

Abstract
Activating FLT3 internal tandem duplication (FLT3-ITD) mutations in acute myeloid leukemia (AML) associate with inferior outcomes. We determined that pacritinib, a JAK2/FLT3 inhibitor, has in vitro activity against FLT3-ITD and tyrosine kinase domain (TKD) mutations. Therefore, we conducted a phase I study of pacritinib in combination with chemotherapy in AML patients with FLT3 mutations to determine the pharmacokinetics and preliminary toxicity and clinical activity. Pacritinib was administered at a dose of 100 mg or 200 mg twice daily following a 3+3 dose-escalation in combination with cytarabine and daunorubicin (cohort A) or with decitabine induction (cohort B). A total of thirteen patients were enrolled (five in cohort A; eight in cohort B). Dose limiting toxicities include hemolytic anemia and grade 3 QTc prolongation in two patients who received 100 mg. Complete remission was achieved in two patients in cohort A, one of whom had a minor D835Y clone at baseline. One patient in cohort B achieved morphologic leukemia free state. Seven patients (two in cohort A; five in cohort B) had stable disease. In conclusion, pacritinib, an inhibitor of FLT3-ITD and resistant-conferring TKD mutations, was well tolerated and demonstrated preliminary anti-leukemic activity in combination with chemotherapy in patients with FLT3 mutations.
Introduction
Approximately 25–30% of adults and 13–16% of pediatric patients with acute myeloid leukemia (AML) harbor a constitutively active internal tandem duplication (ITD) mutation in FMS-like tyrosine kinase 3 (FLT3) or FLT3-ITD+ AML. Such activating mutations in FLT3 have been associated with a shorter remission period and lower overall survival[1, 2]. Small molecule FLT3 tyrosine kinase inhibitors (TKI) have been under active clinical development either as single agents or in combination with chemotherapy. However, intrinsic and acquired drug resistance, such as the emergence of secondary tyrosine kinase domain mutations (TKD) or activation of alternative signaling pathways, including JAK/STAT pathway, remains a challenge in FLT3 TKI therapy[3, 4].
Pacritinib, an oral TKI with activity against Janus Associated Kinase 2 (JAK2), is undergoing clinical development for myelofibrosis. Two phase III studies of pacritinib in adults with high-risk myelofibrosis were recently reported[5, 6], and a phase II dose finding study of pacritinib (100 mg once daily, 100 mg twice daily, versus 200 mg once daily) is ongoing ( NCT03165734).
In addition to JAK2 inhibition, pacritinib has demonstrated to have anti-leukemic activity in AML based on its inhibition of IRAK1[7] as well as FLT3 signaling[8, 9]. Here, we present the preclinical activity of pacritinib in models harboring FLT3-ITD and FLT3 TKD mutations and results from a pilot phase I study evaluating the toxicity profile, pacritinib pharmacokinetics, and preliminary clinical activity of pacritinib administered in combination with cytarabine and daunorubicin or with decitabine in adults with FLT3-ITD+ AML.
Materials and Methods
Biochemical evaluation of pacritinib activity
The activity of pacritinib against FLT3 wildtype, FLT3-ITD, and FLT3 tyrosine kinase domain (TKD) mutations was determined in a competitive binding assay (KdELECT by Eurofins, San Diego, CA). Drug and DMSO control were tested in the concentration range of 0.003–30 000 nM. The binding constants (Kds) were calculated with a standard dose-response curve using the Hill equation and a non-linear square fit with the Levenberg-Marquardt algorithm. Activity against FLT3-ITD and FLT3 D835Y was also evaluated in a kinase assay (HotSpot Kinase assay, Reaction Biology, Malvern, PA). Drug and control were tested in the concentration range of 2.5–50 000 nM. Enzymatic activity was measured relative to DMSO control, and a standard dose response with the Hill equation was used to calculate IC50.
Cell culture
Ba/F3, MV4–11, MOLM13, and MOLM13-Res cells were cultured and maintained as previously described[10].
Cell viability assays
The activity of pacritinib and in Ba/F3 cells transfected with vector control (VC) or different FLT3 mutants and pacritinib and midostaurin in three FLT3-ITD+ AML cell lines was evaluated using MTT assay (Roche Diagnostics, Mannheim, Germany) as described previously[10, 11]. The activity of midostaurin and pacritinib in FLT3-ITD+/−/IDH2-R140Q+/− double knock-in murine leukemia were assessed using CellTiter-Glo assay (Promega, Madison, WI) according to manufacturer’s instructions. Murine blast cells were incubated with RPMI supplemented with 10% FBS, GlutaMAX (Life Technologies, Grand Island, NY) and murine cytokines IL3 (10ng/mL), SCF (10ng/mL), and GM-CSF (20ng/mL) from PeproTech (Rocky Hills, NJ) to support continuous propagation of cells during treatment. All assays were conducted at 72 hours after addition of drugs. Results were measured as a mean percentage of DMSO-treated control cells at each concentration (performed in 3–6 replicates). Pretreated bone marrow blast samples collected during screening were treated with DMSO or increasing concentrations of pacritinib or midostaurin in RPMI supplemented with 10% FBS, hFLT3 ligand (10ng/mL), hIL3 (10ng/mL), hGM-CSF (10ng/mL), and hSCF (10ng/mL) from PeproTech for 72 hours. Viability was monitored by trypan blue in an untreated well every 24 hours. The dose response was measured at 72 hours or when viability reached 50% or less, whichever came earlier. The blasts viability to dose response was measured using CellTiter-Glo assay (Promega) according to the manufacturer’s instructions (performed in 3–6 replicates).
Western blot analysis
Cells were treated with pacritinib or DMSO for 4 hours and lysed in RIPA buffer supplemented with protease and phosphatase inhibitors. Immunoprecipitation was carried out overnight with anti-FLT3 antibodies (Santa Cruz Biotechnology, Dallas, TX). Dynabeads (ThermoFisher, Waltham, MA) were added the next day and incubated for 4 hours. Samples were then prepared according to manufacturer’s instructions. Eluted lysates or total cell lysates were separated by SDS-polyacrylamide gel electrophoresis and transferred to PVDF membranes followed by western blot analysis using the indicated primary antibodies: FLT3, phospho-FLT3, STAT5, and phospho-STAT5 (Cell Signaling Technology, Danvers, MA).
Patient population
Patients >18 years old with a histologically confirmed diagnosis of untreated AML (excluding acute promyelocytic leukemia), or those with relapsed or refractory AML (cohort B only), Eastern Cooperative Oncology Group (ECOG) status <2, with the presence of FLT3 mutations were eligible for enrollment. Patients were required to have adequate organ function defined as total bilirubin of 2.0 mg/dL or less unless due to Gilbert’s disease, aspartate aminotransferase and alanine aminotransferase of 2.5x the upper limit of normal or less, creatinine clearance of 50 mL/min or greater by Cockcroft-Gault, New York Heart Association (NYHA) congestive heart failure (CHF) class II or better, and left ventricular ejection fraction of 50% or greater. Patient life expectancy was required to be greater than 6 months when present with co-morbid illnesses.
Exclusion criteria included patients who had core-binding factor AML with (inv(16), t(8;21)); patients with uncontrolled intercurrent illness including but not limited to symptomatic CHF, unstable angina, myocardial infarction within 6 months, severe uncontrolled ventricular arrhythmias or electrocardiographic evidence of acute ischemia or active conduction system abnormalities; pregnant or breastfeeding women; baseline QTc greater than 450ms or patients taking medications that prolong QTc interval; patients who received potent cytochrome P450 3A4 (CYP3A4) inhibitor 1 week prior to treatment; and use of concomitant potent CYP3A4 inducers. A full list is available in the supplementary material. This protocol was approved by The Ohio State University (OSU) IRB and registered at ClinicalTrials.gov (Identifier: NCT02323607). Informed consent was obtained from all patients.
Treatment plan
Patients were treated in parallel in one of two cohorts (Supplementary Figure 1). Fit patients who were eligible for intensive chemotherapy were assigned to cohort A and received pacritinib on days 1–4 and days 8–21, cytarabine 100mg/m2 days 5–11, and daunorubicin 60mg/m2 days 5–7. Patients > 60 years, who were considered unfit for intensive therapy, and those with relapsed/refractory disease, were assigned to cohort B receiving pacritinib days 1–21 and decitabine 20mg/m2 days 5–14. Initially, patients were treated with pacritinib 200mg twice daily (dose level 1). However, due to safety concerns, pacritinib was temporarily placed on a full clinical hold by the Food and Drug Administration. At the time of the clinical hold, two patients had been enrolled in cohort B. Once the hold was lifted, the protocol was amended to change the study design to a standard 3+3 dose-escalation design. Following the amendment, patients treated on dose level −2 received 200 mg of pacritinib per day (100 mg by mouth twice a day). Patients on dose level −1 received 300 mg per day (200 mg in the morning and 100 mg in the evening) and patients on dose level 1 received 400 mg of pacritinib per day (200 mg twice a day). Treatment consisted of 1–2 cycles of induction therapy (cohort A) or up to 4 induction cycles (cohort B). Patients in cohort A who achieved complete remission (CR) were evaluated for hematopoietic stem cell transplant or additional courses of chemotherapy at the discretion of the clinician. Patients in cohort B who achieved CR were able to proceed with transplant, if eligible, or 5-day maintenance courses of decitabine and pacritinib. Patients received full supportive care including transfusions of blood and blood products, antibiotics, and antiemetics when appropriate. Trial enrollment was prematurely terminated following loss of financial support from the drug manufacturer.
Safety Assessments
This study utilized the National Cancer Institute Common Terminology Criteria for Adverse Events version 4.03 to characterize toxicities. Dose-limiting toxicity (DLT) was evaluated in the first course and included any grade ≥3 non-hematological toxicities not resulting from active leukemia except alopecia; line associated venous thrombosis; infection not resulting from unexpectedly complicated myelosuppression; grade 3 fatigue, anorexia, constipation; grade 3 nausea or vomiting not requiring tube feedings, total parental nutrition or hospitalization; or grade 3 or higher transaminases that resolve to grade 1 or baseline within 5 days. Hematological DLTs included failure to recover neutrophil count (ANC>500 cells/μL) by day 32 in patients with <5% blasts in the bone marrow, absence of myelodysplastic changes, and/or absence of evidence of disease by flow cytometry in the bone marrow. For patients with >5% blasts, these were not considered DLT. Grade 4 thrombocytopenia with clinically significant bleeding was also considered a DLT. The maximum tolerated dose (MTD) was defined as the dose level at which one or fewer of six patients in a dose level experienced DLT.
Pharmacokinetic studies
Serial blood samples for pacritinib plasma pharmacokinetic studies were obtained on days 1 and 21 at pretreatment and after pacritinib administration at 1, 2, 3, 5, 24 hours; a pretreatment sample of blood and bone marrow was obtained on day 5. Pacritinib was quantitated in plasma and bone marrow lysates using a validated LC/MS-MS assay in The Ohio State University Comprehensive Cancer Center (CCC) Pharmacoanalytical Shared Resource. Bone marrow concentrations were normalized to amount of protein from each sample based on a BCA assay (Thermo Scientific, Rockford, IL). It was assumed that 20% of total wet weight of human cells is protein, and the density of tissue was assumed as 1g/mL for the calculation of bone marrow tissue concentrations of pacritinib[12].
Mutation analysis
Analysis of mutations in FLT3 exons 17 and 20 was conducted by deep amplicon sequencing as previously described[13] with few modifications. Additional mutations co-occurring with FLT3-ITD were determined using a targeted gene panel in the OSU Department of Pathology. Details are provided in the Supplementary Material.
Response criteria and definitions
Assessment of clinical response was made according to the International Working Group criteria[14]. Bone marrow response to treatment was evaluated by morphologic and flow cytometry studies.
Results
Biochemical, in vitro and ex vivo cellular activity of pacritinib
Binding affinities and kinase inhibitory activity of pacritinib to FLT3 wildtype, FLT3-ITD and FLT3 TKD mutants were examined (Fig. 1A, Supplementary Table 1). Pacritinib demonstrated similar or more potent binding to TKD mutants (Kd range, 0.69 to 17 nM) compared to FLT3-ITD (Kd = 8.2 nM). Pacritinib Kd values were in the range of those observed for midostaurin, an FDA approved FLT3 inhibitor (Supplementary Table 2). In a kinase assay, pacritinib inhibited FLT3-ITD and D835Y with IC50 values of 9 nM and 3.1 nM, respectively (Fig. 1B, Supplementary Table 2). In a cellular context, pacritinib showed midrange nM activity against Ba/F3 cells expressing FLT3-ITD (IC50 = 133 nM) and TKD mutants (IC50 range, 97–434 nM) (Fig. 1C, Supplementary Table 3). Western blot analysis of lysates from pacritinib-treated Ba/F3 cells expressing FLT3-ITD/D835Y or ITD/F691L mutants showed inhibition of phospho-FLT3 and phospho-STAT5 at concentrations of 0.25–1 µM (Fig. 1D). We further evaluated pacritinib in the FLT3-ITD+ AML cell lines, MV4–11 and MOLM13, as well as a FLT3 inhibitor resistant form of MOLM13 that contains a D835Y mutation (MOLM13-Res). Pacritinib had activity with mean IC50 values of 33 nM, 73 nM, and 173 nM in MV4–11, MOLM13, and MOLM13-Res, respectively (Fig 1D, Supplementary Table 4). We assessed the ex vivo activity of pacritinib in primary FLT3-ITD+ AML samples. In murine primary leukemia cells with double knock-in of FLT3-ITD+/−/IDH2-R140Q+/−, mutations that co-occur in AML, pacritinib inhibited cell viability with an IC50 value of 8.7 µM, while midostaurin had an IC50 value of 10.7 µM (Fig. 1F, Supplementary Table 5). Pacritinib also inhibited the viability of patient samples with IC50 values ranging from 152 to 302 nM, while midostaurin inhibited with IC50 values ranging from 250 to 470 nM in the same samples (Fig. 1G, Supplementary Table 5).
Fig 1. In vitro activity of pacritinib in various FLT3-ITD+ models.

(A) Activity of pacritinib against different FLT3 mutants in a binding assay. (B) Activity of pacritinib against FLT3-ITD and FLT3 D835Y in a kinase assay. (C) Activity of pacritinib in Ba/F3 cells transfected with different FLT3 mutants in a cell viability assay (mean ± SEM, n=12, two experiments). (D) Signaling inhibition with pacritinib in Ba/F3 cells expressing different FLT3 double mutants via Western blot (representative images from two experiments). (E) Activity of pacritinib in AML cell lines in a viability assay (mean ± SD, n=12). Activity of pacritinib in (F) murine primary FLT3-ITD+/−/IDH2-R140Q+/− leukemia cells and (G) pre-treatment patient primary blast samples in a viability assay (mean ± SD, n=3). Baseline mutations in each patient are denoted in parentheses.
Patient characteristics
Between December 2014 and January 2017 five patients were enrolled in cohort A and eight patients were enrolled in cohort B. Eight patients (62%) had newly diagnosed untreated AML and five patients (38%) had relapsed/refractory AML. All patients were positive for a FLT3-ITD mutation. One patient (patient 9) had a baseline FLT3 D835Y mutation (VAF, 2.3%) (Supplementary Table 7). Other co-occurring mutations were present at baseline, and these include NPM1, IDH2, and TET2 as well as other mutations (Supplementary Table 8). Other baseline characteristics are summarized in Table 1. Of thirteen patients, all patients were evaluable for toxicity, and ten patients were evaluable for response.
Table 1.
Patient demographics and baseline characteristics
| Characteristics | All Patients (n=13) | Cohort A (n=5) | Cohort B (n=8) |
|---|---|---|---|
| Age | |||
| Median (Range) | 60 (33 – 76) | 60 (34 – 66) | 56 (33–76) |
| Gender | |||
| Female | 5 (38) | 1 (20) | 4 (50) |
| Male | 8 (62) | 4 (80) | 4 (50) |
| Race | |||
| Asian | 1 (7.7) | 0 (0) | 1 (12.5) |
| White | 11 (85) | 5 (100) | 6 (75) |
| African American | 1 (7.7) | 0 (0) | 1 (12.5) |
| Type of AML | |||
| Refractory | 2 (15) | 0 (0) | 2 (25) |
| Relapsed | 3 (23) | 0 (0) | 3 (38) |
| Untreated | 8 (62) | 5 (100) | 3 (38) |
| History of CNS disease | |||
| Yes | 0 (0) | 0 (0) | 0 (0) |
| No | 13 (100) | 5 (100) | 8 (100) |
| History of extramedullary disease | |||
| Yes | 0 (0) | 0 (0) | 0 (0) |
| No | 12 (92) | 5 (100) | 7 (88) |
| unknown | 1 (8) | 0 (0) | 1 (12) |
| ELN 2011 Genetic Group | |||
| Favorable | 2 (15) | 0 (0) | 2 (25) |
| Intermediate I | 6 (46) | 1 (20) | 5 (62.5) |
| Intermediate II | 5 (38) | 4 (80) | 1 (12.5) |
| FLT3 ITD mutation | |||
| Yes | 13 (100) | 5 (100) | 8 (100) |
| No | 0 (0) | 0 (0) | 0 (0) |
| FLT3 TKD mutation | |||
| Yes | 1 (8) | 1 (20) | 0 (0) |
| No | 12 (92) | 4 (80) | 8 (100) |
| CEBPA Mutation | |||
| Yes | 0 (0) | 0 (0) | 0 (0) |
| No | 13 (100) | 5 (100) | 8 (100) |
| NPM1 mutation | |||
| Yes | 4 (31) | 0 (0) | 4 (50) |
| No | 9 (69) | 5 (100) | 4 (50) |
| ECOG at baseline (Performance Status) | |||
| 0 | 3 (23) | 1 (20) | 2 (25) |
| 1 | 9 (69) | 4 (80) | 5 (62) |
| 2 | 1 (8) | 0 (0) | 1 (13) |
| Prior MDS | |||
| Yes | 0 (0) | 0 (0) | 0 (0) |
| No | 13 (100) | 5 (100) | 8 (100) |
| Prior MPN | |||
| Yes | 1 (8) | 1 (20) | 0 (0) |
| No | 10 (77) | 4 (80) | 6 (75) |
| unknown | 2 (15) | 0 (0) | 2 (25) |
| Bone Marrow Blast | |||
| Median (Range) | 40 (2 – 88) | 40 (23–65) | 40 (2–88) |
| Peripheral Blood Blast | |||
| Median (Range) | 15 (0 – 88) | 15 (7 – 32) | 13 (0 – 88) |
| WBC | |||
| Median (Range) | 8.3 (0.4 – 126) | 5.8 (1.4 – 9.1) | 11 (0.4–126) |
| ANC | |||
| Median (Range) | 0.4 (0 – 5.5) | 0.6 (0.1 – 2.6) | 0.3 (0.0 – 5.5) |
| HGB | |||
| Median (Range) | 7.5 (7 – 13) | 7.4 (7 – 8.7) | 8.7 (7.1 – 13) |
| Platelets | |||
| Median (Range) | 44 (11 – 340) | 57 (29 – 340) | 26 (11 – 330) |
Efficacy
One patient in cohort A was not evaluable for treatment response due to early death and 2 patients in cohort B were not evaluable for response due to early discontinuation of therapy. Of four evaluable patients in cohort A, two patients (5 and 9; Fig. 2) achieved CR, and two patients had stable disease. Patient 9, whom achieved a CR, had a minor clone with a FLT3 D835Y mutation at baseline. In cohort B, one patient achieved morphologic leukemia free state (MLFS), and five patients had stable disease. Median survival in both cohorts was 292 days (95% CI: 36–580), and response rate defined as CR or MLFS was 23.1% (95% CI: 5.0–53.8%). Kaplan-Meier analysis for overall survival of patients who received pacritinib is shown in Figure 3.
Fig 2. Clinical course of pacritinib in patients achieving complete response in cohort A.
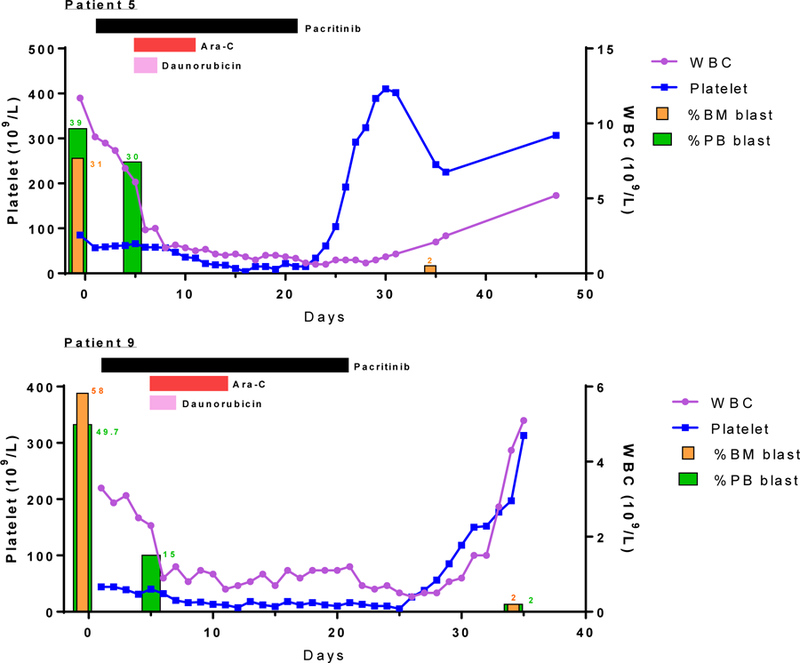
Clinical course of patient 5 and 9 who received pacritinib in combination with cytarabine and daunorubicin. Pacritinib was started on day 1 and horizontal bars indicate duration of the treatment.
Fig 3. Kaplan-Meier curve for overall survival of patients who received pacritinib.
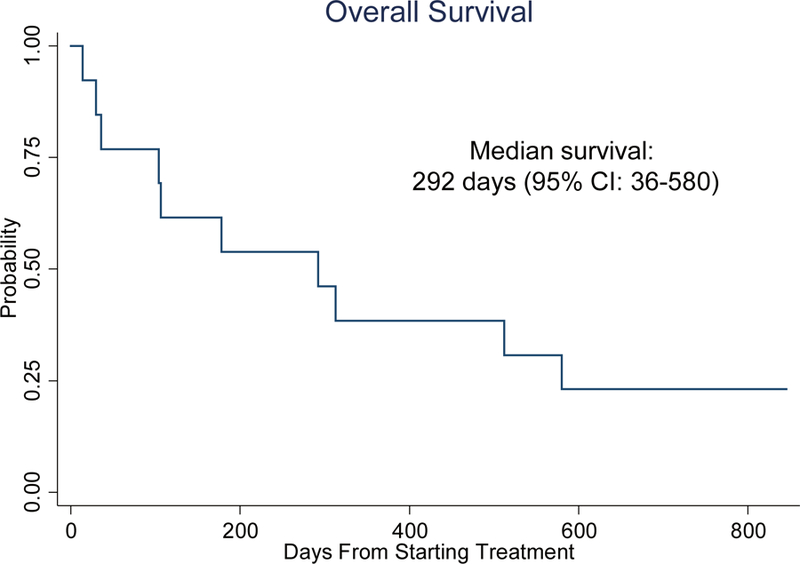
All patients in both cohorts were included in the analysis
Safety
In cohort A, five patients were treated with pacritinib 100mg twice daily, with one patient having grade 3 hemolytic anemia (patient 8) as a DLT. In cohort B, six patients were treated with pacritinib 100mg twice daily, with one patient having grade 3 QTc prolongation (patient 3) as a DLT. Two additional patients were enrolled to cohort B at the next pacritinib dose level of 200 mg twice daily. A summary of toxicity grade observed in each treatment cohort is listed in Table 2; a complete list of toxicity is provided in Supplementary Table 6. Four patients in cohort A and one patient in cohort B had grade 1 or 2 QTc prolongation.
Table 2.
A summary table of toxicity grade observed in each treatment cohort
| Cohort A: Pacaritinib 200mg, cytarabine, 100mgm2, Daunorubicin 60mg/m2 (n = 5) | Cohort B dose 1: Pacritinib 200mg, Decitabine 20mg/m2 (n = 6) | Cohort B Dose 2: Pacritinib 400mg, Decitabine 20mg/m2 (n = 2) | Overall (n = 13) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1&2 | 3+ | Total (n = 5) | 1&2 | 3+ | Total (n = 6) | 1&2 | 3+ | Total (n = 2) | 1&2 | 3+ | Total (n = 13) | |
| Blood and lymphatic system disorders | ||||||||||||
| ANEMIA | 0 | 3 (60) | 3 (60) | 1 (16.7) | 5 (83.3) | 6 (100) | 1 (50) | 1 (50) | 2 (100) | 2 (15.4) | 9 (69.2) | 11 (84.6) |
| ACTIVATED PARTIAL THROMBOPLASTIN TIME PROLONGED | 1 (20) | 0 | 1 (20) | 1 (16.7) | 0 | 1 (16.7) | 0 | 0 | 0 | 2 (15.4) | 0 | 2 (15.4) |
| FEBRILE NEUTROPENIA | 0 | 3 (60) | 3 (60) | 0 | 1 (16.7) | 1 (16.7) | 0 | 0 | 0 | 0 | 4 (30.8) | 4 (30.8) |
| LYMPHOCYTE COUNT DECREASED | 1 (20) | 2 (40) | 3 (60) | 1 (16.7) | 2 (33.3) | 3 (50) | 0 | 1 (50) | 1 (50) | 2 (15.4) | 5 (38.5) | 7 (53.8) |
| NEUTROPHIL COUNT DECREASED | 0 | 2 (40) | 2 (40) | 0 | 3 (50) | 3 (50) | 0 | 1 (50) | 1 (50) | 0 | 6 (46.2) | 6 (46.2) |
| PLATELET COUNT DECREASED | 0 | 5 (100) | 5 (100) | 0 | 5 (83.3) | 5 (83.3) | 0 | 1 (50) | 1 (50) | 0 | 11 (84.6) | 11 (84.6) |
| WHITE BLOOD CELL DECREASED | 0 | 4 (80) | 4 (80) | 0 | 3 (50) | 3 (50) | 0 | 1 (50) | 1 (50) | 0 | 8 (61.5) | 8 (61.5) |
| Cardiac disorders | ||||||||||||
| ELECTROCARDIOGRAM QT CORRECTED INTERVAL PROLONGED | 4 (80) | 0 | 4 (80) | 1 (16.7) | 1 (16.7) | 2 (33.3) | 0 | 0 | 0 | 5 (38.5) | 1 (7.7) | 6 (46.2) |
| Gastrointestinal disorders | ||||||||||||
| CONSTIPATION | 0 | 0 | 0 | 1 (16.7) | 0 | 1 (16.7) | 1 (50) | 0 | 1 (50) | 2 (15.4) | 0 | 2 (15.4) |
| DIARRHEA | 3 (60) | 0 | 3 (60) | 1 (16.7) | 0 | 1 (16.7) | 0 | 0 | 0 | 4 (30.8) | 0 | 4 (30.8) |
| MUCOSITIS ORAL | 2 (40) | 0 | 2 (40) | 1 (16.7) | 1 (16.7) | 2 (33.3) | 0 | 0 | 0 | 3 (23.1) | 1 (7.7) | 4 (30.8) |
| NAUSEA | 3 (60) | 0 | 3 (60) | 3 (50) | 0 | 3 (50) | 0 | 1 (50) | 1 (50) | 6 (46.2) | 1 (7.7) | 7 (53.8) |
| VOMITING | 0 | 0 | 0 | 2 (33.3) | 0 | 2 (33.3) | 1 (50) | 0 | 1 (50) | 3 (23.1) | 0 | 3 (23.1) |
| General disorders and administration site conditions | ||||||||||||
| EDEMA LIMBS | 1 (20) | 0 | 1 (20) | 1 (16.7) | 0 | 1 (16.7) | 0 | 0 | 0 | 2 (15.4) | 0 | 2 (15.4) |
| FATIGUE | 3 (60) | 0 | 3 (60) | 2 (33.3) | 0 | 2 (33.3) | 0 | 0 | 0 | 5 (38.5) | 0 | 5 (38.5) |
| Infections and infestations | ||||||||||||
| LUNG INFECTION | 0 | 1 (20) | 1 (20) | 0 | 1 (16.7) | 1 (16.7) | 0 | 0 | 0 | 0 | 2 (15.4) | 2 (15.4) |
| Metabolism and nutrition disorders | ||||||||||||
| ANOREXIA | 0 | 0 | 0 | 2 (33.3) | 0 | 2 (33.3) | 0 | 0 | 0 | 2 (15.4) | 0 | 2 (15.4) |
| HYPERGLYCEMIA | 1 (20) | 0 | 1 (20) | 1 (16.7) | 0 | 1 (16.7) | 1 (50) | 0 | 1 (50) | 3 (23.1) | 0 | 3 (23.1) |
| HYPOALBUMINEMIA | 2 (40) | 0 | 2 (40) | 1 (16.7) | 0 | 1 (16.7) | 0 | 0 | 0 | 3 (23.1) | 0 | 3 (23.1) |
| HYPOKALEMIA | 1 (20) | 0 | 1 (20) | 2 (33.3) | 0 | 2 (33.3) | 0 | 1 (50) | 1 (50) | 3 (23.1) | 1 (7.7) | 4 (30.8) |
| HYPOPHOSPHATEMIA | 0 | 1 (20) | 1 (20) | 0 | 1 (16.7) | 1 (16.7) | 0 | 0 | 0 | 0 | 2 (15.4) | 2 (15.4) |
| Respiratory, thoracic and mediastinal disorders | ||||||||||||
| DYSPNEA | 1 (20) | 0 | 1 (20) | 0 | 0 | 0 | 1 (50) | 0 | 1 (50) | 2 (15.4) | 0 | 2 (15.4) |
| HYPOXIA | 0 | 2 (40) | 2 (40) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 (15.4) | 2 (15.4) |
| PLEURAL EFFUSION | 1 (20) | 0 | 1 (20) | 1 (16.7) | 0 | 1 (16.7) | 0 | 0 | 0 | 2 (15.4) | 0 | 2 (15.4) |
| RESPIRATORY FAILURE | 0 | 2 (40) | 2 (40) | 0 | 1 (16.7) | 1 (16.7) | 0 | 0 | 0 | 0 | 3 (23.1) | 3 (23.1) |
| Skin and subcutaneous tissue disorders | ||||||||||||
| PRURITUS | 4 (80) | 0 | 4 (80) | 0 | 0 | 0 | 0 | 0 | 0 | 4 (30.8) | 0 | 4 (30.8) |
| RASH MACULO-PAPULAR | 1 (20) | 1 (20) | 2 (40) | 0 | 0 | 0 | 0 | 0 | 0 | 1 (7.7) | 1 (7.7) | 2 (15.4) |
| RASH | 2 (40) | 0 | 2 (40) | 1 (16.7) | 0 | 1 (16.7) | 0 | 0 | 0 | 3 (23.1) | 0 | 3 (23.1) |
| Vascular disorders | ||||||||||||
| HYPERTENSION | 1 (20) | 1 (20) | 2 (40) | 0 | 0 | 0 | 1 (50) | 0 | 1 (50) | 2 (15.4) | 1 (7.7) | 3 (23.1) |
Pacritinib pharmacokinetics
All thirteen patients completed plasma pharmacokinetic studies, and pacritinib bone marrow concentrations were measured in four patients. Pacritinib concentrations on days 1, 5 and 21 are summarized in Table 3. On day 1, mean ± standard deviation plasma concentrations were similar at the 100 mg twice daily dose level in both cohorts A and B at 5 h (3.7 ± 1.1 µg/mL and 4.0 ± 3.1 µg/mL, respectively) and 24 h (6.8 ± 2.5 µg/mL and 6.5 µg/mL, respectively) after drug administration. Maximum steady-state concentrations were reached on day 21 (9.6 ± 4.7 µg/mL in both cohorts A and B combined). Pacritinib concentrations in bone marrow samples were similar to corresponding plasma concentrations.
Table 3.
Pacritinib plasma and bone marrow concentrations.
| Patients | Cohort | Dose | Pacritinib (µg/mL) |
|||||
|---|---|---|---|---|---|---|---|---|
| D1–5h | D1–24h | D5-pre-dose | D5-BM | D21–5h | D21–24h | |||
| 4 | A | 100mg BID | 3.0 | 5.5 | 5.7 | 4.1 | 6.5 | 7.4 |
| 5 | A | 100mg BID | 2.2 | 4.8 | 5.2 | 5.2 | 7.3 | 7.2 |
| 8 | A | 100mg BID | 3.7 | 7.9 | 5.0 | |||
| 9 | A | 100mg BID | 5.3 | 11 | 11 | 9.4 | 8.5 | |
| 13 | A | 100mg BID | 2.5 | 4.8 | 5.3 | 17 | 17 | |
| Mean | 3.7 | 6.8 | 6.4 | 10 | 10 | |||
| SD | 1.1 | 2.5 | 2.3 | 4.1 | 4.2 | |||
| 3 | B | 100mg BID | 3.6 | 4.5 | 4.8 | |||
| 6 | B | 100mg BID | 1.9 | 2.1 | 1.8 | 3.8 | 5.3 | 5.1 |
| 7 | B | 100mg BID | 1.8 | 4.0 | 3.1 | 3.7 | 6.0 | |
| 10 | B | 100mg BID | 7.4 | 12 | 7.5 | 12 | 16 | |
| 11 | B | 100mg BID | 7.4 | 12 | 28 | 15 | 14 | |
| 12 | B | 100mg BID | 1.4 | 6.1 | 4.6 | 5.6 | 5.2 | |
| Mean | 4.0 | 6.5 | 8.3 | 8.4 | 9.2 | |||
| SD | 3.1 | 4.2 | 10 | 4.9 | 5.3 | |||
| All Patients | 100 mg BID | Mean | 3.7 | 6.7 | 7.4 | 9.1 | 9.6 | |
| SD | 2.3 | 3.5 | 7.2 | 9.6 | 4.7 | |||
| 1 | B | 200mg BID | 3.3 | 4.8 | 3.5 | |||
| 2 | B | 200mg BID | 4.6 | 7.0 | 9.0 | |||
Abbreviations: D1, day 1; D5, day 5; D21, day 21; BM, bone marrow; BID, twice daily.
Discussion
FLT3-ITD+ AML remains a challenging molecular subtype of AML, and TKIs directed at inhibition of FLT3 signaling is an active area of clinical research. To our knowledge, this is the first combination study of pacritinib and chemotherapy in FLT3-ITD+ AML patients to report safety, preliminary clinical activity and pacritinib pharmacokinetics in this population.
Previous reports have shown pacritinib to be active against de novo FLT3-ITD+ AML models in cell lines, primary cells, and a cell line xenograft[9]. We wanted to further assess its anti-leukemic activity in various models of drug resistant FLT3-ITD+ AML. In binding and kinase assays, pacritinib retained activity against FLT3 TKD mutants compared to FLT3-ITD. In Ba/F3 cells expressing FLT3 mutant proteins, sensitivity of pacritinib remained similar between the ITD or TKD mutants and dual ITD/TKD mutants and inhibited phospho-FLT3 and its downstream mediator, phospho-STAT5. Furthermore, in MOLM13-Res (ITD/D835Y+) AML cells, the pacritinib IC50 value did not show a large shift compared to that in parental MOLM13 (ITD+) cells. This is in contrast to the type II FLT3 inhibitors sorafenib and quizartinib, to which AML cells with TKD mutants become highly resistant[13, 15]. Given the frequent co-occurrence of additional mutations with FLT3-ITD in AML patients[16–18], it is worth highlighting that pacritinib had similar ex vivo activity to midostaurin against murine primary leukemia cells harboring FLT3-ITD along with a IDH2-R140Q mutation[19] and was ~2-fold more potent than midostaurin in a patient sample harboring an IDH2 mutation along with FLT3-ITD. Collectively, our in vitro assessment of pacritinib showed that pacritinib has type I inhibitor properties with activity against FLT3 inhibitor resistant forms of FLT3-ITD+ AML.
In total, two patients in cohort A achieved CR and one patient in cohort B achieved MLFS. The two patients who attained CR were able to receive allogeneic stem cell transplants. One of these two patients (patient 9) had a minor baseline FLT3 TKD D835Y clone. While it is possible that the two patients in cohort A who achieved CR would have done so with standard 7+3 induction alone, it is notable that the percentage of peripheral blood blasts in patient 9 was reduced by 35% after the first 5 days of pacritinib therapy without receiving cytarabine and daunorubicin.
Many patients had other co-occurring mutations at baseline. Both patient 5 and 9 who achieved CR had NPM1 mutations, which is associated with a better prognosis in FLT3-ITD+ AML. However, patient 5 who also had a IDH2 mutation did not respond to pacritinib single therapy in first 5 days as patient 9 did who had the NPM1 mutation alone indicated by their blast count reduction from pre-treatment to day 5 of therapy.
Anemia and QTc prolongation were DLTs seen at pacritinib 100mg twice daily. However, the MTD of pacritinib with chemotherapy or decitabine was not defined in either cohort A or cohort B due to early study termination by the trial sponsor. While dose escalation to 200 mg twice daily may have been possible after the completion of enrollment to the 100 mg twice daily dose level, we were not able to enroll additional patients. Due to the truncation of the study, the potential for increased efficacy at higher doses could not be evaluated. Bone marrow concentrations of pacritinib were measured in two patients from each cohort. Pacritinib exposure in bone marrow was similar to that of plasma in each patient indicating pacritinib was well accumulated at the site of action.
In conclusion, pacritinib was relatively well tolerated at a dose of 100mg twice daily in combination with intensive or non-intensive chemotherapy and demonstrated preliminary activity in patients newly diagnosed and relapsed/refractory FLT3-ITD+ AML. Our preclinical evaluation demonstrated that pacritinib was active against drug resistant forms of FLT3-ITD+ AML and may provide clinical benefit in this setting.
Supplementary Material
Acknowledgement
We would like to acknowledge CTI BioPharma for providing the drug and financial support for the trial and the service provided by The Ohio State University Comprehensive Cancer Center Shared Resources and Cores.
Funding
This work was supported by CTI BioPharma. It was also supported by the National Institute of Health grants R01 CA138744 to Sharyn D. Baker, R35 CA197734 to John C. Byrd and Cancer Center Support Grant P30 CA021765, funds from The Ohio State University Comprehensive Cancer Center Pelotonia foundation, and Eli Lilly fellowship to Jae Yoon Jeon.
Footnotes
Conflict of interest
Research support was provided by CTI BioPharma to Bhavana Bhatnagar, and all other authors declare that they have no conflict of interest
Ethical approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards
Informed consent
Informed consent was obtained from all individual participants included in the study.
Reference
- 1.Larrosa-Garcia M and Baer MR, FLT3 Inhibitors in Acute Myeloid Leukemia: Current Status and Future Directions. Mol Cancer Ther, 2017. 16(6): p. 991–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Annesley CE and Brown P, The Biology and Targeting of FLT3 in Pediatric Leukemia. Front Oncol, 2014. 4: p. 263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Daver N, et al. , Secondary mutations as mediators of resistance to targeted therapy in leukemia. Blood, 2015. 125(21): p. 3236–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lim SH, Dubielecka PM, and Raghunathan VM, Molecular targeting in acute myeloid leukemia. J Transl Med, 2017. 15(1): p. 183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mesa RA, et al. , Pacritinib versus best available therapy for the treatment of myelofibrosis irrespective of baseline cytopenias (PERSIST-1): an international, randomised, phase 3 trial. Lancet Haematol, 2017. 4(5): p. e225–e236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mascarenhas J, et al. , Pacritinib vs Best Available Therapy, Including Ruxolitinib, in Patients With Myelofibrosis: A Randomized Clinical Trial. JAMA Oncol, 2018. 4(5): p. 652–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hosseini MM, et al. , Inhibition of interleukin-1 receptor-associated kinase-1 is a therapeutic strategy for acute myeloid leukemia subtypes. Leukemia, 2018. 32(11): p. 2374–2387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.William AD, et al. , Discovery of the macrocycle 11-(2-pyrrolidin-1-yl-ethoxy)-14,19-dioxa-5,7,26-triaza-tetracyclo[19.3.1.1(2,6). 1(8,12)]heptacosa-1(25),2(26),3,5,8,10,12(27),16,21,23-decaene (SB1518), a potent Janus kinase 2/fms-like tyrosine kinase-3 (JAK2/FLT3) inhibitor for the treatment of myelofibrosis and lymphoma. J Med Chem, 2011. 54(13): p. 4638–58. [DOI] [PubMed] [Google Scholar]
- 9.Hart S, et al. , Pacritinib (SB1518), a JAK2/FLT3 inhibitor for the treatment of acute myeloid leukemia. Blood Cancer J, 2011. 1(11): p. e44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zimmerman EI, et al. , Crenolanib is active against models of drug-resistant FLT3-ITD-positive acute myeloid leukemia. Blood, 2013. 122(22): p. 3607–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jarusiewicz JA, et al. , Discovery of a Diaminopyrimidine FLT3 Inhibitor Active against Acute Myeloid Leukemia. ACS Omega, 2017. 2(5): p. 1985–2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wisniewski JR, et al. , Absolute Proteome Analysis of Colorectal Mucosa, Adenoma, and Cancer Reveals Drastic Changes in Fatty Acid Metabolism and Plasma Membrane Transporters. J Proteome Res, 2015. 14(9): p. 4005–18. [DOI] [PubMed] [Google Scholar]
- 13.Baker SD, et al. , Emergence of polyclonal FLT3 tyrosine kinase domain mutations during sequential therapy with sorafenib and sunitinib in FLT3-ITD-positive acute myeloid leukemia. Clin Cancer Res, 2013. 19(20): p. 5758–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cheson BD, et al. , Revised recommendations of the International Working Group for Diagnosis, Standardization of Response Criteria, Treatment Outcomes, and Reporting Standards for Therapeutic Trials in Acute Myeloid Leukemia. J Clin Oncol, 2003. 21(24): p. 4642–9. [DOI] [PubMed] [Google Scholar]
- 15.Smith CC, et al. , Validation of ITD mutations in FLT3 as a therapeutic target in human acute myeloid leukaemia. Nature, 2012. 485(7397): p. 260–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Patel JP, et al. , Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. N Engl J Med, 2012. 366(12): p. 1079–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Papaemmanuil E, et al. , Genomic Classification and Prognosis in Acute Myeloid Leukemia. N Engl J Med, 2016. 374(23): p. 2209–2221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McMahon CM, et al. , Mechanisms of acquired resistance to gilteritinib therapy in relapsed and refractory FLT3-mutated acute myeloid leukemia. Blood, 2017. 130. [Google Scholar]
- 19.Shih AH, et al. , Combination Targeted Therapy to Disrupt Aberrant Oncogenic Signaling and Reverse Epigenetic Dysfunction in IDH2- and TET2-Mutant Acute Myeloid Leukemia. Cancer Discov, 2017. 7(5): p. 494–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.