

Abstract
A set of novel Kv7.2/7.3 (KCNQ2/3) channel blockers was synthesized to address several liabilities of the known compounds XE991 (metabolic instability and CYP inhibition) and the clinical compound DMP 543 (acid instability, insolubility, and lipophilicity). Using the anthrone scaffold of the prior channel blockers, alternative heteroarylmethyl substituents were installed via enolate alkylation reactions. Incorporation of a pyridazine and a fluorinated pyridine gave an analog (compound 18, JDP-107) with a promising combination of potency (IC50 = 0.16 μM in a Kv7.2 thallium flux assay), efficacy in a Kv7.2/7.3 patch clamp assay, and drug-like properties.
Keywords: Kv7 blocker, voltage-gated potassium channel, schizophrenia, DMP 543, JDP-107
New and more efficacious treatments for schizophrenia are greatly needed, and this need has not yet been diminished by enormous investments in the clinical investigation of new drug targets.1–3 One approach to schizophrenia drug discovery involves the repurposing of compounds that have at least demonstrated safety in clinical trials.4 With this in mind, our interest in the chemical modification and redesign of bioactive natural and unnatural scaffolds led us to reexamine blockers of specific voltage-gated potassium channels of the family Kv7 (KCNQ). These are expressed in the nervous system, and two subtypes, KCNQ2 and KCNQ3, combine to form heteromeric channels that regulate the excitability of neurons via the so-called M-current.5 Previously, certain compounds possessing a rigid carbo- or heterocyclic core with two pendant Lewis basic sites were demonstrated to enhance the release of acetylcholine (ACh) in neuronal tissue and were under clinical investigation as cognitive enhancers for the treatment of dementia.6–11 Linopirdine (1)6,12–14 was well-tolerated but ineffective as a cognitive enhancer in a phase 3 trial with Alzheimer’s patients.15 Subsequent work led to the characterization of these compounds as blockers of Kv7 (KCNQ) potassium channels that control neuronal M-currents.16–20 More recently, a patent by Ghasemzadeh reported that the KCNQ2/3 channel blockers XE991 (2)7 and DMP 543 (3),10,11 a compound also previously in clinical trials, could reverse the effects of phencyclidine (PCP) in a mouse model of schizophrenia, as measured by prepulse inhibition, social interaction time, and forced delayed alternation task.21 The patent also claimed that such compounds could be used to attenuate the negative symptoms of schizophrenia or treat drug addiction. Very recently, Shi and Xie reported that XE991 acts as a neuroprotective agent in a mouse model of Parkinson’s disease.22 These findings prompted us to reexamine the use of KCNQ2/3 channel blockers for the potential treatment of neurological disorders, and to identify novel compounds with improved properties.
Beginning with patent applications first submitted in the 1980s,23 researchers at DuPont Pharmaceuticals disclosed examples of scaffolds with α,α-bispyridylmethyl substituents as neuronal ACh releasers for potential use as cognitive enhancers (compounds 1 to 4, Figure 1). With dozens of analogs previously disclosed in patents and publications, the pharmacophore for this compound class is now well established, with a rigid central scaffold flanked by two pendant hydrogen bond accepting moieties. Linopirdine and XE991 suffer from a lack of metabolic stability due to pyridine N-oxidation, which can be ameliorated by fluorination at the 2-position of the pyridine (DMP 543).10 Two more recent examples of KCNQ2 blockers are UCL207724,25 and ML252,26 at least one of which suffers from low metabolic stability.26 Based on the promising safety profile reported for linopirdine in the clinic,15 and the potency and stability of DMP 543,10 we focused our efforts on identifying novel anthrone-based KCNQ2/3 blockers that may have improved properties. In particular, DMP 543 has several liabilities, including low aqueous solubility (6 μM) and a lack of stability under acidic conditions.27,28 Herein are described our efforts at addressing these issues with novel analogs (Figure 1, bottom).
Figure 1.

Previous and planned KCNQ channel blockers
The acidic instability of DMP 543 is likely due to a SNAr mechanism whereby the pyridine is reversibly protonated prior to attack by water at the 2-position, followed by elimination of fluoride.28 We reasoned that replacement of the 2-fluoro pyridine substituents with 2-trifluoromethyl (5) or 3,5-difluoro (6) could provide compounds that maintained KCNQ2/3 channel blocking activity and stability to pyridine N-oxidation while greatly improving stability under acidic conditions. We also reasoned that improved solubility may be feasible without compromising metabolic stability by using alternative heterocycles. A 4-pyrimidine analog of XE991 was reported, but it was more than tenfold less potent (EC50 = 4.4 vs 0.4 μM), and possessed less than 1% of the efficacy of XE991 in an assay measuring ACh release.7 Extensive SAR studies have indicated that heteroatoms at the termini of both side chains are required for ACh release activity,6,7,14 and we hypothesized that the pyridazine analog 7 could act as a hydrogen-bond acceptor and perhaps maintain potency and efficacy. We would expect it to have improved metabolic stability relative to XE991, improved solubility and acid stability relative to DMP 543,29 and a decreased volume of distribution, as suggested by its calculated logP (ClogP) that is 3 units lower than DMP 543 (2.9 versus 5.9), using ChemAxon MarvinSketch v.18.3. The 2-methylpyridine 8 was prepared as a control compound to compare in particular with the 2-trifluoromethyl analog 5. Calculated pKa values of 5 to 8 suggest that they could all potentially undergo salt formation with HCl (not possible with DMP 543), which could lead to improved kinetic solubilities and thus potentially improved oral bioavailabilities.
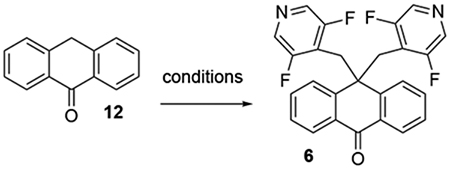
Analogs 5–8 were synthesized by the alkylation of the anthrone scaffold with mesylate electrophiles, as reported by Pesti for the synthesis of DMP 543.30 To provide suitable controls, we also synthesized DMP 543 itself. A representative synthesis is given in Scheme 1 for 6; synthetic details for the preparation of the other compounds is provided in the Supplemental data. Pyridine carboxylic acid 9 was converted to a mixed anhydride, then reduced with NaBH4 to yield alcohol 10, which was converted to the benzylic mesylate 11. The alkylation of anthrone with 11 to yield the final compound 6 was investigated by varying the base, additive, and solvent (Table 1). Initial optimization attempts explored different counterions for the tert-butoxide base with lithium and potassium tert-butoxide (entries 1–3) both giving similarly low yields even upon further heating. These reactions also showed the formation of monalkylated anthrone 13 by LC-MS as a prominent byproduct. Addition of 18-crown-6 ether (entry 4) to KOt-Bu did not improve the yield. Increasing amounts of NaI (entries 5 and 6) gave only a slight improvement in yield over entry 1. The use of NaH as the base (entry 7) was shown to significantly improve the conversion and yield of the second alkylation, as measured by remaining 13 after completion of the reaction.
Scheme 1.

Representative synthesis of symmetrical bis-alkylated anthrone
Table 1.
Optimization of Anthrone Alkylationa
| entry | base | additive | solvent/temp. | % yieldb,c |
|---|---|---|---|---|
| 1 | LiOt-Bu | – | THF/60 °C | 3b |
| 2 | KOt-Bu | – | THF/60 °C | 7b |
| 3 | LiOt-Bu | – | THF/90 °Cd | 12c |
| 4 | KOt-Bu | 18-crown-6 (2.4 eq.) | THF/60 °C | 3b |
| 5 | LiOt-Bu | Nal (1.5 eq.) | THF/60 °C | 7c |
| 6 | LiOt-Bu | Nal (2.0 eq.) | THF/60 °C | 21c |
| 7 | NaH | – | THF/60 °C | 46b,c |
General conditions: (0.055 mmol) and base (0.132 mmol) were mixed in THF (4.0 mL). The anthrone/base solution was added to a solution of the mesylate (0.110 mmol) and NaI (varying equivalents) in THF (4.0 mL), which had been heated at 50°C for 3 h prior to addition. The reactions were then stirred at 60°C for 12–16 h.
Isolated yield.
Yield measured by 1H NMR using pentachloroethane as internal standard.
Reaction run in sealed tube in an oil bath heated to 90°C.
Compounds 5–8 were tested for their ability to block currents at a concentration of 1.5 μM in tsA201 cells expressing Kv7.2/7.3, using a manual patch clamp protocol. The inhibition values for these preliminary screens are summarized in Table 2, along with XE 991 and DMP 543. Compounds 6 and 8 were the only novel compounds of the initial group that showed significant inhibition of Kv7.2/7.3 channel currents. 6 (65 ± 9% inhibition) showed superior efficacy to DMP 543 (31 ± 13%), and was comparable to the best-in-class compound XE991 (81 ± 6%), at a concentration of 1.5 μM. Representative current traces are given for XE991, DMP 543 and 6 in Figure 2.
Table 2.
K+ channel and CYP 3A4 inhibition dataa
| compound | Kv7.2/7.3 patch clamp inhib.b (@1.5 μM) | Kv7.2 flux IC50 (μM)c | hERG flux IC50 (μM)d | CYP3A4 inhib.e (@50 μM) |
|---|---|---|---|---|
| XE991 (2) | 81 ± 6% | 0.055 | >32 | NT |
| DMP 543 (3) | 31 ± 13% | 0.048 | 32 | 82% |
| 5 | 6% | 5.6 | inactive | 95% |
| 6 | 65 ± 9% | 0.20 | 12 | 97% |
| 7 | 12% | 10 | inactive | 28% |
| 8 | 33% | 0.54 | inactive | 92% |
| JDP-107 (18) | 73 ± 7% | 0.16 | >32 | N.T. |
See Supplementary data for assay protocols. N.T. = not tested.
Patch clamp assay using tsA201 cells expressing Kv7.2/7.3. For active compounds, means are provided for n=4 or 5, ± SEM.
With HEK 293 cells expressing Kv7.2 channels.
With HEK 293 cells expressing hERG channels.
CYP 3A4 activities were determined using the Vivid CYP450 screening kit (Life Technologies, Carlsberg, CA, USA) according to the manufacturer’s instructions.
Figure 2.

Electrophysiology traces of XE991 (A), DMP 543 (B), and 6 (C), in tsA201 cells expressing KCNQ2/3. XE991 (20 μM) was added late in each run to determine maximal channel inhibition.
Prior to in vitro metabolic stability studies, we measured the ability of our compounds to inhibit cytochromes P450 (CYPs), specifically the subtype 3A4 most associated with drug metabolism (Table 2). Using high concentrations (50 μM), all compounds inhibited CYP 3A4 to a significant degree, which is not surprising given the presence of pendant heterocycles in each compound capable of coordinating to heme iron centers. The bispyridazine 7 showed the lowest level of inhibition (28%), which is not unexpected given that it is a less basic heterocycle and a weaker metal ligand.
To attempt to avoid the liability of CYP inhibition that could introduce drug-drug interactions upon clinical use, we considered the preparation of unsymmetrically alkylated anthrones. The pharmacophore for these channel blockers (ACh releasers) is well established to possess two pendant hydrogen bond acceptors, but we wished to determine if Kv7.2/7.3 could be effectively blocked by compounds possessing one stronger and one weaker hydrogen bond acceptor. Wilkerson reported that compounds in the oxindole series (e.g., linopirdine) can be highly efficacious acetylcholine release enhancers with one of the 4-pyridinylmethyl substituents replaced with an aliphatic ester or nitrile.6 We reasoned that a compound containing one pyridazine could maintain efficacy and resistance to metabolic N-oxide formation, while potentially attenuating CYP inhibition. Such a compound is also expected to possess improved solubility and decreased volume of distribution relative to DMP 543, which is likely a desirable clinical profile if brain penetrance can be maintained.10
The synthesis of the pyridine-pyridazine 18 (JDP-107) is outlined in Scheme 2. Pyridazine-4-carboxylic acid (14) was converted to the methyl ester 15, then reduced with NaBH4 to generate alcohol 16. Anthrone (12) was sequentially alkylated with mesylate 11, then the mesylate 17 generated in situ from 16, thus generating the desired analog 18.
Scheme 2.

Synthesis of pyridine-pyridazine JDP-107 (18)
Next, the compounds were tested in a thallium flux assay using HEK 293 cells expressing Kv7.2 or hERG channels (Table 2).31 Compounds with sub-micromolar potencies showed similar maximal inhibition of Kv7.2, including XE991. DMP 543 was the most potent compound tested, with IC50 = 0.048 μM, very similar to XE991 (0.055 μM). The bis-trifluoromethylpyridine analog 5 was ~100-fold less potent in the Kv7.2 assay, and showed minimal inhibition in the patch clamp KCNQ2/3 assay. The bis-pyridazine 7 also showed only moderate inhibition of Kv7.2 (IC50 = 10 μM). The bis-methylpyridine analog 8 showed better potency in the Kv7.2 assay (IC50 = 0.54 μM). Pleasingly, the bis-(3,5-difluoropyridine) 6 showed good inhibition of Kv7.2 (IC50 = 0.20 μM) to go with its promising electrophysiology results. Comparable inhibition was also measured with the mixed pyridine/pyridazine analog JDP-107 (18) (Figure 3, IC50 = 0.18 μM), within approximately 3-fold the potency of DMP 543. This confirmed our hypothesis that effective inhibition may be obtained with one more basic and one less basic moiety which presumably act as H-bond acceptors within the channel. 18 also showed only minimal inhibition of the hERG channel, with no inhibition observed at 1 μM and an IC50 > 30 μM. 6 showed a significantly higher level of hERG inhibition, with IC50 = 12 μM. Importantly, 18 was also the most efficacious of the new analogs in the Kv7.2/7.3 patch clamp assay, with a maximal inhibition of 73 ± 7%, significantly better than DMP 543 (31%).
Figure 3.

Concentration-response for JDP-107 (18) in Kv7.2 thallium flux assay. Error bars indicate standard error of the mean (SEM) for 3 wells.
The success of the inhibitors 6 and 18 prompted us to profile their drug-like properties in comparison to DMP 543 (Table 3). Both DMP 543 and 18 inhibit CYP 3A4 substantially at a concentration of 10 μM, which could make for possible drug-drug interactions in human subjects. Both compounds also show decent liver microsomal stability, with DMP 543 showing the better stability after a 1 h incubation (97% vs 84% remaining). To compare the acid stability of compounds, DMP 543, 6, and 18 were subjected to strongly acidic conditions (CH3CN/0.2N aq. HCl). Compounds 6 and 18 showed no degradation, and DMP 543 showed some degradation (83% remaining after 24 h), based on HPLC analysis.
Table 3.
Comparison of XE991, DMP 543, 6, and JDP-107 (18)a
| XE991 | DMP 543 | 6 | JDP-107 (18) | |
|---|---|---|---|---|
| Kv7.2/7.3 patch clamp inhib. (@ 1.5 pM)b | 81% | 31% | 65% | 73% |
| Kv7.2 flux IC50c | 0.055 μM | 0.048 μM | 0.20 μM | 0.16 μM |
| hERG flux IC50d | >32 μM | 32 μM | 12 μM | >32 μM |
| CYP 3A4 inhib. (@ 10 μM)e | N.T. | 79% | N.T. | 90% |
| metabolic stability (human liver microsomes)f | N.T. | 97% | N.T. | 84% |
| acid stabilityg | N.T. | 83% | >99% | >99% |
| ClogPh | 4.86 | 5.93 | 5.43 | 4.17 |
| calc. pKah | 5.77 | −0.16 | 1.47 | 2.91 |
| tPSAi | 41.79 | 41.79 | 41.79 | 54.15 |
| CNS MPO scorej | 4.0 | 3.6 | 3.4 | 4.0 |
| kinetic solubilityk | N.T. | 22 μM | 30 μM | 27 μM |
| cytotoxicity (CC50, hepG2 and HEK 293 cells)l | N.T. | 132 μM; 77 μM | 114 μM; 79 μM | >150 μM |
| Off-target activitiesm | N.T. | 6 (α1A Ki = 1.1–3.1 μM; σ1 Ki = 3.9 μM) | 2 (PBR Ki = 2.5 μM; σ1 Ki = 1.8 μM) | 5 (D3 Ki > 10 μM; σ1 Ki > 10 μM) |
See Supplementary data for assay protocols. N.T. = not tested.
Patch clamp assay using tsA201 cells expressing Kv7.2/7.3.
With HEK 293 cells expressing Kv7.2 channels.
With HEK 293 cells expressing hERG channels.
CYP 3A4 activities were determined using the Vivid CYP450 screening kit (Life Technologies, Carlsberg, CA, USA) according to the manufacturer’s instructions.
With human liver microsomes; percent remaining after 1 h.
Percent remaining based on relative HPLC peak areas (254 nm) after heating at 60°C in 1:1 MeCN:0.2N aq. HCl for 24 h at a concentration of 0.3 mM.
Calculated with ChemAxon MarvinSketch v.18.3.
Calculated with ChemDraw Prime v.16.0.
Central Nervous System Multiparameter Optimization desirability score, calculated according to the spreadsheet published by Wager et al.32
Using 2.5% DMSO/water.
Measured using CellTiter-Glo® assay.
Defined as the number of targets (of 40, mostly GPCRs) which show >30% binding at a concentration of 10 μM. These studies were performed by the National Institute of Mental Health Psychoactive Drug Screening Program (NIMH PDSP).
To estimate the potential for our compounds to be safe CNS-active compounds, we used Pfizer’s CNS multiparameter optimization desirability tool (CNS MPO), a freely available spreadsheet.32 For each molecule, ClogP, ClogD, total polar surface area (tPSA), molecular weight, number of hydrogen-bond donors, and calculated pKa were determined and input into the spreadsheet to determine the CNS MPO scores. ClogP, ClogD, and pKa were calculated with ChemAxon MarvinSketch v.18.3, and tPSA was calculated with ChemDraw Prime v.16.0. The clinical compound DMP 543 possesses a score of 3.6 out of a possible 6, with 6 being the score for compounds with the most desirable properties for CNS drug development. The attenuated lipophilicity of 18 gave it an improved score of 4.0. However, there was not a substantial increase in kinetic aqueous solubility with the addition of a pyridazine to the scaffold (22 μM for DMP 543 vs 28 μM for 18). Both DMP 543 and 18 showed low cytotoxicity with hepG2 cells using the CellTiter-Glo® assay, with 18 showing no measurable cytotoxicity.
Finally, DMP 543, 6, and 18 were submitted to the Psychoactive Drug Screening Program for off-target screening against a panel of mostly GPCRs, using competitive radioligand binding assays.33 JDP-107 showed >30% inhibition of 5 of 40 targets (mostly GPCRs); Kis were measured to be >10 μM for the D3 and σ1 receptors. We conclude that at this stage, it has a clean receptor off-target binding profile for in vivo CNS studies.
We have determined that the anthrone scaffold of Kv7 inhibitors can tolerate replacements of pendant pyridine moieties with alternative heterocycles, and that such compounds could have an improved range of properties more suitable for in vivo and clinical studies. Specifically, we identified the pyridazine-containing analog 18 (JDP-107) that possesses comparable potency to DMP 543 in a Kv7.2 assay, superior efficacy in the Kv7.2/7.3 patch clamp assay, superior acid stability, and attenuated lipophilicity, though solubility and CYP inhibition continue to be liabilities of this compound class. We anticipate that these liabilities could be addressed in future compounds via alternative heterocycles and/or the judicious incorporation of pyridazine substituents.
Supplementary Material
ACKNOWLEDGMENT
We thank Dr. M. Behnam Ghasemzadeh (Marquette University) for helpful discussions; Dr. Sheng Cai (Marquette University) for assistance with LC-MS and NMR instruments, and ACD Labs (NMR processing software) and ChemAxon Inc. (NMR and physicochemical property prediction software). Off-target binding data was generously provided by the National Institute of Mental Health’s Psychoactive Drug Screening Program, Contract # HHSN-271–2013-00017-C (NIMH PDSP). The NIMH PDSP is directed by Bryan L. Roth MD, PhD at the University of North Carolina at Chapel Hill and Project Officer Jamie Driscoll at NIMH, Bethesda MD, USA.
Funding Sources
C.D. thanks Marquette University for startup funding. E.J.D. was supported by R01GM127513 and University of California funds.
ABBREVIATIONS
- ACh
acetylcholine
- ClogP
calculated logarithm of ([n-octanol]/[water]) partition coefficient
- CNS
central nervous system
- CYP
cytochrome P450
- DCM
dichloromethane
- DMAP
4-dimethylaminopyridine
- hERG
human ether-à-go-go-related gene, a voltage-gated potassium channel equivalent to Kv11.1
- KCNQ
voltage-gated potassium channels now identified as the family Kv7
- Kv
voltage-gated potassium channel
- LAH
lithium aluminum hydride
- NIMH
National Institute of Mental Health
- MPO
multi-parameter optimization
- PCP
phencyclidine
- Ms
methanesulfonyl or mesyl
- PCP
phencyclidine
- PDSP
Psychoactive Drug Screening Program
- pKa
negative logarithm of the acidity equilibrium constant
- SAR
structure-activity relationship
- THF
tetrahydrofuran
- tPSA
topological polar surface area
Footnotes
Supplementary data
Synthetic protocols, assay protocols, and select compound characterization data.
A preliminary version of this manuscript was submitted to the preprint server ChemRxiv.34 C.D.W. is an owner of WaveFront Biosciences and ION Biosciences, manufacturers of the Panoptic plate reader and thallium-sensitive dyes used for the thallium flux assays described in this manuscript.
REFERENCES
- (1).Azmanova M; Pitto-Barry A; Barry NPE Schizophrenia: Synthetic Strategies and Recent Advances in Drug Design. MedChemComm 2018, 9 (5), 759–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Stępnicki P; Kondej M; Kaczor AA Current Concepts and Treatments of Schizophrenia. Molecules 2018, 23 (8), 2087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Conn PJ; Lindsley CW; Meiler J; Niswender CM Opportunities and Challenges in the Discovery of Allosteric Modulators of GPCRs for Treating CNS Disorders. Nat Rev Drug Discov 2014, 13 (9), 692–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Lago SG; Bahn S Clinical Trials and Therapeutic Rationale for Drug Repurposing in Schizophrenia. ACS Chem Neurosci 2019, 10 (1), 58–78. [DOI] [PubMed] [Google Scholar]
- (5).Brown DA; Passmore GM Neural KCNQ (Kv7) Channels. British Journal of Pharmacology 2009, 156 (8), 1185–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Wilkerson WW; Kergaye AA; Tam SW 3-Substituted, 3-(4-Pyridinylmethyl)-1,3-Dihydro-1-Phenyl-2H-Indol-2-Ones as Acetylcholine Release Enhancers: Synthesis and SAR. J. Med. Chem 1993, 36 (20), 2899–2907. [DOI] [PubMed] [Google Scholar]
- (7).Wilkerson WW; Earl RA; Calabrese JC; Drammond S; Teleha CA; Voss ME; Zaczek R Acetylcholine Release Enhancers Related to Linopirdine: a Structure—Activity Relationship Study. II. European Journal of Medicinal Chemistry 1996, 31 (4), 319–330. [Google Scholar]
- (8).Teleha CA; Wilkerson WW; Earl RA. Polycyclic Systems, and Derivatives Thereof, as Neurotransmitter Release Enhancers Useful in the Treatment of Cognitive Disorders. U.S. 5,990,132.
- (9).Brown BS; Aiken SP; Zaczek R; Hartig PR; Teleha CA; Wilkerson WW; Earl RA. Blockade of Neuronal M-Channels as a Therapeutic Approach to the Treatment of Neurological Disease. U.S. 5,750,528.
- (10).Earl RA; Zaczek R; Teleha CA; Fisher BN; Maciag CM; Marynowski ME; Logue AR; Tam SW; Tinker WJ; Huang SM; Chorvat RJ 2-Fluoro-4-Pyridinylmethyl Analogues of Linopirdine as Orally Active Acetylcholine Release-Enhancing Agents with Good Efficacy and Duration of Action. J. Med. Chem 1998, 41 (23), 4615–4622. [DOI] [PubMed] [Google Scholar]
- (11).Zaczek R; Chorvat RJ; Saye JA; Pierdomenico ME; Maciag CM; Logue AR; Fisher BN; Rominger DH; Earl RA Two New Potent Neurotransmitter Release Enhancers, 10,10-Bis(4-Pyridinylmethyl)-9(10H)-Anthracenone and 10,10-Bis(2-Fluoro-4-Pyridinylmethyl)-9(10H)-Anthracenone: Comparison to Linopirdine. J. Pharmacol. Exp. Ther 1998, 285 (2), 724–730. [PubMed] [Google Scholar]
- (12).Nickolson VJ; William Tam S; Myers MJ; Cook L DuP 996 (3,3-Bis(4-Pyrindinylmethyl)-1-Phenylindolin-2-One) Enhances the Stimulus-Induced Release of Acetylcholine From Rat Brain in Vitro and in Vivo. Drug Dev. Res 1990, 19 (3), 285–300. [Google Scholar]
- (13).Cook L; Nickolson VJ; Steinfels GF; Rohrbach KW; Denoble VJ Cognition Enhancement by the Acetylcholine Releaser DuP 996. Drug Dev. Res 1990, 19 (3), 301–314. [Google Scholar]
- (14).Earl RA; Myers MJ; Johnson AL; Scribner RM; Wuonola MA; Boswell GA; Wilkerson WW; Nickolson VJ; William Tam S; Brittelli DR; Chorvat RJ; Zaczek R; Cook L; Wang C; Zhang X; Lan R; Mi B; Wenting H Acetylcholine-Releasing Agents as Cognition Enhancers. Structure-Activity Relationships of Pyridinyl Pendant Groups on Selected Core Structures. Bioorg. Med. Chem. Lett 1992, 2 (8), 851–854. [Google Scholar]
- (15).Rockwood K; Beattie BL; Eastwood MR; Feldman H; Mohr E; Pryse-Phillips W; Gauthier S A Randomized, Controlled Trial of Linopirdine in the Treatment of Alzheimer’s Disease. Can J Neurol Sci 1997, 24 (2), 140–145. [DOI] [PubMed] [Google Scholar]
- (16).Lamas JA; Selyanko AA; Brown DA Effects of a Cognition-Enhancer, Linopirdine (DuP 996), on M-Type Potassium Currents (IK(M)) and Some Other Voltage- and Ligand-Gated Membrane Currents in Rat Sympathetic Neurons. Eur. J. Neurosci 1997, 9 (3), 605–616. [DOI] [PubMed] [Google Scholar]
- (17).Costa AM; Brown BS Inhibition of M-Current in Cultured Rat Superior Cervical Ganglia by Linopirdine: Mechanism of Action Studies. Neuropharmacology 1997, 36 (11–12), 1747–1753. [DOI] [PubMed] [Google Scholar]
- (18).Schnee ME; Brown BS Selectivity of Linopirdine (DuP 996), a Neurotransmitter Release Enhancer, in Blocking Voltage-Dependent and Calcium-Activated Potassium Currents in Hippocampal Neurons. J. Pharmacol. Exp. Ther 1998, 286 (2), 709–717. [PubMed] [Google Scholar]
- (19).Wang HS; Pan Z; Shi W; Brown BS; Wymore RS; Cohen IS; Dixon JE; McKinnon D KCNQ2 and KCNQ3 Potassium Channel Subunits: Molecular Correlates of the M-Channel. Science 1998, 282 (5395), 1890–1893. [DOI] [PubMed] [Google Scholar]
- (20).Wang HS; Brown BS; McKinnon D; Cohen IS Molecular Basis for Differential Sensitivity of KCNQ and I(Ks) Channels to the Cognitive Enhancer XE991. Molecular Pharmacology 2000, 57 (6), 1218–1223. [PubMed] [Google Scholar]
- (21).Ghasemzadeh MB Modulation of KCNQ Potassium Channel Activity for Treatment of Psychiatric Disorders and the Symptoms Thereof. U.S. 2010/0310681.
- (22).Liu H; Jia L; Chen X; Shi L; Xie J The Kv7/KCNQ Channel Blocker XE991 Protects Nigral Dopaminergic Neurons in the 6-Hydroxydopamine Rat Model of Parkinson’s Disease. Brain Research Bulletin 2018, 137, 132–139. [DOI] [PubMed] [Google Scholar]
- (23).Earl RA; Myers MJ; Nickolson VJ a,a-Disubstituted Aromatics and Heteroaromatics as Cognition Enhancers. U.S. 5,173,489.
- (24).Shah MM; Javadzadeh-Tabatabaie M; Benton DCH; Ganellin CR; Haylett DG Enhancement of Hippocampal Pyramidal Cell Excitability by the Novel Selective Slow-Afterhyperpolarization Channel Blocker 3-(Triphenylmethylaminomethyl)Pyridine (UCL2077). Molecular Pharmacology 2006, 70 (5), 1494–1502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Soh H; Tzingounis AV The Specific Slow Afterhyperpolarization Inhibitor UCL2077 Is a Subtype-Selective Blocker of the Epilepsy Associated KCNQ Channels. Molecular Pharmacology 2010, 78 (6), 1088–1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Cheung Y-Y; Yu H; Xu K; Zou B; Wu M; McManus OB; Li M; Lindsley CW; Hopkins CR Discovery of a Series of 2-Phenyl- N-(2-(Pyrrolidin-1-Yl)Phenyl)Acetamides as Novel Molecular Switches That Modulate Modes of K v7.2 (KCNQ2) Channel Pharmacology: Identification of (S)-2-Phenyl- N-(2-(Pyrrolidin-1-Yl)Phenyl)Butanamide (ML252) as a Potent, Brain Penetrant K v7.2 Channel Inhibitor. J. Med. Chem 2012, 55 (15), 6975–6979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Rabel SR; Shinwari MK; Nemeth GA; Blom KF; Maurin MB Characterization of the Solution Stability and Degradation Products of the Novel Neurotransmitter Release Enhancer 10, 10-Bis (2-Fluoro-4-Pyridinylmethyl)-9(10H)-Anthracenone. Drug Stability 1997, 1 (4), 224–230. [Google Scholar]
- (28).Chen JG; Markovitz DA; Yang AY; Rabel SR; Pang J; Dolinsky O; Wu LS; Alasandro M Degradation of a Fluoropyridinyl Drug in Capsule Formulation: Degradant Identification, Proposed Degradation Mechanism, and Formulation Optimization. Pharm Dev Technol 2000, 5 (4), 561–570. [DOI] [PubMed] [Google Scholar]
- (29).Wermuth CG Are Pyridazines Privileged Structures? MedChemComm 2011, 2 (10), 935–941. [Google Scholar]
- (30).Pesti JA; Huhn GF; Yin J; Xing Y; Fortunak JM; Earl RA Efficient Pyridinylmethyl Functionalization: Synthesis of 10,10-Bis[(2-Fluoro-4-Pyridinyl)Methyl]-9(10 H)-Anthracenone (DMP 543), an Acetylcholine Release Enhancing Agent. J. Org. Chem 2000, 65 (23), 7718–7722. [DOI] [PubMed] [Google Scholar]
- (31).Weaver CD; Harden D; Dworetzky SI; Robertson B; Knox RJ A Thallium-Sensitive, Fluorescence-Based Assay for Detecting and Characterizing Potassium Channel Modulators in Mammalian Cells. J Biomol Screen 2004, 9 (8), 671–677. [DOI] [PubMed] [Google Scholar]
- (32).Wager TT; Hou X; Verhoest PR; Villalobos A Central Nervous System Multiparameter Optimization Desirability: Application in Drug Discovery. ACS Chem Neurosci 2016, 7 (6), 767–775. [DOI] [PubMed] [Google Scholar]
- (33).Besnard J; Ruda GF; Setola V; Abecassis K; Rodriguiz RM; Huang X-P; Norval S; Sassano MF; Shin AI; Webster LA; Simeons FRC; Stojanovski L; Prat A; Seidah NG; Constam DB; Bickerton GR; Read KD; Wetsel WC; Gilbert IH; Roth BL; Hopkins AL Automated Design of Ligands to Polypharmacological Profiles. Nature 2012, 492 (7428), 215–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Porter J; Vivas-Rodriguez O; Weaver CD; Dickson E; Alsafran A; DiMilo E; Arnold L; Dockendorff C An Anthrone-Based Kv7.2/7.3 Channel Blocker with Improved Properties for the Investigation of Psychiatric and Neurodegenerative Disorders. ChemRxiv 2019. (preprint): 10.26434/chemrxiv.8246417.v1 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.