

Abstract
The G-protein-coupled receptor (GPCR) accessory protein MRAP2 is implicated in energy control in rodents, notably via melanocortin-4 receptor (MC4R)1. Although some MRAP2 mutations have been described in people with obesity1–3, their functional consequences on adiposity remain elusive. Using large-scale sequencing of MRAP2 in 9,418 people, we identified 23 rare heterozygous variants associated with increased obesity risk in both adults and children. Functional assessment of each variant shows that loss-of-function MRAP2 variants are pathogenic for monogenic hyperphagic obesity, with hyperglycemia and hypertension. This contrasts with other monogenic forms of obesity characterized by excessive hunger, including MC4R deficiency, that present with low blood pressure and normal glucose tolerance4. The pleiotropic metabolic effect of loss-of-function mutations in MRAP2 might be due to the failure of different MRAP2-regulated GPCRs in various tissues including pancreatic islets.
According to the World Health Organization, the worldwide prevalence of obesity nearly tripled between 1975 and 2016. About 2 billion people are currently overweight, and their co-morbidities represent a major medical burden. The biological link between obesity and type 2 diabetes is still debated as most individuals with obesity never develop diabetes. If classic views postulate that insulin resistance eventually exhaust insulin-producing cells, alternatively genetically-driven abnormal pathways may lead to both appetite dysregulation and insulin secretion defects, and also to other abnormalities such as hypertension. While common obesity is seen as a multifactorial disorder, we and others have found that rare mutations in more than 15 genes cause monogenic obesity, including MC4R (encoding the melanocortin-4 receptor) that is the most frequently mutated gene in monogenic obesity5. Importantly, the characterization of these mutations has enabled the development of new drugs (e.g. the MC4R agonist setmelanotide in patients deficient for POMC or LEPR)6,7. Recently, a study has shown that the loss of function of Mrap2 (encoding melanocortin-2 receptor accessory protein 2) is associated with rodent obesity1. The authors demonstrated that MRAP2 interacted directly with MC4R, and enhanced MC4R downstream signaling in response to a MC4R agonist, suggesting that MC4R signaling was a mechanism linking Mrap2 loss of function and obesity1. When sequencing MRAP2 in 976 people with obesity and controls, the authors identified four rare variants in four participants with severe obesity, that were not present in the controls, and suggested MRAP2 as a new gene causing monogenic obesity1. The authors did not perform statistical or functional analyses of these variants. Another rare non-synonymous variant was described in a patient with obesity associated with the Prader-Willi-like syndrome, but still without functional assays2. Furthermore, Schonnop et al. described a rare MRAP2 mutation (p.Q174R) decreasing MC4R activity in vitro, in a girl with severe obesity3. Here, we performed a large-scale resequencing study of MRAP2, in combination with functional assays of detected variants, to accurately decipher the functional link between MRAP2 signaling and obesity (and possibly other phenotypes) in humans.
The coding exons of MRAP2 were sequenced in 9,418 participants including 7,239 adults, and 2,179 children or adolescents (Supplementary Table 1). We detected 23 rare heterozygous variants (with a minor allele frequency [MAF] between 0.053 and 1.65‰), 14 of which were novel (Table 1, Fig. 1). The cluster of rare MRAP2 variants was significantly associated with an increased risk of obesity in adults (nvariant = 14; P = 8.04×10-4 with an odds ratio [OR] of 3.80, 95% confidence interval [CI]: 1.71–9.26) and in children or adolescents (nvariant = 13; P = 0.0148 with an OR of 2.91, 95% CI: 1.23–7.32). When we added the participants with overweight in the adult case-control study, the cluster of rare MRAP2 variants was still significantly associated with an increased risk of adiposity (nvariant = 17; P = 2.25×10-3 with an OR of 3.13, 95% CI: 1.53–7.27). These data are in line with exome sequencing data from 42,992 participants included in the Accelerating Medicine Partnership (AMP) Type 2 Diabetes knowledge portal, where protein-truncating or missense MRAP2 variants (with a MAF < 1% in each ancestry) were significantly associated with increased body mass index (BMI; nvariant = 46; P = 3.49×10-4 with β = 0.0364 kg/m2, 95% CI: 0.0165–0.0564). This association was even stronger when focusing on protein-truncating or probably deleterious missense MRAP2 variants (nvariant = 9; P = 2.36×10-5 with β = 0.154 kg/m2, 95% CI: 0.0828–0.226).
Table 1. Rare MRAP2 variants detected in the present study including 1,991 adults with obesity, 2,465 adults with overweight, 2,783 adults with normal weight, 1,137 children/adolescents with obesity, and 1,042 children/adolescents with normal weight.
| Chr | Pos (Hg19) | Mutation* | MAF (‰) | PVS1 | PS1 | PS2 | PS3 | PM1 | PM2 | PM4 | PM5 | PP1 | PP2 | PP3 | Pathogenicity | Number and status of carriers |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 6 | 84765033 | c.-5_5del, p.? | 0.053 | 1 | 0 | NA | NA | 0 | 1 | 1 | 0 | NA | 1 | NA | Pathogenic (LOF) | 1 ob child |
| 6 | 84765035 | c.-3_7del, p.? | 0.053 | 1 | 0 | NA | 1 (LOF) | 0 | 1 | 1 | 0 | NA | 1 | NA | Pathogenic (LOF) | 1 ob adult |
| 6 | 84765044 | c.7G>A, p.A3T | 0.053 | 0 | 0 | NA | 0 | 0 | 0 | 0 | 0 | NA | 1 | 0 | VUS | 1 ovw adult |
| 6 | 84765044 | c.7G>T, p.A3S | 0.16 | 0 | 0 | NA | 0 | 0 | 1 | 0 | 0 | NA | 1 | 0 | VUS | 2 ob children, 1 ob adult |
| 6 | 84765074 | c.37C>G, p.Q13E | 0.053 | 0 | 0 | NA | 0 | 0 | 1 | 0 | 0 | NA | 1 | 0 | VUS | 1 nw child |
| 6 | 84765129 | c.92G>T, p.G31V | 0.053 | 0 | 0 | NA | 1 (LOF) | 0 | 1 | 0 | 0 | NA | 1 | 0 | Lik. pathogenic (LOF) | 1 ob adult |
| 6 | 84765132 | c.95C>T, p.P32L | 0.053 | 0 | 0 | NA | 0 | 0 | 1 | 0 | 0 | NA | 1 | 1 | VUS | 1 nw child |
| 6 | 84772669 | c.185T>G, p.F62C | 0.053 | 0 | 0 | NA | 1 (LOF) | 0 | 1 | 0 | 0 | NA | 1 | 1 | Lik. pathogenic (LOF) | 1 ovw adult |
| 6 | 84798812 | c.230A>G, p.N77S | 0.11 | 0 | 0 | 0 | 1 (LOF) | 0 | 0 | 0 | 0 | 1 | 1 | 0 | Lik. pathogenic (LOF) | 1 ob child, 1 ob adult |
| 6 | 84798854 | c.272T>C, p.V91A | 0.053 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 1 | 0 | VUS | 1 nw child |
| 6 | 84798877 | c.295G>C, p.E99Q | 0.053 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | VUS | 1 ob adult |
| 6 | 84798886 | c.304A>T, p.K102* | 0.053 | 1 | 0 | NA | 1 (LOF) | 0 | 1 | 1 | 0 | NA | 1 | NA | Pathogenic (LOF) | 1 ovw adult |
| 6 | 84798919 | c.337A>G, p.R113G | 0.11 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 1 | VUS | 1 ob child, 1 ob adult |
| 6 | 84798922 | c.340T>G, p.S114A | 0.053 | 0 | 0 | NA | 0 | 0 | 1 | 0 | 0 | NA | 1 | 1 | VUS | 1 ob child |
| 6 | 84798944 | c.362A>G, p.N121S | 0.053 | 0 | 0 | NA | 0 | 0 | 1 | 0 | 0 | NA | 1 | 0 | VUS | 1 ob adult |
| 6 | 84798955 | c.373C>T, p.R125C | 0.69 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | VUS | 4 ob / 1 nw children, 2 ob / 4 ovw / 2 nw adults |
| 6 | 84798956 | c.374G>A, p.R125H | 1.65 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | VUS | 6 ob / 3 nw children, 9 ob / 10 ovw / 3 nw adults |
| 6 | 84798979 | c.397C>T, p.H133Y | 0.053 | 0 | 0 | NA | 0 | 0 | 0 | 0 | 0 | NA | 1 | 0 | VUS | 1 nw child |
| 6 | 84798991 | c.409G>A, p.A137T | 0.11 | 0 | 0 | NA | 0 | 0 | 0 | 0 | 0 | NA | 1 | 0 | VUS | 1 nw child, 1 nw adult |
| 6 | 84799067 | c.485T>C, p.M162T | 0.11 | 0 | 0 | NA | 0 | 0 | 1 | 0 | 0 | NA | 1 | 0 | VUS | 1 ob / 1 nw adult |
| 6 | 84799159 | c.577A>G, p.T193A | 0.053 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 1 | 0 | VUS | 1 ob adult |
| 6 | 84799166 | c.584C>T, p.P195L | 0.16 | 0 | 0 | 0 | 1 (LOF) | 0 | 1 | 0 | 0 | 1 | 1 | 0 | Lik. pathogenic (LOF) | 1 ob child, 1 ob / 1 ovw adult |
| 6 | 84799189 | c.607G>T, p.D203Y | 0.053 | 0 | 0 | NA | 0 | 0 | 0 | 0 | 0 | NA | 1 | 0 | VUS | 1 nw adult |
Chr, chromosome; Lik., likely; LOF, loss-of-function; MAF, minor allele frequency in the present study; NA, not available; nw, normal weight; ob, obese; ovw, overweight; PM1, PM2, PM4 or PM5, ‘moderate’ pathogenicity ACMG criteria; Pos, position (according to the human alignment hg19/GRCh37); PP1, PP2 or PP3, ‘supporting’ pathogenicity ACMG criteria; PS1, PS2 or PS3, ‘strong’ pathogenicity ACMG criteria; PVS1, ‘very strong’ pathogenicity ACMG criterion; VUS, variant of uncertain significance.
All these mutations were heterozygous.
Figure 1. Location of the MRAP2 variants detected in the present sequencing study.

Blue bubbles represent amino acids in loci of high sequence homology (in Human versus Chimp, Northern white-cheeked gibbon, Macaque, Olive baboon, Rat, Mouse, Dog, Platypus, Chicken, Frog and Zebrafish) according to Ensembl. Yellow bubbles represent loci of the transmembrane domain16, which are highly conserved. Other amino acids are represented by green bubbles. Mutations are written in red. The pathogenic, loss-of-function mutations are bold and underlined. The purple triangle pinpoints the putative N-linked glycosylation site21.
Here, we used the standards and guidelines of the American College of Medical Genetics and Genomics (ACMG) to assess the pathogenicity of each detected variant. To address the strong pathogenic ACMG criterion PS3, we analyzed the functional effect of each MRAP2 variant on MC4R activity. MC4R and mutant or wild-type MRAP2 were overexpressed in Chinese hamster ovary (CHO) cells and the cyclic adenosine monophosphate (cAMP)–dependent protein kinase (PKA) signaling was analyzed through luciferase reporter assays in response to α-melanocyte-stimulating hormone (αMSH; the canonical agonist of MC4R) and adrenocorticotropic hormone (ACTH; another MC4R agonist in the presence of MRAP2) (Extended data Fig. 1)8–10. When compared with wild-type MRAP2, we found that six MRAP2 variants (i.e., c.-3_7del, p.G31V, p.F62C, p.N77S, p.K102* and p.P195L) significantly decreased cAMP–PKA signaling downstream of MC4R in response to αMSH and ACTH (Extended data Fig. 2). These loss-of-function variants were mostly located in highly conserved loci (Fig. 1). According to ACMG criteria (now including the functional-based criterion PS3), we identified seven pathogenic or likely pathogenic, loss-of-function variants (i.e., c.-5_5del, c.-3_7del, p.G31V, p.F62C, p.N77S, p.K102* and p.P195L; Table 1. These variants were identified in seven adults of European origin with obesity or overweight, and in three European adolescents with obesity (Table 2). Therefore, they were completely penetrant for obesity or overweight. Two mutations (p.N77S and p.P195L) cosegregated with obesity in two families that were available for segregation analysis (Extended data Fig. 3). The majority (75%) of the carriers reported abnormal eating behavior, i.e. overeating, snacking and/or bulimia (Table 2). According to the serum levels of 15 steroid hormones (including cortisol, cortisone, testosterone, aldosterone), none of the carriers had dysfunction of the hypothalamic-pituitary-adrenal axis (Supplementary Table 2). We found that all carriers except one adolescent (participant #10) presented with the phenotypes of metabolic syndrome (Table 2). Apart from high adiposity, the two most frequent metabolic features were hyperglycemia and hypertension (Table 2, Fig. 2). This is noteable as the prevalence of the aggregation of these phenotypes that constitute the so-called metabolic syndrome among people with (polygenic) overweight or obesity usually ranges from 20 to 60% according to our present cohort analysis (Supplementary Table 3) and the literature11. Importantly, when compared to other monogenic forms of obesity including those due to the deficiency of LEP, LEPR, MC4R, PCSK1, POMC or SIM1, MRAP2 deficiency is singular as it leads to a markedly higher rate of hyperglycemia and hypertension (Fig. 2, Supplementary Table 4). Among MRAP2-deficient participants, we did not find any other (likely) pathogenic variant in 48 genes known to be involved in monogenic obesity or in monogenic diabetes, except for the 17-year old participant #10. In this subject, we indeed also found a likely pathogenic mutation in ABCC8 (NM_000352.4: c.647G>A, p.R216H). Pathogenic gain-of-function mutations in ABCC8 cause monogenic diabetes in infancy, while loss-of-function mutations cause hypoglycemic episodes due to inappropriate hyperinsulinism12,13. In view of her low fasting glucose levels (4.33 mmol/l) with surprisingly elevated fasting insulin levels (88.9 pmol/l), it is possible that this presumably inactivating ABCC8 mutation increases insulin secretion, thus normalizing glycaemia and masking the metabolic abnormalities caused by the pathogenic MRAP2 mutation.
Table 2. Clinical data of participants carrying a pathogenic, loss-of-function MRAP2 variant.
| Participants | #1 | #2 | #3 | #4 | #5 | #6 | #7 | #8 | #9 | #10 |
|---|---|---|---|---|---|---|---|---|---|---|
| MRAP2 mutation | c.-3_7del | p.G31V | p.F62C | p.N77S | p.K102* | p.P195L | p.P195L | c.-5_5del | p.N77S | p.P195L |
| Ancestry | Eur | Eur | Eur | Eur | Eur | Eur | Eur | Eur | Eur | Eur |
| Age at investigation (yrs) | 44 | 48 | 44 | 43 | 61 | 61 | 49 | 13 | 12 | 17 |
| Sex (M/F) | F | F | M | M | F | F | F | F | M | F |
| BMI | 49.3 | 30.1 | 25.3 | 49.6 | 25.3 | 32.81 | 27.9 | 34.8 | 30.9 | 29.7 |
| BMI-for-age (percentile) | - | - | - | - | - | - | - | 99th | 99th | 95th |
| Obesity | Class III | Class I | Overweight | Class III | Overweight | Class I | Overweight | Severe obesity | Severe Obesity | Obesity |
| Age of obesity onset (yrs) | - | - | - | - | - | - | - | - | 2 | 8 |
| Obesity during childhood or adolescence | Yes | - | - | Yes | - | - | - | Yes | Yes | Yes |
| Eating behavior | Continuous diet | None | Snacking | Continuous diet | None | - | Bulimia - Snacking | - | Overeating - Snacking | Overeating - Bulimia |
| Treatment | Antidepressant | AHT | None | None | Cholesterol lowering drug | AHT / Cholesterol lowering drug | None | - | None | None |
| Fasting glucose (mmol/L) | 7.21 | 6.14 | 6.05 | 5.50 | 5.77 | 6.16 | 6.59 | 5.30 | 5.70 | 4.33 |
| 2h glucose during an OGTT (mmol/L) | 11.4 | - | - | 5.70 | - | 12.5 | - | - | 7.20 | 5.66 |
| Fasting insulin (pmol/L) | 122 | 149 | 35.9 | 25.0 | 57.7 | 97.2 | 74.5 | 37.0 | 181 | 88.9 |
| HOMA2-%B | 83.0 | 129 | 49.6 | 38.8 | 75.4 | 95.1 | 69.4 | 66.0 | 170 | 178 |
| HOMA2-IR | 2.44 | 2.85 | 0.710 | 0.490 | 1.12 | 1.89 | 1.48 | 0.710 | 3.38 | 1.58 |
| Waist-hip ratio | 1.01 | 0.902 | 0.918 | 0.947 | 0.960 | - | 0.852 | - | 1.06 | 0.866 |
| Waist circumference (cm) | 131 | 101 | 90 | 142 | 95 | - | 92 | - | 103 | 103 |
| SBP (mmHg) | - | 140 | 138 | 160 | 143 | 150 | 128 | - | 120 | - |
| DBP (mmHg) | - | 85 | 80 | 100 | 93 | 90 | 80 | - | 80 | - |
| TC (mmol/L) | 5.28 | 6.66 | 7.07 | 3.66 | 5.40 | 9.00 | 6.53 | - | 5.06 | 4.62 |
| HDL (mmol/L) | 0.820 | 1.19 | 1.21 | 1.08 | 1.63 | 1.44 | 1.83 | - | 1.08 | 1.39 |
| TG (mmol/L) | 1.19 | 1.62 | 0.980 | 1.77 | 1.00 | 4.67 | 1.72 | - | 2.27 | 0.900 |
| Hypertension | - | Yes | Yes | Yes | Yes | Yes | No | - | No | - |
| Diabetes | T2D | PD | PD | NGT | PD | T2D | PD | NGT | PD | NGT |
| Metabolic syndrome | Yes | Yes | Yes | Yes | Yes | Yes | Yes | - | Yes | No |
AHT, antihypertensive drug; BMI, body mass index; DBP, diastolic blood pressure; Eur, European; F, female; HDL, high-density lipoprotein; HOMA2-%B, homeostasis model assessment of steady state beta-cell function; HOMA2-IR, homeostasis model assessment of insulin resistance; M, male; NGT, normal glucose tolerance; OGTT, oral glucose tolerance test; PD, prediabetes; SBP, systolic blood pressure; T2D, type 2 diabetes; TC, total cholesterol; TG, triglycerides; yrs, years.
Figure 2. Rate of hyperglycemia, hypertension, low HDL and high TG in patients deficient for LEP, LEPR, MC4R, PCSK1, POMC, SIM1 or MRAP2.

HDL, high-density lipoprotein; NA, not available; TG, triglyceride.
Orange areas show rates of hyperglycemia, hypertension, low HDL and high TG, while blue areas show rates of normal glucose, normal blood pressure, high HDL and low TG.
This figure was generated from Table 2 (for MRAP2 deficiency) and Supplementary Table 4 (for deficiencies of LEP, LEPR, MC4R, PCSK1, POMC and SIM1).
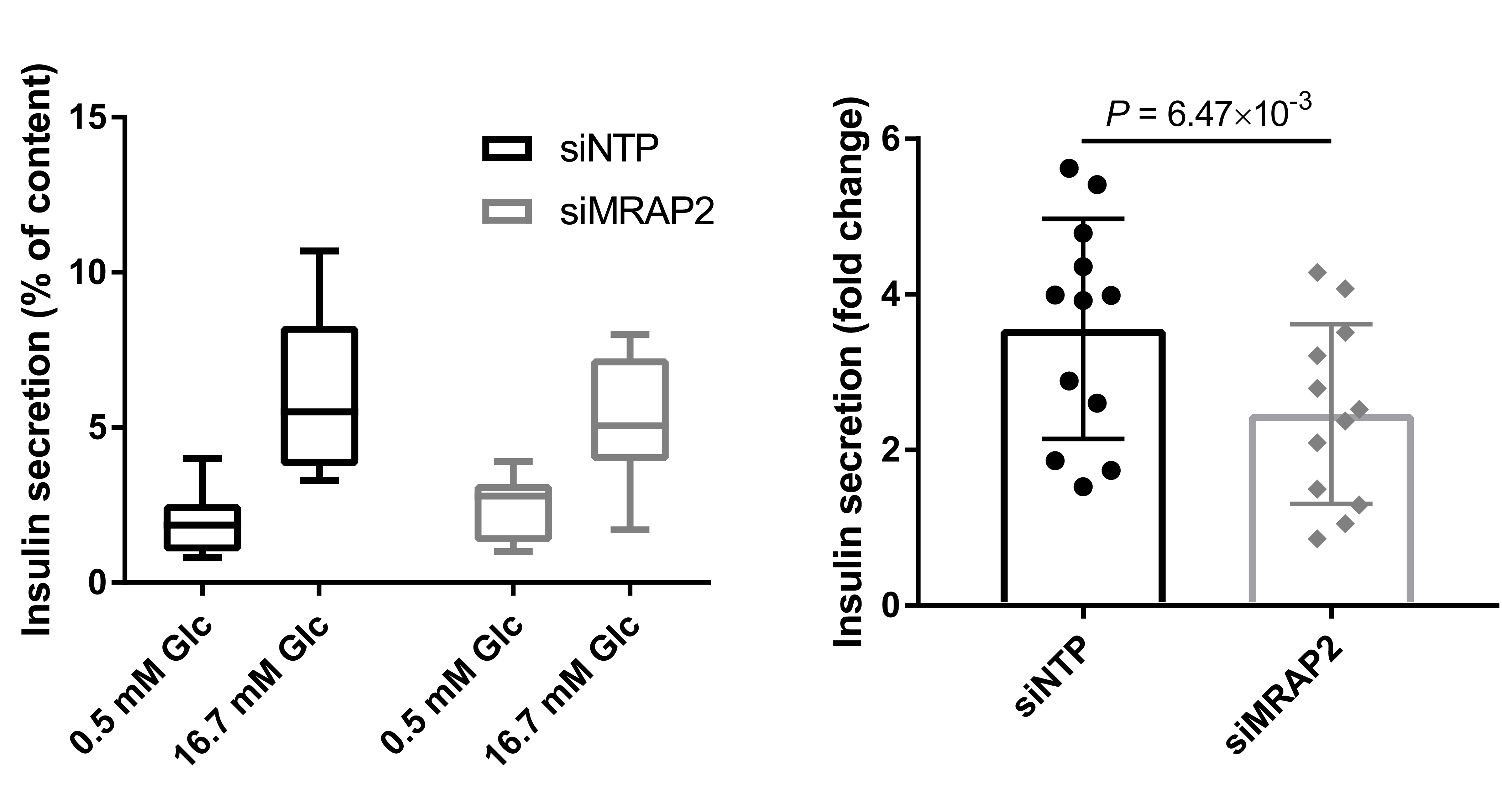
As the MRAP2-deficient participants mostly presented with hyperglycemia in addition to high adiposity, we assessed MRAP2 expression in a panel of human tissues using a PCR-free technology that we previously described and validated14. We found that MRAP2 expression level was similarly high in human pancreatic islets and beta cells, the human beta-cell line EndoC-βH1, and brain regions (Extended data Fig. 4.a). We confirmed MRAP2 protein expression in human islets and EndoC-βH1 by Western blot (Extended data Fig. 4.b). Based on these data, we assessed the involvement of MRAP2 in beta-cell function by performing glucose-stimulated insulin secretion assay in EndoC-βH1 cells15, in which MRAP2 expression was decreased with a specific siRNA. We found that the fold change of insulin concentration was significantly decreased in EndoC-βH1 cells treated with the siRNA targeting MRAP2 compared to the control cells (Extended data Fig. 5). These results were in line with previous data obtained from mouse models. Indeed, although Asai et al. showed that Mrap2-null mice had normal fasting insulin and normal tolerance to intraperitoneal glucose injection1, two subsequent studies reported that Mrap2-null mice (with different genetic backgrounds from the previous Mrap2-null mice) had impaired glucose homeostasis under fasting or after a glucose or insulin tolerance test16,17. Rouault et al. also demonstrated that impaired glucose tolerance of Mrap2 KO mice occurred before they developed obesity16. These results suggest that MRAP2 mutations could have a direct functional deleterious effect on beta cells.
In conclusion, we describe a new monogenic form of hyperphagic obesity due to pathogenic, loss-of-function mutations in MRAP2, which is also associated with hyperglycemia and hypertension. This is phenotypically very different from other forms of monogenic obesity with appetite dysfunction described so far, including MC4R deficiency, where patients with obesity along with high plasma glucose and high blood pressure have been scarcely reported (Fig. 2, Supplementary Table 4 and ref.4). In addition to brain and pancreatic islets, MRAP2 is expressed in several human metabolic tissues including the gut, kidney, adipose tissue and skeletal muscle (Extended data Fig. 4.a). MRAP2 not only regulates the activity of melanocortin receptors but is also involved in the regulation of other GPCRs including prokineticin receptors and the growth hormone secretagogue receptor 1a (GHSR1a), namely the ghrelin receptor18,19. Therefore, we suggest that the spectrum of metabolic phenotypes observed in people carrying a pathogenic, loss-of-function MRAP2 mutation is due to the impaired regulatory effect of MRAP2 on metabolically active GPCRs across various key tissues, explaining the pleiotropic effect of this protein. In this regard, ghrelin and its receptor have been involved in blood pressure regulation20, which could explain the high blood pressure observed in the people deficient for MRAP2. As MRAP2 deficiency partly impacts the MC4R pathway, the eating behavior problems in MRAP2-deficient subjects might be treated by the MC4R agonist setmelanotide7.
Methods
Study participants
We investigated 9,418 blood DNA samples from several population studies: 1/3,766 participants from the D.E.S.I.R. 9-year prospective study including middle-aged men and women from western France22. 2/ 3,594 participants who were recruited and followed-up either by the CNRS UMR8199 (Lille, France), by the Department of Nutrition of Hotel-Dieu Hospital (Paris, France), or by the Centre d'Etude du Polymorphisme Humain (CEPH, Saint-Louis Hospital, Paris, France)23,24. 3/ 929 participants who were recruited from the Department of Endocrinology of the Corbeil-Essonnes Hospital (Corbeil-Essonnes, France)23. 4/ 672 participants from the French Haguenau regional cohort study25. 5/ 457 participants from the French Fleurbaix-Laventie Ville Santé study26. Clinical data of participants are shown in Supplementary Table 1. The study protocols were approved by local ethics committees. All participants older than 18 years signed an informed consent form. Oral assent from children or adolescents was obtained and parents (or legal guardian) signed an informed consent form.
In participants older than 18 years, class I obesity was defined as 30 ≤ BMI < 35 kg/m2, class II as 35 ≤ BMI < 40 kg/m2, class III as BMI ≥ 40 kg/m2, overweight as 25 ≤ BMI < 30 kg/m2 and normal weight as BMI < 25 kg/m2. In children and adolescents younger than 18 years, obesity was defined as 95th ≤ BMI-for-age < 99th percentile, severe obesity was defined as BMI-for-age ≥ 99th percentile, while normal weight was defined as BMI-for-age < 85th percentile according to the Centers for Disease Control and Prevention (CDC) growth charts. Type 2 diabetes was defined as fasting plasma glucose ≥ 7.0 mmol/L, plasma glucose measured 2 hours during an oral glucose tolerance test (OGTT) ≥ 11.1 mmol/L, and/or use of hyperglycemia treatment; hyperglycemia or pre-diabetes was defined as 5.6 ≤ fasting plasma glucose < 7.0 mmol/l without hyperglycemia treatment, and normal glucose was defined as fasting plasma glucose < 5.6 mmol/l without hyperglycemia treatment27.
Metabolic syndrome was defined following the criteria of the third report of the National Cholesterol Education Program (NCEP) expert panel on detection, evaluation, and treatment of high blood cholesterol in adults (ATP III)28: a waistline that measured at least 88 cm for women and 102 cm for men or a waist-hip ratio ≥ 0.9 for men and ≥ 0.8 for women, triglyceride levels ≥ 1.70 mmol/l, high-density lipoprotein (HDL) levels ≤ 1.04 mmol/l in men and HDL levels ≤ 1.30 mmol/l in women, systolic blood pressure (SBP) ≥ 130 mmHg or diastolic blood pressure (DBP) ≥ 85 mmHg, and hyperglycemia. A patient with metabolic syndrome had at least three criteria28.
In the carriers of a pathogenic, loss-of-function MRAP2 mutation, steroid hormones were measured in serum samples through liquid chromatography-tandem mass spectrometry (LC-MS/MS; Waters ACQUITY UPLC technology on the Xevo TQ-XS Mass Spectrometer), by Lille hospital (Lille, France).
MRAP2 sequencing
DNA sequencing of MRAP2 (NM_138409.3) was performed either by Sanger sequencing (n = 809) or by next-generation sequencing (NGS; n = 8,609). Regarding Sanger sequencing, the three coding exons of MRAP2 were analyzed in three fragments. Primer sequences and PCR conditions are available upon request. Fragments were sequenced in both directions, and subsequently analyzed using the 3730xl DNA Analyzer (Applied Biosystems). Electrophoregram reads were assembled and examined using the Variant Reporter software (Applied Biosystems). Regarding NGS, target enrichment was performed according to the manufacturer's protocol (NimbleGen SeqCap EZ) for Illumina sequencing. Briefly, 1 μg DNA was fragmented through sonication (Covaris E220 Focused-ultrasonicator). The fragmented DNA samples were end-repaired and ligated to the adapters using the KAPA HTP Library Preparation Kits, on the Hamilton Microlab STARlet automate. These samples were subsequently amplified by PCR. After size selection and sample quantification (Perkin Elmer LabChip GX), 24 samples were combined in a single pool of at least 1 μg, and hybridized to the biotin-labeled SeqCap EZ probe pool. After 72 hours at 47 °C, the captures were purified using the SeqCap Hybridization and Wash Kit on the Agilent Bravo Automated Liquid Handling Platform. Captures were subsequently amplified using the KAPA HiFi HotStart ReadyMix and quantified by both Perkin Elmer LabChip GX and Thermo Fisher Scientific Qubit fluorometric quantitation assays. Then, the samples were sequenced on the Illumina HiSeq 4000 system (with a throughput of one pool per lane), using a paired-end 2×150 bp protocol. The demultiplexing of sequence data was performed using bcl2fastq Conversion Software (Illumina; version 2.17). Subsequently, sequence reads were mapped to the human genome (hg19/GRCh37) using Burrows-Wheeler Aligner (version 0.7.13)29. The variant calling was performed using Genome Analysis ToolKit (GATK; version 3.3)30. Only variants with a coverage higher than 8 reads were kept for further analyses. The annotation of variants was performed using the Ensembl Perl Application Program Interfaces (version 75) and custom Perl scripts to include data from both dbSNP (version 135) and dbNSFP (version 3.0) databases31,32. All detected variants in MRAP2 had a QUAL score higher than 50. Furthermore, no variant had more than 5% of missing genotype (with a coverage below 8 reads or a QUAL score below 50) across the participants, and no participant had more than 5% of missing genotypes (with a coverage below 8 reads or a QUAL score below 50) across MRAP2.
The location of each variant (either detected by Sanger sequencing or NGS) was determined relative to the translation initiation codon using Human Genome Variation Society (HGVS) nomenclature for the description of sequence variations. The positions of mutations were indicated according to the human genome build hg19/GRCh37.
Sequencing of genes involved in monogenic obesity or monogenic diabetes
In the 10 carriers of a pathogenic, loss-of-function MRAP2 mutation, we analyzed the putative presence of a pathogenic or likely pathogenic variant in 48 genes known to be involved in monogenic obesity or monogenic diabetes via NGS: ABCC8, ADCY3, APPL1, BDNF, BLK, CEL, CEP19, DNAJC3, DYRK1B, FOXP3, GATA4, GATA6, GCK, GLIS3, GNAS, HNF1A, HNF1B, HNF4A, IER3IP1, INS, KCNJ11, KLF11, KSR2, LEP, LEPR, MAGEL2, MC4R, MNX1, NEUROD1, NEUROG3, NKX2-2, NTRK2, PAX4, PAX6, PCBD1, PCSK1, PDX1, POMC, PTF1A, RFX6, SH2B1, SIM1, SLC19A2, SLC2A2, STAT3, TRMT10A, TUB and WFS1.
Ancestry assessment
Ancestry of mutated individuals was assessed using the first two genotypic principal components (PC1 and PC2) calculated from at least 10,000 single nucleotide polymorphisms (SNPs; with a MAF >5%) present in each mutated individual (where genotypes were obtained from DNA arrays or NGS data) and in the 1,000 Genomes project.
Design of variant analysis
We only analyzed rare variants of potential interest (i.e., missense variants, stop gained variants, frameshift variants, inframe variants, initiator codon variants, stop retained variants, stop lost variants, splice donor variants and splice acceptor variants) with a MAF below 1%. We assessed the pathogenicity of the variants using the ACMG criteria for the interpretation of rare variants (Supplementary Tables 5 & 6)33. To address the strong pathogenic criterion PS3, we used our in-house in vitro functional analyses (see below).
In vitro functional analyses
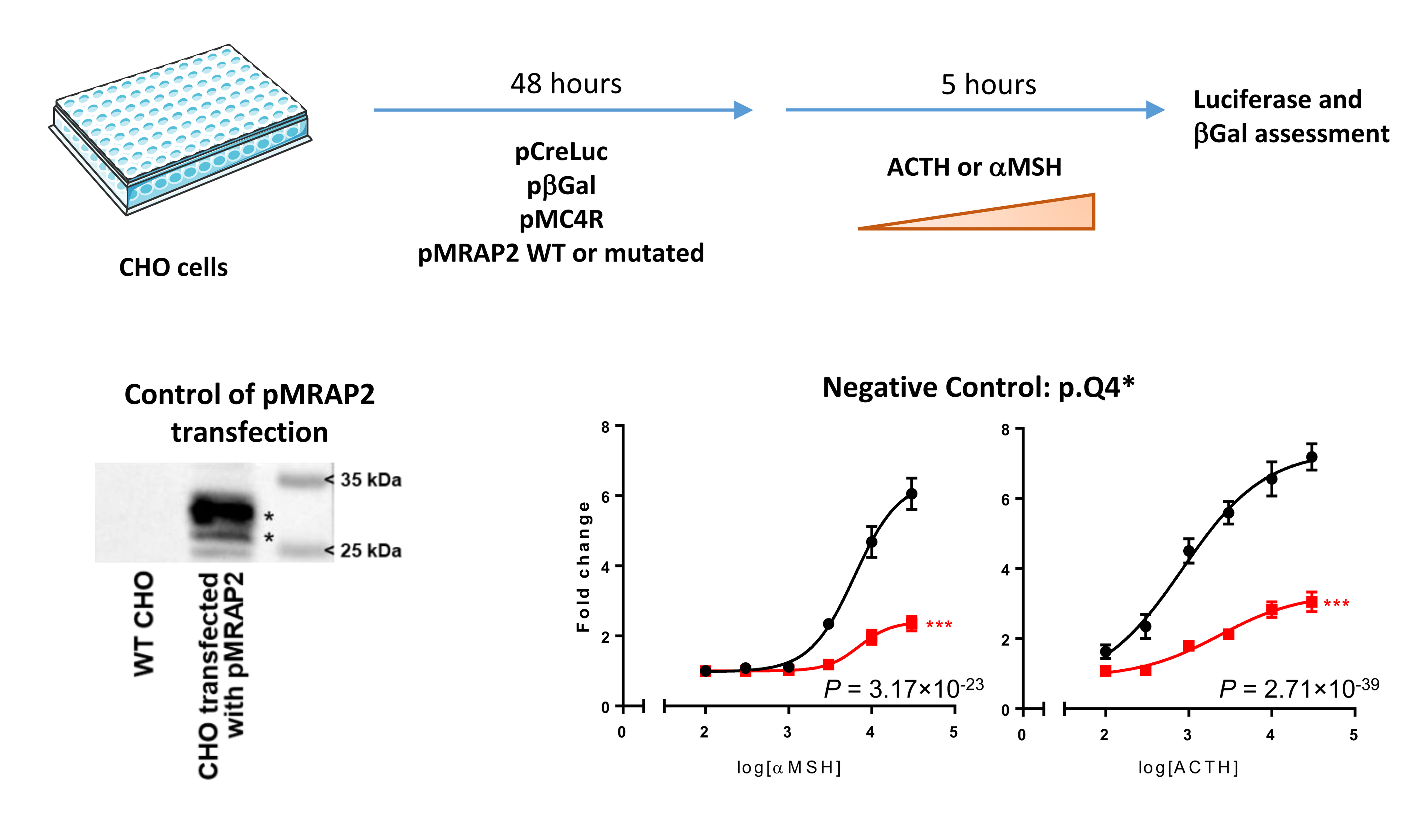
Twenty-two variants (including one frameshift variant and 21 missense variants) detected in the participants and one negative control variant (c.10C>T, p.Q4*) were generated in 23 different plasmids (pcDNA3.1 MRAP2) using the QuikChange site-directed mutagenesis kit (Stratagene). Each novel plasmid was checked by Sanger sequencing. The effect of each MRAP2 variant on MC4R activity was assessed in CHO cells in response to different concentrations (0 – 30 nM) of αMSH and of ACTH. For this purpose, CHO cells were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) F12 with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin (Gibco) at 37 °C and 5% CO2. Cells were transfected in suspension with 225 ng/ml of plasmid including the firefly luciferase gene under the control of cAMP response element (CRE), 150 ng/ml of plasmid including the β galactosidase gene, 41.25 ng/ml of MC4R plasmid and either 333.75 ng/ml wild-type MRAP2 plasmid or 333.75 ng/ml mutated MRAP2 plasmid using Fugene 6 (Promega), and they were seeded in 48-well plates at a concentration of 600,000 cells/ml in a volume of 200 μl. The day after, the medium was replaced by medium with 0.1% FBS for overnight serum starvation; 48 hours after the transfection, the cells were treated with increasing doses of αMSH (Abcam) or ACTH (Sigma) from 0 to 30,000 pM. Five hours after the treatment, the cells were lysed with 100 μl of Luciferase Cell Culture Lysis buffer (Promega). The luminescence was assessed by adding 25 μl of the Luciferase Assay System reagent (Promega) in 40 μl of the lysate and was read under a Glomax luminometer (Promega). The β-galactosidase activity was measured after incubation of 40 μl of the lysate for 5 minutes with 100 μl of home-made buffer (NaHPO4 0.479 g, NAH2PO4 – 2 H2O 1.060 g, KCl 2M 0.5 ml, MgCl2 1M 0.1 ml, Q.S. to 100 ml water; extemporaneous addition of β-mercaptoethanol [3.25 μl/ml] and ortho-nitrophenyl-β-galactoside [ONPG] 4 mM [250 μl/ml]) and was read at 450 nm. Of note, we primarily found that the best profile of MC4R activation in response to αMSH was obtained using one volume of MC4R plasmid in addition to eight volumes of MRAP2 plasmid, when compared to 1:2, 1:3 and 1:6 MC4R:MRAP2 ratios (data not shown). The experiments were done in technical triplicates, and each experiment was repeated three times. Luciferase measurements in relative luminescence unit (RLU) were normalized using β-galactosidase measures. Fold change was computed by dividing normalized luciferase (L*) by the mean of the baseline luciferase measures. This normalized luciferase fold-change (FCL*) was analyzed using a linear regression model. The mutation (M), the agonist concentration (C), as an orthogonal polynomial function of degree 3 (PC) to enable possible non-linear relations between (FCL*) and (C), and the interaction term (MPC) between M and PC was included in the model as a covariate.
The model was defined as follows:
with,
Our functional assay was validated using a negative control (p.Q4*; Supplementary Figure 2).
MRAP2 Expression analyses
The expression analysis of MRAP2 was performed using the NanoString technology, that is a multiplex digital quantification of nucleic acids (without PCR-based amplification), in a large panel of human tissues, as previously described34. The panel included human RNA from the colon, small intestine, liver, kidney, adipose tissue, placenta, lung, skeletal muscle, heart, brain, substantia nigra, hippocampus, dorsal root ganglion, insula, hypothalamus, pituitary gland, caudate nucleus, frontal lobe, pancreatic islets, pancreatic beta cells (obtained by laser capture microdissection [LCM beta cells] or sorted by flow cytometry [FACS sorted beta cells]), exocrine pancreas and the pancreatic beta cell line EndoC-βH135.
Western Blot analyses
Proteins were extracted from CHO cells transfected or not with 450 ng of MRAP2 plasmid, human pancreatic islets (pool of 2 donors) and EndoC-βH1 cells, using Pierce RIPA Buffer (Thermo Scientific) according to manufacturer’s instructions. Proteins were quantified by Bradford technique using BioRad Protein Assay (BioRad). 50 μg (for islets and EndoC-βH1) or 10 μg (for CHO cells) of proteins with Laemmli buffer 4X (Alfa Aesar) were denaturated at 95 °C for 5 minutes and loaded for migration on a concentration gel (Acrylamide 4% [Dutscher], Tris 125 mM [Dutscher], Sodium Dodecyl Sulfate [SDS] 0.04% [Sigma], Ammonium Persulfate 0.08% [Sigma], Temed 0.004% [Dutscher]) followed by a separation gel (Acrylamide 10%, Tris 375 mM, SDS 0.2%, Ammonium Persulfate 0.1%, Temed 0.05%). The migration was performed in Tris-Glycine-SDS Buffer (Eurobio-Ingen). After migration, the proteins were transferred on a nitrocellulose membrane (BioRad) in Tris-Glycine Buffer (Eurobio-Ingen). The non-specific sites were blocked with a 1 hour-incubation in TBS Buffer (Euromedex) + 0.1% Tween 20 (Biosolve) + 5% skimmed milk (Dutscher) under agitation. Then, the membrane was incubated overnight at 4 °C under agitation with the MRAP2 antibody (Bioss; 1/300 in TBS Buffer + 0.1% Tween 20 + 5% Bovine Serum Albumin [Sigma Aldrich]). After the incubation, the membrane was washed 3 times in TBS Buffer + 0.1% Tween 20 and then incubated for 1 hour at room temperature under agitation with anti-rabbit HRP antibody (Cell signaling; 1/2500 in TBS Buffer + 0.1% Tween 20 + 5% skimmed milk). After the incubation, the membrane was washed 3 times in TBS Buffer + 0.1% Tween 20. ECL Prime (Amersham) was used to reveal the protein of interest and the reading was performed using a Chemidoc (BioRad) after 120 seconds of exposure.
MRAP2 knockdown and insulin secretion assays in EndoC-βH1
EndoC-βH1 were cultured at 37 °C, 5% CO2 with Optiβ1 medium (Univercell) in T25 flasks coated with β-coat (Univercell) according to the manufacturer’s instructions35. Cells (500,000 cells/ml) were transfected with either an ON-TARGETplus non-targeting pool for controls (siNTP) or small interfering RNA (siRNA) targeting MRAP2 (siMRAP2; 20 μM; Horizon Discovery) using Lipofectamine 3000 (Invitrogen) in OptiMEM (Gibco) complemented with 50 μM 2-mercaptoethanol, 10 mM nicotinamide (Calbiochem, Merck Millipore), 5.5 mg/ml human transferrin (Sigma–Aldrich), 6.7 ng/ml sodium selenite (Sigma–Aldrich), 100 U/ml penicillin, and 100 mg/ml streptomycin (Life Technologies) and seeded (50,000 cells/well) in 96-wells plate coated with β-coat. After 6 hours, the medium was replaced by Optiβ1 medium. The day after, the medium was renewed, and 2 days after transfection, the medium was replaced by DMEM no glucose (Gibco) supplemented with 2% BSA fraction V (Roche Diagnostics), 50 μM 2-mercaptoethanol, 10 mM nicotinamide (Calbiochem, Merck Millipore), 5.5 mg/ml human transferrin (Sigma–Aldrich), 6.7 ng/ml sodium selenite (Sigma–Aldrich), 100 U/ml penicillin, and 100 mg/ml streptomycin (Life Technologies) and 2.8 mM of D-glucose (Sigma). Three days after transfection, cells were rinsed with β-Krebs buffer (Univercell) and 100 μl of Krebs buffer containing 0.5 mM glucose was added to each well. After 1 hour, supernatants were collected and 100 μl of Krebs buffer containing 16.7 mM glucose was added. After 1 hour, supernatants were collected and cells were lysed using TETG buffer (20 mM Tris–HCl pH 8.0, 137 mM NaCl, 1% Triton X-100, 10% Glycerol, 2 mM EGTA with protease inhibitors; Roche). Supernatants and lysates were centrifuged at 700 g, 4 °C for 5 minutes and insulin was assessed after proper dilution of the samples in water (1:16 for supernatants; 1:200 for lysates), using ELISA Human Insulin Kit (Mercodia, Uppsala, Sweden) according to the manufacturer's instructions. Absorbance data were measured, with technical duplicates of experimental triplicates. Biological experiments were repeated four times leading to 12 measurements for each condition. In these experiments, the MRAP2 knockdown was 68.5 ± 7.9% using the siMRAP2. To ensure a reduced technical bias from the absorbance data, the technical duplicates' average absorbance was kept when the relative error was lower than 20% among the technical duplicates (this threshold being based on the observed distribution of the technical relative errors over 100 experiments). Fold changes of insulin secretion (i.e. secretion at stimulatory glucose levels divided by secretion at basal glucose levels) were then computed. Fold change of insulin secretion for siMRAP2 was analyzed using a linear regression adjusted for experimental conditions (operator and date).
Statistical analyses for association studies
Among adults, we performed a case-control study for ‘obesity’ or ‘overweight and obesity’, adjusted for age, sex, and the presence of type 2 diabetes (controls were adults with normal weight). Among children and adolescents, we performed a case-control study for ‘obesity’, adjusted for age and sex (controls were children/adolescents with normal weight). Using the MiST method36, the rare variants were analyzed as a single cluster. MiST provided a score statistic S(π) for the mean effect (π) of the cluster, and a score statistic S(τ) for the heterogeneous effect (τ) of the cluster.
Let the equation of the model be: Y = αX + πGZ, where Y and X were the phenotype and the matrix of covariates, respectively.
Z was a vector of ones repeated n times, with n the number of rare variants in MRAP2, so .
As none of the association studies was significantly heterogeneous (Pτ > 0.25), we only showed the p-value associated with the mean effect (π) of the cluster.
Extended Data
Extended Data Fig. 1. Design of the functional assessment of each MRAP2 mutation and its validation.

αMSH, α-melanocyte-stimulating hormone; ACTH, adrenocorticotropic hormone; βGal, β-galactosidase; CHO, Chinese hamster ovary cells; pβGal, plasmid including the β-galactosidase gene; pCreLuc, plasmid including the firefly luciferase (Luc) gene under the control of cAMP response element (CRE); pMC4R, plasmid including MC4R; pMRAP2, plasmid including MRAP2; WT, wild-type.
For the control of pMRAP2 transfection into CHO cells, we performed Western blot analyses in wild-type CHO cells and CHO cells transfected with pMRAP2. We confirmed that wild-type CHO cells did not endogenously express MRAP2, while CHO cells transfected with pMRAP2 expressed MRAP2 protein (27 kDa, bottom asterisk), as well as its glycosylated form (29 kDa, top asterisk).
For the negative control, data are cAMP reporter activity (CRE-Luc normalized with β-galactosidase), expressed as fold change after 0 to 30,000 pM αMSH (on the left) or ACTH (on the right), relative to 0 pM. Data are the mean ± SEM of three independent experiments in technical triplicates. Fold change was computed by dividing normalized luciferase (L*) by the mean of the baseline luciferase measures. This normalized luciferase fold-change (FCL*) was analyzed using a linear regression model. The mutation (M), the agonist concentration (C), as an orthogonal polynomial function of degree 3 (PC) to enable possible non-linear relations between FCL* and C, and the interaction term (MPC) between M and PC was included in the model as a covariate.
***P < 0.001, MC4R + p.Q4* MRAP2 (red) versus MC4R + wild-type MRAP2 (black).
Extended Data Fig. 2. Effect of MRAP2 variants on MC4R activity in response to αMSH and ACTH, in CHO cells.

Data are cAMP reporter activity (CRE-Luc normalized with β-galactosidase) in CHO cells cotransfected with MC4R plasmid and wild-type or mutated MRAP2 plasmid, expressed as fold change after 0 to 30,000 pM αMSH or ACTH, relative to 0 pM. Data are the mean ± SEM of three independent experiments in technical triplicates. Fold change was computed by dividing normalized luciferase (L*) by the mean of the baseline luciferase measures. This normalized luciferase fold-change (FCL*) was analyzed using a linear regression model. The mutation (M), the agonist concentration (C), as an orthogonal polynomial function of degree 3 (PC) to enable possible non-linear relations between (FCL*) and C, and the interaction term (MPC) between M and PC was included in the model as a covariate.
*P < 0.05, **P < 0.01, ***P < 0.001, MC4R + mutated MRAP2 (colors) versus MC4R + wild-type MRAP2 (black).
Extended Data Fig. 3. Co-segregation of p.N77S (carried by participants #4 and 9) and p.P195L (carried by participants #6 and 10) with obesity in two families.

HT, hypertension; MS, metabolic syndrome; MS, no metabolic syndrome; NBP, normal blood pressure; NG, normal fasting glucose; NM, mutation carrier; NN, wild-type; NW, normal weight; Ob, obese; PD, pre-diabetes; SOb, severely obese; TG, triglycerides.
Extended Data Fig. 4. Expression of MRAP2 in human pancreatic islets and beta cells through a) PCR-free quantification of MRAP2 mRNA levels in a panel of human tissues, and b) Western blot analyses of human islets and EndoC-βH1 cells using MRAP2 antibody.

FACS sorted beta cell, pancreatic beta cells sorted by flow cytometry; LCM beta cell, pancreatic beta cells obtained by laser capture microdissection. Three independent experiments showed similar results of Extended data Fig. 4b.
Extended Data Fig. 5. Impaired insulin secretion from EndoC-βH1 cells treated with siRNA targeting MRAP2.

EndoC-βH1 cells were transfected with control non-targeting pool siRNA (siNTP) or MRAP2 siRNA (siMRAP2) and were analyzed 72 h thereafter. (Left) Insulin secretion (percentage of secretion of the total insulin content) was analyzed in response to 60 min incubation with 0.5 mM glucose, followed by 60 min incubation with 16.7 mM glucose. Data are box and whisker plots (with the minimum and the maximum) of four independent experiments. (Right) Fold change data are mean values ± SEM of four independent experiments. Fold change of insulin secretion for siMRAP2 was analyzed using a linear regression adjusted for experimental conditions (operator and date). Glc, glucose.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
We are grateful to all individuals included in the different cohort studies. We thank A. Abderrahmani (University of Lille, Lille, France) for his technical advice. We thank L. Chan (Queen Mary University of London, London, UK) for providing the plasmids including MRAP2 and MC4R. We thank F. Allegaert and N. Larcher for DNA collection and storage. We thank Endocells for providing the pancreatic beta cell line, EndoC-βH1. We thank the Type 2 Diabetes Knowledge Portal and the groups that provided data to this resource. We are grateful to the reviewers for their fruitful comments.
This work was supported by grants from the French-speaking society of Diabetes (Société Française du Diabète; to AB), from the European Foundation for the Study of Diabetes / Lilly (to AB), from the French National Research Agency (ANR-10-LABX-46 [European Genomics Institute for Diabetes] and ANR-10-EQPX-07-01 [LIGAN-PM], to PF), from the European Research Council (ERC GEPIDIAB – 294785, to PF; ERC Reg-Seq – 715575, to AB), from FEDER (to PF) and from the ‘Région Nord Pas-de-Calais’ (to PF). AB was supported by Inserm.
Footnotes
URLs: Type 2 Diabetes Knowledge Portal. MRAP2. type2diabetesgenetics.org. 2019 July 7; http://www.type2diabetesgenetics.org/gene/geneInfo/MRAP2.
Life Sciences Reporting Summary: Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
All relevant data have been included in the manuscript and/or in its supplementary tables and figures. Source data of Extended Data Figures 1, 2, 4 and 5 were provided. Targeted DNA-seq data of MRAP2-deficient patients were deposited in the NCBI Sequence Read Archive (SRA) under PRJNA564478.
Code availability
Code to perform analyses in this manuscript are available from the authors upon request (A.B., M.D. and M.C.).
Author Contributions
P.F. and A.B. conceived the idea for the study and supervised the analyses. M.B., J.M., M.H., A.D., R.B., H.L., E.D., B.T., E.V., J.P., J.T., A.G., M.B., M.D., S.G., M.C. and A.B. performed the experiments and/or analyses. M.B. and A.B. wrote the first draft of the paper. P.F. revised the paper. S.F., G.C., J.-M.B., C.L.-M., M.T., R.S., J.W., C.A., J.K.-C., F.P., R.B., B.B., M.M. and P.F. contributed data (i.e. cohort studies or beta-cell models). Furthermore, all authors critically reviewed the paper and approved the report for submission.
Competing interests
No conflicts were reported.
References
- 1.Asai M, et al. Loss of function of the melanocortin 2 receptor accessory protein 2 is associated with mammalian obesity. Science. 2013;341:275–278. doi: 10.1126/science.1233000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Geets E, et al. Copy number variation (CNV) analysis and mutation analysis of the 6q14.1-6q16.3 genes SIM1 and MRAP2 in Prader Willi like patients. Mol Genet Metab. 2016;117:383–388. doi: 10.1016/j.ymgme.2016.01.003. [DOI] [PubMed] [Google Scholar]
- 3.Schonnop L, et al. Decreased melanocortin-4 receptor function conferred by an infrequent variant at the human melanocortin receptor accessory protein 2 gene. Obesity (Silver Spring) 2016;24:1976–1982. doi: 10.1002/oby.21576. [DOI] [PubMed] [Google Scholar]
- 4.Greenfield JR, et al. Modulation of blood pressure by central melanocortinergic pathways. N Engl J Med. 2009;360:44–52. doi: 10.1056/NEJMoa0803085. [DOI] [PubMed] [Google Scholar]
- 5.El-Sayed Moustafa JS, Froguel P. From obesity genetics to the future of personalized obesity therapy. Nat Rev Endocrinol. 2013;9:402–413. doi: 10.1038/nrendo.2013.57. [DOI] [PubMed] [Google Scholar]
- 6.Kühnen P, et al. Proopiomelanocortin Deficiency Treated with a Melanocortin-4 Receptor Agonist. New England Journal of Medicine. 2016;375:240–246. doi: 10.1056/NEJMoa1512693. [DOI] [PubMed] [Google Scholar]
- 7.Clément K, et al. MC4R agonism promotes durable weight loss in patients with leptin receptor deficiency. Nature Medicine. 2018;24:551–555. doi: 10.1038/s41591-018-0015-9. [DOI] [PubMed] [Google Scholar]
- 8.Soletto L, et al. Melanocortin Receptor Accessory Protein 2-Induced Adrenocorticotropic Hormone Response of Human Melanocortin 4 Receptor. J Endocr Soc. 2019;3:314–323. doi: 10.1210/js.2018-00370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Josep Agulleiro M, et al. Melanocortin 4 receptor becomes an ACTH receptor by coexpression of melanocortin receptor accessory protein 2. Mol Endocrinol. 2013;27:1934–1945. doi: 10.1210/me.2013-1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang J, et al. The interaction of MC3R and MC4R with MRAP2, ACTH, α-MSH and AgRP in chickens. J Endocrinol. 2017;234:155–174. doi: 10.1530/JOE-17-0131. [DOI] [PubMed] [Google Scholar]
- 11.Bradshaw PT, Monda KL, Stevens J. Metabolic syndrome in healthy obese, overweight, and normal weight individuals: the Atherosclerosis Risk in Communities Study. Obesity (Silver Spring) 2013;21:203–209. doi: 10.1002/oby.20248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stanley CA. Perspective on the Genetics and Diagnosis of Congenital Hyperinsulinism Disorders. J Clin Endocrinol Metab. 2016;101:815–826. doi: 10.1210/jc.2015-3651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bonnefond A, Froguel P. Rare and common genetic events in type 2 diabetes: what should biologists know? Cell Metab. 2015;21:357–368. doi: 10.1016/j.cmet.2014.12.020. [DOI] [PubMed] [Google Scholar]
- 14.Ndiaye FK, et al. Expression and functional assessment of candidate type 2 diabetes susceptibility genes identify four new genes contributing to human insulin secretion. Mol Metab. 2017;6:459–470. doi: 10.1016/j.molmet.2017.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ravassard P, et al. A genetically engineered human pancreatic β cell line exhibiting glucose-inducible insulin secretion. J Clin Invest. 2011;121:3589–3597. doi: 10.1172/JCI58447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rouault AAJ, Srinivasan DK, Yin TC, Lee AA, Sebag JA. Melanocortin Receptor Accessory Proteins (MRAPs): Functions in the melanocortin system and beyond. Biochim Biophys Acta Mol Basis Dis. 2017;1863:2462–2467. doi: 10.1016/j.bbadis.2017.05.008. [DOI] [PubMed] [Google Scholar]
- 17.Novoselova TV, et al. Loss of Mrap2 is associated with Sim1 deficiency and increased circulating cholesterol. J Endocrinol. 2016;230:13–26. doi: 10.1530/JOE-16-0057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chaly AL, Srisai D, Gardner EE, Sebag JA. The Melanocortin Receptor Accessory Protein 2 promotes food intake through inhibition of the Prokineticin Receptor-1. Elife. 2016;5 doi: 10.7554/eLife.12397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Srisai D, et al. MRAP2 regulates ghrelin receptor signaling and hunger sensing. Nat Commun. 2017;8:713. doi: 10.1038/s41467-017-00747-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mao Y, Tokudome T, Kishimoto I. Ghrelin and Blood Pressure Regulation. Curr Hypertens Rep. 2016;18:15. doi: 10.1007/s11906-015-0622-5. [DOI] [PubMed] [Google Scholar]
- 21.Chan LF, et al. MRAP and MRAP2 are bidirectional regulators of the melanocortin receptor family. PNAS. 2009;106:6146–6151. doi: 10.1073/pnas.0809918106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Balkau B. [An epidemiologic survey from a network of French Health Examination Centres, (D.E.S.I.R.): epidemiologic data on the insulin resistance syndrome] Rev Epidemiol Sante Publique. 1996;44:373–375. [PubMed] [Google Scholar]
- 23.Sladek R, et al. A genome-wide association study identifies novel risk loci for type 2 diabetes. Nature. 2007;445:881–885. doi: 10.1038/nature05616. [DOI] [PubMed] [Google Scholar]
- 24.Meyre D, et al. Genome-wide association study for early-onset and morbid adult obesity identifies three new risk loci in European populations. Nat Genet. 2009;41:157–159. doi: 10.1038/ng.301. [DOI] [PubMed] [Google Scholar]
- 25.Leger J, et al. Reduced final height and indications for insulin resistance in 20 year olds born small for gestational age: regional cohort study. BMJ. 1997;315:341–347. doi: 10.1136/bmj.315.7104.341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Romon M, et al. Relationships between physical activity and plasma leptin levels in healthy children: the Fleurbaix-Laventie Ville Santé II Study. Int J Obes Relat Metab Disord. 2004;28:1227–1232. doi: 10.1038/sj.ijo.0802725. [DOI] [PubMed] [Google Scholar]
- 27.American Diabetes Association. 2. Classification and Diagnosis of Diabetes: Standards of Medical Care in Diabetes-2019. Diabetes Care. 2019;42:S13–S28. doi: 10.2337/dc19-S002. [DOI] [PubMed] [Google Scholar]
- 28.Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults. Executive Summary of the Third Report of the National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III) JAMA: The Journal of the American Medical Association. 2001;285:2486–2497. doi: 10.1001/jama.285.19.2486. [DOI] [PubMed] [Google Scholar]
- 29.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McKenna A, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sherry ST, et al. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 2001;29:308–311. doi: 10.1093/nar/29.1.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu X, Wu C, Li C, Boerwinkle E. dbNSFP v3.0: A One-Stop Database of Functional Predictions and Annotations for Human Nonsynonymous and Splice-Site SNVs. Hum Mutat. 2016;37:235–241. doi: 10.1002/humu.22932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Richards S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–424. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ndiaye FK, et al. Expression and functional assessment of candidate type 2 diabetes susceptibility genes identify four new genes contributing to human insulin secretion. Mol Metab. 2017;6:459–470. doi: 10.1016/j.molmet.2017.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ravassard P, et al. A genetically engineered human pancreatic β cell line exhibiting glucose-inducible insulin secretion. J Clin Invest. 2011;121:3589–3597. doi: 10.1172/JCI58447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sun J, Zheng Y, Hsu L. A unified mixed-effects model for rare-variant association in sequencing studies. Genet Epidemiol. 2013;37:334–344. doi: 10.1002/gepi.21717. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.