | 155.7 |
| 25 | 33.8 | 33.7 | 33.7 | 35.3 | 33.7 |
| 26 | 20.9 | 20.9 | 20.8 | 22.4 | 20.9 |
| 27 | 20.9 | 20.9 | 20.8 | 22.4 | 20.9 |
| 28 | 105.6 | 105.8 | 105.8 | 106.8 | 105.6 |
Measured on 400 MHz NMR
Measured on 700 MHz NMR
Assigned from the observed HMBC correlation
Figure 4.

Key TOCSY ( ) and HMBC (
) and HMBC ( ) correlations of 2
) correlations of 2
In order to determine the relative configuration of compound 2, a series of 1D NOESY experiments was performed (Figure 5). Irradiation of the oxygen-bearing methine at δH 3.58 (ddd, J = 15.8, 10.9, 4.7 Hz, H-3) resulted in the collapse of the signal at δH 1.68 (H-5). Referencing other compounds with C-3 hydroxylation, compound 2 bore greater similarity in the 1H and 13C NMR chemical shifts and splitting patterns with OH-3β15 rather than those with OH-3α.16 When the OH-3 is alpha-oriented, the 1H and 13C chemical shifts of the oxygen-bearing methine (H-3 and C-3) are ca. δH 4.0 ppm and ca. δC 66 ppm respectively. In contrast, those that are beta-oriented resonate at ca. δH 3.4 ppm and δC 74 ppm, respectively. As such, the hydroxy group at C-3 has been assigned as β-oriented. Next, when the oxygen-bearing methine at δH 3.74 (H-7) was irradiated, the signals at δH 2.23 (H-14) and δH 4.34 (H-15) were detected (Figure 5), in similar manner to previous literature.9 The relative configuration at C-20 was determined by the irradiation of the methyl proton signal at δH 1.37 (CH3-21), which resulted in the collapse of signals at H-17 (δH 2.15) but not the acetal proton at H-18 (δH 5.54). Moreover, irradiation of the acetal proton at H-18 (δH 5.54) did not affect any signals. This evidence allowed confirmation that the acetal proton (H-18) and the methyl group (CH3-21) are oriented to face away from each other (Figure 5). Owing to structural similarities between compounds 2 and 6, compound 2 was thus assigned as (20R)-7,8-dihydroxypaxisterol.
Figure 5.

Key NOESY ( ) correlations of 2
) correlations of 2
Compound 3 was obtained as white crystalline needles. The HRESIMS of 3 was consistent with C28H44O5 from the sodiated molecular ion peak and its 13C NMR spectrum, and hence was identical to that of compound 2. Furthermore, from the 1H and 13C NMR spectra, it was evident that compounds 2 and 3 are close structural analogues (Tables 2 and 3). Inspection of the 1H and 13C NMR data of compounds 2 and 3, however, revealed differences in the chemical shifts of H-7 (δH 3.74 and δH 4.56), C-8 (δC 73.9 and δC 93.9), C-13 (δC 57.2 and δC 63.8), and H-14 (δH 2.23 and δH 1.83). These differences were due to changes in the carbons flanking the acetal bridge from C-15 to C-8 in 3, as the HMBC spectrum showed cross-peaks from H-18 (δH 5.47) to C-8 (δC 94.2) but not to C-15 (δC 74.3) (Figure 6), as seen in 2.
Table 2.
1H NMR Data for 2−5 in MeOD
| δH, m, (J in Hz) | ||||
|---|---|---|---|---|
| position | 2a | 3a | 4b | 5b |
| 1 | 1.77, m | 1.67, m | 1.82, m | 1.86, m |
| 1.02, m | 1.02, m | 1.16, m | 1.10, m | |
| 2 | 1.75, m | 1.74, ddd (12.9, 7.9, 2.2) | 1.79, m | 1.78, dd (12.0, 6.3) |
| 1.49, m | 1.42, m | 1.55, m | 1.40, m | |
| 3 | 3.58, ddd (15.8, 10.9, 4.7) | 3.53, ddd (15.9, 10.9, 4.8) | 3.51, m | 3.53, m |
| 4 | 1.48, dd(12.0, 5.5) | 1.53, dd (12.4, 4.8) | 2.36, m | 1.70, dd (13.0, 6.3) |
| 1.37, m | 1.37, dd (12.4, 10.9) | 1.27, dd (13.0, 2.6) | ||
| 5 | 1.68, m | 1.76, m | 1.44, m | |
| 6 | 2.06, m | 1.75, m | 5.52, d (4.7) | 2.09, m |
| 1.26, m | 1.35, m | 1.27, m | ||
| 7 | 3.74, t (2.6) | 4.56, m | 4.49, d (4.7) | 5.89, brs |
| 9 | 1.11, m | 1.46, m | 1.64, m | 1.67, m |
| 11 | 1.80, m | 1.90, m | 1.94, m | 1.61, m |
| 1.63, m | 1.74, m | 1.71, m | 1.56, m | |
| 12 | 2.27, m | 2.15, m | 2.22, m | 1.84, m |
| 1.71, m | 1.86, m | 1.96, m | ||
| 14 | 2.23, brs | 1.83, d (5.2) | 1.89, d (5.0) | 1.72, d (6.8) |
| 15 | 4.34, brs | 4.38, dd (5.2, 4.9) | 4.46, t (5.0) | 4.46, t (5.1) |
| 16 | 1.95, m | 2.07, d (14.1) | 2.13, m | 2.27, ddd (14.5, 9.8, 7.4) |
| 1.83, m | 1.91, ddd (14.1, 8.6, 4.9) | 1.87, m | 1.83, m | |
| 17 | 2.15, d (9.5) | 2.18, d (8.6) | 2.21 | 1.60, d (9.8) |
| 18 | 5.54, s | 5.47, s | 5.51, s | 1.04, s |
| 19 | 0.98, s | 1.01, s | 1.20, s | 0.88, s |
| 21 | 1.37, s | 1.33, s | 1.34, s | 1.31, s |
| 22 | 1.79, m | 1.89, m | 1.89, m | 1.65, m |
| 1.77, m | 1.78, m | 1.57, m | ||
| 23 | 2.01, m | 2.08, m | 2.08, m | 2.13, m |
| 2.06, m | ||||
| 25 | 2.28, sept (6.8) | 2.29, sept (6.9) | 2.30, sept (6.8) | 2.28, sept (6.9) |
| 26 | 1.06, d (6.8) | 1.06, d (6.9) | 1.07, d (6.8) | 1.06, d (6.9) |
| 27 | 1.06, d (6.8) | 1.06, d (6.9) | 1.07, d (6.8) | 1.06, d (6.9) |
| 28 | 4.79, brs | 4.80, brs | 4.79, brs | 4.76, brs |
| 4.73, brs | 4.74, brs | 4.74, brs | 4.70, brs | |
Measured on 400 MHz NMR
Measured on 700 MHz NMR
Figure 6.

Comparison of key HMBC ( ) correlations from the acetal proton H-18 to the carbons flanking the acetal bridge of 2 and 3
) correlations from the acetal proton H-18 to the carbons flanking the acetal bridge of 2 and 3
Crystals of 3 were obtained by a vapor diffusion method from a mixture of n-hexanes and ethyl acetate, and analyzed by a single-crystal X-ray diffraction. The X-ray structure of 3 corroborated the proposed planar structure and allowed for the unambiguous assignment of the absolute configuration of 3: 3S, 7R, 8S, 15R, and 20S (Figure 7). Compound 3 was assigned the trivial name auransterol.
Figure 7.

ORTEP plot of 3 delineating atom numbers and drawn with anisotropic displacement ellipsoids at the 50% probability level
Compound 4 was isolated as a white amorphous solid. Its molecular formula was determined by the sodiated molecular ion peak. The IR and the NMR spectroscopic data suggested that 4 is also a derivative of auransterol. The molecular formula of 4 (C28H42O5) indicated two hydrogen atoms less than that in compound 3 (C28H44O5), suggesting the possibility of an additional unsaturation in the molecule. The 1H and 13C NMR spectra of 4 (Tables 2 and 3) were comparable to those of 3, except for the absence of signals at δH 1.76 (H-5) and δH 1.75/δH 1.35 (H-6a/H-6b) in the 1H NMR spectrum of 4 and its respective 13C NMR resonances at C-5 (δC 35.85) and C-6 (δC 33.3) in the 13C NMR spectrum. Furthermore, an additional olefinic proton at δH 5.52, along with two corresponding olefinic 13C NMR resonances at δC 121.4 and δC 143.7, indicated an unsaturation across the C-5−C-6 bond in compound 4. The position of this olefin was subsequently confirmed by HMBC correlations (Figure 8) from H-6 (δH 5.52) to C-4 (δC 41.5) and C-8 (δC 93.4), as well as from H-4 (δH 2.36), H-7 (δH 4.49) and CH3-19 (δH 1.20) to C-5 (δC 143.7). Compound 4 was assigned structurally as (7S)-5-en-auransterol.
Figure 8.

Key HMBC ( ) correlations in the vicinity of the C5-C6 double bond in 4
) correlations in the vicinity of the C5-C6 double bond in 4
Notable differences in the 13C NMR chemical shifts at positions C-7, C-9 and C-20 (Table 3) of compounds 2−4 prompted the relative configurations at C-7 and C-20 of all three compounds to be examined by selective 1D NOE irradiation of the proton signals of H-7 and CH3-21, respectively. Signals corresponding to H-14 of 2 (δH 2.23) and 4 (δH 1.89) were affected following irradiation of the H-7 proton signal, but not for compound 3, indicating that the configuration at C-7 in 3 was different from 2 and 4. As for the stereocenter at C-20, compound 2 was established to have an inverse stereochemical relationship compared to those in compounds 3 and 4, as irradiation of the methyl proton CH3-21 affected the acetal proton signal, H-18, only in the latter two compounds. The most stable conformations of the 3D structures of compounds 2−4, with the lowest energies calculated by MM2 in Chem3D ver. 17.1 software, substantiated the relative configurations of the three compounds determined by 1D NOESY data (Figure 9).
Figure 9.

Stable 3D conformations of (a) compound 2, (b) compound 3 and (c) compound 4 depicting distances between non-bonded protons in green after energies were minimized using MM2 on Chem3D software.
Compound 5 was isolated as a white amorphous solid. The structure of the compound was identified by comparison of the 1H and 13C NMR spectroscopic data with those reported.17 The complete unambiguous assignments of its NMR signals are shown in Tables 2 and 3. As the stereochemical configuration at C-15 was not defined in a previous report, the signal at δH 4.46 (H-15) was irradiated in a selective 1D NOESY experiment and the signal at H-14 (δH 1.72) was affected as a result. As such, the hydroxy group at C-15 was assigned as a β-OH in 5 as [(15R*,20S*)-15,20-dihydroxyepisterol].
Cytotoxicity Assays.
Compounds 1−3 were evaluated for their in vitro antiproliferative activities against a panel of human cancer cell lines (Table 4) using the previously described sulforhodamine B cytotoxicity assay method.18 Compound 1 showed selective cytotoxicity towards MCF-7 and A2780 cell lines. The selective cytotoxicity profile of compound 3 towards HT-29 cells suggests the possibility of a structure-activity relationship that could be further explored via the synthesis of additional analogues.
Table 4.
Biological Evaluation (IC50 in μM) of Compounds 1−3 Isolated from Penicillium aurantiacobrunneum.
| compound | MCF-7 | A2780 | HT-29 | HeLa | DU-145 |
|---|---|---|---|---|---|
| 1 | 6.0±1.6b | 8.2±2.7b | >10 | >10 | >10 |
| 2 | >10 | >10 | >10 | >10 | >10 |
| 3 | >10 | >10 | 9.8 | >10 | >10 |
| paclitaxela | 0.018 | 0.001 | 0.001 | 0.001 | |
| 4-hydroxytamoxifena | 17.5 |
Used as positive control
Standard deviations were calculated based on three independent experiments, with triplicates each
EXPERIMENTAL SECTION
General Experimental Procedures.
All chemical solvents were acquired from Fisher Scientific. Column chromatography was carried out with SiliaFlash® P60 (230–400 mesh) purchased from Parc-Technologique BLVD (Quebec City, Canada) as the normal stationary phase and octadecyl-functionalized (C18) silica gel (200–400 mesh) purchased from Sigma-Aldrich as reversed stationary phase. Analytical thin-layer chromatography was performed on aluminum-backed TLC (250 μm thickness) purchased from Dynamic Adsorbent Inc. Preparative HPLC was carried out on Hitachi Primaide HPLC equipped with Primaide 1430 diode array detector, a Primaide 1210 autosampler, a Primaide 1110 pump with degasser, and a semi-preparative Cogent Bidentate C18™ HPLC column (4 μm, 250 × 10 mm, i.d). Specific rotation data were taken with an Anton Paar polarimeter. UV spectra were obtained from Hitachi U-2910 UV/vis double-beam spectrophotometer with Fisher Scientific silica cuvettes (catalog number: 14-385-910C). IR spectra were measured on a Thermo-Nicolet 6700 Fourier-transform IR spectrophotometer on a KBr round crystal window from Sigma-Aldrich (catalog number: Z267635–1EA). NMR spectra were taken at 300 K on Bruker AVIII400HD NMR and on Bruker Ascend 700 MHz. High-resolution electrospray ionization mass spectra were recorded using Thermo LTQ Orbitrap™ Mass Analyzer.
Lichen Collection and Isolation of P. aurantiacobrunneum.
Niebla homalea (Ach.) Runder & Bowler (Ramalinaceae) was collected on October 30, 2017 from coastal scrub on sand with rock outcrops, just east of a lighthouse on a narrow peninsula with strong cross winds in Marin County, Point Reyes, CA, U.S.A. (37°53.09.50, 122°37.34.83, 553 ft.), by one of the authors (R. W. S.). Voucher specimens of the lichen (Spjut 17806) were deposited in a herbaria at the Université de Liège Département de Botanique, Belgium (LG), where DNA was also extracted from fresh material (Sérusiaux, LG 6251) for a phylogenetic study of Ramalinaceae,19 and at the World Botanical Associates (WBA). An associated voucher specimen of the lichen was also deposited at the Division of Medicinal Chemistry and Pharmacognosy, College of Pharmacy, The Ohio State University as A1–3.
A small portion of the lichen was placed on a lid of a petri dish containing PDA. The petri dish culture was inverted such that the lichen and agar did not have any contact with each other. Spores from unknown mycobiont associates of the lichen were allowed to settle on the agar and develop into fungal colonies. Each distinctive fungal colony established on the agar plate was isolated by streaking on another plate with PDA until an axenic culture of P. aurantiacobrunneum was obtained.
The fungal strain was identified by morphological inspection and sequencing of the internal transcribed spacers (ITS1 and ITS2) of the rRNA locus. DNA from a pure culture of the fungus was prepared by mechanical disruption of the mycelia followed by phenol/chloroform extraction and ethanol precipitation of the aqueous phase. The ITS region was amplified by Polymerase Chain Reaction (PCR) using two primers that flank ITS region (ITS1: TCCGTAGGTGAACCTGCGG; ITS4: TCCTCCGCTTATTGATATGC). The ITS sequence of the isolated fungus (SI) was 100% identical to that of P. aurantiacobrunneum (GenBank accession: MF281340.2). A voucher specimen of this fungus was stored at −80 °C at the Division of Medicinal Chemistry and Pharmacognosy, College of Pharmacy, The Ohio State University as RAK_A16.
Culture Conditions of P. aurantiacobrunneum and Extraction of Cultured Contents.
Axenic cultures of P. aurantiacobrunneum were initially fermented on PDA and International Streptomyces Project 2 (ISP2) agar, and periodically maintained by subculturing on either ISP2 or PDA at 20 °C. Three extracts of the fungus, namely RAK-16 PDA E, RAK-16 BR E and RAK-16 PDB E, that had been cultured on PDA, brown rice and PDB respectively, were prepared as described in the following paragraphs.
RAK-16 PDA E: Ten PDA culture plates of P. aurantiacobrunneum, which had been cultured for seven days, were extracted with ethyl acetate (250 mL × 2) to yield a yellowish-brown extract, RAK-16 PDA E (232 mg) after drying in vacuo.
RAK-16 BR E: The fungal strain was added into a brown rice medium (ca. 100 g) that was previously sterilized by adding 100 mL distilled water and autoclaving in a 500-mL Erlenmeyer flask with a Tuttnauer EZ10. The culture was allowed to ferment at room temperature in a fume hood away from sunlight for 17 days. Thereafter, the fungal culture was transferred to a 2-L Erlenmeyer flask and ca. 1.6 L of ethyl acetate was added to the culture for extraction. The mixture was sonicated for 1 h and allowed to stand at room temperature overnight. This extraction process was repeated with roughly the same volume of fresh ethyl acetate twice and the combined extract, was dried under reduced pressure with a rotary evaporator to afford ca. 2.7 g of brown oily residue (RAK-16 BR E).
RAK-16 PDB E: P. aurantiacobrunneum was grown in PDB at 20 °C, shaken at 150 rpm, for 10 days. The resulting culture was filtered and then extracted twice, each with 250 mL of ethyl acetate. The two ethyl acetate extracts were then combined and dried under reduced pressure to obtain ca. 50 mg of the brown extract, RAK-16 PDB E.
Isolation of Secondary Metabolites from P. aurantiacobrunneum.
The dried extract of P. aurantiacobrunneum cultured on PDA was dissolved in HPLC-grade methanol and chromatographed by HPLC using a gradient solvent system of 70% to 75% methanol in water for the first 20 min, before conducting a methanol wash for the next 8 min and re-equilibration of the column with 70% methanol in water for another 8 min to yield 3.3 mg of compound 1 (tR: 16.3 min).
The dried extract of P. aurantiacobrunneum cultured on brown rice (RAK-16 BR E) was dissolved in methanol and defatted with n-hexanes to yield two fractions, which were dried under reduced pressure with a rotary evaporator to yield ca. 2.0 g of a yellow oily residue from the n-hexanes fraction and ca. 0.5 g of a brown residue from methanol fraction. The bioactive methanol fraction (MCF-7, IC50 = 4.9 μg/mL and A2780, IC50 = 2.6 μg/mL) was further fractionated over reversed-phase silica gel open column using a mixed solvent system of acetonitrile with 0.1% trifluoroacetic acid, and water, in the ratio of 7:3. This afforded a total of 21 fractions of which fractions 5, 8 and 9 were subjected to further separation. Fractions were selected for further purification based on their 1H NMR spectra and cytotoxicity assay results.
Fraction 5 was allowed to elute by gravity over a normal-phase silica gel open glass column (100 × 15 mm, i.d) with a solvent system consisting of chloroform and methanol in the ratio 20:1, and a total of 19 pooled fractions were obtained. Of these 19 fractions, subfraction 7 was further purified over another silica gel glass column (30 × 15 mm, i.d) using the solvent system ethyl acetate and n-hexanes in the ratio 2:1 to afford 0.78 mg of compound 4. The constituents in fraction 8 were separated using a silica gel open glass column (100 × 15 mm, i.d), eluted with ethyl acetate and n-hexanes in the ratio 4:1 to yield 1.8 mg of compound 2 and 17.7 mg of compound 3. Compound 5 (0.4 mg) was obtained from a two-step purification process, first from fraction 9 over C18 open glass column (50 × 15 mm, i.d) with 70% methanol in water to give 11 fractions, followed by elution of subfraction 11 with ethyl acetate and n-hexanes (4:1) over silica gel open glass column (20 × 15 mm, i.d).
Compound 6 (1.7 mg) was purified from an extract of P. aurantiacobrunneum grown in PDB (RAK-16 PDB E) using a C18 open column (35 × 15 mm, i.d) with 70% methanol in water under medium pressure.
4-epi-Citreoviridin (1): Yellow oil; [α]D25 +8 (c 0.2, MeOH); UV (MeOH) λmax (log ε) 285 (4.23), 293 (4.22), 385 (4.25) nm; IR (KBr) νmax 3398 (br), 2925, 2654, 1691, 1625, 1560, 1457, 1406, 1250, 1095 cm−1; 1H NMR (400 MHz, CD3OD) and 13C NMR (100 MHz, CD3OD), see Table 1; HRESIMS m/z 425.19431 [M+Na]+ (calcd for C23H30O6Na, 425.19346).
(20R)-7,8-Dihydroxypaxisterol (2): White amorphous solid; [α]D25 −16 (c 0.2, MeOH); IR (KBr) νmax 3437, 2925, 2854, 1583, 1451, 1377, 1260, 1052, 1030 cm−1; 1H NMR (400 MHz, CD3OD) and 13C NMR (100 MHz, CD3OD), see Tables 2 and 3; HRESIMS m/z 483.30838 [M+Na]+ (calcd for C28H44O5Na, 483.30810).
Auransterol (3): White crystalline needles; [α]D25 −26 (c 0.6, MeOH); IR (KBr) νmax 3377, 2930, 2864, 1448, 1377, 1276 cm−1; 1H NMR (400 MHz, CD3OD) and 13C NMR (100 MHz, CD3OD), see Tables 2 and 3; HRESIMS m/z 483.30859 [M+Na]+ (calcd for C28H44O5Na, 483.30810).
(7S)-5-en-Auransterol (4): White amorphous solid; [α]D25 −11 (c 0.07, MeOH); IR (KBr) νmax 3445, 2924, 2853 cm−1; 1H NMR (700 MHz, CD3OD) and 13C NMR (175 MHz, CD3OD), see Tables 2 and 3; HRESIMS m/z 481.29214 [M+Na]+ (calcd for C28H42O5Na, 481.29245).
(15R*,20S*)-Dihydroxyepisterol (5): White amorphous powder; [α]D25 −2 (c 0.06, MeOH); IR (KBr) νmax 3390, 2956, 2923, 2851, 1644, 1557, 1463, 1379, 1261, 1098 cm−1; 1H NMR (700 MHz, CD3OD) and 13C NMR (175 MHz, CD3OD), see Tables 2 and 3; HRESIMS m/z 453.33459 [M+Na]+ (calcd for C28H46O3Na, 453.33392).
X-ray Crystallography of Auransterol (3).
Crystals of compound 3 were formed from n-hexanes and ethyl acetate by a vapor diffusion method. The data collection crystal was a clear, colorless plate. Examination of the diffraction pattern on a Bruker D8 Venture diffractometer system with a Photon II detector indicated a monoclinic crystal system. All work was done at 200 K using an Oxford Cryosystems Cryostream Cooler. The data collection strategy was set up to measure a quadrant of reciprocal space with a redundancy factor of 6, which means that 90% of the reflections were measured at least 6 times. Omega and phi scans with a frame width of 0.5° and a frame time of 15 seconds were used. The data frames were collected using the program APEX3 and processed with the SAINT program within APEX3.20 Absorption and beam corrections were made with the multiscan technique in SADABS.21
The structure was solved by the direct methods procedure in SHELXT22 in P21. There are four independent molecules in the asymmetric unit (labeled as A, B, C and D). Full-matrix least-squares refinements based on F2 were performed in SHELXL-2014/7,23 as incorporated in the WinGX package.24 The correct enantiomer was chosen based on the known chiral centers in the molecule. There are also four solvent molecules of water in the asymmetric unit.
For the methyl groups, the hydrogen atoms were added at calculated positions using a riding model with U(H) = 1.5 * Ueq(bonded carbon atom). The torsion angle, which defines the orientation of the methyl group about the C-C bond, was refined. The hydroxy group hydrogen atoms were refined isotropically. The remaining hydrogen atoms were added at calculated positions using a riding model with U(H) = 1.2 * Ueq(bonded carbon atom). Hydrogen atoms could be located for only three out of the four water molecules. These positions of these hydrogen atoms were fixed and their Uiso values were refined.
The final refinement cycle was based on 24056 intensities and 1293 variables and resulted in agreement factors of R1(F) = 0.050 and wR2(F2) = 0.112. For the subset of data with I > 2*sigma(I), the R1(F) value is 0.042 for 21338 reflections. The final difference electron density map contains maximum and minimum peak heights of 0.52 and −0.24 e/Ǻ3. Neutral atom scattering factors were used and include terms for anomalous dispersion.25
The CIF file for 3 has been deposited in the Cambridge Crystallographic Data Centre [deposition number: CCDC 1876026].
Sulphorhodamine B (SRB) Cytotoxicity Assay.
SRB was purchased from Sigma Aldrich (ACS). A2780 cells were purchased from Sigma-Aldrich (ECACC catalog no. 93112519) and MCF-7 cells were obtained from the U.S. National Cancer Institute. Both cell lines were grown in Roswell Park Memorial Institute (RPMI)-1640 medium with l-glutamine purchased from Fisher Scientific, supplemented with 10% fetal bovine serum purchased from Atlanta Biologicals (GA, USA) and 1% w/v Gibco® Antibiotic-Antimycotic solution purchased from Life Technologies, and incubated in humidified incubator with an atmosphere of 95% air and 5% CO2 at 37 °C. The cells were sub-cultured when they reached 80–90% confluence. The SRB cytotoxicity assay was performed based on the protocol previously described.18 In 96-well plates, the cell seeding concentration for MCF-7 was 6000 cells per mL, and that for A2780 was 25000 cells per mL while the seeding density of 100,000 cells per mL was used for HT-29, HeLa, and DU-145. The cells were incubated for 72 h at 37 °C in an atmosphere of 95% air and 5% CO2.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by a supplement of the program grant from the National Cancer Institute (3P01CA125066). We would also like to thank the Raymond W. Doskotch Graduate Fellowship from the Division of Medicinal Chemistry and Pharmacognosy for supporting one of the authors (C. Y. T.). We would also like to recognize the assistance obtained from the instrumentation facility of the College of Pharmacy and the CCIC in acquiring the spectroscopic data of the compounds.
Footnotes
Supporting Information
Spectroscopic data of compounds 1−5 (UV, IR, NMR, HRESIMS), and crystallographic data of auransterol (3) are available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
REFERENCES
- (1).Singh A Herbalism, Phytochemistry and Ethnopharmacology. CRC Press: Boca Raton, FL; 2011. [Google Scholar]
- (2).Vartia KO In The Lichens, Ahmadjian V; Hale ME, Eds.; Academic Press: New York, 1973; pp 547–561. [Google Scholar]
- (3).Okuyama E; Umeyama K; Yamazaki M; Kinoshita Y; Yamamoto Y Planta Med. 1995, 61, 113–115. [DOI] [PubMed] [Google Scholar]
- (4).Ingólfsdóttir K Phytochemistry 2002, 61, 729–736. [DOI] [PubMed] [Google Scholar]
- (5).Ingolfsdottir K; Gissurarson SR; Müller-Jakic B; Breu W; Wagner H Phytomedicine 1996, 2, 243–246. [DOI] [PubMed] [Google Scholar]
- (6).Millot M; Tomasi S; Studzinska E; Rouaud I; Boustie JJ Nat. Prod 2009, 72, 2177–2180. [DOI] [PubMed] [Google Scholar]
- (7).Yang Y; Bhosle SR; Yu YH; Park S-Y; Zhou R; Taş İ; Gamage CDB; Kim KK; Pereira I; Hur J-S; Ha HH; Kim H Molecules 2018, 23, 2968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Hamada N; Miyagawa H The Lichenologist 1995, 27, 201–205. [Google Scholar]
- (9).Yasuzawa T; Yoshida M; Sano H J. Chem. Soc. Perkin. Trans. I 1990, 3145–3149. [Google Scholar]
- (10).Sakabe N; Goto T; Hirata Y Tetrahedron 1977, 33, 3077–3081. [Google Scholar]
- (11).da Rocha MW; Resck IS; Caldas ED Food Addit. Contam. Part A Chem. Anal. Control Expo. Risk Assess 2015, 32, 584–595. [DOI] [PubMed] [Google Scholar]
- (12).Steyn PS; Vleggaar R; Wessels PL; Woudenberg MJ Chem. Soc. Perkin. Trans. I 1982, 2175–2178. [Google Scholar]
- (13).Suh H; Wilcox CS J. Am. Chem. Soc 1988, 110, 470–481. [Google Scholar]
- (14).Auckloo BN; Pan C; Akhter N; Wu B; Wu X; He S Front. Microbiol 2017, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Wang W; Li F; Park Y; Hong J; Lee C-O; Kong JY; Shin S; Im KS; Jung JH J. Nat. Prod 2003, 66, 384–391. [DOI] [PubMed] [Google Scholar]
- (16).Holland IP; McCluskey A; Sakoff JA; Gilbert J; Chau N; Robinson PJ; Motti CA; Wright AD; van Altena IA J. Nat. Prod 2009, 72, 102–106. [DOI] [PubMed] [Google Scholar]
- (17).Onodera H; Ichimura M; Baba K; Agatsuma T; Sasho S; Suzuki M; Iwamoto S; Kakita S WO2009096445A1, January 29, 2009. [Google Scholar]
- (18).Tan CY; Inagaki M; Chai H-B; Lambrechts MK; Önder A; Kiremit HÖ; Rakotondraibe LH Phytochem. Lett 2017, 19, 77–82. [Google Scholar]
- (19).Spjut RW; Simon A; Guissard M; Sérusiaux E Phylogenetic relationships and diversity of the lichen genera Niebla, Ramalina and Vermilacinia (Ramalinaceae). Botanical Society of America Conference, Botany 2019, Tucson, AZ, July 27–31, 2019. [Google Scholar]
- (20).APEX3 v2017.3–0 and SAINT v8.38A, Bruker Analytical X-ray Instruments, Inc.: Madison, WI. [Google Scholar]
- (21).Krause L; Herbst-Irmer R; Sheldrick GM; Stalke DJ Appl. Crystallogr 2015, 48, 3–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Sheldrick GM Acta Crystallogr. A Found. Adv 2015, 71, 3–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Sheldrick GM Acta Crystallogr. C Struct. Chem 2015, 71, 3–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Farrugia LJ J. Appl. Crystallogr 2012, 45, 849–854. [Google Scholar]
- (25).Prince E International Tables for Crystallography, Volume C: Mathematical, Physical and Chemical Tables; Springer: Heidelberg, 2004. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.