

Abstract
Since the serendipitous discovery of the first class of modern antidepressants in the 1950’s, all pharmacotherapies approved by the Food and Drug Administration for major depressive disorder (MDD) have shared a common mechanism of action, increased monoaminergic neurotransmission. Despite the widespread availability of antidepressants, as many as 50% of depressed patients are resistant to these conventional therapies. The significant length of time required to produce meaningful symptom relief with these medications, 4–6 weeks, indicates that other mechanisms are likely involved in the pathophysiology of depression and these mechanisms may yield more viable targets for drug development. For decades, no viable candidate target with a different mechanism of action to that of conventional therapies proved successful in clinical studies. Now several exciting avenues for drug development are under intense investigation. One of these emerging targets is modulation of endogenous opioid tone. This review will evaluate preclinical and clinical evidence pertaining to opioid dysregulation in depression focusing on the role of the endogenous ligands endorphin, enkephalin, dynorphin, and nociceptin/orphanin FQ (N/OFQ) and their respective receptors, mu (MOR), delta (DOR), kappa (KOR), and the N/OFQ receptor (NOP) in mediating behaviors relevant to depression and anxiety. Finally, putative opioid based antidepressants that are being tested in clinical trials, ALKS5461, JNJ–67953964 (formerly LY2456302 and CERC–501) and BTRX–246040 (formerly LY–2940094) will be discussed. This review will illustrate the potential therapeutic value of targeting opioid dysregulation in developing novel therapies for major depression disorder.
Keywords: Depression, MOR –mu opioid receptor, KOR – kappa opioid receptor, DOR – delta opioid receptor, NOP – nociceptin/orphaninFQ receptor, JNJ–67953964, Buprenorphine, ALKS–5461, BTRX–246040
1. Introduction
Major depressive disorder (MDD) is one of the most prevalent psychiatric disorders in the world WHO (2017). Despite the widespread use of medications to treat depression, only 35% of patients achieve full remission of symptoms with their first antidepressant trial (Kautzky, et al., 2019). Conventional antidepressants require 4–6 weeks of administration prior to the onset of therapeutic efficacy, during which time patients continue to experience incapacitating levels of depression and in some cases unrelenting suicidal ideation (Cipriani, et al., 2018; Duman & Aghajanian, 2012; Trivedi, 2006). Treatment of depression is further complicated by the co–occurrence of other disorders, including anxiety, post–traumatic stress disorder, substance abuse and chronic pain (Campbell, et al., 2007; Fava, et al., 2008; Lai, et al., 2015; Manning & Jackson, 2013; Stubbs, et al., 2017). Overall, 30–50% of patients are resistant to drug therapies, or exhibit partial relief of depressive symptoms despite continuing treatment and adjunct therapy with other treatment strategies (Fava, et al., 2008; van Bronswijk, et al., 2019). There is an urgency for psychiatric medicine to discover novel therapeutic strategies for treating depression to address a growing population of treatment–resistant patients and the lengthy treatment period prior to the emergence of clinical efficacy.
At present, nearly all pharmacotherapies for depression approved by the Food and Drug Administration (FDA) share a common mechanism of action, increased monoaminergic neurotransmission of norepinephrine (NE), dopamine (DA) and serotonin (5–HT) (Ramaker & Dulawa, 2017). One emerging avenue for novel drug development is modulation of endogenous opioid tone. Natural opioid derivatives have been used to alleviate melancholia for centuries (Pecina, et al., 2018). The development of selective ligands for key opioid receptors and significant advances in understanding endogenous opioid signaling and behavior have provided a framework for considering the potential roles of different opioid signaling pathways in endophenotypes relevant to depression. At the time of writing this article, one of the few antidepressants with a novel mechanism of action is being considered by the FDA is ALKS–5461, with antagonist activity at both kappa (KOR) and mu opioid receptors (MOR), which has shown considerable efficacy in treatment resistant depressed patients (Ehrich, et al., 2015a; Yovell, et al., 2016). Other selective opioid antagonists are now in phase 1 and 2 clinical trials and those results will yield important findings for the field. This review will serve two primary functions: 1) to highlight findings that support the importance of opioid signaling in the pathophysiology of depression. Yet, depression is a heterogeneous disorder (Akil, et al., 2018), with patients exhibiting a range of endophenotypes including negative affect, dysphoria, anhedonia, social withdrawal, cognitive impairment, sleep disturbances, changes in appetite and general activity. As MDD encompasses a heterogeneous cluster of behavioral symptoms, section 2 of this review will critically evaluate the contribution of opioid receptors in modulating behavioral domains as defined by the NIMH research domain criterion (RDoC) (Insel, 2014), which target specific endophenotypes shared across multiple disorders. Table 1 – 4 outline the behavioral domains relevant to MDD, the behavioral constructs used to model these domains in preclinical studies and the impact of opioid receptors within these constructs. 2) Section 3 will then discuss the most promising opioid compounds currently in clinical trials for MDD.
Table 1. MOR dysregulation in depression.
These data are compiled from preclinical and clinical studies that implicate MOR signaling dysregulation in behavioral constructs used to investigate the five key domains of negative valence, positive valence, cognitive systems, systems for social processes and arousal/regulatory systems. MOR –mu opioid receptor, CeA – central nucleus of the amygdala, NAc – nucleus accumbens, VLPO –ventrolateral preoptic nucleus, ENK – enkephalin, NIH – novelty induced hypophagia, BP – binding potential, REM – rapid eye movement.
| Domain | Constructs | Behavioral effects | Reference |
|---|---|---|---|
| Negative Valence: | Acute threat (Fear) | Oprm1−/−mice exhibited reductions in freezing behavior | (Sanders, et al., 2005) |
| Systemic and intra-amygdalar injection of MOR agonists impaired cued and contextual fear. Intra-NAc administration of MOR agonist impaired contextual fear only. | (Cole & McNally, 2009; Good & Westbrook, 1995; Szczytkowski-Thomson, et al., 2013; Szklarczyk, et al., 2015; Westbrook, et al., 1997) | ||
| MOR antagonists enhanced the acquisition of fear in rodents. | (Fanselow, et al., 1991; Halladay & Blair, 2012; Helmstetter & Fanselow, 1987; Szklarczyk, et al., 2015) | ||
| MOR antagonists enhanced the acquisition of fear in humans. | (Eippert, et al., 2008; Haaker, et al., 2017) | ||
| Potential threat (Anxiety) | MOR antagonists reduced latencies in the NIH | (Almatroudi, et al., 2015; Almatroudi, et al., 2018; Browne, et al., 2017; Robinson, et al., 2017) | |
| Sustained threat (Aversive emotional state) | Prolonged exposure to chronic stress changed Oprm1−/−and ENK mRNA expression, and MOR BP in the cortex, striatum and amygdala. | (Berube, et al., 2013; Berube, et al., 2014; Browne, et al., 2018; Falcon, et al., 2016; Johnston, et al., 2015; Miczek, et al., 2011; Nikulina, et al., 2008; Nikulina, et al., 1999; Nikulina, et al., 2005) | |
| Oprm1−/−mice are resistant to behavioral deficits induced following chronic swim and chronic restraint stress. | (Contet, et al., 2006; Ide, et al., 2010; Wang, et al., 2002) | ||
| Loss | Decreased MOR BP in corticoamygdalar structures and posterior thalamus during a sustained sadness challenge | (Kennedy, et al., 2006) | |
| Positive Valence: | Reward Responsiveness | Juvenile Oprm1−/−mice find social interactions less salient | (Cinque, et al., 2012) |
| Cognitive Systems: | Attention | Attentional set shifting was enhanced by morphine administration in healthy controls | (Quednow, et al., 2008) |
| Systems for social processes | Affiliation and Attachment | Juvenile Oprm1−/−mice find social interactions less salient | (Cinque, et al., 2012) |
| Oprm1−/−mice do not exhibit reductions in social interaction following stress | (Komatsu, et al., 2011) | ||
| Social Communication | MOR agonist administration promoted attention to faces and eyes of others. MOR antagonism reduced attention to these social cues in healthy male subjects. |
(Chelnokova, et al., 2016) | |
| Perception and Understanding of Self | Decreased MOR BP in corticoamygdalar structures and posterior thalamus during a sustained sadness challenge | (Kennedy, et al., 2006) | |
| Greater magnitude of change in subjective self-esteem in depressed subjects in a social rejection challenge, was associated with reduced corticoamygdalar MOR BP | (Hsu, et al., 2015) | ||
| Arousal/Regulatory systems | Arousal | Sensorimotor gating was enhanced by morphine administration in healthy controls. | (Quednow, et al., 2008) |
| Sleep and Wakefulness | Sleep deprivation decreases MOR BP | (Fadda, et al., 1991) | |
| MOR agonists inhibit firing of neurons in VLPO, increasing wakefulness | (Greco, et al., 2008; Wang, et al., 2013) | ||
| Activation of MORs disrupts REM sleep | (Cronin, et al., 1995) |
Table 4. NOP dysregulation in depression.
These data are compiled from preclinical and clinical studies that implicate NOP and N/OFQ in behavioral constructs that relate to depression under the domains of negative valence, positive valence, and arousal/regulatory systems. CeA – central nucleus of the amygdala, SNP – single nucleotide polymorphism, N/OFQ – nociceptin/orphaninFQ, NOP – nociceptin/orphaninFQ receptor, EPM – elevated plus maze, LDB – light /dark box, FST – forced swim test, LH – learned helplessness, LPS – lipopolysaccharide, SCN – suprachiasmatic nucleus.
| Domain | Constructs | Behavioral effects | Reference |
|---|---|---|---|
| Negative Valence: | Acute threat (Fear) | Systemic or intra-CeA administration of NOP agonists decreased freezing to the conditioned stimulus | (Andero, et al., 2013; Witkin, et al., 2016) |
| G allele carriers of the rs6010719 SNP in the OPRL1 gene exhibited increased physiological startle measures of fear discrimination and greater functional connectivity between the amygdala and posterior insula. | (Andero, et al., 2013) | ||
| Potential threat (Anxiety) | N/OFQ enhanced thigmotaxis in the open field. | (Fernandez, et al., 2004) | |
| N/OFQ induced anxiogenic effects in rats on the EPM and LDB | (Fernandez, et al., 2004) | ||
| NOP agonists produced anxiolytic effects | (Duzzioni, et al., 2011) | ||
| NOP−/−mice exhibit reductions in anxiety like behavior compared to wildtype controls | (Gavioli, et al., 2007) | ||
| Sustained threat (Aversive emotional state) | NOP antagonists produce antidepressant-like effects in the FST, LH and LPS-induced depressive-like behavior | (Asth, et al., 2016; Gavioli, et al., 2003; Gavioli, et al., 2004; Goeldner, et al., 2010; Holanda, et al., 2016; Medeiros, et al., 2015) | |
| NOP−/−mice show reductions in depressive like behavior compared to their wildtype littermates. | (Gavioli, et al., 2007) | ||
| Positive Valence: | Reward Valuation | NOP agonists stimulate feeding behavior | (Ciccocioppo, et al., 2014; Nicholson, et al., 2002) |
| Arousal/Regulatory systems | Circadian rhythm | N/OFQ reduces neuronal activation in the SCN and can induce sedation | (Gompf, et al., 2005) |
| NOP ligands were more efficacious when administered during the nadir of corticosterone secretion | (Leggett, et al., 2007) |
2. Opioid dysregulation and the pathophysiology of depression
Expressed throughout the central and peripheral nervous system, MOR, KOR and delta opioid receptors (DOR) modulate a range of physiological processes and behaviors, including pain sensation, gastrointestinal function, immunity, reward, aversion and mood (Lutz & Kieffer, 2013). In addition to reviewing the potential impact of these opioid receptors in the pathophysiology and treatment of MDD, the utility of endogenous and synthetic ligands of the nociceptin/orphanin FQ (NOP) receptor (formerly opioid like receptor (ORL1)) will also be discussed.
2.1. Opioid signaling
Activation of opioid receptors by their endogenous ligands endorphin, enkephalin (ENK), dynorphin (DYN), and nociceptin/orphanin FQ (N/OFQ) decreases neurotransmitter release in a cell–type and pathway specific manner in discrete brain nuclei implicated in the pathophysiology of neuropsychiatric disorders. Opioid receptors belong to the superfamily of 7–transmembrane–spanning G–protein–coupled receptors (GPCRs). Coupled to pertussis toxin sensitive G–proteins including Gαi, the activation of opioid receptors results in the inhibition of adenylate cyclase activity, Figure 1. The dissociation of the Gα and Gβγ subunits rapidly activates inwardly rectifying potassium channels resulting in hyperpolarization of the cell, and can block calcium conductance, thereby reducing calcium dependent neurotransmitter release (Gompf, et al., 2005; Hjelmstad & Fields, 2003; Pennock & Hentges, 2016; Rawls & McGinty, 2000; Ronken, et al., 1993b; Rutz, et al., 2007; Weiss, et al., 2007). Typically, ligand bound opioid receptors are phosphorylated, desensitized and internalized, and eventually recycled back to the cell surface (Al–Hasani & Bruchas, 2011). However, not all ligands induce equivalent internalization and many arrestin–bound internalized GPCRs still signal through mitogen activated protein kinase (MAPK) pathways (Schmid & Bohn, 2009) such as extracellular signal–regulated kinase (ERK), c–Jun N–terminal Kinase (JNK) and p38. These kinases are integral in the transfer of neurotrophic signals from the cell surface to the nucleus, inducing cell directed gene transcription that ultimately modulates synaptic plasticity and neuronal survival. ERK translocates to the nucleus to phosphorylate transcription factors that regulate gene expression required for growth and differentiation. ERK can also regulate targets in the cytosol. JNK phosphorylates nuclear transcription factors involved in growth, survival, differentiation and apoptosis, and p38 MAPK phosphorylation regulates transcription of genes involved in cytokine production and apoptosis. A growing number of animal studies have highlighted the potential importance of these signaling pathways in the development and alleviation of depression (Galeotti & Ghelardini, 2012). Indeed, ERK signaling is necessary for the reversal of depressive–like behaviors produced following administration of conventional antidepressants and more rapid acting therapeutics such as electroconvulsive shock and ketamine (Bravo, et al., 2009; Hansen, et al., 2007; Leskiewicz, et al., 2013; Musazzi, et al., 2010; Ramaker & Dulawa, 2017; Ren, et al., 2018; Tang, et al., 2017). Conversely, p38 MAPK activation is associated with stress–induced dysphoria and aversion (Bruchas, et al., 2007; Ehrich, et al., 2015b; Land, et al., 2009). Developing compounds that exhibit arrestin–dependent biased agonism, and preferential activation of one MAPK pathway over another, may yield promising therapeutics for multiple disorders where opioid dysregulation is evident. Moreover, aberrant neuronal firing and synaptic plasticity deficits are characteristic features of rodent models of stress and depression (Duman & Aghajanian, 2012; Howe & Kenny, 2018; Lutz & Kieffer, 2013; Ota & Duman, 2013). As opioid signaling stimulates cellular processes involved in facilitating stress adaptation and resilience across many cell types, including neurons and glia, the normalization of aberrant opioidergic tone may be recognized as a mechanism through which opioid compounds can reestablish normal neuronal function and reverse depressive behaviors. The specific kinases and signaling pathways modulated by the four opioid receptors discussed in this review are identified will be discussed in detail in the following sections.
Figure 1.
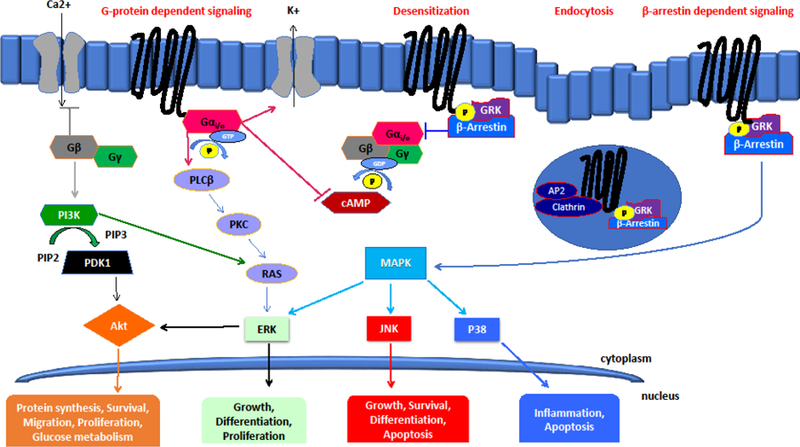
Agonist binding to opioid receptors induces pertussis toxin sensitive G protein coupling and activation, followed by rapid phosphorylation of the receptor by G–protein–coupled receptor kinases (GRKs). Subsequently, the Gα and Gβγ subunits dissociate to modulate ion channel conductance and several secondary messengers. Gα rapidly activates inwardly rectifying potassium channels resulting in hyperpolarization of the cell. The Gα subunit also inhibits adenylate cyclase activity and induces phospholipase C /protein kinase C (PLCβ/PKC) signaling. Inhibition of calcium conductance and the subsequent reduction in calcium dependent neurotransmitter release is regulated by the Gβγ subunit, which can also induce phosphatidylinositol–4, 5–bisphosphate 3–kinase (PI3K)/AKT pathway. Desensitization of phosphorylated opioid receptors is dependent on β–arrestin, which interferes with further G protein coupling. Following β–arrestin desensitization, the AP2 adaptor complex facilitates clathrin–mediated endocytosis into vesicles. Receptor internalization is then followed by recycling or lysosomal degradation. Agonist–stimulated β–arrestin also scaffolds mitogen activated protein kinase (MAPK) kinases, which effect robust activation of downstream signaling pathways including extracellular signal–regulated kinase (ERK), c–Jun N–terminal Kinase (JNK) and p38.
2.2. Mu Opioid Receptor (MOR)
Extracted from the poppy plant, Papaver somniferum, the MOR agonist morphine and other opium derivatives have been used for millennia to treat a wide variety of ailments, from pain and insomnia to diarrhea. Acting at MORs, the inherent euphorigenic properties of these agonists are thought to exert their influence on mood through modulation of glutamatergic and dopaminergic neurotransmission (Chartoff & Connery, 2014). Densely expressed in the neocortex, throughout the mesencephalon and subcortical regions including the striatopallidal pathway, amygdala, hippocampus, thalamus, and insula (Mansour, et al., 1987; Zubieta, et al., 1999; Zubieta, et al., 2001), MORs are preferentially activated by the endogenous opioid peptides β–endorphin and ENK in a region–dependent manner (Beleslin, et al., 1982; Hughes, et al., 1977; Nicoll, et al., 1977; Rossier, et al., 1977). The OPRM1 gene encodes at least three receptor isotypes, with multiple splice variants reported across species, some of which only possess 6 transmembrane domains, but remain functional (Pasternak, et al., 2004; Pasternak & Snyder, 1975; Wang, et al., 1994; Wolozin & Pasternak, 1981). A large body of evidence exists detailing the complex, ligand specific effects of MOR activation and β–arrestin dependent internalization particularly in relation to analgesic tolerance (Dang & Christie, 2012; Melief, et al., 2010; Raehal & Bohn, 2011). Despite the effectiveness of MOR agonists in the alleviation of pain, the emergence of tolerance, dependence and substance abuse mitigate against the continued use of MOR agonists for most diseases (Charbogne, et al., 2014). Yet, MORs are heavily implicated in the pathophysiology of depression and, as the following sections will show, modulation of opioidergic tone is critical to the remediation of core endophenotypes of depression (see Table 1) including social withdrawal, negative and positive valence.
2.2.1. MOR and systems for social processes
PET imaging studies of MOR binding potential (BP), with the selective radiotracer [11C] carfentanil, illustrates the extent of altered MOR signaling in MDD patients. Utilizing a sustained sadness challenge, whereby patients recounted an event that evoked sadness, female subjects diagnosed with MDD exhibited decreased MOR BP in the anterior insular cortex, anterior and posterior thalamus, ventral basal ganglia, amygdala, and periamygdalar cortex, compared to controls (Kennedy, et al., 2006). In a neutral state, reduced MOR BP was still observed in the right posterior thalamus of MDD subjects. This region stands out because depressed subjects who exhibited no symptom relief following 10 weeks of fluoxetine treatment, exhibited even greater reductions in MOR BP in the posterior thalamus (Kennedy, et al., 2006). Thus, MOR binding in the posterior thalamus may be a potential biomarker for treatment response and depression severity. Such examples of aberrant MOR signaling may underlie social interaction deficits, impaired stress adaptation, and poor cognitive flexibility. Poor social function has been described as a trait of many individuals diagnosed with MDD, causing withdrawal from loved ones and social avoidance behaviors (Kupferberg, et al., 2016). Avoidance of attachment in adulthood was negatively correlated with MOR availability in the thalamus, anterior cingulate cortex (ACC), amygdala, and insula in depressed subjects (Nummenmaa, et al., 2015), whereas, greater trait resilience to rejection was positively correlated with MOR activation in the amygdala, periaqueductal gray (PAG) and ACC (Hsu, et al., 2013). Depressed individuals also exhibited greater increases in subjective well–being following acceptance and lowering of self–esteem after rejection compared to healthy controls, exaggerated bivalent emotional responses that were sustained over a longer period than that exhibited by controls (Hsu, et al., 2015). These data outline the importance of modulating MOR tone in depressed patients to facilitate improved resilience to negative social stimuli and hedonic response to social stimuli.
Preclinical studies recapitulate the clinical finding that aberrant MOR function is involved in mediating social anhedonia (Table 1). Mice with genetic deletion of MORs, Oprm1–/–, do not display social avoidance following stress exposure (Komatsu, et al., 2011). Moreover, juvenile Oprm1–/–mice and wild type mice treated with MOR antagonists during early life (naltrexone 1 mg/kg, SC, on post–natal day 1–4) find social interactions less salient than their controls (Cinque, et al., 2012). In addition, reports indicate that decreased Oprm1 and ENK expression occurs in the amygdala of mice and rats exposed to social defeat stress paradigms, but in contrast the expression of these genes was elevated in the ventral tegmental area (VTA), NAc and cortical regions suggesting region–specific effects of stress on MOR signaling. Specifically, Oprm1 mRNA levels were elevated in the VTA following just one single exposure to social defeat in rats (Nikulina, et al., 1999). VTA Oprm1 mRNA expression was upregulated by exposure to chronic social stress for up to 21 days after the final stress exposure, suggesting persistent activation of striatal MOR signaling within the VTA following chronic stress (Nikulina, et al., 2008). Furthermore, knockdown of MORs within the VTA blocked the behavioral and molecular alterations induced by social defeat in rats (Johnston, et al., 2015). In mice exposed to 10 days of social defeat stress, a stress susceptible phenotype, measured as significant social avoidance, was associated with robustly elevated Oprm1 mRNA in the frontal cortex and ventral striatum relative to control and defeated mice that exhibit a stress resilient phenotype (Browne, et al., 2018). In contrast, Oprm1 expression was dramatically reduced in the amygdala of defeated mice (Browne, et al., 2018), mirroring the decrease in ENK reported in the BLA of stress susceptible rats relative to controls and stress resilient defeated rats (Berube, et al., 2014). Similar region–specific changes in Oprm1 expression were found following exposure to unpredictable chronic mild stress, where Oprm1 was markedly diminished in the basolateral nucleus of the amygdala in rats (Berube, et al., 2013) and mice (Falcon, et al., 2016). Remarkably, reducing the expression of Oprm1 improved abnormal social behavior exhibited by mice with genomic doubling of methyl CpG binding protein 2 (MECP2), which is necessary for transcriptional repression of genes, and specifically this murine model is used to investigate the development of behaviors and neurochemistry underlying the development of autism and anxiety (Samaco, et al., 2012). Overall, these preclinical data confirm that regional alterations in MOR signaling are implicated in social interaction deficits. Social anhedonia may serve as a potential prognostic indicator of treatment resistance in subjects with MDD (McMakin, et al., 2012). Thus, utilizing constructs of social processes in rodents may provide a translationally relevant behavioral domain in which to screen novel antidepressant medications.
2.2.2. MOR and positive valence
Given that remediation of reward processing is a critical factor in achieving sustained relief from symptoms of depression in humans, it is important to understand the role of MORs in regulating incentive salience and hedonic tone, two critical components of reward processing (Admon & Pizzagalli, 2015; Calabrese, et al., 2014). Tasks that engage positive valence systems require the mesolimbic dopamine (DA) circuitry, although most of this information has been obtained from preclinical studies (Table 1). In stress–naïve rats, treatment with the MOR agonist DAMGO can enhance signal tracking and conditioned incentive behavior (DiFeliceantonio & Berridge, 2016). However, stress–induced activation of MORs in the VTA reduced DA neurotransmission in the nucleus accumbens (NAc), a major site of reward processing in the brain (Latagliata, et al., 2014). Conversely, local administration of MOR antagonists into the VTA increased striatal DA concentrations countering the response to stressful stimuli (Devine, et al., 1993). Thus, MOR blockade may produce beneficial behavioral effects in the presence of aversive stimuli. Another example involves the novelty induced hypophagia (NIH) paradigm, where the increased latency to approach and consume palatable food in a novel environment is attenuated by chronic antidepressant treatment (Dulawa & Hen, 2005). Similarly, administration of the selective MOR antagonist cyprodime, the opioid antagonist naltrexone, the mixed opioid analgesic buprenorphine and its KOR/MOR antagonist derivative BU10119 counteracted the impact of the novel environment at suppressing approach latencies for food (Almatroudi, et al., 2015; Almatroudi, et al., 2018; Falcon, et al., 2015; Robinson, et al., 2017). In addition to tests conducted in naïve mice, the effects of buprenorphine in the NIH test were blocked in mice with genetic deletion of Oprm1–/–(Robinson, et al., 2017). Results from knockout animals should be interpreted with caution, as these mice can exhibit aberrant developmental patterns and may have unknown compensatory mechanisms that could potentially confound the outcomes of these pharmacological studies. However, we subsequently determined that in a murine model (A112G Oprm1) of the highly penetrant non–synonymous human A118G single–nucleotide polymorphism (SNP), mice that possessed the G allele were unresponsive to buprenorphine’s anxiolytic action in the NIH test, antinociception in the hot plate test and hyperlocomotion (Browne, et al., 2017). This is important as this SNP confers a dramatic reduction in the binding affinity of endogenous opioids at MORs and the general function of MORs in these mice (Bond, et al., 1998; Mague, et al., 2009; Zhang, et al., 2005b). Indeed, human carriers of the G allele have higher subjective pain scores, require greater quantities of opioid analgesics to relieve their pain and exhibit greater rewarding effects of alcohol and nicotine compared to with carriers of the A allele, indicating aberrant MOR function (Bach, et al., 2015; Bonenberger, et al., 2015; Chou, et al., 2006; Ray, et al., 2006; Sia, et al., 2008). Together, these data support the importance of MORs at mediating behavioral investigations in response to novel stimuli. Although more empirical evidence is required, the emerging data suggest that in the context of stress, MOR antagonists may positively modulate the performance of motivated behaviors and positive valence.
2.2.3. MOR and negative valence
Preclinical evidence has also established the importance of MORs in the emergence of stress resilience in the context of acute threat (fear) and potential threat (anxiety) (Bowers, et al., 2012; Bowers & Ressler, 2015). Most of the information regarding the importance of MORs in these behavioral constructs of negative valence has emerged from studies conducted in knockout mice (Table 1). Genetic deletion of MORs not only protected mice from stress–induced behavioral deficits but also blocked immune dysfunction following stress exposure, although increases in circulating levels of adrenocorticotropic hormone (ACTH), corticosterone, and proopiomelanocortin (POMC) mRNA expression in the pituitary remained intact (Contet, et al., 2006; Ide, et al., 2010; Wang, et al., 2002). In addition, Oprm1–/–mice exhibited a slight reduction in freezing behavior following re–exposure to the context in which mice were previously shocked (Sanders, et al., 2005). In contrast to the global knockdown of Oprm1, pharmacological blockade of MORs by naloxone and CTOP enhanced acquisition of conditioned fear, increased freezing in response to the conditioned stimulus and impaired extinction (Helmstetter & Fanselow, 1987; Westbrook, et al., 1991). Impaired contextual fear memory and a failure to extinguish fear memories is used as a rodent analog of intrusion memories, a core feature of posttraumatic stress disorder (PTSD), a psychiatric disorder with high levels of comorbidity with depression. Clinical findings indicated that morphine administered during the peritrauma period may attenuate the development of PTSD in the months following trauma (Bryant, et al., 2009; Holbrook, et al., 2010). This finding agrees with preclinical studies in rats and mice that show impaired acquisition of fear memory following morphine treatment (Good & Westbrook, 1995; Szczytkowski–Thomson, et al., 2013; Szklarczyk, et al., 2015; Westbrook, et al., 1997). This may be due to morphine impairing consolidation of information within the treatment context. In humans and rodents, MOR mediated disruption and enhancement of conditioned fear occurs at the level of PAG and amygdala (Cole & McNally, 2009; Eippert, et al., 2008; Haaker, et al., 2017). Stimulation of MORs located on GABAergic intercalated neurons of the central amygdala (CeA), which gate local (basolateral amygdala (BLA)) and distal (infralimbic cortex) inputs, attenuates BLA feedforward inhibition during extinction training, ultimately maintaining fear expression (Blaesse, et al., 2015; Winters, et al., 2017). It has also been suggested that CeA intercalated neurons may actually facilitate basal anxiety without exposure to a threatening or aversive stimulus (Palomares–Castillo, et al., 2012), as local infusion of morphine and the MOR antagonist CTAP into the CeA enhanced and decreased anxiety like behavior on the elevated plus maze (EPM), respectively. However, in response to predator odor, another model used to induce physiological and behavioral characteristics of PTSD, DAMGO infusions facilitated exploration and reduced defensive burying (Wilson & Junor, 2008). These intriguing findings point to context–dependent effects of MOR activation in response to specific constructs of negative valence, i.e. acute or sustained threat.
2.2.4. MOR, arousal and cognitive systems
The locus coeruleus (LC)–norepinephrine (NE) system is a major arousal system that also regulates cognitive processes through its forebrain projections (Mather & Harley, 2016). LC activity is co–regulated during stress by the stress–related neuropeptide, corticotropin–releasing factor (CRF) acting at CRF1 and enkephalin (ENK) acting at MOR. ENK axon terminals deriving from the cells in the nucleus paragigantocellularis (PGi) and CRF axon terminals from cells of the central nucleus of the amygdala converge onto common LC dendrites that co–localize CRF1 and MOR immunolabeling (Tjoumakaris, et al., 2003). Activation of CRF1 and MOR has opposing excitatory and inhibitory effects on LC neurons, respectively. In response to acute stress, CRF afferents are engaged to activate LC neurons but there is also evidence for ENK release, which may restrain this activation and promote recovery of activity to baseline when the stressor is terminated. For example, administration of an opioid antagonist results in a greater LC activation by stressors and slower recovery to baseline activity (Curtis, et al., 2002; Curtis, et al., 2001). This would also be predicted in subjects that were tolerant to opioids and could explain enhanced sensitivity to stress in individuals that chronically use opioids.
The degree to which LC activity is regulated by CRF or ENK afferents is related to coping strategy. For example, after a single exposure to resident–intruder stress, most intruder rats readily assume a submissive posture and in these animals LC neurons, ENK–LC–projecting neurons and CRF–LC–projecting neurons are all activated as indicated by c–fos expression (Reyes et al., 2015). With repeated exposures two populations of rats emerge defined by their degree of subordination as quantified by the onset to assume a submissive posture (Wood, et al., 2010). In submissive rats, the ENK inhibitory influence is lost and CRF afferents remain activated. In contrast, for rats that resist defeat, ENK afferents to the LC remain activated by the stressor while CRF afferents are no longer activated (Reyes, et al., 2015). The loss of an inhibitory counterbalance in subjects with a subordinate coping strategy may increase vulnerability to opioid abuse in an effort to substitute for a diminished endogenous opioid response.
Notably, in rats with a history of repeated social stress, administration of the opioid antagonist, naloxone, robustly increases LC discharge rates in a manner reminiscent of that seen after naloxone administration to opioid dependent rats (Chaijale, et al., 2013). This finding suggests that the stress can elicit sufficient ENK release to produce a similar plasticity as that produced by opioid dependence.
Finally, sex differences in CRF1 and MOR function in the LC are speculated to underscore the high prevalence of stress–related disorders in women compared to their male counterparts (Valentino & Bangasser, 2016). Specifically, LC neurons of female rats are more sensitive to activation by CRF compared to males. This has been attributed to a bias in CRF1 coupling to the GTP–binding protein, Gs that would result in enhanced signaling and decreased association with β–arrestin, which would result in decreased receptor internalization (Valentino & Bangasser, 2016). MOR receptor protein and mRNA are greater in male compared to female rat LC (Guajardo, et al., 2017). This translated to a greater efficacy of MOR agonists in inhibiting LC neurons in male compared to female rats. Together, the sex differences in CRF1 and MOR in the LC would favor over activation of this system in response to stress in females. At a behavioral level, MOR activation within the LC modulated cognitive processing in an operant strategy–shifting task in distinct patterns for male and female rats. Thus, whereas intra–LC DAMGO administration increased the number of total errors, premature responses, regressive errors, and random errors in males, it only increased perseverative errors in female rats (Guajardo, et al., 2017). The implications of such findings raise questions regarding sex specific effects of opioid therapeutics on cognitive processes. This will be an important aspect of drug development going forward as cognitive impairment remains one of the key untreated symptoms of MDD (Jacobson, et al., 2018)
2.2.5. MOR implications
Overall, these studies suggest that modulating opioidergic tone at MORs has beneficial effects in models of aberrant emotional behavior. Antagonism at MORs could be useful for subjects displaying behavioral suppression due to anhedonia, social withdrawal and anxiety. However, MOR activation around the peritrauma period may prove therapeutic as MOR agonists could impair memory consolidation and prevent the later emergence of PTSD. Much more work is required to fully delineate the beneficial effects of selective MOR ligands on behaviors relevant to depression.
2.3. Kappa Opioid Receptor (KOR)
Originally named for the agonist ketocyclazocine, (Pasternak, 1980), KORs are distributed in regions of the brain that are critical for motivation, reward, pain and emotional valence. In situ hybridization studies in rodents, (Hiller, et al., 1992; Mansour, et al., 1987; Mansour, et al., 1986), and later in humans (Simonin, et al., 1995), confirmed dense expression of KORs in the parietal and temporal cortex, basal forebrain, thalamus, endopiriform cortex and amygdala. This pattern of expression is established by the time the late prenatal stages develop (Zhu, et al., 1998) and parallels that of the endogenous ligand DYN (DePaoli, et al., 1994; Mansour, et al., 1987; Mansour, et al., 1986), one of the opioid peptides derived from preprodynorphin (Akil, et al., 1984). Two subtypes of KORs have been identified to date, KOR1 and KOR2. KOR1 preferentially binds arylacetamide–like agonists such as U–50488H and U–69539 and the antagonist norbinaltorphimine (nor–BNI), whereas KOR2 has a 100–fold lower binding affinity for nor–BNI and is entirely insensitive to U–69539. The KOR agonists bremazocine and GR–89696 are typically used to investigate KOR–2 mediated signaling and behavior. Theoretically, 6 possible RNA isoforms of the KOR have been proposed, as the Orpk1 gene has two promoter sites and two polyadenylation sites (Wei, et al., 2000).
2.3.1. Stress induced aberrant KOR signaling – relevance to depression
In contrast to the euphoric effects of MOR agonism, humans (Pfeiffer, et al., 1986; Ranganathan, et al., 2012) and rodents exhibit dysphoria and aversion following KOR activation (Bals–Kubik, et al., 1993; Bruchas, et al., 2007; Chefer, et al., 2013; del Rosario Capriles & Cancela, 2002; Land, et al., 2008; Mori, et al., 2002; Shippenberg & Herz, 1986; Zhang, et al., 2005a). Stress has repeatedly been shown to modulate DYN and KOR protein and mRNA levels in rodents. However, different stressors produce varied region–specific alterations. Acute (3h) immobilization stress and exposure to the more severe learned helplessness paradigm, increased DYN A and DYN B immunoreactivity in the hippocampus and NAc of rats; however a 15 min forced swim stress exposure elevated DYN A levels only in the hippocampus (Shirayama, et al., 2004). A later study which evaluated the expression of Pdyn and Oprk1 by in situ hybridization following 2–or 9–days recovery from immobilization stress established that single, or repeated exposure to immobilization elevated Oprk1–mRNA levels in striatum and NAc, but these effects diminished by day 9 of recovery (Lucas, et al., 2011). Conversely, Pdyn mRNA expression was unchanged after the shorter recovery period but was elevated following both single and repeated immobilization stress at day 9 (Lucas, et al., 2011), indicating a neuroplastic change within the DYN/KOR circuit that could sensitize these brain regions to stress in the future. Following exposure to a resident intruder paradigm, no alterations in DYN expression, as measured by radioimmunoassay, were noted in the mPFC, VTA or NAc (Nocjar, et al., 2012). However, these rats did exhibit a significant reduction in DYN expression within the hypothalamus (Nocjar, et al., 2012). In contrast, when defeated animals were segregated into stress–susceptible and resilient groups, DYN mRNA was increased within the dorsal and medial shell of the NAc of susceptible rats and in the striatum of both resilient and susceptible rats compared to controls (Berube, et al., 2013). In mice exposed to acute (1 day) or chronic (10 days) of social defeat stress, DYN mRNA expression was augmented in the NAc following acute stress, but decreased following chronic social defeat (Donahue, et al., 2015). Reversal of the stress induced decrease in NAc Pdyn was produced following chronic administration of the antidepressant imipramine (Donahue, et al., 2015). No alterations were detected in Oprk1 expression in this study. However, ablation of KORs specifically on NAc DA transporter–expressing neurons promoted stress resilience in mice exposed to defeat (Donahue, et al., 2015). In a separate study, it was established that Oprk1 mRNA expression within the frontal cortex of stress susceptible defeated mice was robustly elevated relative to non–stress controls and stress–resilient mice one week following cessation of chronic social defeat stress (Browne, et al., 2018). Additionally, following 3 weeks of chronic mild stress, stressed mice exhibit significant reductions in Pdyn mRNA expression in the amygdala (Falcon, et al., 2016). Moreover, these stress exposed mice exhibited a marked elevation in Oprk1 mRNA expression in the striatum and decreased expression within the frontal cortex, which were normalized following one week of treatment with the mixed opioid compound buprenorphine (Falcon, et al., 2016). Together these studies highlight the diverse regional alterations induced following different stress paradigms and highlight potential long–term alterations that occur in DYN/KOR signaling that are often overlooked as studies do not always investigate these genes at later time points following recovery from stress.
Post transcriptional and epigenetic regulation of KOR isoforms was also evident following stress exposure. C57BL/6J mice subjected to forced swim stress exhibited enhanced mRNA expression of KOR isoform B in the sensorimotor cortex, hippocampus and brainstem, and isoform A in the medial prefrontal cortex (mPFC) (Flaisher–Grinberg, et al., 2012). In all regions examined, increased expression of KORs was associated with polyadenylation site 1 (PA1) upregulation and epigenetic changes selective for KOR transcripts controlled by promoter 1 (Pr1), including reduced HDAC1 recruitment and elevated levels of histone 4 acetylation for the transcription factor c–Myc (Flaisher–Grinberg, et al., 2012). Differential regulation of KOR has been reported in stress sensitive strains of rodents, WKY rats, and BALB/cJ and DBA/2J mice, compared to their normosensitive control strains (Pearson, et al., 2006; Saito, et al., 2003), suggesting that epigenetic regulation of KORs may have a significant phenotypic impact on the behavioral expression of stress. However, it should be noted that other genetic differences in regulatory regions may account for some of these strain differences reported. These data highlight the dynamic sensitivity of transcriptional regulation of KORs to the physiological impact of stress across multiple situations in rodents.
Postmortem studies in suicide victims with major depression revealed increased expression of prodynorphin (PDYN) in the patch compartment of the caudate, but not in the dorsolateral prefrontal or cingulate cortices. Conversely, PDYN expression was decreased in depressed subjects within the periamygdaloid complex (Anderson, et al., 2013; Hurd, 2002; Hurd, et al., 1997; Peckys & Hurd, 2001). Subsequent neuroimaging studies have highlighted low KOR availability in amygdala–ACC–ventral striatal circuit in the phenotypic expression of dysphoria in patients diagnosed with depression, anhedonia and PTSD (Pietrzak, et al., 2014). This study also identified low KOR availability in the insula, caudate, and frontal cortex were negatively associated with the severity of dysphoria/emotional numbing expressed by subjects (Pietrzak, et al., 2014). Furthermore, a history of child abuse has been associated with downregulation of the KOR in the anterior insula and epigenetic changes resulting in long–term enhancement of glucocorticoid receptor interactions with endogenous opioids (Lutz, et al., 2018). These findings highlight the importance of brain region specific regulation of KOR expression and binding. For example, within the insula, a severe stressor such as child abuse was sufficient to epigenetically downregulate KOR expression as a compensatory or protective mechanism during development that results in an increased risk for multiple disorders in later life. Equally, severe stressors such as trauma later in life may enhance dynorphin binding of KOR in the aversion network including the insula and amygdala, promoting a more fearful and dysphoric state. Thus, aberrant KOR signaling has emerged as a potential transdiagnostic marker common across multiple psychiatric disorders with translational confirmation provided using the constructs specific to negative valence, specifically following exposure to chronic stress.
2.3.2. KOR and negative valence
Global knockdown of KORs by genetic deletion of exon 1 in mice did not produce a measurable change in phenotypic behavior, notably no changes in depressive–like behavior (Filliol, et al., 2000), impairment in spatial memory (Jamot, et al., 2003), or alterations in stress–reactivity (Contet, et al., 2006). Given that KOR activity promotes a stress–like behavioral phenotype it would be logical to hypothesize that global knockout of KOR would result in a stress–resilient phenotype. However, the importance of KOR activation in immune regulation should not be overlooked. Unlike the stress–protective effects reported in Oprm1–/–mice, constitutive deletion of Oprk1 in mice enhanced humoral activity and exacerbated autoimmune disorders (Du, et al., 2016; Gaveriaux–Ruff, et al., 2003), indicating that KORs are important in immune function.
Negative valence as per RDoC constructs can be assessed under several categories, acute threat (fear), potential threat (anxiety) and sustained threat (aversive emotional state, potentially produced by stress exposure). It has been repeatedly demonstrated that measures of potential threat are augmented by KOR deletion (Table 2). Ablation of KORs on neurons that express the dopamine transporter (DAT) produced robust reductions in anxiety compared to wildtype controls (Van’t Veer, et al., 2013). In line with these findings, bilateral intra–mPFC administration of the KOR antagonist nor–BNI increased center time in the open field test (Van’t Veer, et al., 2013). Underlying this behavioral effect, it was proposed that nor–BNI attenuated BLA mediated inhibition of PFC cell firing (Dilgen, et al., 2013). Furthermore, KOR activation in response to a stressful stimulus preferentially regulated BLA to mPFC inputs (Tejeda, et al., 2015). Within the BLA, anxiogenic–like effects produced by stress or pharmacological activation of CRF receptor 1 (CRF–R1) were shown to trigger dynorphin release and were blocked by administration of KOR antagonists (Bruchas, et al., 2009). In agreement with these findings, exposure of rats to a fear–conditioning paradigm resulted in a dramatic upregulation of Oprk1 mRNA levels within the BLA, but not in the CeA or hippocampus (Knoll, et al., 2011). Moreover, phosphorylation of KORs was dramatically upregulated by local CRF injection into the BLA, dorsal raphe nucleus and dorsal hippocampus and to a lesser degree in the ventral pallidum, ventral tegmental area, nucleus accumbens and bed nucleus of the stria terminalis (Land, et al., 2008). The ability of CRF to activate KORs was blocked by administration of nor–BNI and in Pdyn knockout mice (Land, et al., 2008). The effects of CRF on KOR mediated conditioned place aversion were specifically produced by CRF–R2 activation within the BLA (Land, et al., 2008). More recently, it was shown that within the CeA, CRF facilitates the release of DYN which in turn activates KORs that effectively attenuate CRF induced increases in presynaptic GABA release within the nucleus (Kang–Park, et al., 2015). The functional relevance of KOR signaling within the CeA at a behavioral level has not been explored in depth, but these data clearly indicate the important regulatory function of KORs on amygdalar neurotransmission, a key region in the emergence of negative valence. Thus, CRF induced KOR activation is an important consideration in exploring the detrimental effects of acute and chronic stress. Indeed, there is a body of work that suggests the aversive quality of KOR agonists is diminished or unaffected following chronic stress exposure relative to acute stress. Specifically, acute restraint stress enhanced the aversive quality of low dose bremazocine, a dose that did not evoke conditioned place aversion in normal animals, but chronic stress did not facilitate conditioned place aversion to low–dose bremazocine (del Rosario Capriles & Cancela, 2002). In the context of the reward effects of drugs of abuse, a single exposure to swim stress and administration of U50488 (5 mg/kg) 5 min post swim was sufficient to reinstate cocaine and nicotine place preference (Al–Hasani, et al., 2013). However, exposure to sub–chronic social defeat stress and chronic mild stress did not evoke KOR mediated reinstatement of cocaine place preference (Al–Hasani, et al., 2013). These data are important as they demonstrate the ability of KORs to modulate positive and negative valence under different stress conditions.
Table 2. KOR dysregulation in depression.
These data are compiled from preclinical and clinical studies that implicate KOR and dynorphin in depression using behavioral constructs that relate to domains of negative valence, positive valence, cognitive systems, systems for social processes and arousal/regulatory systems. KOR – kappa opioid receptor, PDYN – prodynorphin, CeA – central nucleus of the amygdala, NAc – nucleus accumbens, ACC – anterior cingulate cortex, KO – knockout, DAT–KOR KO – KOR knockdown in neurons expressing the dopamine transporter, EPM – elevated plus maze, OF – open field, LDB – light/dark box, NIH – novelty induced hypophagia, FST – forced swim test, LH – learned helplessness, nor–BNI – nor–binaltorphimine, NREM – non–rapid eye movement. PTSD – post traumatic stress disorder.
| Domain | Constructs | Behavioral effects | Reference |
|---|---|---|---|
| Negative Valence: | Acute threat (Fear) | KOR antagonists reduced acquisition and expression of conditioned fear behavior and fear potentiated startle. | (Fanselow, et al., 1991; Knoll, et al., 2007; Knoll, et al., 2011; Rogala, et al., 2012; Szklarczyk, et al., 2015) |
| Intra-dorsal hypothalamus injection of nor-BNI potentiated freezing behavior in contextual fear, Injection of the KOR2 agonist GR 89696, but not the KOR1 agonist U-69593 reduced freezing | (Vanz, et al., 2018) | ||
| Potential threat (Anxiety) | DAT-KOR KO mice display lower levels of baseline anxiety compared to their wildtype controls on the EPM and open field. | (Van’t Veer, et al., 2013) | |
| KOR antagonists produce anxiolytic effects in naïve and stressed animals on the EPM, OF, LDB, NIH and defensive withdrawal/burying paradigms. | (Browne, et al., 2018; Bruchas, et al., 2009; Carr & Lucki, 2010; Jackson, et al., 2015; Knoll, et al., 2007; Knoll, et al., 2011; Rogala, et al., 2012; Tejeda, et al., 2015; Valenza, et al., 2017; Van’t Veer, et al., 2013) | ||
| Sustained threat (Aversive emotional state) | KOR agonists produce aversion and dysphoria in humans. | (Pfeiffer, et al., 1986; Ranganathan, et al., 2012) | |
| KOR agonists produce aversion and dysphoria in rodents. | (Bals-Kubik, et al., 1993; Bruchas, et al., 2007; Chefer, et al., 2013; del Rosario Capriles & Cancela, 2002; Land, et al., 2008; Mori, et al., 2002; Zhang, et al., 2006) | ||
| Increased PDYN and Oprk1 mRNA expression persist for days to weeks following the cessation of stress. | (Berube, et al., 2013; Berube, et al., 2014; Browne, et al., 2018; Donahue, et al., 2015; Falcon, et al., 2016; Lucas, et al., 2011; Nocjar, et al., 2012; Shirayama, et al., 2004) | ||
| Oprk1−/−and PDYN−/−KO are resilient to the prodepressive effects of stress. | (Donahue, et al., 2015) | ||
| KOR antagonist produce antidepressant activity in naïve and stress exposed rodents. | (Beardsley, et al., 2005; Browne, et al., 2018; Carr, et al., 2010; Huang, et al., 2016; Land, et al., 2008; Mague, et al., 2003; McLaughlin, et al., 2003; Reed, et al., 2012; Takahashi, et al., 2018; Valenza, et al., 2017) | ||
| Loss | Low KOR availability in amygdala-ACC-ventral striatal circuit is associated with loss and dysphoria in patients diagnosed with depression, anhedonia and PTSD | (Pietrzak, et al., 2014) | |
| Positive Valence: | Reward Responsiveness | KOR activation reduces DA release with NAc | (De Vries, et al., 1990; Di Chiara & Imperato, 1988; Margolis, et al., 2003; Mulder, et al., 1984; Ronken, et al., 1993a) |
| Reward Valuation | DAT-KOR KO mice are resilient to stress induced anhedonia. | (Donahue, et al., 2015) | |
| Cognitive Systems: | Perception | KOR agonists are hallucinogenic and produce psychotomimetic effects | (Butelman & Kreek, 2015; Maqueda, et al., 2015) |
| Working Memory | KOR antagonists blocked agonist induced disruptions in 5 choice serial reaction time task | (Nemeth, et al., 2010) | |
| Aged Pdyn−/−mice did not develop the spatial and object recognition deficits that occurred in wildtype controls. | (Menard, et al., 2013) | ||
| Systems for social processes | Affiliation and Attachment | DAT-KOR KO mice are resilient to stress induced social interaction deficits. | (Donahue, et al., 2015) |
| Arousal/Regulatory systems | DYN release in ventrolateral preoptic nucleus increased NREM sleep by 51% | (Greco, et al., 2008) |
A compelling body of evidence has demonstrated the robust anti–stress effects of KOR antagonists in rodent behavioral tests relevant to depression, anxiety and anhedonia. Central and systemic injections of KOR antagonists and genetic deletion of either KOR or PDYN produced antidepressant–like effects in behavioral tests, such as the FST and learned helplessness (LH) paradigms (Beardsley, et al., 2005; Browne, et al., 2018; Carr, et al., 2010; Huang, et al., 2016; Land, et al., 2008; Mague, et al., 2003; McLaughlin, et al., 2003; Reed, et al., 2012; Valenza, et al., 2017), and consistently reduced anxiety–like and fear–related behaviors across a number of tasks, including the EPM, open field, NIH, conditioned burying and fear conditioning (Browne, et al., 2018; Bruchas, et al., 2009; Carr & Lucki, 2010; Jackson, et al., 2015; Knoll, et al., 2007; Knoll, et al., 2011; Rogala, et al., 2012; Valenza, et al., 2017; Van’t Veer, et al., 2013). Behavioral effects produced in response to repeated stress are also sensitive to KOR antagonists. The increase in immobility scores in the FST following repeated swim stress was prevented by nor–BNI (10 mg/kg, IP) pretreatment (McLaughlin, et al., 2003). Additionally, co–treatment with either nor–BNI (10 mg/kg, IP) or PF–04455242, (1–10 mg/kg, SC) reduced the time intruder rats spent in a submissive or defeated posture over the course of a three–day social defeat stress paradigm (Grimwood, et al., 2011; McLaughlin, et al., 2006). Exposure to a more stressful 10–day social defeat paradigm produced robust alterations in sleep architecture and disrupted circadian regulation of temperature and locomotor activity that were ameliorated by JDTic (30 mg/kg, IP) treatment during the stress (Wells, et al., 2017). Moreover, DAT–KOR knockout mice exhibited stress resilience by failing to develop stress–induced anhedonia following exposure to a similar social defeat paradigm (Donahue, et al., 2015). Furthermore, in the stress sensitive and highly anxious Wistar Kyoto rat, nor–BNI, DIPPA, and buprenorphine produced robust antidepressant–like effects but had no effect in normosensitive Sprague Dawley or Wistar rats (Browne, et al., 2015; Carr, et al., 2010). Overall, these data highlight a strong body of evidence demonstrating the potential of KOR antagonists to target multiple constructs under the domain of negative valence (Table 2).
2.3.3. KOR mediated molecular alterations
Molecular mediators identified with KORs have been examined and functionally selective signaling pathways have been associated with their behavioral effects. To date, some of the most pertinent findings have been found in relation to GRK3 phosphorylation of serine 369 in the carboxyl–terminal domain of KOR, which initiates arrestin–dependent receptor desensitization and internalization (Jordan, et al., 2000; Reyes, et al., 2010; Trapaidze, et al., 2000). Bruchas and colleagues established that arrestin dependent p38 MAPK signaling mediated KOR induced dysphoria, as inhibition of p38 MAPK blocked DYN–mediated increases in immobility in the forced swim stress paradigm and prevented conditioned place aversion produced by KOR agonists (Bruchas, et al., 2007). They confirmed that p38 MAPK was the primary mediator in vitro, showing that activation of KOR induced phosphorylation of p38 MAPK was blocked 1) by a receptor mutation that prevented GRK/arrestin–dependent desensitization, 2) by GRK3 gene knock–out, and 3) via arrestin3 suppression (Bruchas, et al., 2006). Similarly, GRK3 dependent activation of ERK½ signaling persists for several hours following KOR agonist treatment (Bruchas, et al., 2008). In line with KOR mediated induction of ERK½ phosphorylation and the subsequent upregulation of cAMP response element binding protein (CREB), this robust molecular characteristic has been observed following exposure to a wide variety of stressors. Rodents subjected to mild footshocks, acute and chronic restraint, and chronic mild stress all exhibited persistent ERK½ hyperphosphorylation in PFC dendrites and a reduction of phospho–CREB expression in several cortical and subcortical regions (Kuipers, et al., 2003; Trentani, et al., 2002). Pronounced alterations in ERK and CREB are also evident in the NAc and hippocampus following chronic stress and even diet–induced obesity (Gur, et al., 2007; Kreibich, et al., 2009; Lee, et al., 2012; Moron, et al., 2010; Schmidt & Duman, 2010; Sharma & Fulton, 2013). KOR antagonists reversed stress–induced ERK½ hyperphosphorylation and the subsequent CREB–mediated induction of PDYN gene expression (Bruchas, et al., 2008; Jamshidi, et al., 2016; Pliakas, et al., 2001; Potter, et al., 2011). The importance of examining KOR mediated intracellular signaling in the context of stress and drug treatments can’t be overemphasized as KOR agonists and antagonists may show different patterns of signaling after exposure to stress or in stress–sensitive subject compared with stress–naïve subjects.
2.3.4. KOR mediated circuit–based dysfunction
Under normal conditions, KOR agonism is an important modulator of GABAergic, glutamatergic and monoaminergic neurotransmission (Halasy, et al., 2000; Hjelmstad & Fields, 2003; Land, et al., 2009; Lemos, et al., 2011; McFadzean, et al., 1987; Reyes, et al., 2010; Wagner, et al., 2001). Within the dorsal raphe nucleus (DRN), KORs are located on GABAergic interneurons that inhibit serotonin (5–HT) firing. Thus, activation of KORs results in an overall increase in 5–HT release from raphe cell bodies from their terminals in the forebrain. Utilizing local injections of the KOR antagonist nor–BNI and lentiviral knockdown of KORs in the DRN, it was shown that KOR–evoked release of 5–HT in NAc terminals was necessary for KOR agonist–induced aversion (Land, et al., 2009). Subsequently, it was established that although acute KOR activation inhibited excitatory synaptic transmission presynaptically and postsynaptically activated G–protein–gated inwardly rectifying potassium channels (GIRKs), chronic stress exposure downregulated the intensity of postsynaptic KOR–mediated GIRK currents, but did not modulate the ability of KORs to presynaptically inhibit excitatory transmission (Lemos, et al., 2012). These data highlight the importance of conducting circuit–based evaluations under pathologically relevant conditions. Another potentially important facet of KOR regulation of the serotonin system is the ability of agonists to downregulate expression of the serotonin transporter (SERT). A recent in vitro study demonstrated that U–69593 (5–20 µM) and U–50488 (5–20 µM) agonism produced dose–dependent decreases in 5–HT uptake 24 h post treatment in EM4 T cells transfected with SERT. Long–term reductions in 5–HT uptake were mediated by attenuated SERT exocytosis and enhanced SERT endocytosis and phosphorylation, ultimately reducing the functional availability of surface SERT, all of which could be blocked by nor–BNI pretreatment (Sundaramurthy, et al., 2017). As most conventional antidepressants exert their effects through blockade of serotonin reuptake at the synapse, it would be of interest to explore whether drugs that modulate KOR could be given with conventional antidepressants to enhance their therapeutic effects.
Mesolimbic DA projections from the VTA to the NAc and PFC regulate reinforcement and motivation. Persistent activation of KOR by DYN within this stress sensitive pathway is proposed not only as a key mediator of drug seeking behavior (Chavkin & Koob, 2016; Kreek & Koob, 1998; Lalanne, et al., 2014), but is also implicated in the development of two clinical hallmarks of depression, blunted hedonic response and cognitive impairment (Jacobson, et al., 2018; Pizzagalli & Carlezon, 2017). A large body of evidence has demonstrated that DA neurotransmission in the ventral striatum is tightly regulated by D2 autoreceptors and also by presynaptically located KORs that robustly decrease DA release and neuronal firing rates (De Vries, et al., 1990; Di Chiara & Imperato, 1988; Margolis, et al., 2003; Mulder, et al., 1984; Ronken, et al., 1993a). At the level of the NAc, KORs are co–localized with DAT, on DA terminals, where they can control the intensity of DA reuptake (Fuentealba, et al., 2006). Initially, it was shown that administration of KOR agonists into both the VTA and NAc elicited robust conditioned place aversion in rats (Bals–Kubik, et al., 1993). Moreover, systemic administration of the KOR agonist salvinorin A produced similar effects to that of intra–VTA injections, promoting immobility in the FST and increased intracranial self–stimulation thresholds in Sprague Dawley rats that correlated with decreased extracellular DA release within the NAc in a dose–dependent manner (Carlezon, et al., 2006). Interestingly, under normal conditions, KOR agonists decrease the phasic release of DA within the NAc, yet exposure to acute restraint stress (Anstrom & Woodward, 2005) and chronic social defeat stress (Cao, et al., 2010; Krishnan, et al., 2008; Razzoli, et al., 2011; Wook Koo, et al., 2016) have been shown to induce persistent increases in phasic DA release from VTA–NAc projecting neurons. Moreover, these physiological changes were reversed by chronic administration of the selective serotonin reuptake inhibitor fluoxetine (Cao, et al., 2010), suggesting that stress–induced alterations in phasic activity of DA release within the ventral striatum may serve as a biomarker of stress that is amenable to treatment.
Recent work evaluating DYN/KOR signaling on DA neurotransmission within the NAc has moved the field to consider a more complex picture of local and pathway specific inhibition of neurotransmission by KORs. A subpopulation of DYN positive neurons that is responsible for KOR mediated aversion has been identified within the NAc shell (Al–Hasani & Bruchas, 2011). It has been proposed that abnormal KOR function at the level of the NAc may produce negative affect and negative reinforcement of salient stimuli. Such complex KOR modulation is also thought to occur in other nuclei where GABAergic interneurons fine–tune excitation–inhibition balance to modulate network activity. Mimicking the pharmacological effect of KOR agonists, a 5–minute exposure to a cold swim stress was sufficient to induce long lasting activation of KORs located on GABAergic synapses within the VTA. At this site, KORs acted to block LTPGABA (Graziane, et al., 2013; Polter, et al., 2014). A follow up study established that the transient activation of KORs by KOR agonist infusion and acute cold swim stress resulted in a sustained blockade of LTPGABA for up to at least 5 days post agonist exposure (Polter, et al., 2017). Although the exact mechanism mediating sustained suppression of LTPGABA requires further study, this is an intriguing finding and highlights the need for further investigation of KOR modulation of GABA in different nuclei that regulate the mesolimbic DA system. Similarly, KORs robustly inhibited excitatory glutamatergic synapses projecting from the BLA onto dopamine D1 receptor expressing medium spiny neurons (MSNs), but not those from the ventral hippocampus. KORs also indirectly promoted dopamine D2 receptor drive, as KORs inhibit GABAergic collaterals from D1 MSN onto D2 expressing MSNs (Tejeda, et al., 2017). Thus, KORs fine tune glutamatergic evoked long–term potentiation (LTP), via DA D1, and long–term depression (LTD), via DA D2, to consequently regulate synaptic strength. Further investigation of the ability of KORs to fine–tune LTPGlut and synaptic plasticity are warranted, especially in light of the recent development of glutamatergic–based compounds as potential antidepressant compounds (Henter, et al., 2018).
DA neurotransmission is a key neurotransmitter system altered in the context of aversion and reward and is robustly modulated by opioid receptors. Under normal physiological circumstances KOR agonists directly inhibit a subpopulation of VTA DA neurons through activation of GIRKs (Margolis, et al., 2003). Subsequent studies determined that KORs in the VTA were located selectively on a subpopulation of DA neurons that project to the mPFC, (Margolis, et al., 2006), where they are involved in modulating cognitive processes (Lammel, et al., 2014) and aversion. Intra–mPFC administration of KOR agonists produced conditioned place aversion in rats (Bals–Kubik, et al., 1993), which may be mediated by local inhibition of DA overflow (Tejeda, et al., 2013). Correspondingly, intra–mPFC administration of the KOR antagonist nor–BNI by reverse dialysis increased basal DA dialysate within the PFC and blocked the development of aversion to a KOR agonist (Tejeda, et al., 2013). In support of the hypothesis that KOR–mPFC DA projections are involved in aversion, mice with selective deletion of KORs on these DA neurons failed to exhibit KOR–induced aversion and reductions in DA release within the PFC (Chefer, et al., 2013; Tejeda, et al., 2013). Rescue of KOR agonist behavioral effects was observed following intra–VTA delivery of KORs using an adeno–associated viral gene construct administered to DATCre–KOR KO mice (Chefer, et al., 2013). As levels of PFC DA release are selectively regulated (decreased) by KOR activation in the VTA (Margolis, et al., 2006), it has been suggested that decreased VTA D2 receptor function induced by repeated KOR activation may reduce overall DA release in the PFC during stress exposure, and inhibit presynaptic glutamate release onto pyramidal neurons within the PFC, ultimately reducing the activity of mPFC projections to other regions (Tejeda, et al., 2013). Overall these data demonstrate the robust effects of KORs on mPFC neurotransmission in modulating aversion in response to stressful stimuli.
2.3.5. KOR Implications
Endogenous DYN/KOR signaling regulates glutamate, GABA and DA at a local (within nuclei) and network level. Ultimately these effects assume important biological significance by modulating a range of behavioral endophenotypes that pertain to increased anxiety, depression and aversion in animal models. Very little is known about the impact of KORs on the PFC mediated cognitive processes that are impaired in depressed patients (Baune, et al., 2018; McIntyre, et al., 2017). It has been difficult to separate blunted motivation and cognition processes. As translational tests that are independent of intact hedonic responses (Der–Avakian, et al., 2016) are utilized more regularly in preclinical studies, the impact of opioid receptors on the important cognitive facet of MDD can be explored in detail (Jacobson, et al., 2018). Finally, the consideration of sex differences in sensitivity to KOR ligands is emerging as an important concern for the field. Female rodents require higher doses of KOR agonists and antagonists than their male counterparts to achieve comparable behavioral effects in relation to analgesia, anxiety and depression (Chartoff & Mavrikaki, 2015; Laman–Maharg, et al., 2018; Liu, et al., 2013; Robles, et al., 2014; Russell, et al., 2014; Williams, et al., 2018). It has been suggested that differential KOR signaling may underlie this behavioral change. In contrast with male C57BL/6J mice, female mice treated with nor–BNI failed to exhibit a reduction in immobility in the FST across a wide range of doses nor did they exhibit an elevation in JNK signaling (Laman–Maharg, et al., 2018). These findings are important because human studies have determined greater KOR availability in males (Vijay, et al., 2016), implying important sex–differences in relation to KOR ligand sensitivity. Further research is required to delineate metabolic or gonadal dependent effects on KOR ligand signaling. These considerations highlight the importance of sex differences in the development of novel KOR ligands for major depression
2.4. Delta Opioid Receptor (DOR)
Initially isolated from mouse vas deferens (Lord, et al., 1977), the DOR was the first opioid receptor to be sequenced (Evans, et al., 1992; Kieffer, et al., 1992). This 372 amino acid, 7 transmembrane GPCR (Kieffer, et al., 1992; Knapp, et al., 1994; Quock, et al., 1999; Simonin, et al., 1994) has high binding affinity for β–endorphin and leu–enkephalin (Evans, et al., 1992; Kieffer, et al., 1992). Brain regions in rats found with high Oprd1 mRNA expression included the frontal cortex, hippocampus, NAc and amygdalar complex, regions of interest for depression and anxiety, (Mansour, et al., 1987). Localization of DORs was later confirmed by immunohistochemical studies (Alvira–Botero & Garzon, 2006; Cahill, et al., 2001a) and fluorescently tagged DOR–eGFP mice (Erbs, et al., 2012; Scherrer, et al., 2006). Furthermore, the regional distribution of DORs is conserved in humans. PET ligand binding using [(11)C]methylnaltrindole identified the highest levels of DOR binding in the temporal, insular, occipital, frontal, and cingulate cortices (Arvidsson, et al., 1995; Madar, et al., 1996; Smith, et al., 1999), and the amygdala and putamen (Weerts, et al., 2011). Although no splice variants of the Oprd1 gene have been identified (Pasternak, 2004), two functionally distinct DOR isoforms have been characterized, DOR1 and DOR2 (Jiang, et al., 1991; Sofuoglu, et al., 1991, 1992; Thorat & Hammond, 1997), both of which can form heteromers with MORs (Gomes, et al., 2000; Rothman, et al., 1992; Rothman, et al., 1991). Unlike the DOR monomer/homomer, the DOR/MOR heteromer induces a distinct cellular signal transduction pathway (Hasbi, et al., 2007; Rozenfeld & Devi, 2011). Although DOR1 and DOR2 ligands have similar affinity for MORs (van Rijn, et al., 2013), selective DOR1 and DOR2 ligands produced divergent effects on the same behavioral endpoint. For a thorough review of the DOR1 and DOR2 selective ligands, see (Saitoh & Nagase, 2018; van Rijn, et al., 2013).
2.4.1. DOR, pain and negative valence
Depression and anxiety are common comorbid disorders in patients with chronic pain (Stubbs, et al., 2017). Agonists of DORs are remarkably effective in models of chronic pain (Abdallah & Gendron, 2018). Frequently conventional antidepressants are used to treat pain and their effects may involve DORs. The beneficial effects of chronic imipramine on neuropathic allodynia in mice required DOR activation (Benbouzid, et al., 2008a; Benbouzid, et al., 2008b). Conversely, the antidepressant effects of chronic imipramine were accompanied by marked reductions in [3H]–DPDPE binding of DORs in the frontal cortex (Varona, et al., 2003). A role for DORs in treating depression and anxiety was supported by finding systemic administration of the DOR agonist (+)–4–[(aR)–a–((2S,5R)–4–allyl–2,5–dimethyl–1–piperazinyl)–3–methoxybenzyl]–N, N–diethylbenzamide (SNC80), reduced immobility in the rat FST, increased exploration of the open arms on the EPM, and attenuated the conditioned suppression of locomotor activity (Jutkiewicz, et al., 2004; Jutkiewicz, et al., 2005a; Jutkiewicz, et al., 2005b; Jutkiewicz, et al., 2003; Jutkiewicz, et al., 2006; Saitoh, et al., 2004; Torregrossa, et al., 2006). The beneficial effect of DOR agonists on anxiety agreed with evidence from Oprd1–/–mice (Table 3), which exhibited increased depressive and anxiety–like behaviors (Filliol, et al., 2000). Although SNC80 produced positive effects on tests relevant to depression in rodents, the convulsant effects of SNC80 presented a limitation for clinical development. New derivatives of SNC80 and TAN–67 were generated to dissociate the convulsant and sedating effects of DOR agonists from their antidepressant effects and improve permeability across the blood brain barrier. These drugs included some morphinan derivatives and other alkaloid diarylmethylpiperazines. One such DOR1 selective agonist was a TAN–67 derivative, KNT–127, that produced comparable antidepressant and anxiolytic effects as those of earlier DOR agonists without any side effects (Nozaki, et al., 2014; Saitoh, et al., 2011).
Table 3. DOR dysregulation in depression.
These data are compiled from preclinical and clinical studies that implicate DOR and ENK in depression using behavioral constructs that relate to the domains of negative valence, positive valence, systems for social processes and arousal/regulatory systems. DOR – delta opioid receptor, ENK – enkephalin, ppENK – preproenkephalin, CeA – central nucleus of the amygdala, NAc – nucleus accumbens, EPM – elevated plus maze, FST – forced swim test, BP – binding potential.
| Domain | Constructs | Behavioral effects | Reference |
|---|---|---|---|
| Negative Valence: | Acute threat (Fear) | DOR agonists attenuated conditioned suppression of activity. | (Jutkiewicz, et al., 2004; Jutkiewicz, et al., 2005a; Jutkiewicz, et al., 2003; Saitoh, et al., 2004; Torregrossa, et al., 2006) |
| Increased efficacy of DOR agonists on conditioned suppression of activity in stressed rats. | (Hebb, et al., 2005) | ||
| Knockdown of ENK in the CeA reduced freezing during the training phase of fear conditioning but did not alter freezing during testing. | (Poulin, et al., 2013) | ||
| Potential threat (Anxiety) | DOR agonists produce anxiolytic effects | (Jutkiewicz, et al., 2004; Saitoh, et al., 2004; Torregrossa, et al., 2006) | |
| DOR antagonists produce anxiogenic effects in mice | (Saitoh, et al., 2011) | ||
| Oprd1−/−mice exhibit increased levels of anxiety | (Filliol, et al., 2000) | ||
| Sustained threat (Aversive emotional state) | DOR agonists produce antidepressant-like activity in the FST. | (Jutkiewicz, et al., 2005a; Jutkiewicz, et al., 2005b; Jutkiewicz, et al., 2003) | |
| Oprd1−/−mice exhibit increased levels of immobility in the FST. | (Filliol, et al., 2000) | ||
| Increased trafficking of vesicles containing DORs to the cell surface of mice exposed to cold swim stress. | (Commons, 2003) | ||
| Acute stress increases ENK release. | (Henry et al., 2017) | ||
| Chronic or severe stress exposure decreases ENK levels. | (Berube, et al., 2013; Berube, et al., 2014; Poulin, et al., 2013) | ||
| Enkephalinase inhibitors reduced immobility scores in the FST. | (Jutkiewicz, et al., 2006) | ||
| ENK knockdown in the CeA increased exploration on the EPM. | (Poulin, et al., 2013) | ||
| Positive Valence: | Reward Responsiveness | Chronic restraint stress decreased ppENK in the NAc of rats that exhibited decreased sucrose preference | (Poulin, et al., 2013) |
| Systems for social processes | Affiliation and Attachment | Following stress exposure, there is increased trafficking of DOR containing vesicles and increased efficacy of DOR agonists in reducing suppression of activity following social instability and social defeat. | (Commons, 2003) |
| Perception and Understanding of Others | DOR activation produces stimulant effects in socially dominant and singly housed rats, but elicited depressant effects in subdominant rats | (Pohorecky, et al., 1999) | |
| Arousal/Regulatory systems | Sleep and Wakefulness | Sleep deprivation decreases DOR BP | (Fadda, et al., 1991) |
2.4.2. DOR Isoform specific effects on behavior
Despite the fact that no distinct genetic or molecular mechanisms distinguish between DOR1 and DOR2 subtypes (van Rijn, et al., 2013), DOR1 and DOR2 selective ligands have been identified based on differences in their pharmacological effects and these selective ligands produce divergent and opposing effects on the behavior of animals. For example, the anxiolytic effect of the DOR agonist KNT–127 on the EPM was blocked by pretreatment with the DOR2 selective antagonist naltriben but not by the DOR1 selective antagonist 7–benzylidenenaltrexone (BNTX), yet BNTX robustly blocked KNT–127’s antinociceptive effects (Sugiyama, et al., 2014). Earlier studies reported that the DOR2 antagonists naltrindole and naltriben produced anxiogenic effects in tests for anxiety–like behavior (Saitoh, et al., 2011). In line with these findings, naltrindole blocked the dose–dependent increase in time spent in the open arms of the plus–maze and reversal of conditioned suppression of locomotor activity produced by SCN80 (Saitoh, et al., 2005). Overall, this would suggest a DOR2 selective role in the modulation of anxiety behavior. However, as newer ligands have been developed, it has become apparent that DOR1 may also regulate anxiety–like behavior. Direct infusion of the DOR1 selective agonist DPDPE into the amygdala has been shown to decrease anxiety on the EPM (Randall–Thompson, et al., 2010). Such effects are even more robust in the context of stress, where Tan–67’s anxiolytic activity on the EPM was evident only in ethanol–withdrawn mice but not in controls (van Rijn, et al., 2010). Overall these studies highlight the need to continue the thorough examination of the complex interaction of DOR isotypes and their selective ligands in the context of stress and behavior.
2.4.3. Molecular mechanisms underlying DOR’s effects on anxiety and stress resilience
The somewhat ambiguous effects of DOR ligands on anxiety may be due in part to the dynamic regulation of DOR translocation. DORs are transported throughout axon terminals in large dense core vesicles (Zhang, et al., 2006). Migration of these sequestered vesicles to the cell surface can be dramatically upregulated in response to inflammation, (Cahill, et al., 2003), following which the effects of endogenous and exogenous DOR ligands are amplified. Increased expression of DORs in the cell membrane and enhanced behavioral effects of DOR agonists have been shown in studies following stress exposure, including foot shock, social instability and social defeat (Commons, 2003; Hebb, et al., 2005; Margolis, et al., 2011; Pohorecky, et al., 1999) and withdrawal from chronic alcohol (Margolis, et al., 2008; van Rijn, et al., 2010), cocaine (Perrine, et al., 2008) and morphine (Cahill, et al., 2001b). Other stressors may have differential effects on DOR expression that can be augmented by enkephalinergic tone, which can lead to promoting stress resilience (Henry, et al., 2017). In sleep deprived rats, increased ENK binding of DOR was postulated to lead to an upsurge in endocytosis and degradation of DORs (Commons, 2003). This concurs with previous studies where the effects of dysregulated sleep are negated by naltrindole, resulting in increased wakefulness (Moss, et al., 1993; Reinoso–Barbero & de Andres, 1995). Although acute stress augments ENK (Henry, et al., 2017), chronic or severe stress exposure decreased ENK levels that are concurrent with elevated DOR expression and activity (Berube, et al., 2013; Berube, et al., 2014; Poulin, et al., 2013; Poulin, et al., 2014). Modulating enkephalinergic tone, possibly with enkephalinase inhibitors (Dripps & Jutkiewicz, 2018; Jutkiewicz, et al., 2006), could be useful in downregulating DOR activity and promoting stress resilience (Table 3).
One mechanism of regulating stress responses and the emergence of disorders such as depression occurs at the level of the HPA axis. Just as with other opioid receptors, DORs are frequently colocalized with CRF positive neurons in the hypothalamus and on somatostatin positive GABAergic neurons in CA1 region of the hippocampus (Williams, et al., 2011). Extrahypothalamic DOR/CRF colocalized neurons are also apparent in the BLA, where 55 % of the CRF neurons are DOR immunoreactive and in the CeA where 67 % of the CRF labeled neurons are DOR positive (Reyes, et al., 2017). Although proestrus females had similar levels of CRF receptor density in in the stratum radiatum of CA1 as males, a greater number of their receptors were dual labeled for DOR and CRF than males (Williams, et al., 2011). Given that females are twice as likely as males to develop stress–related disorders such as anxiety and depression, DORs represent a strategically placed, sex–specific target for therapeutic development, where DOR ligands can counteract the effects of CRF on negative affective states.
2.4.4. DOR –treating comorbid psychiatric and substance use disorders
Anxiety is frequently comorbid with substance use disorders and is a critical factor in relapse to drug taking (Goodwin & Stein, 2013; Lai, et al., 2015; McHugh, 2015; Vorspan, et al., 2015). Emerging evidence suggests that DORS can target these comorbid disorders. DORs have been associated with alcohol and nicotine use because of robust alterations in the salience of alcohol and nicotine in Oprd1–/–mice (Filliol, et al., 2000; Lutz & Kieffer, 2013). Intra–VTA injection of the DOR agonist DPDPE attenuated ethanol drinking in low–drinking rats, but not in high drinking rats (Margolis, et al., 2008). GABAA mediated evoked and spontaneous IPSCs were correspondingly inhibited in DPDPE treated low–drinking animals only (Margolis, et al., 2008). The impact of DORs on mesolimbic DA neurotransmission under stressful conditions may provide an important link between the comorbidity of psychiatric disorders and substance use disorders. A follow up study found that DPDPE increased the amplitude of evoked VTA IPSCs in a subset of stressed animals exposed to footshock that exhibited high corticosterone levels (Margolis, et al., 2011). These neurons were TH positive, but no other anatomical or physiological properties differentiated these neurons from other neuronal subsets. In contrast, stressed rats with lower corticosterone levels exhibited the expected inhibition of GABAA evoked IPSCs by DORs (Margolis, et al., 2011). These divergent DOR agonist effects on VTA DA transmission are consistent with the differential behavioral effects of DOR agonists under basal conditions and in response to stress. Additional studies may delineate the effects of DOR agonists on DA transmission under stressful conditions that are relevant to affective behavior.
2.4.5. DOR implications
The complex ligand specific effects of DOR agonists is an interesting facet of DOR pharmacology. Moreover, the stress–specific effect of DOR isoforms warrants further exploration. Given the strong analgesic effects of DORs, ligands of this receptor may be most beneficial in patients with chronic pain and comorbid depression. Similarly, as DOR antagonists produce dramatic reductions in anxiety like behavior and addiction, the utility of these compounds in treating comorbid anxiety and substance use disorder may be a significant application. Clinical trials evaluating DOR ligands specifically for MDD or anxiety are few in number, but following the successful preclinical data obtained from assays of defeat, learned helplessness and anxiety in rodents (Hudzik, et al., 2011), the highly selective DOR agonist 4–(R)–(3–aminophenyl)[4–(4–fluorobenzyl)–piperazin–1–yl]methyl}–N,N–diethylbenzamide (AZD2327), was assessed in subjects diagnosed with anxious major depressive disorder, and identified positive effects on the endpoints which included decreased vascular endothelial growth factor (VEGF) levels and elevated EEG gamma power compared to non–responders (Richards, et al., 2016). Although there are no ongoing clinical trials of DOR ligands in MDD and anxiety, continued investigation of these compounds for psychiatric disorders is justified.
2.5. Nociceptin/OrphaninFQ (N/OFQ) and NOP
NOP (formerly ORL1) was first isolated in 1994 (Mollereau, et al., 1994). Emerging evidence supports the use of NOP antagonists as a potential therapeutic for substance use disorders, obesity, Parkinson’s disease and pain (Kallupi, et al., 2017; Witkin, et al., 2014; Zaveri, 2016). Development of drugs that modulate the N/OFQ system initially focused on introducing modifications of the peptide bond between the Phe1 and Gly2 of N/OFQ. These efforts resulted in the successful generation of the first NOP partial agonist [F/G]N/OFQ(1–13)–NH2 in 1998 (Guerrini, et al., 1998). Shortly after this, the first NOP antagonist [Nphe1]N/OFQ(1–13)–NH2 was synthesized. See the following reviews for an excellent synopsis of the various endogenous and synthetic agonists and antagonists of NOP (Gavioli & Calo, 2013; Zaveri, 2016).
2.5.1. N/OFQ and nocistatin
N/OFQ and nocistatin are derived from the precursor prepronociceptin/orphanin FQ (ppN/OFQ). N/OFQ shares a high degree of homology with DYN but is 1000–fold more selective for NOP compared to KORs, and has no activity at MOR or DOR (Meunier, et al., 1995; Mollereau, et al., 1999; Reinscheid, et al., 1995). Conversely, nocistatin behaves as a functional N/OFQ antagonist, producing effects opposite to those of N/OFQ (Gavioli, et al., 2008), but less is known about this ppN/OFQ derivative. Activation of NOP receptors triggers the same pattern of Gi/Go coupled signal transduction cascades as other GPCRs (Hawes, et al., 2000), and like the other opioid receptors, N/OFQ mRNA is localized in brain regions implicated in the development of stress–related psychiatric disorders. N/OFQ mRNA expression in rat was detected in the cortex, hippocampus, amygdala, thalamus, hypothalamus and DRN (Lachowicz, et al., 1995). The high levels of N/OFQ expression in limbic structures were confirmed in subsequent studies in rodents (Neal, et al., 1999a; Neal, et al., 1999b), rhesus monkeys (Kimura, et al., 2011) and humans (Lohith, et al., 2012).
Functionally, N/OFQ acts as an inhibitory neurotransmitter suppressing neuronal activity and subsequent release of other neurotransmitters (Yu, et al., 1997). N/OFQ inhibited K+ induced release of serotonin in neocortex–derived synaptosomes, an effect that was diminished by NOP antagonists (Mela, et al., 2004). N/OFQ also inhibited serotonin release at the level of the DRN in a GIRK–dependent manner (Mogil & Pasternak, 2001; Nazzaro, et al., 2010; New & Wong, 2002; Vaughan & Christie, 1996). Norepinephrine release in rat neocortex was also reduced following N/OFQ treatment (Mela, et al., 2004; Okawa, et al., 2001; Siniscalchi, et al., 2002). Additionally, NOP agonists reduced basal and drug–induced release of DA in the NAc (Di Giannuario & Pieretti, 2000; Di Giannuario, et al., 1999; Murphy, et al., 1999; Murphy, et al., 1996; Murphy & Maidment, 1999). Overall, these data show that N/OFQ, like the other endogenous opioids, can regulate the tone of monoaminergic neurotransmission.
2.5.2. N/OFQ’s anti–stress effects
A complicated picture exists regarding N/OFQ’s anti–stress effects and its promotion of stress–related dysfunction. In general, the positive anti–stress effects of N/OFQ occur at the level of the hypothalamus. Glucocorticoids are necessary for the increased production of N/OFQ, as adrenalectomized rats failed to show stress–induced elevations in N/OFQ release, whereas supplementation with corticosterone rescued N/OFQ release (Nativio, et al., 2012). Interestingly, the NOP antagonist UFP–101 had no effect on HPA axis activation in stress naïve states but blocked N/OFQ induced elevation of circulating levels of corticosterone and expression of CRF and POMC mRNA in the hypothalamus (Leggett, et al., 2006). Furthermore, NOP ligands were more efficacious when administered during the nadir of corticosterone secretion (Leggett, et al., 2007), pointing to a modulatory role of N/OFQ on the HPA axis (Table 4). This hypothesis was strengthened by the finding that N/OFQ reduces neuronal activation in the suprachiasmatic nucleus, where it can act as a regulator of the circadian cycling of the HPA axis (Gompf, et al., 2005). Activation of the N/OFQ system at the level of the hypothalamus is necessary for adaptation to novelty or mild stressors. Central administration of N/OFQ and other NOP agonists elevated circulating levels of corticosterone and ACTH in stress naïve rodents in a dose–dependent manner (Devine, et al., 2001; Fernandez, et al., 2004; Leggett, et al., 2006; Nicholson, et al., 2002). Furthermore, NOP agonists enhanced the secretion of these stress hormones in rats following exposure to novelty novel environment, but not in rats exposed to restraint, a more severe stressor (Devine, et al., 2001). Consistent with an increase in stress hormones were the pronounced anxiogenic effects of N/OFQ in rats when tested on the EPM (Vitale, et al., 2006) and the reductions in anxiety–like behavior in NOP knockout mice compared to their wildtype controls (Gavioli, et al., 2007).
Extrahypothalamic colocalization of N/OFQ and CRF in monoaminergic nuclei and limbic structures regulate this anxiogenic phenotype which may underlie the emergence of stress–related disorders such as PTSD, eating disorders and of course MDD. Within the DRN, CRF (1–100 nM), dose–dependently inhibited [(3)H]5–hydorxytrypamine ([(3)H]–5–HTP) outflow in a CRF–R1 dependent, bicuculline sensitive manner. Indicating that CRF–R1 activation inhibits GABA interneurons within the DRN. Conversely N/OFQ exerted a CRF–R1 independent and bicuculline–insensitive inhibition [(3)H]–5–HTP outflow, specifically modulated DRN 5–HT neurons. In the context of stress, reduced 5–[(3)H]–5–HTP outflow in DRN slices from rats exposed to a 15 min forced swim stress was partially reversed by CRF–R1 antagonism with antalarmin, but inhibited further by N/OFQ administration (Nazzaro, et al., 2010; Nazzaro, et al., 2009). N/OFQ acts as an anxiolytic–like agent in the rat and behaves as a functional antagonist of CRF, requiring activation and inhibition of 5–HT neurotransmission across multiple brain regions. In stress–naïve rats exposed to the anxiogenic provoking conditions of the elevated plus maze and defensive burying test, N/OFQ showed anxiolytic–like effects while CRF displayed anxiogenic–like effects. Moreover, pretreatment with N/OFQ blocked CRF’s anxiogenic effects. Under these anxiogenic conditions, N/OFQ significantly decreased 5–HT levels in the frontal cortex and increased 5HT1A receptor density, but CRF did modify these parameters in this region. Conversely, in the pons, N/OFQ failed to modulate 5–HT turnover, whereas CRF decreased 5–HT levels and increased 5–HIAA content and decreased 5HT1A Bmax and KD (Filaferro, et al., 2014). Together these findings illustrate the interplay of CRF and N/OFQ in modulating behaviors regulated by 5–HT neurotransmission.
Similar, region–dependent changes in N/OFQ have been demonstrated to oppose the actions of CRF in the context of intermittent food restriction, where a 15 min binge eating session decreased hypothalamic mRNA levels of CRF–R1, N/OFQ and NOP. In contrast, CRF mRNA expression was upregulated both in the hypothalamus and VTA in a frustrated food reward task in food restricted rats. The changes in NOP and CRF–R1 expression were shown to be dependent on the DNA methylation at gene promoters produced by this binge eating model, epigenetic effects that were differentially regulated in in the hypothalamus and VTA (Pucci, et al., 2016). Eating disorders are also associated with high levels of anxiety and depression and in the context of stress–related disorders the divergent functions of the amygdala are of significance. N/OFQ opposition of CRF stimulation in the discrete nuclei of the amygdala is apparent following stress exposure.
Restraint stress produced selective upregulation of NOP and downregulation of the CRF–R1 mRNA in the CeA and BLA. More specifically, acute application of CRF significantly increased GABAA–mediated IPSPs in CeA, which was blocked by N/OFQ (Ciccocioppo, et al., 2014). Remarkably, the authors of this study determined that in stressed rats only, baseline CeA GABAergic responses were elevated and N/OFQ exerted a larger inhibition of IPSPs relative to non–stressed rats. Moreover, NOP antagonism increased IPSP amplitudes only in rats exposed to restraint (Ciccocioppo, et al., 2014), suggesting a functional recruitment of the N/OFQ system after acute stress.
Interestingly, in naïve and chronic stressed rats, acute restraint stress decreased levels of N/OFQ in the basal forebrain independent of prior stress exposure, but exposure to chronic restraint alone did not change N/OFQ content within the basal forebrain (Devine, et al., 2003). In contrast, chronic stress increased N/OFQ in the hippocampus, specifically in the dentate gyrus (Nativio, et al., 2012). These data suggest that stress recruits the N/OFQ system in a region–specific manner. Moreover, in the context of stress, where elevated levels of CRF are maladaptive, extrahypothalamic N/OFQ may produce anti–stress effects and reduce anxiety. N/OFQ administration into the CeA blocked CRF stimulation of GABA release (Cruz, et al., 2012). This blockade of CRF was also thought to underlie the capacity of N/OFQ to abolish the anorectic effect of restraint following injection into the BNST (Ciccocioppo, et al., 2014). These findings are consistent and highlight the complexity of region–specific alterations in the N/OFQ NOP system. The preclinical data support the hypothesis that NOP agonists have anxiolytic activity during exposure to aversive stimuli (Vitale, et al., 2006). However, development of N/OFQ as an anxiolytic is limited by N/OFQ’s effects on processes outside the CNS. N/OFQ inhibits gastric motility, produces antitussive effects, vasodilation, and negative effects on cardiac tissue; it can also stimulate inflammation and in some cases sepsis (Armstead, 2011; Gavioli & Romao, 2011; Lambert, 2008; Leggett, et al., 2009; Serrano–Gomez, et al., 2011). Therefore, a great deal of work is required to develop safer N/OFQ–like compounds for anxiety.
2.5.3. NOP and acute threat
Emerging evidence also points to a role for NOP in fear consolidation (Table 4). Microarray studies determined that Oprl1 mRNA expression was the most differentially gene regulated in response to immobilization stress in mice. Administration of the NOP agonist SR–8993 directly into the CeA, where stress–induced Oprl1 levels of expression were highest, impaired consolidation of fear memory and decreased freezing to the conditioned stimulus in stressed mice (Andero, et al., 2013). The same group then explored the impact of OPRL1 in humans, where they identified a SNP in the OPRL1 gene, rs6010719, which was associated with a self–reported history of childhood trauma and PTSD symptoms after a traumatic event. G allele carriers, who were at increased risk for PTSD, positively correlated with progressive trauma exposures. Subjects with the G allele exhibited increased physiological startle measures of fear discrimination and greater functional connectivity between the amygdala and posterior insula (Andero, et al., 2013). Recent work has also shown that impaired cue induced fear memory consolidation occurred in the presence of NOP antagonism, suggesting a key modulatory role of N/OFQ neurotransmission in the context of stress, and PTSD in particular (Tollefson, et al., 2017). These data suggest that Oprl1 is associated with amygdala function, fear processing, and PTSD symptoms.
2.5.4. NOP agonists and negative valence
As outlined above, NOP agonists produced anxiolytic effects during stress exposure and inhibited the consolidation of fear memory. In contrast, NOP antagonists selectively produced antidepressant–like activity in behavioral tests relevant to depression but were not active on tests relevant to anxiety (Gavioli & Calo, 2013; Witkin, et al., 2014). NOP–/–mice exhibited significant reductions in immobility scores in the FST compared to the wild type littermates, and did not show differences in motoric activity (Gavioli, et al., 2007). This is in line with the significant antidepressant activity of NOP antagonists. Across Wistar rats and the CD–1, Swiss and C57BL/6N mouse strains, UFP–101, J–113397, SB–612111 all produced significant reductions in immobility time in the FST and tail suspension test (TST) (Asth, et al., 2016; Gavioli, et al., 2003; Gavioli, et al., 2004; Goeldner, et al., 2010; Medeiros, et al., 2015). Importantly, antidepressant activity of NOP ligands is associated with β–arrestin 2 mediated signaling (Asth, et al., 2016). Few studies have evaluated the antidepressant potential of NOP antagonists in rodent models of depression, but Wistar rats exposed to chronic mild stress did show increased sucrose preference scores following 21 days of UFP–101 administration (Vitale, et al., 2009). Recent evidence from the same group shows that UPF–101 (10 nmol/i.c.v) can produce reversals of anhedonia (sucrose preference deficits) by the second week of treatment (Vitale, et al., 2009). These data clearly show the potential of NOP antagonists to rapidly alleviate negative affective states and enhance hedonic responding to palatable food (Table 4).
2.5.5. NOP implications
Most of the information pertaining to the pharmacological action and function of N/OFQ in disease states has come from rodent studies, but emerging clinical findings support the hypothesis that increased levels of N/OFQ may modulate negative emotional states. Significant species differences in N/OFQ expression, localization and density of NOPs are apparent across rodents (Florin, et al., 1997; Florin, et al., 2000), primates (Bridge, et al., 2003; Kimura, et al., 2011) and humans (Berthele, et al., 2003; Lohith, et al., 2012). These differences could lead to differential regulation of physiological responses by the N/OFQ NOP system across species. Despite these limitations and based on the strong preclinical evidence that demonstrate a consistent pattern of antidepressant–like effect of NOP antagonists, progress has been made in translating these compounds into the clinic. NOP antagonists have been shown to be safe and well tolerated in humans and critically has been shown to enhance positive emotional processing (Post, et al., 2016b). These studies will be covered comprehensively in the next section.
3. Opioid compounds in development for depression
The “opium cure” for depression was the first well defined therapeutic for a psychiatric illness, when Kraepelin (approximately 1891) recommended guidelines for using increasing, then decreasing, doses of opioids in tinctures to treat severe bouts of depression (Weber & Emrich, 1988). Unfortunately, this therapy relied heavily on the euphorigenic action of MOR agonists and was limited by concern for opioid abuse. Moreover, the serendipitous discovery of the antidepressant effects of monoamine oxidase inhibitors and tricyclic antidepressants in the 1950’s changed the standards of treatment for MDD (Lopez–Munoz, et al., 2007). Six decades later, however, despite their refinement, a substantial portion of patients do not respond to monoamine–based therapies. Preclinical studies have laid a more informative foundation by showing how modulation of different opioid receptors can normalize many of the core endophenotypes of depression. The clinical evidence for this hypothesis is growing, with many studies describing rapid and sustained alleviation of severe and unremitting depression in treatment resistant patients by multimodal opioid–based compounds. This next section will appraise the current status of opioid–based compounds that have shown positive effects in clinical studies.
3.1. Buprenorphine
Buprenorphine is an FDA–approved opioid analgesic currently used for the treatment of opioid addiction and chronic pain (Lutfy & Cowan, 2004). The analgesic effects of buprenorphine are mediated by MOR partial agonism but modulated by KORs, DORs and NOP receptors (Grinnell, et al., 2016; Ide, et al., 2004; Lutfy, et al., 2003). As one of the key therapies for opioid use disorder, buprenorphine differs from methadone because its partial agonist activity at MORs reduces the effects of other opioids on euphoria and respiratory depression (Davis, 2012). However, methadone remains the most widely used medication for opioid maintenance therapy due to legal restrictions on prescribing buprenorphine (Cicero, et al., 2014; Manhapra, et al., 2016; Manhapra, et al., 2017; Tsui, et al., 2018). The contribution of MOR, DOR and NOP receptors to buprenorphine’s antidepressant action is only just emerging. In the context of MDD, it is the high affinity and efficacy of buprenorphine at KORs that is hypothesized to underlie the marked alterations in mood. Initial observations in patients with comorbid opioid use and depression, suggested that buprenorphine and buprenorphine/naltrexone treatment improved mood and reduced negative affect (Kosten, et al., 1990; Resnick, et al., 1991). Treatment of opioid use disorder with comorbid depression or a recent history of depression had significantly better outcomes when treated with buprenorphine than methadone (Gerra, et al., 2006b; Weiss, et al., 2011). Similarly, multidrug users with depression displayed better adherence to treatment, reduced dysphoria, drug craving and improved global functioning when treated with a buprenorphine/naloxone combination compared to buprenorphine or methadone alone (Gerra, et al., 2006a). Thus, the effects of buprenorphine on affect may be an important mediator of its efficacy in treating opioid use disorder.
These positive observations on mood from patients with opioid use disorder also emerge when treating opioid naïve patients with depression. However, it should be noted that the doses of buprenorphine used in individuals with substance use disorder (16–32 mg/day) are nearly 10–fold higher than the doses used for treating depression in opioid–naïve patients (0.2–4 mg/day). In the early 1980’s, Emrich detailed the effects of buprenorphine (0.2 mg morning and evening, sublingual) in 10 depressed patients, with 5 patients showing significant improvements in mood over the course of only 1 week and a return of symptoms following discontinuation (Emrich, et al., 1982). The next clinical report of buprenorphine’s antidepressant action came over a decade later when Bodkin evaluated buprenorphine’s effects in severely ill treatment–resistant depressed patients. All patients exhibited a rapid response to treatment, measured by a significant reduction in Hamilton Depression Scale (HAM–D) scores within the first week of treatment (0.45 mg/day to 3.6 mg/day). Six out of seven patients achieved remission of symptoms at the end of the 4–6 week treatment period (Bodkin, et al., 1995). A more recent report documented remission from severe, chronic unremitting depression following just 1 week of buprenorphine treatment (sublingual 0.8–2.0 mg/day), and complete remission at the end of the study in 5/6 patients as measured by HAM–D scores and in 4/6 patients using self–rated Beck Depression Inventory (BDI) scores (Nyhuis, et al., 2008). Additional evidence in support of buprenorphine’s antidepressant effects came from an open label, 8–week trial conducted in elderly treatment–resistant depressed patients (Karp, et al., 2014). Of fifteen patients examined in the study, 5 patients had already completed a 12–week trial with venlafaxine treatment and were deemed non–responders with a Montgomery–Åsberg Depression Rating Scale (MADRS) score of ≥10. At baseline for the Karp study, the mean MADRS score was 27 (SD=7.3, range 18–42). These severely depressed patients were administered a 0.2 mg/day sublingual buprenorphine dose for the first week, following which the dose was increased if MADRS scores remained greater than 10. The mean daily dose was 0.4 mg/day (SD=0.21, range 0.12 –0.83 mg). At the end of the 8–week trial, the average MADRS score was 9.5 (SD=9.5, range 0–33). This robust reduction in depression was apparent as early as week 3, where the mean change from baseline was –15 (SD=7.9, range –25–2). Interestingly, these effects were primarily driven by dramatic reductions in ratings of sadness and pessimistic thoughts (Karp, et al., 2014). Elevated mood was accompanied by increased psychomotor speed and engagement in the performance of cognitive tasks. Patients exhibited improved learning, delayed recall and word discrimination, suggesting an overall enhancement in cognitive abilities. During the 4–week follow up period post discontinuation, subjects showed no signs of withdrawal, but depressive symptoms did return (Karp, et al., 2014). Another noteworthy attribute of short–term, low–dose buprenorphine treatment is the significant and rapid attenuation of suicidal ideation (Striebel & Kalapatapu, 2014; Yovell, et al., 2016).
Until recently, preclinical evaluation of buprenorphine’s behavioral effects was primarily focused on pain and substance use disorders (Cowan, 2007; Cowan, et al., 1977a). Our laboratory published the first preclinical studies demonstrating antidepressant–like and anxiolytic–like effects of low dose buprenorphine (Falcon, et al., 2015), results consistent with the clinical findings. Rodent tests relevant to depression and anxiety are dependent on motor activity. Unfortunately, the hyperlocomotion produced immediately following buprenorphine administration (Marquez, et al., 2007) necessitates testing buprenorphine at time points when the motor effects are no longer apparent, i.e. ≥ 8 h post treatment. Remarkably, selecting a 24 h post treatment interval, buprenorphine (0.25–0.5 mg/kg, IP) produced an inverted U–shaped dose–response curve in the mouse FST, reducing immobility scores at a time following injection when desipramine no longer exerted its antidepressant activity (Falcon, et al., 2015). Moreover, morphine had no activity at this time point. In comparison, the long–lasting KOR antagonist nor–BNI effectively reduced immobility in the FST 24 h post treatment. In the NIH test, low–dose buprenorphine (0.25 mg/kg, IP) decreased the latencies to approach and consume a palatable food in a novel environment 24 h hours post treatment (Falcon, et al., 2015). This result is important as the NIH test was reported to be sensitive only to chronic, but not acute, treatment with conventional antidepressants (Dulawa & Hen, 2005). This pattern of behavioral effects occurs following a single administration on tests that usually require chronic treatment with classical antidepressants and its long–term persistent effects, are reminiscent of the rapid activity of the compound reported in clinical studies. Another group in the United Kingdom also reported the anxiolytic activity of buprenorphine in CD–1 mice at a slightly higher dose (1 mg/kg, IP) (Almatroudi, et al., 2015). Furthermore, subchronic treatment (0.25 mg/kg daily for 6 days, IP) did not produce tolerance to the behavioral effects of buprenorphine in the FST or NIH tests (Falcon, et al., 2015). Similarly, in rats, buprenorphine (0.75 –2.25 mg/kg, SC) significantly attenuated immobility and increased swimming in the FST and increased exploration in a novel environment when tested in two substrains of stress hyperreactive WKY rats 24 h post treatment. Interestingly, two other rat strains that are not stress hyperreactive, the Sprague Dawley (SD) and Wistar rat, were insensitive to the effects of buprenorphine (Browne, et al., 2015). This replicated previous findings with other KOR antagonists, nor–BNI and DIPPA, which produced antidepressant–like activity in WKY rats but failed to induce behavioral change in SD rats (Carr, et al., 2010; Carr & Lucki, 2010). In subsequent studies that utilized rodent models of chronic stress, buprenorphine treatment (0.25 mg/kg, IP for 7–14 days) effectively reversed behavioral deficits. Anhedonia measured using sucrose preference, anxiety–like behavior in the light dark box and depressive–like behavior in the FST, induced following chronic mild stress were reversed by buprenorphine treatment yet no behavioral effects were noted for buprenorphine treated non–stressed controls (Falcon, et al., 2016). Further, mice exposed to 10 days of chronic social defeat exhibited improvements in social interaction scores following 1 week of buprenorphine treatment (0.25 mg/kg, IP), but buprenorphine did not alter behavior in non–stressed controls (Browne, et al., 2018). This stress by treatment interaction was also apparent at a molecular level, where stress–induced alterations of mRNA expression of Oprk1 and Oprm1 genes in cortical and limbic structures were normalized following buprenorphine treatment (Falcon, et al., 2016).
As buprenorphine has activity at multiple opioid receptors (Lutfy et al, 2004), an important goal of these initial preclinical studies was to determine which opioid receptors were associated with buprenorphine’s behavioral effects. In these studies, pharmacological blockade of KORs with the long–lasting antagonist nor–BNI (10 mg/kg, IP) and genetic deletion of KORs (Oprk1–/–mice) prevented the antidepressant–like effects of buprenorphine (0.25 mg/kg, IP) in the FST (Falcon, et al., 2016). In contrast, genetic deletion of MORs (Oprm1–/–mice) produced greater sensitivity to buprenorphine in the FST, where a typically inactive low dose of buprenorphine (0.125 mg/kg, IP) reduced immobility scores by 40%. Genetic deletion of DORs (Oprd1–/–mice), and blockade of ORL1 receptors with JTC–801 (1 mg//kg, IP) did not affect buprenorphine’s activity in the FST. Taken together, these data suggest that KOR antagonism is the key mediator of buprenorphine’s antidepressant–like effects in the FST. In contrast, Oprm1–/–mice failed to respond to buprenorphine (0.25 mg/kg, IP) in the NIH test, and a greater magnitude of effect was detected in Oprk1–/–mice at this low dose (Robinson, et al., 2017). Following the early phase of action where MORs are partially activated, buprenorphine exhibits a second prolonged phase of slow dissociation from the receptor resulting in a period of functional blockade of MORs (Cowan, et al., 1977b; Walker, et al., 1995). It is the second, latent phase of MOR blockade rather than the initial activation of these receptors that mediates buprenorphine’s activity in the NIH test. Corroboration of this functional antagonism was obtained using the hot plate test, where morphine (10 mg/kg IP) antinociception was blocked 24 h after buprenorphine pretreatment at the dose and time used in NIH testing (Robinson, et al., 2017). In contrast to buprenorphine, the selective MOR antagonist cyprodime (10 mg/kg, IP) and pan–opioid antagonist naltrexone (1 mg/kg, IP) reduced approach latencies in the NIH test at 1 h but not 24 h post treatment because they are short–acting and lack a protracted phase of MOR blockade. Activation of MORs using morphine (10 mg/kg, IP) and the KOR antagonist nor–BNI (10 mg/kg, IP) were ineffective in the NIH test. Furthermore, only the behavioral effects of buprenorphine that are associated with MORs are blocked in mice that possess the hyporesponsive G allele of the murine A112G Oprm1 model of the human A118G SNP (Browne, et al., 2017). In contrast, mice with the AA, AG, and GG genotypes responded equally well to buprenorphine’s effects in the FST assay (Browne, et al., 2017), a behavioral effect associated with KORs. These studies using acute behavioral assays confirm that blockade of MORs and KORs produce complimentary effects on measures of anxiety and depression. Overall, these findings show how preclinical assays can be used to clarify some of the complex pharmacological properties of buprenorphine and support the use of multi–opioid antagonist compounds for the treatment of depression.
As the preclinical literature indicates, KOR blockade, followed by latent MOR antagonism appears to mediate the behavioral effects of the low doses of buprenorphine on tests associated with antidepressant drugs. However, there are concerns about the safety of buprenorphine, such as the potential for abuse liability in this vulnerable patient population (Cicero, et al., 2014). Although prolonged use can produce physical dependence or a risk of abuse, there have not been sufficiently controlled comparisons of risk of buprenorphine between opioid naïve and opioid experienced depressed patients. In fact, blunted reward or anhedonia is a core endophenotype of depression and whether depressed patients would use buprenorphine for its mild euphorigenic properties has never been established in a controlled setting. Indeed, clinical studies of chronic pain patients describe withdrawal from low doses of buprenorphine as “relatively mild” compared to heroin or morphine. It has been shown that extracellular levels of dopamine within the NAc, measured using in vivo microdialysis, were unchanged by low–dose buprenorphine treatment in stress–naïve mice, but this dose successfully blocked reductions in DA release induced by systemic administration of the KOR agonist U–50488 (Falcon, et al., 2016). This is a critical finding, as buprenorphine can effectively mitigate the stress–like reductions in DA transmission post U–50488 at low doses that do not negatively alter normal DA neurotransmission. These data also agree with evidence obtained from healthy human subjects, where the anti–stress effects and improved emotional processing of stimuli following buprenorphine treatment occurred in the absence of a subjective high (Bershad, et al., 2015; Bershad, et al., 2016). Medicinal chemists are generating buprenorphine derivatives that can harness the beneficial effects and negate any abuse liability associated with the compound. The first of these studies has detailed the potential antidepressant activity of the buprenorphine derivative BU10119 (Almatroudi, et al., 2018). In the interim, buprenorphine can be administered for treatment of depression through a skin patch, depot injection, or a subcutaneous implant, routes of administration that would have minimal diversion or abuse potential. The question of abuse liability in a vulnerable patient population is a constant concern. However, as the evidence for the antidepressant efficacy of buprenorphine becomes more compelling, the risk/benefits for treatment resistant patients will be reassessed.
3.2. ALKS–5461
ALKS–5461, a combination of buprenorphine with the MOR antagonist samidorphan (Wentland, et al., 2009a; Wentland, et al., 2009b; Wentland, et al., 2005), represents the best attempt yet to harness the antidepressant effects of buprenorphine and mitigate its abuse potential. The feasibility of ALKS–5461 for use in depressed patients was demonstrated in a placebo–controlled trial conducted in healthy opioid–experienced subjects and individuals with a current depressive episode that were unresponsive to treatment (Ehrich, et al., 2015a). Firstly, the study determined the most effective combination ratio of buprenorphine/samidorphan for the alleviation of depression and blockade of MOR agonist activity. Maximal blockade of MORs, measured using pupillometry, was achieved with a 1:1 ratio of buprenorphine/samidorphan. No change in the subjective hedonic value of the drug combination, or sedation, and significantly fewer side effects were reported for the 1:1 dosing regimen. A robust reduction of ratings on HAM–D and a trend towards significant reductions on the MADRS were detected at the end of 1 week of treatment, with no withdrawal symptoms observed following discontinuation of the opioid antagonist combination (Ehrich, et al., 2015a). Subsequently, ALKS–5461 was evaluated as an adjunct therapy for MDD diagnosed subjects with inadequate/partial response to treatment with SSRI or SNRIs (Fava, et al., 2016). Following 4 weeks of low dose, 2mg/2mg, buprenorphine/samidorphan daily, outcomes across three depression–rating scales, HAM–D, MADRS and the Clinical Global Impressions severity scale (CGI–s) were significantly improved (Fava, et al., 2016). ALKS–5461 was granted approval from the FDA as a Fast Track Designated Medicine in October 2017. However, a review by the FDA’s Psychopharmacologic Drugs Advisory Committee recommended that the drug’s benefit–risk profile was not adequate to support approval. Additional trials to establish efficacy may be needed. Moving forward, ALKS–5641 represents an important progressive development of opioid therapeutics for the treatment of depression by targeting multiple opioid receptors to offer optimal results in alleviating depression. With that in mind, it is logical to evaluate the full potential of buprenorphine alone and to design other analogs that have a better profile at MORs.
3.3. JNJ–67953964
The selective KOR antagonist JNJ–67953964 (formerly LY2456302 and CERC–501) is under development for depression and substance abuse disorders. JNJ–67953964 was one of a series of aminobenzyloxyarylamide KOR antagonists produced by Eli Lily (Mitch, et al., 2011). Unlike the long–lasting KOR antagonists, JDTic and norBNI, JNJ–67953964 was absorbed rapidly following oral administration and was eliminated within 48 h of administration when administered at the low KOR–selective doses used in human and preclinical studies (Lowe, et al., 2014). JNJ–67953964 is 6.3 and 34–fold more selective for KORs compared to MORs and DORs respectively (Rorick–Kehn, et al., 2014b; Wang, et al., 2017). Although PET imaging measured substantial binding of JNJ–67953964 in the striatum of rats and mice, where a large number of KORs are expressed (Zheng, et al., 2013), no occupancy of MOR or DORs were detected for doses up to 30 mg/kg (Rorick–Kehn, et al., 2014a). A follow up study in rats demonstrated that JNJ–67953964 saturated occupancy for KORs at all doses tested (3–300 mg/kg, PO), and achieved 50% occupancy of MOR and DOR at 84.4 and 214.6 mg/kg PO respectively (Rorick–Kehn, et al., 2014a). The behavioral consequences of receptor engagement at MORs and KORs were assessed on various tasks. JNJ–67953964 (0.3–3 mg/kg PO) administered 1 h prior to the KOR agonist, U–69593 (1 mg/kg SC), blocked KOR–mediated analgesia in the rat formalin test, but had no effect on morphine analgesia, even at doses as high as 17 mg/kg SC. In comparison with the long–lasting KOR antagonist JDTic, JNJ–67953964 failed to block KOR–mediated analgesia one–week post administration, suggesting a shorter duration of activity. KOR–mediated disruption of prepulse inhibition (U–69593, 3 mg/kg SC) was similarly blocked by JNJ–67953964 (0.1 – 1 mg/kg PO), but MOR–mediated (morphine 20 mg/kg IP) disruption of prepulse inhibition was unaffected by JNJ–67953964 treatment. In a later report, blockade of MORs by JNJ–67953964 at various doses was tested using pupil diameter measurements. Morphine–induced mydriasis in rats and fentanyl–induced miosis in healthy humans (25 and 60 mg/kg) were attenuated by JNJ–67953964 (100–300 mg/kg SC), at doses that were 100–fold greater than those required to produce KOR–specific effects (Rorick–Kehn, et al., 2014a). Together, these data advocate for the KOR–selective antagonist activity of JNJ–67953964.
PET imaging in rhesus monkeys revealed the highest binding of JNJ–67953964) in the putamen, followed by the globus pallidus, caudate, cingulate cortex, thalamus, insular, cerebellum and the frontal and temporal cortices (Zheng, et al., 2014). In rodents, dose–dependent increases in receptor occupancy were noted for JNJ–67953964 administered PO, with 90% of KORs in the striatum occupied at the 10 mg/kg dose for a period of up to 8 h which declined to 50% occupancy by 48 h (Rorick–Kehn, et al., 2014b). These findings correspond to data obtained from healthy human controls. PET imaging of KORs conducted 2.5 hours post dosing revealed dose–dependent receptor occupancy, with 35% and 95% of receptors occupied at 0.5 mg/kg and 10 mg/kg, respectively (Naganawa, et al., 2016). JNJ–67953964 still occupied KORs 24 h post administration, when 19% of receptors remained occupied at 0.5 mg/kg and 72% were occupied at 10 mg/kg. Interestingly, the highest level of binding at 0.5 mg/kg JNJ–67953964 was in the hippocampus, whereas binding of KORs after 10 mg/kg JNJ–67953964 was more evenly distributed across the caudate, cingulate cortex, hippocampus and amygdala (Naganawa, et al., 2016). Overall, these data demonstrate KOR selective binding in brain regions implicated in the pathophysiology of depression. A Proof of Concept trial utilizing MRI evaluation of ventral striatal activation and clinical anhedonia, following 8 weeks of treatment with JNJ–67953964 has been completed, NCT02218736. The data available on clinicaltrials.gov confirm that JNJ–67953964 engages with the neural circuitry involved in reward, as patients treated with JNJ–67953964 exhibited greater ventral striatal activation during the Monetary Incentive Delay Task compared with placebo. In addition, clinical anhedonia as measured by the Snaith–Hamilton Pleasure Scale (SHAPS) was reduced following JNJ–67953964 relative to placebo treated subjects. Overall, JNJ–67953964 demonstrated proof of mechanism and engaged RDoC reward–related subdomains, meeting the go criterion for the Fast–Fail Trials Program for continued clinical evaluation of the compound (Krystal, et al., 2018).
Support for the use of JNJ–67953964 to treat negative affect has been largely demonstrated in rodent tests of stress used to study antidepressants and addiction models. The first thorough examination of JNJ–67953964’s behavioral effects were reported by Rorick–Kehn et al., where JNJ–67953964 (10 mg/kg PO) reduced FST immobility scores of NIH–Swiss mice to levels comparable with the antidepressant imipramine (15 mg/kg IP) (Rorick–Kehn, et al., 2014b). Based on the FST assay, optimal doses of JNJ–67953964 differed between strains from 1 –3 mg/kg for C57BL/6J mice (Browne, et al., 2018) to 30 mg/kg for ICR mice (Wang, et al., 2017). In addition, a single dose of JNJ–67953964 (3 mg/kg, IP) reduced latencies to approach and consume a palatable food in a novel environment 24 h post administration (Browne, et al., 2018). The combination of low dose JNJ–67953964 (1 and 3 mg/kg, PO) with low dose citalopram (5 mg/kg, IP) produced a greater magnitude of effect than JNJ–67953964 or imipramine alone in NIH–Swiss mice (Rorick–Kehn, et al., 2014b). However, JNJ–67953964 (1 mg/kg IP for 7 days) failed to reverse social interaction deficits following 10 days of chronic social defeat in C57BL/6J mice at a dose that was effective in tests of antidepressant activity in stress naïve mice (Browne, et al., 2018).
The depression and anxiety emerge during withdrawal or abstinence is increasingly recognized as a contributor to relapse, it is important to consider the treatment of these psychiatric disorders in substance use disorder to prevent relapse and enhance abstinence. Evidence for the beneficial effects of JNJ–67953964 in rodent models of substance use disorders has led to the suggestions that JNJ–67953964 may be useful in remediating the effects of withdrawal from cocaine, alcohol and nicotine (Domi, et al., 2018; Jackson, et al., 2015; Lowe, et al., 2014; Reed, et al., 2018). Just like other KOR antagonists (Deehan, et al., 2012; Doyon, et al., 2006; Walker & Koob, 2008), JNJ–67953964 (3 and 10 mg/kg PO) treatment markedly reduced the number of drinking bouts and volume of ethanol consumed in alcohol preferring rats (Rorick–Kehn, et al., 2014b). Likewise, signs of spontaneous nicotine withdrawal in ICR mice were significantly ameliorated by JNJ–67953964 pretreatment, so too were elevated anxiety on the EPM and hyperalgesia on the hot plate test. JNJ–67953964 pretreatment also attenuated mecamylamine precipitated condition place aversion (Jackson, et al., 2015). These data agree with previous reports examining other KOR antagonists, nor–BNI and JDTic, on nicotine withdrawal effects (Jackson, et al., 2010). These suggestions led to a recent clinical study showing that JNJ–67953964 can modulate cue induced craving in cocaine use disorder (Reed, et al., 2018). In addition to providing evidence for sustained abstinence from substance use, it will be important for the field to discern whether selective KOR antagonists alleviate depressive symptoms in patients with comorbid substance use disorders.
3.4. BTRX–246040
The NOP receptor antagonist BRTX–246040 is under development for depression, eating disorders and alcohol abuse. PET imaging confirmed that the NOP receptor antagonist BTRX–246040 (formerly LY–2940094) readily penetrates the human brain, with peak drug concentrations in plasma observed 2 to 6 h post administration. At an EC50 of 2.94 to 3.46 ng/ml, >80% NOP receptors were occupied across the prefrontal cortex, occipital cortex, putamen, and thalamus, (Raddad, et al., 2016); the pattern of receptor occupancy of BTRX–246040 was comparable between rodents and humans. Preclinical evaluation of BTRX–246040 has shown significant effects on rodent tests for antidepressants. Comparable reductions in FST immobility to that produced by imipramine were measured in both mice (Witkin, et al., 2016) and rats (Post, et al., 2016b) treated with BTRX–246040 (30 mg/kg, PO). Moreover, the antidepressant–like effect of BTRX–246040 in the FST was blocked in NOP–/–mice (Witkin, et al., 2016), supporting the hypothesis that NOP receptor antagonism mediates the antidepressant activity of BTRX–246040. These data are in line with in vitro studies in CHO and human cell lines, where BTRX–246040 exhibited 1000–fold higher selectivity for NOP compared to MOR, DOR and KORs (Statnick, et al., 2016). Just as other NOP antagonists failed to modulate behavior in tests relevant to anxiety, BTRX–246040 did not modulate anxiety–like behavior in rodents. In contrast to the anxiolytic benzodiazepine chlordiazepoxide, BTRX–246040 did not increase the number of punished licks in the rat Vogel conflict assay (Post, et al., 2016b). Similar effects were noted in mice where BTRX–246040 was inactive in the marble burying test (Post, et al., 2016b). Moreover, BTRX–246040 failed to modulate operant behavior of rats maintained under a DRL 72s schedule (Witkin, et al., 2016); conditioned suppression of palatable food intake (Witkin, et al., 2016), or novelty suppressed feeding behavior (Witkin, et al., 2016). Furthermore, no effects on cognition or motoric effects were noted following treatment with BTRX–246040 (Witkin, et al., 2016). However, BTRX–246040 (30 mg/kg, PO) significantly reduced fear conditioned freezing in response to a conditioned stimulus in C57BL/6 mice and blocked stress–induced hyperthermia in rats (Witkin, et al., 2016).
Several studies have shown that BTRX–246040 can modulate endogenous monoaminergic tone leading to suggestions that this compound can enhance the antidepressant–like effects of SSRIs and other antidepressants (Witkin, et al., 2016). Extracellular levels of monoamines in the prefrontal cortex of Sprague Dawley rats were significantly altered following BTRX–246040, where a rapid 50% rise in DA levels occurred during the first 30 min following administration, and then gradually decreased to baseline levels after 3 h. In contrast, 5–HT levels gradually increased by 50% compared to baseline 2 h post administration and remained elevated at the end of the 4–hour period of testing (Post, et al., 2016b). Cotreatment of BTRX–246040 augmented the antidepressant–like effects of SSRIs in the mouse FST. Conversely, the ability of ethanol to increase extracellular dopamine in the nucleus accumbens of male Sprague Dawley rats was blocked by BTRX–246040 (30 mg/kg, PO) (Rorick–Kehn, et al., 2016) leading to suggestions that the NOP antagonist may be useful in treating ethanol use disorder. This idea was supported by BTRX–246040 reducing ethanol self–administration and stress–induced reinstatement in Indiana Alcohol–Preferring (P) and Marchigian Sardinian Alcohol–Preferring (msP) Rats (Rorick–Kehn, et al., 2016). In addition, the potential clinical utility of BTRX–246040 in feeding disorders was shown in a rodent model of binge eating (Statnick, et al., 2016). Hyperphagia was induced using consumption of a highly palatable diet in lean Long Evans rats, or mild calorie restriction diet under a diet induced obese (DIO) model. BTRX–246040 (10 and 30 mg/kg) normalized caloric intake to that of control lean Long Evans rats in both experiments. Similar effects were observed in DIO in C57BL/6J mice following food restriction, where BTRX–246040 treatment (20 and 30 mg/kg) dramatically reduced food intake in a free access food paradigm (Statnick, et al., 2016). Overall, these preclinical studies provided compelling suggestions for the further investigation of BTRX–246040 for a wide range of neuropsychiatric disorders.
Given the preclinical support for BTRX–246040 as an antidepressant, a proof of concept study for BTRX–246040 in depressed human patients reported a reduction in HAMD–17 scores following 8 weeks of oral treatment. Although the magnitude of reduction did not meet the pre–defined proof of concept criterion, these patients exhibited a shift in emotional processing towards more positive stimuli and large reductions in depressed mood, suggesting that BTRX–246040 could have potential therapeutic value for key domains of MDD (Post, et al., 2016b). Preclinical studies also emphasized the potential utility of BTRX–246040 in alcohol use disorder. Concurrent to the study in depressed subjects, another proof of concept study evaluating the efficacy of BTRX–246040 in alcohol dependence was conducted (Post, et al., 2016a). In this study the primary endpoint, mean number of drinks per day (change from baseline), was not altered following 8 weeks of treatment. However, a significant reduction in the number heavy of drinking days and an increased percentage of days abstinent was also reported, and these effects were more pronounced in females (Post, et al., 2016a). Of great interest in the study was the 90% probability of a greater improvement in the Hospital Anxiety and Depression Scale (HADS) score in BRTX–246040 treated individuals compared to placebo. Blackthorn Therapeutics is now recruiting for a Phase 2 trial with BTRX–246040 in major depressed patients following the completion of several clinical trials, NCT01724112, NCT01404091 and NCT01263236. In addition, given the strong preclinical evidence demonstrating the efficacy of BTRX–246040 in animal models of alcohol dependence, an ongoing clinical trial (NCT01798303), is investigating the efficacy of BTRX–246040 for treating alcohol use disorder.
4. Conclusions
Strong preclinical evidence has supported the evaluation of opioid–based compounds in clinical trials for depression. Going forward it is apparent that multimodal compounds, compounds involving positive effects on depression from a combination of opioid receptors, may ultimately yield the best outcomes in those individuals with treatment resistant depression or other comorbid disorders. In developing new therapeutics that show efficacy in clinical trials, it is important to be mindful of the increasing use of the RDoC developed by the NIMH. This framework may help to demonstrate biological effects of opioid compounds that are pertinent for multiple domains of depression or similar deficits in multiple psychiatric disorders. Table 1–4 outline the involvement of all opioid receptors in domains that are relevant to depression. These tables highlight the importance of KORs in mediated behaviors that reflect negative valence, MORs in social processing, and emphasizes the need for more comprehensive investigation of all opioid receptors in constructs of cognitive processes. Although this is an arduous process, it will ultimately improve the translation of therapeutics to humans. Overall, the positive outcomes associated with opioid–based compounds in clinical trials confirm that multimodal opioid–based compounds can have potential to be used to normalize many of the core endophenotypes of depression.
Figure 2.
Expression of MOR, KOR, DOR and NOP in brain nuclei implicated in affective states, including the monoaminergic nuclei, ventral tegmental area (VTA), locus coeruleus (LC), and dorsal raphe nucleus (DRN). These receptors are colocalized in the anterior hypothalamus (Ant. Hypo) where they modulate neuroendocrine secretion, limbic structures and cortical regions required for mood and cognitive function and densely expressed in the nucleus of the periaqueductal grey (PAG) where they are required for central pain processing. Basal nucleus of the stria terminals (BNST), septum (Sep), habenula (Hb), hippocampus (Hipp), amygdala (Amy), thalamus (Thal), nucleus accumbens (NAc), anterior cingulate cortex (ACC), prelimbic cortex (PrL), infralimbic cortex (IL), and anterior insular cortex (AIC). The darker shading indicates higher levels of opioid receptor expression.
Acknowledgements:
This research was supported by US Public Health Service (USPHS) grants R01 MH92412 and R01 MH105623.
Footnotes
Conflict of interest:
No conflict of interest.
References
- Abdallah K, & Gendron L (2018). The Delta Opioid Receptor in Pain Control. Handb Exp Pharmacol, 247, 147–177. [DOI] [PubMed] [Google Scholar]
- Admon R, & Pizzagalli DA (2015). Dysfunctional Reward Processing in Depression. Curr Opin Psychol, 4, 114–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akil H, Gordon J, Hen R, Javitch J, Mayberg H, McEwen B, et al. (2018). Treatment resistant depression: A multi–scale, systems biology approach. Neurosci Biobehav Rev, 84, 272–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akil H, Watson SJ, Young E, Lewis ME, Khachaturian H, & Walker JM (1984). Endogenous opioids: biology and function. Annu Rev Neurosci, 7, 223–255. [DOI] [PubMed] [Google Scholar]
- Al–Hasani R, & Bruchas MR (2011). Molecular mechanisms of opioid receptor–dependent signaling and behavior. Anesthesiology, 115, 1363–1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al–Hasani R, McCall JG, & Bruchas MR (2013). Exposure to chronic mild stress prevents kappa opioid–mediated reinstatement of cocaine and nicotine place preference. Front Pharmacol, 4, 96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almatroudi A, Husbands SM, Bailey CP, & Bailey SJ (2015). Combined administration of buprenorphine and naltrexone produces antidepressant–like effects in mice. J Psychopharmacol, 29, 812–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almatroudi A, Ostovar M, Bailey CP, Husbands SM, & Bailey SJ (2018). Antidepressant–like effects of BU10119, a novel buprenorphine analogue with mixed kappa/mu receptor antagonist properties, in mice. Br J Pharmacol, 175, 2869–2880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvira–Botero MX, & Garzon M (2006). Cellular and subcellular distributions of delta opioid receptor activation sites in the ventral oral pontine tegmentum of the cat. Brain Res, 1123, 101–111. [DOI] [PubMed] [Google Scholar]
- Andero R, Brothers SP, Jovanovic T, Chen YT, Salah–Uddin H, Cameron M, et al. (2013). Amygdala–dependent fear is regulated by Oprl1 in mice and humans with PTSD. Sci Transl Med, 5, 188ra173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson SA, Michaelides M, Zarnegar P, Ren Y, Fagergren P, Thanos PK, et al. (2013). Impaired periamygdaloid–cortex prodynorphin is characteristic of opiate addiction and depression. J Clin Invest, 123, 5334–5341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anstrom KK, & Woodward DJ (2005). Restraint increases dopaminergic burst firing in awake rats. Neuropsychopharmacology, 30, 1832–1840. [DOI] [PubMed] [Google Scholar]
- Armstead WM (2011). Nociceptin/orphanin phenylalanine glutamine (FQ) receptor and cardiovascular disease. Cardiovasc Ther, 29, 23–28. [DOI] [PubMed] [Google Scholar]
- Arvidsson U, Dado RJ, Riedl M, Lee JH, Law PY, Loh HH, et al. (1995). delta–Opioid receptor immunoreactivity: distribution in brainstem and spinal cord, and relationship to biogenic amines and enkephalin. J Neurosci, 15, 1215–1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asth L, Ruzza C, Malfacini D, Medeiros I, Guerrini R, Zaveri NT, et al. (2016). Beta–arrestin 2 rather than G protein efficacy determines the anxiolytic–versus antidepressant–like effects of nociceptin/orphanin FQ receptor ligands. Neuropharmacology, 105, 434–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bach P, Vollsta Dt–Klein S, Kirsch M, Hoffmann S, Jorde A, Frank J, et al. (2015). Increased mesolimbic cue–reactivity in carriers of the mu–opioid–receptor gene OPRM1 A118G polymorphism predicts drinking outcome: a functional imaging study in alcohol dependent subjects. Eur Neuropsychopharmacol, 25, 1128–1135. [DOI] [PubMed] [Google Scholar]
- Bals–Kubik R, Ableitner A, Herz A, & Shippenberg TS (1993). Neuroanatomical sites mediating the motivational effects of opioids as mapped by the conditioned place preference paradigm in rats. J Pharmacol Exp Ther, 264, 489–495. [PubMed] [Google Scholar]
- Baune BT, Malhi GS, Morris G, Outhred T, Hamilton A, Das P, et al. (2018). Cognition in depression: Can we THINC–it better? J Affect Disord, 225, 559–562. [DOI] [PubMed] [Google Scholar]
- Beardsley PM, Howard JL, Shelton KL, & Carroll FI (2005). Differential effects of the novel kappa opioid receptor antagonist, JDTic, on reinstatement of cocaine–seeking induced by footshock stressors vs cocaine primes and its antidepressant–like effects in rats. Psychopharmacology (Berl), 183, 118–126. [DOI] [PubMed] [Google Scholar]
- Beleslin DB, Samardzic R, Krstic SK, & Micic D (1982). Differences in central effects of beta–endorphin and enkephalins: beta–endorphin. A potent psychomotor stimulant. Neuropharmacology, 21, 99–102. [DOI] [PubMed] [Google Scholar]
- Benbouzid M, Choucair–Jaafar N, Yalcin I, Waltisperger E, Muller A, Freund–Mercier MJ, et al. (2008a). Chronic, but not acute, tricyclic antidepressant treatment alleviates neuropathic allodynia after sciatic nerve cuffing in mice. Eur J Pain, 12, 1008–1017. [DOI] [PubMed] [Google Scholar]
- Benbouzid M, Gaveriaux–Ruff C, Yalcin I, Waltisperger E, Tessier LH, Muller A, et al. (2008b). Delta–opioid receptors are critical for tricyclic antidepressant treatment of neuropathic allodynia. Biol Psychiatry, 63, 633–636. [DOI] [PubMed] [Google Scholar]
- Bershad AK, Jaffe JH, Childs E, & de Wit H (2015). Opioid partial agonist buprenorphine dampens responses to psychosocial stress in humans. Psychoneuroendocrinology, 52, 281–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bershad AK, Seiden JA, & de Wit H (2016). Effects of buprenorphine on responses to social stimuli in healthy adults. Psychoneuroendocrinology, 63, 43–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berthele A, Platzer S, Dworzak D, Schadrack J, Mahal B, Buttner A, et al. (2003). [3H]–nociceptin ligand–binding and nociceptin opioid receptor mrna expression in the human brain. Neuroscience, 121, 629–640. [DOI] [PubMed] [Google Scholar]
- Berube P, Laforest S, Bhatnagar S, & Drolet G (2013). Enkephalin and dynorphin mRNA expression are associated with resilience or vulnerability to chronic social defeat stress. Physiol Behav, 122, 237–245. [DOI] [PubMed] [Google Scholar]
- Berube P, Poulin JF, Laforest S, & Drolet G (2014). Enkephalin knockdown in the basolateral amygdala reproduces vulnerable anxiety–like responses to chronic unpredictable stress. Neuropsychopharmacology, 39, 1159–1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaesse P, Goedecke L, Bazelot M, Capogna M, Pape HC, & Jungling K (2015). mu–Opioid Receptor–Mediated Inhibition of Intercalated Neurons and Effect on Synaptic Transmission to the Central Amygdala. J Neurosci, 35, 7317–7325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodkin JA, Zornberg GL, Lukas SE, & Cole JO (1995). Buprenorphine treatment of refractory depression. J Clin Psychopharmacol, 15, 49–57. [DOI] [PubMed] [Google Scholar]
- Bond C, LaForge KS, Tian M, Melia D, Zhang S, Borg L, et al. (1998). Single–nucleotide polymorphism in the human mu opioid receptor gene alters beta–endorphin binding and activity: possible implications for opiate addiction. Proc Natl Acad Sci U S A, 95, 9608–9613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonenberger M, Plener PL, Groschwitz RC, Gron G, & Abler B (2015). Polymorphism in the micro–opioid receptor gene (OPRM1) modulates neural processing of physical pain, social rejection and error processing. Exp Brain Res, 233, 2517–2526. [DOI] [PubMed] [Google Scholar]
- Bowers ME, Choi DC, & Ressler KJ (2012). Neuropeptide regulation of fear and anxiety: Implications of cholecystokinin, endogenous opioids, and neuropeptide Y. Physiol Behav, 107, 699–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowers ME, & Ressler KJ (2015). An Overview of Translationally Informed Treatments for Posttraumatic Stress Disorder: Animal Models of Pavlovian Fear Conditioning to Human Clinical Trials. Biol Psychiatry, 78, E15–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bravo JA, Diaz–Veliz G, Mora S, Ulloa JL, Berthoud VM, Morales P, et al. (2009). Desipramine prevents stress–induced changes in depressive–like behavior and hippocampal markers of neuroprotection. Behav Pharmacol, 20, 273–285. [DOI] [PubMed] [Google Scholar]
- Bridge KE, Wainwright A, Reilly K, & Oliver KR (2003). Autoradiographic localization of (125)i[Tyr(14)] nociceptin/orphanin FQ binding sites in macaque primate CNS. Neuroscience, 118, 513–523. [DOI] [PubMed] [Google Scholar]
- Browne CA, Erickson RL, Blendy JA, & Lucki I (2017). Genetic variation in the behavioral effects of buprenorphine in female mice derived from a murine model of the OPRM1 A118G polymorphism. Neuropharmacology, 117, 401–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Browne CA, Falcon E, Robinson SA, Berton O, & Lucki I (2018). Reversal of Stress–Induced Social Interaction Deficits by Buprenorphine. Int J Neuropsychopharmacol, 21, 164–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Browne CA, van Nest DS, & Lucki I (2015). Antidepressant–like effects of buprenorphine in rats are strain dependent. Behav Brain Res, 278, 385–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruchas MR, Land BB, Aita M, Xu M, Barot SK, Li S, et al. (2007). Stress–induced p38 mitogen–activated protein kinase activation mediates kappa–opioid–dependent dysphoria. J Neurosci, 27, 11614–11623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruchas MR, Land BB, Lemos JC, & Chavkin C (2009). CRF1–R activation of the dynorphin/kappa opioid system in the mouse basolateral amygdala mediates anxiety–like behavior. PLoS One, 4, e8528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruchas MR, Macey TA, Lowe JD, & Chavkin C (2006). Kappa opioid receptor activation of p38 MAPK is GRK3–and arrestin–dependent in neurons and astrocytes. J Biol Chem, 281, 18081–18089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruchas MR, Xu M, & Chavkin C (2008). Repeated swim stress induces kappa opioid–mediated activation of extracellular signal–regulated kinase ½. Neuroreport, 19, 1417–1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryant RA, Creamer M, O’Donnell M, Silove D, & McFarlane AC (2009). A study of the protective function of acute morphine administration on subsequent posttraumatic stress disorder. Biol Psychiatry, 65, 438–440. [DOI] [PubMed] [Google Scholar]
- Butelman ER, & Kreek MJ (2015). Salvinorin A, a kappa–opioid receptor agonist hallucinogen: pharmacology and potential template for novel pharmacotherapeutic agents in neuropsychiatric disorders. Front Pharmacol, 6, 190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahill CM, McClellan KA, Morinville A, Hoffert C, Hubatsch D, O’Donnell D, et al. (2001a). Immunohistochemical distribution of delta opioid receptors in the rat central nervous system: evidence for somatodendritic labeling and antigen–specific cellular compartmentalization. J Comp Neurol, 440, 65–84. [DOI] [PubMed] [Google Scholar]
- Cahill CM, Morinville A, Hoffert C, O’Donnell D, & Beaudet A (2003). Up–regulation and trafficking of delta opioid receptor in a model of chronic inflammation: implications for pain control. Pain, 101, 199–208. [DOI] [PubMed] [Google Scholar]
- Cahill CM, Morinville A, Lee MC, Vincent JP, Collier B, & Beaudet A (2001b). Prolonged morphine treatment targets delta opioid receptors to neuronal plasma membranes and enhances delta–mediated antinociception. J Neurosci, 21, 7598–7607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calabrese JR, Fava M, Garibaldi G, Grunze H, Krystal AD, Laughren T, et al. (2014). Methodological approaches and magnitude of the clinical unmet need associated with amotivation in mood disorders. J Affect Disord, 168, 439–451. [DOI] [PubMed] [Google Scholar]
- Campbell DG, Felker BL, Liu CF, Yano EM, Kirchner JE, Chan D, et al. (2007). Prevalence of depression–PTSD comorbidity: implications for clinical practice guidelines and primary care–based interventions. J Gen Intern Med, 22, 711–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao JL, Covington HE 3rd, Friedman AK, Wilkinson MB, Walsh JJ, Cooper DC, et al. (2010). Mesolimbic dopamine neurons in the brain reward circuit mediate susceptibility to social defeat and antidepressant action. J Neurosci, 30, 16453–16458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlezon WA Jr., Beguin C, DiNieri JA, Baumann MH, Richards MR, Todtenkopf MS, et al. (2006). Depressive–like effects of the kappa–opioid receptor agonist salvinorin A on behavior and neurochemistry in rats. J Pharmacol Exp Ther, 316, 440–447. [DOI] [PubMed] [Google Scholar]
- Carr GV, Bangasser DA, Bethea T, Young M, Valentino RJ, & Lucki I (2010). Antidepressant–like effects of kappa–opioid receptor antagonists in Wistar Kyoto rats. Neuropsychopharmacology, 35, 752–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carr GV, & Lucki I (2010). Comparison of the kappa–opioid receptor antagonist DIPPA in tests of anxiety–like behavior between Wistar Kyoto and Sprague Dawley rats. Psychopharmacology (Berl), 210, 295–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaijale NN, Curtis AL, Wood SK, Zhang XY, Bhatnagar S, Reyes BA, et al. (2013). Social stress engages opioid regulation of locus coeruleus norepinephrine neurons and induces a state of cellular and physical opiate dependence. Neuropsychopharmacology, 38, 1833–1843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charbogne P, Kieffer BL, & Befort K (2014). 15 years of genetic approaches in vivo for addiction research: Opioid receptor and peptide gene knockout in mouse models of drug abuse. Neuropharmacology, 76 Pt B, 204–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chartoff EH, & Connery HS (2014). It’s MORe exciting than mu: crosstalk between mu opioid receptors and glutamatergic transmission in the mesolimbic dopamine system. Front Pharmacol, 5, 116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chartoff EH, & Mavrikaki M (2015). Sex Differences in Kappa Opioid Receptor Function and Their Potential Impact on Addiction. Front Neurosci, 9, 466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavkin C, & Koob GF (2016). Dynorphin, Dysphoria, and Dependence: the Stress of Addiction. Neuropsychopharmacology, 41, 373–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chefer VI, Backman CM, Gigante ED, & Shippenberg TS (2013). Kappa opioid receptors on dopaminergic neurons are necessary for kappa–mediated place aversion. Neuropsychopharmacology, 38, 2623–2631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chelnokova O, Laeng B, Loseth G, Eikemo M, Willoch F, & Leknes S (2016). The micro–opioid system promotes visual attention to faces and eyes. Soc Cogn Affect Neurosci, 11, 1902–1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou WY, Yang LC, Lu HF, Ko JY, Wang CH, Lin SH, et al. (2006). Association of mu–opioid receptor gene polymorphism (A118G) with variations in morphine consumption for analgesia after total knee arthroplasty. Acta Anaesthesiol Scand, 50, 787–792. [DOI] [PubMed] [Google Scholar]
- Ciccocioppo R, de Guglielmo G, Hansson AC, Ubaldi M, Kallupi M, Cruz MT, et al. (2014). Restraint stress alters nociceptin/orphanin FQ and CRF systems in the rat central amygdala: significance for anxiety–like behaviors. J Neurosci, 34, 363–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cicero TJ, Ellis MS, Surratt HL, & Kurtz SP (2014). Factors contributing to the rise of buprenorphine misuse: 2008–2013. Drug Alcohol Depend, 142, 98–104. [DOI] [PubMed] [Google Scholar]
- Cinque C, Pondiki S, Oddi D, Di Certo MG, Marinelli S, Troisi A, et al. (2012). Modeling socially anhedonic syndromes: genetic and pharmacological manipulation of opioid neurotransmission in mice. Transl Psychiatry, 2, e155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cipriani A, Furukawa TA, Salanti G, Chaimani A, Atkinson LZ, Ogawa Y, et al. (2018). Comparative efficacy and acceptability of 21 antidepressant drugs for the acute treatment of adults with major depressive disorder: a systematic review and network meta–analysis. Lancet, 391, 1357–1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole S, & McNally GP (2009). Complementary roles for amygdala and periaqueductal gray in temporal–difference fear learning. Learn Mem, 16, 1–7. [DOI] [PubMed] [Google Scholar]
- Commons KG (2003). Translocation of presynaptic delta opioid receptors in the ventrolateral periaqueductal gray after swim stress. J Comp Neurol, 464, 197–207. [DOI] [PubMed] [Google Scholar]
- Contet C, Gaveriaux–Ruff C, Matifas A, Caradec C, Champy MF, & Kieffer BL (2006). Dissociation of analgesic and hormonal responses to forced swim stress using opioid receptor knockout mice. Neuropsychopharmacology, 31, 1733–1744. [DOI] [PubMed] [Google Scholar]
- Cowan A (2007). Buprenorphine: the basic pharmacology revisited. J Addict Med, 1, 68–72. [DOI] [PubMed] [Google Scholar]
- Cowan A, Doxey JC, & Harry EJ (1977a). The animal pharmacology of buprenorphine, an oripavine analgesic agent. Br J Pharmacol, 60, 547–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowan A, Lewis JW, & Macfarlane IR (1977b). Agonist and antagonist properties of buprenorphine, a new antinociceptive agent. Br J Pharmacol, 60, 537–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cronin A, Keifer JC, Baghdoyan HA, & Lydic R (1995). Opioid inhibition of rapid eye movement sleep by a specific mu receptor agonist. Br J Anaesth, 74, 188–192. [DOI] [PubMed] [Google Scholar]
- Cruz MT, Herman MA, Kallupi M, & Roberto M (2012). Nociceptin/orphanin FQ blockade of corticotropin–releasing factor–induced gamma–aminobutyric acid release in central amygdala is enhanced after chronic ethanol exposure. Biol Psychiatry, 71, 666–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis AL, Bello NT, Connolly KR, & Valentino RJ (2002). Corticotropin–releasing factor neurones of the central nucleus of the amygdala mediate locus coeruleus activation by cardiovascular stress. J Neuroendocrinol, 14, 667–682. [DOI] [PubMed] [Google Scholar]
- Curtis AL, Bello NT, & Valentino RJ (2001). Evidence for functional release of endogenous opioids in the locus ceruleus during stress termination. J Neurosci, 21, RC152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang VC, & Christie MJ (2012). Mechanisms of rapid opioid receptor desensitization, resensitization and tolerance in brain neurons. Br J Pharmacol, 165, 1704–1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis MP (2012). Twelve reasons for considering buprenorphine as a frontline analgesic in the management of pain. J Support Oncol, 10, 209–219. [DOI] [PubMed] [Google Scholar]
- De Vries TJ, Hogenboom F, Mulder AH, & Schoffelmeer AN (1990). Ontogeny of mu–, delta–and kappa–opioid receptors mediating inhibition of neurotransmitter release and adenylate cyclase activity in rat brain. Brain Res Dev Brain Res, 54, 63–69. [DOI] [PubMed] [Google Scholar]
- Deehan GA Jr., McKinzie DL, Carroll FI, McBride WJ, & Rodd ZA (2012). The long–lasting effects of JDTic, a kappa opioid receptor antagonist, on the expression of ethanol–seeking behavior and the relapse drinking of female alcohol–preferring (P) rats. Pharmacol Biochem Behav, 101, 581–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- del Rosario Capriles N, & Cancela LM (2002). Motivational effects mu–and kappa–opioid agonists following acute and chronic restraint stress: involvement of dopamine D(1) and D(2) receptors. Behav Brain Res, 132, 159–169. [DOI] [PubMed] [Google Scholar]
- DePaoli AM, Hurley KM, Yasada K, Reisine T, & Bell G (1994). Distribution of kappa opioid receptor mRNA in adult mouse brain: an in situ hybridization histochemistry study. Mol Cell Neurosci, 5, 327–335. [DOI] [PubMed] [Google Scholar]
- Der–Avakian A, Barnes SA, Markou A, & Pizzagalli DA (2016). Translational Assessment of Reward and Motivational Deficits in Psychiatric Disorders. Curr Top Behav Neurosci, 28, 231–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devine DP, Hoversten MT, Ueda Y, & Akil H (2003). Nociceptin/orphanin FQ content is decreased in forebrain neurones during acute stress. J Neuroendocrinol, 15, 69–74. [DOI] [PubMed] [Google Scholar]
- Devine DP, Leone P, & Wise RA (1993). Mesolimbic dopamine neurotransmission is increased by administration of mu–opioid receptor antagonists. Eur J Pharmacol, 243, 55–64. [DOI] [PubMed] [Google Scholar]
- Devine DP, Watson SJ, & Akil H (2001). Nociceptin/orphanin FQ regulates neuroendocrine function of the limbic–hypothalamic–pituitary–adrenal axis. Neuroscience, 102, 541–553. [DOI] [PubMed] [Google Scholar]
- Di Chiara G, & Imperato A (1988). Opposite effects of mu and kappa opiate agonists on dopamine release in the nucleus accumbens and in the dorsal caudate of freely moving rats. J Pharmacol Exp Ther, 244, 1067–1080. [PubMed] [Google Scholar]
- Di Giannuario A, & Pieretti S (2000). Nociceptin differentially affects morphine–induced dopamine release from the nucleus accumbens and nucleus caudate in rats. Peptides, 21, 1125–1130. [DOI] [PubMed] [Google Scholar]
- Di Giannuario A, Pieretti S, Catalani A, & Loizzo A (1999). Orphanin FQ reduces morphine–induced dopamine release in the nucleus accumbens: a microdialysis study in rats. Neurosci Lett, 272, 183–186. [DOI] [PubMed] [Google Scholar]
- DiFeliceantonio AG, & Berridge KC (2016). Dorsolateral neostriatum contribution to incentive salience: opioid or dopamine stimulation makes one reward cue more motivationally attractive than another. Eur J Neurosci, 43, 1203–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dilgen J, Tejeda HA, & O’Donnell P (2013). Amygdala inputs drive feedforward inhibition in the medial prefrontal cortex. J Neurophysiol, 110, 221–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domi E, Barbier E, Augier E, Augier G, Gehlert D, Barchiesi R, et al. (2018). Preclinical evaluation of the kappa–opioid receptor antagonist CERC–501 as a candidate therapeutic for alcohol use disorders. Neuropsychopharmacology, 43, 1805–1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donahue RJ, Landino SM, Golden SA, Carroll FI, Russo SJ, & Carlezon WA Jr. (2015). Effects of acute and chronic social defeat stress are differentially mediated by the dynorphin/kappa–opioid receptor system. Behav Pharmacol, 26, 654–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyon WM, Howard EC, Shippenberg TS, & Gonzales RA (2006). Kappa–opioid receptor modulation of accumbal dopamine concentration during operant ethanol self–administration. Neuropharmacology, 51, 487–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dripps IJ, & Jutkiewicz EM (2018). Delta Opioid Receptors and Modulation of Mood and Emotion. Handb Exp Pharmacol, 247, 179–197. [DOI] [PubMed] [Google Scholar]
- Du C, Duan Y, Wei W, Cai Y, Chai H, Lv J, et al. (2016). Kappa opioid receptor activation alleviates experimental autoimmune encephalomyelitis and promotes oligodendrocyte–mediated remyelination. Nat Commun, 7, 11120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dulawa SC, & Hen R (2005). Recent advances in animal models of chronic antidepressant effects: the novelty–induced hypophagia test. Neurosci Biobehav Rev, 29, 771–783. [DOI] [PubMed] [Google Scholar]
- Duman RS, & Aghajanian GK (2012). Synaptic dysfunction in depression: potential therapeutic targets. Science, 338, 68–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duzzioni M, Duarte FS, Leme LR, Gavioli EC, & De Lima TC (2011). Anxiolytic–like effect of central administration of NOP receptor antagonist UFP–101 in rats submitted to the elevated T–maze. Behav Brain Res, 222, 206–211. [DOI] [PubMed] [Google Scholar]
- Ehrich E, Turncliff R, Du Y, Leigh–Pemberton R, Fernandez E, Jones R, et al. (2015a). Evaluation of opioid modulation in major depressive disorder. Neuropsychopharmacology, 40, 1448–1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrich JM, Messinger DI, Knakal CR, Kuhar JR, Schattauer SS, Bruchas MR, et al. (2015b). Kappa Opioid Receptor–Induced Aversion Requires p38 MAPK Activation in VTA Dopamine Neurons. J Neurosci, 35, 12917–12931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eippert F, Bingel U, Schoell E, Yacubian J, & Buchel C (2008). Blockade of endogenous opioid neurotransmission enhances acquisition of conditioned fear in humans. J Neurosci, 28, 5465–5472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emrich HM, Vogt P, & Herz A (1982). Possible antidepressive effects of opioids: action of buprenorphine. Ann N Y Acad Sci, 398, 108–112. [DOI] [PubMed] [Google Scholar]
- Erbs E, Faget L, Scherrer G, Kessler P, Hentsch D, Vonesch JL, et al. (2012). Distribution of delta opioid receptor–expressing neurons in the mouse hippocampus. Neuroscience, 221, 203–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans CJ, Keith DE Jr., Morrison H, Magendzo K, & Edwards RH (1992). Cloning of a delta opioid receptor by functional expression. Science, 258, 1952–1955. [DOI] [PubMed] [Google Scholar]
- Fadda P, Tortorella A, & Fratta W (1991). Sleep deprivation decreases mu and delta opioid receptor binding in the rat limbic system. Neurosci Lett, 129, 315–317. [DOI] [PubMed] [Google Scholar]
- Falcon E, Browne CA, Leon RM, Fleites VC, Sweeney R, Kirby LG, et al. (2016). Antidepressant–like Effects of Buprenorphine are Mediated by Kappa Opioid Receptors. Neuropsychopharmacology, 41, 2344–2351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falcon E, Maier K, Robinson SA, Hill–Smith TE, & Lucki I (2015). Effects of buprenorphine on behavioral tests for antidepressant and anxiolytic drugs in mice. Psychopharmacology (Berl), 232, 907–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fanselow MS, Kim JJ, Young SL, Calcagnetti DJ, DeCola JP, Helmstetter FJ, et al. (1991). Differential effects of selective opioid peptide antagonists on the acquisition of pavlovian fear conditioning. Peptides, 12, 1033–1037. [DOI] [PubMed] [Google Scholar]
- Fava M, Memisoglu A, Thase ME, Bodkin JA, Trivedi MH, de Somer M, et al. (2016). Opioid Modulation With Buprenorphine/Samidorphan as Adjunctive Treatment for Inadequate Response to Antidepressants: A Randomized Double–Blind Placebo–Controlled Trial. Am J Psychiatry, 173, 499–508. [DOI] [PubMed] [Google Scholar]
- Fava M, Rush AJ, Alpert JE, Balasubramani GK, Wisniewski SR, Carmin CN, et al. (2008). Difference in treatment outcome in outpatients with anxious versus nonanxious depression: a STAR*D report. Am J Psychiatry, 165, 342–351. [DOI] [PubMed] [Google Scholar]
- Fernandez F, Misilmeri MA, Felger JC, & Devine DP (2004). Nociceptin/orphanin FQ increases anxiety–related behavior and circulating levels of corticosterone during neophobic tests of anxiety. Neuropsychopharmacology, 29, 59–71. [DOI] [PubMed] [Google Scholar]
- Filaferro M, Ruggieri V, Novi C, Calo G, Cifani C, Micioni Di Bonaventura MV, et al. (2014). Functional antagonism between nociceptin/orphanin FQ and corticotropin–releasing factor in rat anxiety–related behaviors: involvement of the serotonergic system. Neuropeptides, 48, 189–197. [DOI] [PubMed] [Google Scholar]
- Filliol D, Ghozland S, Chluba J, Martin M, Matthes HW, Simonin F, et al. (2000). Mice deficient for delta–and mu–opioid receptors exhibit opposing alterations of emotional responses. Nat Genet, 25, 195–200. [DOI] [PubMed] [Google Scholar]
- Flaisher–Grinberg S, Persaud SD, Loh HH, & Wei LN (2012). Stress–induced epigenetic regulation of kappa–opioid receptor gene involves transcription factor c–Myc. Proc Natl Acad Sci U S A, 109, 9167–9172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Florin S, Leroux–Nicollet I, Meunier JC, & Costentin J (1997). Autoradiographic localization of [3H]nociceptin binding sites from telencephalic to mesencephalic regions of the mouse brain. Neurosci Lett, 230, 33–36. [DOI] [PubMed] [Google Scholar]
- Florin S, Meunier J, & Costentin J (2000). Autoradiographic localization of [3H]nociceptin binding sites in the rat brain. Brain Res, 880, 11–16. [DOI] [PubMed] [Google Scholar]
- Fuentealba JA, Gysling K, Magendzo K, & Andres ME (2006). Repeated administration of the selective kappa–opioid receptor agonist U–69593 increases stimulated dopamine extracellular levels in the rat nucleus accumbens. J Neurosci Res, 84, 450–459. [DOI] [PubMed] [Google Scholar]
- Galeotti N, & Ghelardini C (2012). Regionally selective activation and differential regulation of ERK, JNK and p38 MAP kinase signalling pathway by protein kinase C in mood modulation. Int J Neuropsychopharmacol, 15, 781–793. [DOI] [PubMed] [Google Scholar]
- Gaveriaux–Ruff C, Simonin F, Filliol D, & Kieffer BL (2003). Enhanced humoral response in kappa–opioid receptor knockout mice. J Neuroimmunol, 134, 72–81. [DOI] [PubMed] [Google Scholar]
- Gavioli EC, & Calo G (2013). Nociceptin/orphanin FQ receptor antagonists as innovative antidepressant drugs. Pharmacol Ther, 140, 10–25. [DOI] [PubMed] [Google Scholar]
- Gavioli EC, Duarte FS, Guerrini R, Calo G, Rae GA, & TC MDL (2008). GABA(A) signalling is involved in N/OFQ anxiolytic–like effects but not in nocistatin anxiogenic–like action as evaluated in the mouse elevated plus maze. Peptides, 29, 1404–1412. [DOI] [PubMed] [Google Scholar]
- Gavioli EC, Marzola G, Guerrini R, Bertorelli R, Zucchini S, De Lima TC, et al. (2003). Blockade of nociceptin/orphanin FQ–NOP receptor signalling produces antidepressant–like effects: pharmacological and genetic evidences from the mouse forced swimming test. Eur J Neurosci, 17, 1987–1990. [DOI] [PubMed] [Google Scholar]
- Gavioli EC, Rizzi A, Marzola G, Zucchini S, Regoli D, & Calo G (2007). Altered anxiety–related behavior in nociceptin/orphanin FQ receptor gene knockout mice. Peptides, 28, 1229–1239. [DOI] [PubMed] [Google Scholar]
- Gavioli EC, & Romao PR (2011). NOP Receptor Ligands as Potential Agents for Inflammatory and Autoimmune Diseases. J Amino Acids, 2011, 836569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavioli EC, Vaughan CW, Marzola G, Guerrini R, Mitchell VA, Zucchini S, et al. (2004). Antidepressant–like effects of the nociceptin/orphanin FQ receptor antagonist UFP–101: new evidence from rats and mice. Naunyn Schmiedebergs Arch Pharmacol, 369, 547–553. [DOI] [PubMed] [Google Scholar]
- Gerra G, Fantoma A, & Zaimovic A (2006a). Naltrexone and buprenorphine combination in the treatment of opioid dependence. J Psychopharmacol, 20, 806–814. [DOI] [PubMed] [Google Scholar]
- Gerra G, Leonardi C, D’Amore A, Strepparola G, Fagetti R, Assi C, et al. (2006b). Buprenorphine treatment outcome in dually diagnosed heroin dependent patients: A retrospective study. Prog Neuropsychopharmacol Biol Psychiatry, 30, 265–272. [DOI] [PubMed] [Google Scholar]
- Goeldner C, Reiss D, Kieffer BL, & Ouagazzal AM (2010). Endogenous nociceptin/orphanin–FQ in the dorsal hippocampus facilitates despair–related behavior. Hippocampus, 20, 911–916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes I, Jordan BA, Gupta A, Trapaidze N, Nagy V, & Devi LA (2000). Heterodimerization of mu and delta opioid receptors: A role in opiate synergy. J Neurosci, 20, RC110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gompf HS, Moldavan MG, Irwin RP, & Allen CN (2005). Nociceptin/orphanin FQ (N/OFQ) inhibits excitatory and inhibitory synaptic signaling in the suprachiasmatic nucleus (SCN). Neuroscience, 132, 955–965. [DOI] [PubMed] [Google Scholar]
- Good AJ, & Westbrook RF (1995). Effects of a microinjection of morphine into the amygdala on the acquisition and expression of conditioned fear and hypoalgesia in rats. Behav Neurosci, 109, 631–641. [DOI] [PubMed] [Google Scholar]
- Goodwin RD, & Stein DJ (2013). Anxiety disorders and drug dependence: evidence on sequence and specificity among adults. Psychiatry Clin Neurosci, 67, 167–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graziane NM, Polter AM, Briand LA, Pierce RC, & Kauer JA (2013). Kappa opioid receptors regulate stress–induced cocaine seeking and synaptic plasticity. Neuron, 77, 942–954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greco MA, Fuller PM, Jhou TC, Martin–Schild S, Zadina JE, Hu Z, et al. (2008). Opioidergic projections to sleep–active neurons in the ventrolateral preoptic nucleus. Brain Res, 1245, 96–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimwood S, Lu Y, Schmidt AW, Vanase–Frawley MA, Sawant–Basak A, Miller E, et al. (2011). Pharmacological characterization of 2–methyl–N–((2’–(pyrrolidin–1–ylsulfonyl)biphenyl–4–yl)methyl)propan–1–amine (PF–04455242), a high–affinity antagonist selective for kappa–opioid receptors. J Pharmacol Exp Ther, 339, 555–566. [DOI] [PubMed] [Google Scholar]
- Grinnell SG, Ansonoff M, Marrone GF, Lu Z, Narayan A, Xu J, et al. (2016). Mediation of buprenorphine analgesia by a combination of traditional and truncated mu opioid receptor splice variants. Synapse, 70, 395–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guajardo HM, Snyder K, Ho A, & Valentino RJ (2017). Sex Differences in mu–Opioid Receptor Regulation of the Rat Locus Coeruleus and Their Cognitive Consequences. Neuropsychopharmacology, 42, 1295–1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerrini R, Calo G, Rizzi A, Bigoni R, Bianchi C, Salvadori S, et al. (1998). A new selective antagonist of the nociceptin receptor. Br J Pharmacol, 123, 163–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gur TL, Conti AC, Holden J, Bechtholt AJ, Hill TE, Lucki I, et al. (2007). cAMP response element–binding protein deficiency allows for increased neurogenesis and a rapid onset of antidepressant response. J Neurosci, 27, 7860–7868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haaker J, Yi J, Petrovic P, & Olsson A (2017). Endogenous opioids regulate social threat learning in humans. Nat Commun, 8, 15495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halasy K, Racz B, & Maderspach K (2000). Kappa opioid receptors are expressed by interneurons in the CA1 area of the rat hippocampus: a correlated light and electron microscopic immunocytochemical study. J Chem Neuroanat, 19, 233–241. [DOI] [PubMed] [Google Scholar]
- Halladay LR, & Blair HT (2012). The role of mu–opioid receptor signaling in the dorsolateral periaqueductal gray on conditional and unconditional responding to threatening and aversive stimuli. Neuroscience, 216, 82–93. [DOI] [PubMed] [Google Scholar]
- Hansen HH, Rantamaki TP, Larsen MH, Woldbye DP, Mikkelsen JD, & Castren EH (2007). Rapid activation of the extracellular signal–regulated kinase ½ (ERK½) signaling pathway by electroconvulsive shock in the rat prefrontal cortex is not associated with TrkB neurotrophin receptor activation. Cell Mol Neurobiol, 27, 585–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasbi A, Nguyen T, Fan T, Cheng R, Rashid A, Alijaniaram M, et al. (2007). Trafficking of preassembled opioid mu–delta heterooligomer–Gz signaling complexes to the plasma membrane: coregulation by agonists. Biochemistry, 46, 12997–13009. [DOI] [PubMed] [Google Scholar]
- Hawes BE, Graziano MP, & Lambert DG (2000). Cellular actions of nociceptin: transduction mechanisms. Peptides, 21, 961–967. [DOI] [PubMed] [Google Scholar]
- Hebb AL, Drolet G, Mendella PD, Roach SP, Gauthier MS, & Zacharko RM (2005). Intracerebroventricular D–Pen2, D–Pen5–enkephalin administration soon after stressor imposition influences behavioral responsivity to a subsequent stressor encounter in CD–1 mice. Pharmacol Biochem Behav, 82, 453–469. [DOI] [PubMed] [Google Scholar]
- Helmstetter FJ, & Fanselow MS (1987). Effects of naltrexone on learning and performance of conditional fear–induced freezing and opioid analgesia. Physiol Behav, 39, 501–505. [DOI] [PubMed] [Google Scholar]
- Henry MS, Gendron L, Tremblay ME, & Drolet G (2017). Enkephalins: Endogenous Analgesics with an Emerging Role in Stress Resilience. Neural Plast, 2017, 1546125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henter ID, de Sousa RT, & Zarate CA Jr. (2018). Glutamatergic Modulators in Depression. Harv Rev Psychiatry, 26, 307–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiller JM, Fan LQ, & Simon EJ (1992). Age–related changes in kappa opioid receptors in the guinea–pig brain: a quantitative autoradiographic study. Neuroscience, 50, 663–673. [DOI] [PubMed] [Google Scholar]
- Hjelmstad GO, & Fields HL (2003). Kappa opioid receptor activation in the nucleus accumbens inhibits glutamate and GABA release through different mechanisms. J Neurophysiol, 89, 2389–2395. [DOI] [PubMed] [Google Scholar]
- Holanda VA, Medeiros IU, Asth L, Guerrini R, Calo G, & Gavioli EC (2016). Antidepressant activity of nociceptin/orphanin FQ receptor antagonists in the mouse learned helplessness. Psychopharmacology (Berl), 233, 2525–2532. [DOI] [PubMed] [Google Scholar]
- Holbrook TL, Galarneau MR, Dye JL, Quinn K, & Dougherty AL (2010). Morphine use after combat injury in Iraq and post–traumatic stress disorder. N Engl J Med, 362, 110–117. [DOI] [PubMed] [Google Scholar]
- Howe WM, & Kenny PJ (2018). Burst firing sets the stage for depression. Nature, 554, 304–305. [DOI] [PubMed] [Google Scholar]
- Hsu DT, Sanford BJ, Meyers KK, Love TM, Hazlett KE, Walker SJ, et al. (2015). It still hurts: altered endogenous opioid activity in the brain during social rejection and acceptance in major depressive disorder. Mol Psychiatry, 20, 193–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu DT, Sanford BJ, Meyers KK, Love TM, Hazlett KE, Wang H, et al. (2013). Response of the mu–opioid system to social rejection and acceptance. Mol Psychiatry, 18, 1211–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang P, Tunis J, Parry C, Tallarida R, & Liu–Chen LY (2016). Synergistic antidepressant–like effects between a kappa opioid antagonist (LY2444296) and a delta opioid agonist (ADL5859) in the mouse forced swim test. Eur J Pharmacol, 781, 53–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudzik TJ, Maciag C, Smith MA, Caccese R, Pietras MR, Bui KH, et al. (2011). Preclinical pharmacology of AZD2327: a highly selective agonist of the delta–opioid receptor. J Pharmacol Exp Ther, 338, 195–204. [DOI] [PubMed] [Google Scholar]
- Hughes J, Kosterlitz HW, & Smith TW (1977). The distribution of methionine–enkephalin and leucine–enkephalin in the brain and peripheral tissues. Br J Pharmacol, 61, 639–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurd YL (2002). Subjects with major depression or bipolar disorder show reduction of prodynorphin mRNA expression in discrete nuclei of the amygdaloid complex. Mol Psychiatry, 7, 75–81. [DOI] [PubMed] [Google Scholar]
- Hurd YL, Herman MM, Hyde TM, Bigelow LB, Weinberger DR, & Kleinman JE (1997). Prodynorphin mRNA expression is increased in the patch vs matrix compartment of the caudate nucleus in suicide subjects. Mol Psychiatry, 2, 495–500. [DOI] [PubMed] [Google Scholar]
- Ide S, Minami M, Satoh M, Uhl GR, Sora I, & Ikeda K (2004). Buprenorphine antinociception is abolished, but naloxone–sensitive reward is retained, in mu–opioid receptor knockout mice. Neuropsychopharmacology, 29, 1656–1663. [DOI] [PubMed] [Google Scholar]
- Ide S, Sora I, Ikeda K, Minami M, Uhl GR, & Ishihara K (2010). Reduced emotional and corticosterone responses to stress in mu–opioid receptor knockout mice. Neuropharmacology, 58, 241–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Insel TR (2014). The NIMH Research Domain Criteria (RDoC) Project: precision medicine for psychiatry. Am J Psychiatry, 171, 395–397. [DOI] [PubMed] [Google Scholar]
- Jackson KJ, Carroll FI, Negus SS, & Damaj MI (2010). Effect of the selective kappa–opioid receptor antagonist JDTic on nicotine antinociception, reward, and withdrawal in the mouse. Psychopharmacology (Berl), 210, 285–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson KJ, Jackson A, Carroll FI, & Damaj MI (2015). Effects of orally–bioavailable short–acting kappa opioid receptor–selective antagonist LY2456302 on nicotine withdrawal in mice. Neuropharmacology, 97, 270–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson ML, Wulf HA, Browne CA, & Lucki I (2018). Opioid modulation of cognitive impairment in depression. Prog Brain Res, 239, 1–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jamot L, Matthes HW, Simonin F, Kieffer BL, & Roder JC (2003). Differential involvement of the mu and kappa opioid receptors in spatial learning. Genes Brain Behav, 2, 80–92. [DOI] [PubMed] [Google Scholar]
- Jamshidi RJ, Sullivan LC, Jacobs BA, Chavera TA, Berg KA, & Clarke WP (2016). Long–Term Reduction of Kappa Opioid Receptor Function by the Biased Ligand, Norbinaltorphimine, Requires c–Jun N–Terminal Kinase Activity and New Protein Synthesis in Peripheral Sensory Neurons. J Pharmacol Exp Ther, 359, 319–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Q, Takemori AE, Sultana M, Portoghese PS, Bowen WD, Mosberg HI, et al. (1991). Differential antagonism of opioid delta antinociception by [D–Ala2,Leu5,Cys6]enkephalin and naltrindole 5’–isothiocyanate: evidence for delta receptor subtypes. J Pharmacol Exp Ther, 257, 1069–1075. [PubMed] [Google Scholar]
- Johnston CE, Herschel DJ, Lasek AW, Hammer RP Jr., & Nikulina EM (2015). Knockdown of ventral tegmental area mu–opioid receptors in rats prevents effects of social defeat stress: implications for amphetamine cross–sensitization, social avoidance, weight regulation and expression of brain–derived neurotrophic factor. Neuropharmacology, 89, 325–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan BA, Cvejic S, & Devi LA (2000). Kappa opioid receptor endocytosis by dynorphin peptides. DNA Cell Biol, 19, 19–27. [DOI] [PubMed] [Google Scholar]
- Jutkiewicz EM, Eller EB, Folk JE, Rice KC, Traynor JR, & Woods JH (2004). Delta–opioid agonists: differential efficacy and potency of SNC80, its 3–OH (SNC86) and 3–desoxy (SNC162) derivatives in Sprague–Dawley rats. J Pharmacol Exp Ther, 309, 173–181. [DOI] [PubMed] [Google Scholar]
- Jutkiewicz EM, Kaminsky ST, Rice KC, Traynor JR, & Woods JH (2005a). Differential behavioral tolerance to the delta–opioid agonist SNC80 ([(+)–4–[(alphaR)–alpha–[(2S,5R)–2,5–dimethyl–4–(2–propenyl)–1–piperazinyl]–(3–me thoxyphenyl)methyl]–N,N–diethylbenzamide) in Sprague–Dawley rats. J Pharmacol Exp Ther, 315, 414–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jutkiewicz EM, Rice KC, Traynor JR, & Woods JH (2005b). Separation of the convulsions and antidepressant–like effects produced by the delta–opioid agonist SNC80 in rats. Psychopharmacology (Berl), 182, 588–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jutkiewicz EM, Rice KC, Woods JH, & Winsauer PJ (2003). Effects of the delta–opioid receptor agonist SNC80 on learning relative to its antidepressant–like effects in rats. Behav Pharmacol, 14, 509–516. [DOI] [PubMed] [Google Scholar]
- Jutkiewicz EM, Torregrossa MM, Sobczyk–Kojiro K, Mosberg HI, Folk JE, Rice KC, et al. (2006). Behavioral and neurobiological effects of the enkephalinase inhibitor RB101 relative to its antidepressant effects. Eur J Pharmacol, 531, 151–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kallupi M, Scuppa G, de Guglielmo G, Calo G, Weiss F, Statnick MA, et al. (2017). Genetic Deletion of the Nociceptin/Orphanin FQ Receptor in the Rat Confers Resilience to the Development of Drug Addiction. Neuropsychopharmacology, 42, 695–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang–Park M, Kieffer BL, Roberts AJ, Siggins GR, & Moore SD (2015). Interaction of CRF and kappa opioid systems on GABAergic neurotransmission in the mouse central amygdala. J Pharmacol Exp Ther, 355, 206–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karp JF, Butters MA, Begley AE, Miller MD, Lenze EJ, Blumberger DM, et al. (2014). Safety, tolerability, and clinical effect of low–dose buprenorphine for treatment–resistant depression in midlife and older adults. J Clin Psychiatry, 75, e785–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kautzky A, Dold M, Bartova L, Spies M, Kranz GS, Souery D, et al. (2019). Clinical factors predicting treatment resistant depression: affirmative results from the European multicenter study. Acta Psychiatr Scand, 139, 78–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy SE, Koeppe RA, Young EA, & Zubieta JK (2006). Dysregulation of endogenous opioid emotion regulation circuitry in major depression in women. Arch Gen Psychiatry, 63, 1199–1208. [DOI] [PubMed] [Google Scholar]
- Kieffer BL, Befort K, Gaveriaux–Ruff C, & Hirth CG (1992). The delta–opioid receptor: isolation of a cDNA by expression cloning and pharmacological characterization. Proc Natl Acad Sci U S A, 89, 12048–12052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura Y, Fujita M, Hong J, Lohith TG, Gladding RL, Zoghbi SS, et al. (2011). Brain and whole–body imaging in rhesus monkeys of 11C–NOP–1A, a promising PET radioligand for nociceptin/orphanin FQ peptide receptors. J Nucl Med, 52, 1638–1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knapp RJ, Malatynska E, Fang L, Li X, Babin E, Nguyen M, et al. (1994). Identification of a human delta opioid receptor: cloning and expression. Life Sci, 54, PL463–469. [DOI] [PubMed] [Google Scholar]
- Knoll AT, Meloni EG, Thomas JB, Carroll FI, & Carlezon WA Jr. (2007). Anxiolytic–like effects of kappa–opioid receptor antagonists in models of unlearned and learned fear in rats. J Pharmacol Exp Ther, 323, 838–845. [DOI] [PubMed] [Google Scholar]
- Knoll AT, Muschamp JW, Sillivan SE, Ferguson D, Dietz DM, Meloni EG, et al. (2011). Kappa opioid receptor signaling in the basolateral amygdala regulates conditioned fear and anxiety in rats. Biol Psychiatry, 70, 425–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komatsu H, Ohara A, Sasaki K, Abe H, Hattori H, Hall FS, et al. (2011). Decreased response to social defeat stress in mu–opioid–receptor knockout mice. Pharmacol Biochem Behav, 99, 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosten TR, Morgan C, & Kosten TA (1990). Depressive symptoms during buprenorphine treatment of opioid abusers. J Subst Abuse Treat, 7, 51–54. [DOI] [PubMed] [Google Scholar]
- Kreek MJ, & Koob GF (1998). Drug dependence: stress and dysregulation of brain reward pathways. Drug Alcohol Depend, 51, 23–47. [DOI] [PubMed] [Google Scholar]
- Kreibich AS, Briand L, Cleck JN, Ecke L, Rice KC, & Blendy JA (2009). Stress–induced potentiation of cocaine reward: a role for CRF R1 and CREB. Neuropsychopharmacology, 34, 2609–2617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnan V, Han MH, Mazei–Robison M, Iniguez SD, Ables JL, Vialou V, et al. (2008). AKT signaling within the ventral tegmental area regulates cellular and behavioral responses to stressful stimuli. Biol Psychiatry, 64, 691–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krystal AD, Pizzagalli DA, Mathew SJ, Sanacora G, Keefe R, Song A, et al. (2018). The first implementation of the NIMH FAST–FAIL approach to psychiatric drug development. Nat Rev Drug Discov, 18, 82–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuipers SD, Trentani A, Den Boer JA, & Ter Horst GJ (2003). Molecular correlates of impaired prefrontal plasticity in response to chronic stress. J Neurochem, 85, 1312–1323. [DOI] [PubMed] [Google Scholar]
- Kupferberg A, Bicks L, & Hasler G (2016). Social functioning in major depressive disorder. Neurosci Biobehav Rev, 69, 313–332. [DOI] [PubMed] [Google Scholar]
- Lachowicz JE, Shen Y, Monsma FJ Jr., & Sibley DR (1995). Molecular cloning of a novel G protein–coupled receptor related to the opiate receptor family. J Neurochem, 64, 34–40. [DOI] [PubMed] [Google Scholar]
- Lai HM, Cleary M, Sitharthan T, & Hunt GE (2015). Prevalence of comorbid substance use, anxiety and mood disorders in epidemiological surveys, 1990–2014: A systematic review and meta–analysis. Drug Alcohol Depend, 154, 1–13. [DOI] [PubMed] [Google Scholar]
- Lalanne L, Ayranci G, Kieffer BL, & Lutz PE (2014). The kappa opioid receptor: from addiction to depression, and back. Front Psychiatry, 5, 170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laman–Maharg A, Williams AV, Zufelt MD, Minie VA, Ramos–Maciel S, Hao R, et al. (2018). Sex Differences in the Effects of a Kappa Opioid Receptor Antagonist in the Forced Swim Test. Front Pharmacol, 9, 93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert DG (2008). The nociceptin/orphanin FQ receptor: a target with broad therapeutic potential. Nat Rev Drug Discov, 7, 694–710. [DOI] [PubMed] [Google Scholar]
- Lammel S, Lim BK, & Malenka RC (2014). Reward and aversion in a heterogeneous midbrain dopamine system. Neuropharmacology, 76 Pt B, 351–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Land BB, Bruchas MR, Lemos JC, Xu M, Melief EJ, & Chavkin C (2008). The dysphoric component of stress is encoded by activation of the dynorphin kappa–opioid system. J Neurosci, 28, 407–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Land BB, Bruchas MR, Schattauer S, Giardino WJ, Aita M, Messinger D, et al. (2009). Activation of the kappa opioid receptor in the dorsal raphe nucleus mediates the aversive effects of stress and reinstates drug seeking. Proc Natl Acad Sci U S A, 106, 19168–19173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latagliata EC, Valzania A, Pascucci T, Campus P, Cabib S, & Puglisi–Allegra S (2014). Stress–induced activation of ventral tegmental mu–opioid receptors reduces accumbens dopamine tone by enhancing dopamine transmission in the medial pre–frontal cortex. Psychopharmacology (Berl), 231, 4099–4108. [DOI] [PubMed] [Google Scholar]
- Lee JK, Chung J, Druey KM, & Tansey MG (2012). RGS10 exerts a neuroprotective role through the PKA/c–AMP response–element (CREB) pathway in dopaminergic neuron–like cells. J Neurochem, 122, 333–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leggett JD, Dawe KL, Jessop DS, & Fulford AJ (2009). Endogenous nociceptin / orphanin FQ system involvement in hypothalamic–pituitary–adrenal axis responses: relevance to models of inflammation. J Neuroendocrinol, 21, 888–897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leggett JD, Harbuz MS, Jessop DS, & Fulford AJ (2006). The nociceptin receptor antagonist [Nphe1,Arg14,Lys15]nociceptin/orphanin FQ–NH2 blocks the stimulatory effects of nociceptin/orphanin FQ on the HPA axis in rats. Neuroscience, 141, 2051–2057. [DOI] [PubMed] [Google Scholar]
- Leggett JD, Jessop DS, & Fulford AJ (2007). The nociceptin/orphanin FQ antagonist UFP–101 differentially modulates the glucocorticoid response to restraint stress in rats during the peak and nadir phases of the hypothalamo–pituitary–adrenal axis circadian rhythm. Neuroscience, 147, 757–764. [DOI] [PubMed] [Google Scholar]
- Lemos JC, Roth CA, Messinger DI, Gill HK, Phillips PE, & Chavkin C (2012). Repeated stress dysregulates kappa–opioid receptor signaling in the dorsal raphe through a p38alpha MAPK–dependent mechanism. J Neurosci, 32, 12325–12336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemos JC, Zhang G, Walsh T, Kirby LG, Akanwa A, Brooks–Kayal A, et al. (2011). Stress–hyperresponsive WKY rats demonstrate depressed dorsal raphe neuronal excitability and dysregulated CRF–mediated responses. Neuropsychopharmacology, 36, 721–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leskiewicz M, Jantas D, Regulska M, Kaczanowska J, Basta–Kaim A, Budziszewska B, et al. (2013). Antidepressants attenuate the dexamethasone–induced decrease in viability and proliferation of human neuroblastoma SH–SY5Y cells: a involvement of extracellular regulated kinase (ERK½). Neurochem Int, 63, 354–362. [DOI] [PubMed] [Google Scholar]
- Liu NJ, Schnell S, Wessendorf MW, & Gintzler AR (2013). Sex, pain, and opioids: interdependent influences of sex and pain modality on dynorphin–mediated antinociception in rats. J Pharmacol Exp Ther, 344, 522–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohith TG, Zoghbi SS, Morse CL, Araneta MF, Barth VN, Goebl NA, et al. (2012). Brain and whole–body imaging of nociceptin/orphanin FQ peptide receptor in humans using the PET ligand 11C–NOP–1A. J Nucl Med, 53, 385–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez–Munoz F, Alamo C, Juckel G, & Assion HJ (2007). Half a century of antidepressant drugs: on the clinical introduction of monoamine oxidase inhibitors, tricyclics, and tetracyclics. Part I: monoamine oxidase inhibitors. J Clin Psychopharmacol, 27, 555–559. [DOI] [PubMed] [Google Scholar]
- Lord JA, Waterfield AA, Hughes J, & Kosterlitz HW (1977). Endogenous opioid peptides: multiple agonists and receptors. Nature, 267, 495–499. [DOI] [PubMed] [Google Scholar]
- Lowe SL, Wong CJ, Witcher J, Gonzales CR, Dickinson GL, Bell RL, et al. (2014). Safety, tolerability, and pharmacokinetic evaluation of single–and multiple–ascending doses of a novel kappa opioid receptor antagonist LY2456302 and drug interaction with ethanol in healthy subjects. J Clin Pharmacol, 54, 968–978. [DOI] [PubMed] [Google Scholar]
- Lucas LR, Dragisic T, Duwaerts CC, Swiatkowski M, & Suzuki H (2011). Effects of recovery from immobilization stress on striatal preprodynorphin–and kappa opioid receptor–mRNA levels of the male rat. Physiol Behav, 104, 972–980. [DOI] [PubMed] [Google Scholar]
- Lutfy K, & Cowan A (2004). Buprenorphine: a unique drug with complex pharmacology. Curr Neuropharmacol, 2, 395–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lutfy K, Eitan S, Bryant CD, Yang YC, Saliminejad N, Walwyn W, et al. (2003). Buprenorphine–induced antinociception is mediated by mu–opioid receptors and compromised by concomitant activation of opioid receptor–like receptors. J Neurosci, 23, 10331–10337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lutz PE, Gross JA, Dhir SK, Maussion G, Yang J, Bramoulle A, et al. (2018). Epigenetic Regulation of the Kappa Opioid Receptor by Child Abuse. Biol Psychiatry, 84, 751–761. [DOI] [PubMed] [Google Scholar]
- Lutz PE, & Kieffer BL (2013). Opioid receptors: distinct roles in mood disorders. Trends Neurosci, 36, 195–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madar I, Lever JR, Kinter CM, Scheffel U, Ravert HT, Musachio JL, et al. (1996). Imaging of delta opioid receptors in human brain by N1’–([11C]methyl)naltrindole and PET. Synapse, 24, 19–28. [DOI] [PubMed] [Google Scholar]
- Mague SD, Isiegas C, Huang P, Liu–Chen LY, Lerman C, & Blendy JA (2009). Mouse model of OPRM1 (A118G) polymorphism has sex–specific effects on drug–mediated behavior. Proc Natl Acad Sci U S A, 106, 10847–10852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mague SD, Pliakas AM, Todtenkopf MS, Tomasiewicz HC, Zhang Y, Stevens WC Jr., et al. (2003). Antidepressant–like effects of kappa–opioid receptor antagonists in the forced swim test in rats. J Pharmacol Exp Ther, 305, 323–330. [DOI] [PubMed] [Google Scholar]
- Manhapra A, Quinones L, & Rosenheck R (2016). Characteristics of veterans receiving buprenorphine vs. methadone for opioid use disorder nationally in the Veterans Health Administration. Drug Alcohol Depend, 160, 82–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manhapra A, Rosenheck R, & Fiellin DA (2017). Opioid substitution treatment is linked to reduced risk of death in opioid use disorder. BMJ, 357, j1947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manning JS, & Jackson WC (2013). Depression, pain, and comorbid medical conditions. J Clin Psychiatry, 74, e03. [DOI] [PubMed] [Google Scholar]
- Mansour A, Khachaturian H, Lewis ME, Akil H, & Watson SJ (1987). Autoradiographic differentiation of mu, delta, and kappa opioid receptors in the rat forebrain and midbrain. J Neurosci, 7, 2445–2464. [PMC free article] [PubMed] [Google Scholar]
- Mansour A, Lewis ME, Khachaturian H, Akil H, & Watson SJ (1986). Pharmacological and anatomical evidence of selective mu, delta, and kappa opioid receptor binding in rat brain. Brain Res, 399, 69–79. [DOI] [PubMed] [Google Scholar]
- Maqueda AE, Valle M, Addy PH, Antonijoan RM, Puntes M, Coimbra J, et al. (2015). Salvinorin–A Induces Intense Dissociative Effects, Blocking External Sensory Perception and Modulating Interoception and Sense of Body Ownership in Humans. Int J Neuropsychopharmacol, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margolis EB, Fields HL, Hjelmstad GO, & Mitchell JM (2008). Delta–opioid receptor expression in the ventral tegmental area protects against elevated alcohol consumption. J Neurosci, 28, 12672–12681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margolis EB, Hjelmstad GO, Bonci A, & Fields HL (2003). Kappa–opioid agonists directly inhibit midbrain dopaminergic neurons. J Neurosci, 23, 9981–9986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margolis EB, Lock H, Chefer VI, Shippenberg TS, Hjelmstad GO, & Fields HL (2006). Kappa opioids selectively control dopaminergic neurons projecting to the prefrontal cortex. Proc Natl Acad Sci U S A, 103, 2938–2942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margolis EB, Mitchell JM, Hjelmstad GO, & Fields HL (2011). A novel opioid receptor–mediated enhancement of GABAA receptor function induced by stress in ventral tegmental area neurons. J Physiol, 589, 4229–4242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marquez P, Baliram R, Kieffer BL, & Lutfy K (2007). The mu opioid receptor is involved in buprenorphine–induced locomotor stimulation and conditioned place preference. Neuropharmacology, 52, 1336–1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mather M, & Harley CW (2016). The Locus Coeruleus: Essential for Maintaining Cognitive Function and the Aging Brain. Trends Cogn Sci, 20, 214–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McFadzean I, Lacey MG, Hill RG, & Henderson G (1987). Kappa opioid receptor activation depresses excitatory synaptic input to rat locus coeruleus neurons in vitro. Neuroscience, 20, 231–239. [DOI] [PubMed] [Google Scholar]
- McHugh RK (2015). Treatment of co–occurring anxiety disorders and substance use disorders. Harv Rev Psychiatry, 23, 99–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIntyre RS, Best MW, Bowie CR, Carmona NE, Cha DS, Lee Y, et al. (2017). The THINC–Integrated Tool (THINC–it) Screening Assessment for Cognitive Dysfunction: Validation in Patients With Major Depressive Disorder. J Clin Psychiatry, 78, 873–881. [DOI] [PubMed] [Google Scholar]
- McLaughlin JP, Li S, Valdez J, Chavkin TA, & Chavkin C (2006). Social defeat stress–induced behavioral responses are mediated by the endogenous kappa opioid system. Neuropsychopharmacology, 31, 1241–1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaughlin JP, Marton–Popovici M, & Chavkin C (2003). Kappa opioid receptor antagonism and prodynorphin gene disruption block stress–induced behavioral responses. J Neurosci, 23, 5674–5683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMakin DL, Olino TM, Porta G, Dietz LJ, Emslie G, Clarke G, et al. (2012). Anhedonia predicts poorer recovery among youth with selective serotonin reuptake inhibitor treatment–resistant depression. J Am Acad Child Adolesc Psychiatry, 51, 404–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medeiros IU, Ruzza C, Asth L, Guerrini R, Romao PR, Gavioli EC, et al. (2015). Blockade of nociceptin/orphanin FQ receptor signaling reverses LPS–induced depressive–like behavior in mice. Peptides, 72, 95–103. [DOI] [PubMed] [Google Scholar]
- Mela F, Marti M, Ulazzi L, Vaccari E, Zucchini S, Trapella C, et al. (2004). Pharmacological profile of nociceptin/orphanin FQ receptors regulating 5–hydroxytryptamine release in the mouse neocortex. Eur J Neurosci, 19, 1317–1324. [DOI] [PubMed] [Google Scholar]
- Melief EJ, Miyatake M, Bruchas MR, & Chavkin C (2010). Ligand–directed c–Jun N–terminal kinase activation disrupts opioid receptor signaling. Proc Natl Acad Sci U S A, 107, 11608–11613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menard C, Tse YC, Cavanagh C, Chabot JG, Herzog H, Schwarzer C, et al. (2013). Knockdown of prodynorphin gene prevents cognitive decline, reduces anxiety, and rescues loss of group 1 metabotropic glutamate receptor function in aging. J Neurosci, 33, 12792–12804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meunier JC, Mollereau C, Toll L, Suaudeau C, Moisand C, Alvinerie P, et al. (1995). Isolation and structure of the endogenous agonist of opioid receptor–like ORL1 receptor. Nature, 377, 532–535. [DOI] [PubMed] [Google Scholar]
- Miczek KA, Nikulina EM, Takahashi A, Covington HE 3rd, Yap JJ, Boyson CO, et al. (2011). Gene expression in aminergic and peptidergic cells during aggression and defeat: relevance to violence, depression and drug abuse. Behav Genet, 41, 787–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitch CH, Quimby SJ, Diaz N, Pedregal C, de la Torre MG, Jimenez A, et al. (2011). Discovery of aminobenzyloxyarylamides as kappa opioid receptor selective antagonists: application to preclinical development of a kappa opioid receptor antagonist receptor occupancy tracer. J Med Chem, 54, 8000–8012. [DOI] [PubMed] [Google Scholar]
- Mogil JS, & Pasternak GW (2001). The molecular and behavioral pharmacology of the orphanin FQ/nociceptin peptide and receptor family. Pharmacol Rev, 53, 381–415. [PubMed] [Google Scholar]
- Mollereau C, Mouledous L, Lapalu S, Cambois G, Moisand C, Butour JL, et al. (1999). Distinct mechanisms for activation of the opioid receptor–like 1 and kappa–opioid receptors by nociceptin and dynorphin A. Mol Pharmacol, 55, 324–331. [DOI] [PubMed] [Google Scholar]
- Mollereau C, Parmentier M, Mailleux P, Butour JL, Moisand C, Chalon P, et al. (1994). ORL1, a novel member of the opioid receptor family. Cloning, functional expression and localization. FEBS Lett, 341, 33–38. [DOI] [PubMed] [Google Scholar]
- Mori T, Nomura M, Nagase H, Narita M, & Suzuki T (2002). Effects of a newly synthesized kappa–opioid receptor agonist, TRK–820, on the discriminative stimulus and rewarding effects of cocaine in rats. Psychopharmacology (Berl), 161, 17–22. [DOI] [PubMed] [Google Scholar]
- Moron JA, Gullapalli S, Taylor C, Gupta A, Gomes I, & Devi LA (2010). Modulation of opiate–related signaling molecules in morphine–dependent conditioned behavior: conditioned place preference to morphine induces CREB phosphorylation. Neuropsychopharmacology, 35, 955–966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moss IR, Scott SC, & Inman JD (1993). Mu–vs. delta–opioid influence on respiratory and sleep behavior during development. Am J Physiol, 264, R754–760. [DOI] [PubMed] [Google Scholar]
- Mulder AH, Wardeh G, Hogenboom F, & Frankhuyzen AL (1984). Kappa–and delta–opioid receptor agonists differentially inhibit striatal dopamine and acetylcholine release. Nature, 308, 278–280. [DOI] [PubMed] [Google Scholar]
- Murphy NP, Lee Y, & Maidment NT (1999). Orphanin FQ/nociceptin blocks acquisition of morphine place preference. Brain Res, 832, 168–170. [DOI] [PubMed] [Google Scholar]
- Murphy NP, Ly HT, & Maidment NT (1996). Intracerebroventricular orphanin FQ/nociceptin suppresses dopamine release in the nucleus accumbens of anaesthetized rats. Neuroscience, 75, 1–4. [DOI] [PubMed] [Google Scholar]
- Murphy NP, & Maidment NT (1999). Orphanin FQ/nociceptin modulation of mesolimbic dopamine transmission determined by microdialysis. J Neurochem, 73, 179–186. [DOI] [PubMed] [Google Scholar]
- Musazzi L, Mallei A, Tardito D, Gruber SH, El Khoury A, Racagni G, et al. (2010). Early–life stress and antidepressant treatment involve synaptic signaling and Erk kinases in a gene–environment model of depression. J Psychiatr Res, 44, 511–520. [DOI] [PubMed] [Google Scholar]
- Naganawa M, Dickinson GL, Zheng MQ, Henry S, Vandenhende F, Witcher J, et al. (2016). Receptor Occupancy of the kappa–Opioid Antagonist LY2456302 Measured with Positron Emission Tomography and the Novel Radiotracer 11C–LY2795050. J Pharmacol Exp Ther, 356, 260–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nativio P, Pascale E, Maffei A, Scaccianoce S, & Passarelli F (2012). Effect of stress on hippocampal nociceptin expression in the rat. Stress, 15, 378–384. [DOI] [PubMed] [Google Scholar]
- Nazzaro C, Barbieri M, Varani K, Beani L, Valentino RJ, & Siniscalchi A (2010). Swim stress enhances nociceptin/orphanin FQ–induced inhibition of rat dorsal raphe nucleus activity in vivo and in vitro: role of corticotropin releasing factor. Neuropharmacology, 58, 457–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nazzaro C, Marino S, Barbieri M, & Siniscalchi A (2009). Inhibition of serotonin outflow by nociceptin/orphaninFQ in dorsal raphe nucleus slices from normal and stressed rats: Role of corticotropin releasing factor. Neurochem Int, 54, 378–384. [DOI] [PubMed] [Google Scholar]
- Neal CR Jr., Mansour A, Reinscheid R, Nothacker HP, Civelli O, Akil H, et al. (1999a). Opioid receptor–like (ORL1) receptor distribution in the rat central nervous system: comparison of ORL1 receptor mRNA expression with (125)I–[(14)Tyr]–orphanin FQ binding. J Comp Neurol, 412, 563–605. [PubMed] [Google Scholar]
- Neal CR Jr., Mansour A, Reinscheid R, Nothacker HP, Civelli O, & Watson SJ Jr. (1999b). Localization of orphanin FQ (nociceptin) peptide and messenger RNA in the central nervous system of the rat. J Comp Neurol, 406, 503–547. [PubMed] [Google Scholar]
- Nemeth CL, Paine TA, Rittiner JE, Beguin C, Carroll FI, Roth BL, et al. (2010). Role of kappa–opioid receptors in the effects of salvinorin A and ketamine on attention in rats. Psychopharmacology (Berl), 210, 263–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- New DC, & Wong YH (2002). The ORL1 receptor: molecular pharmacology and signalling mechanisms. Neurosignals, 11, 197–212. [DOI] [PubMed] [Google Scholar]
- Nicholson JR, Akil H, & Watson SJ (2002). Orphanin FQ–induced hyperphagia is mediated by corticosterone and central glucocorticoid receptors. Neuroscience, 115, 637–643. [DOI] [PubMed] [Google Scholar]
- Nicoll RA, Siggins GR, Ling N, Bloom FE, & Guillemin R (1977). Neuronal actions of endorphins and enkephalins among brain regions: a comparative microiontophoretic study. Proc Natl Acad Sci U S A, 74, 2584–2588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikulina EM, Arrillaga–Romany I, Miczek KA, & Hammer RP Jr. (2008). Long–lasting alteration in mesocorticolimbic structures after repeated social defeat stress in rats: time course of mu–opioid receptor mRNA and FosB/DeltaFosB immunoreactivity. Eur J Neurosci, 27, 2272–2284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikulina EM, Hammer RP Jr., Miczek KA, & Kream RM (1999). Social defeat stress increases expression of mu–opioid receptor mRNA in rat ventral tegmental area. Neuroreport, 10, 3015–3019. [DOI] [PubMed] [Google Scholar]
- Nikulina EM, Miczek KA, & Hammer RP Jr. (2005). Prolonged effects of repeated social defeat stress on mRNA expression and function of mu–opioid receptors in the ventral tegmental area of rats. Neuropsychopharmacology, 30, 1096–1103. [DOI] [PubMed] [Google Scholar]
- Nocjar C, Zhang J, Feng P, & Panksepp J (2012). The social defeat animal model of depression shows diminished levels of orexin in mesocortical regions of the dopamine system, and of dynorphin and orexin in the hypothalamus. Neuroscience, 218, 138–153. [DOI] [PubMed] [Google Scholar]
- Nozaki C, Nagase H, Nemoto T, Matifas A, Kieffer BL, & Gaveriaux–Ruff C (2014). In vivo properties of KNT–127, a novel delta opioid receptor agonist: receptor internalization, antihyperalgesia and antidepressant effects in mice. Br J Pharmacol, 171, 5376–5386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nummenmaa L, Manninen S, Tuominen L, Hirvonen J, Kalliokoski KK, Nuutila P, et al. (2015). Adult attachment style is associated with cerebral mu–opioid receptor availability in humans. Hum Brain Mapp, 36, 3621–3628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nyhuis PW, Gastpar M, & Scherbaum N (2008). Opiate treatment in depression refractory to antidepressants and electroconvulsive therapy. J Clin Psychopharmacol, 28, 593–595. [DOI] [PubMed] [Google Scholar]
- Okawa H, Kudo M, Kudo T, Guerrini R, Lambert DG, Kushikata T, et al. (2001). Effects of nociceptinNH2 and [Nphe1]nociceptin(1–13)NH2 on rat brain noradrenaline release in vivo and in vitro. Neurosci Lett, 303, 173–176. [DOI] [PubMed] [Google Scholar]
- Ota KT, & Duman RS (2013). Environmental and pharmacological modulations of cellular plasticity: role in the pathophysiology and treatment of depression. Neurobiol Dis, 57, 28–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palomares–Castillo E, Hernandez–Perez OR, Perez–Carrera D, Crespo–Ramirez M, Fuxe K, & Perez de la Mora M (2012). The intercalated paracapsular islands as a module for integration of signals regulating anxiety in the amygdala. Brain Res, 1476, 211–234. [DOI] [PubMed] [Google Scholar]
- Pasternak DA, Pan L, Xu J, Yu R, Xu MM, Pasternak GW, et al. (2004). Identification of three new alternatively spliced variants of the rat mu opioid receptor gene: dissociation of affinity and efficacy. J Neurochem, 91, 881–890. [DOI] [PubMed] [Google Scholar]
- Pasternak GW (1980). Multiple opiate receptors: [3H]ethylketocyclazocine receptor binding and ketocyclazocine analgesia. Proc Natl Acad Sci U S A, 77, 3691–3694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasternak GW (2004). Multiple opiate receptors: deja vu all over again. Neuropharmacology, 47 Suppl 1, 312–323. [DOI] [PubMed] [Google Scholar]
- Pasternak GW, & Snyder SH (1975). Identification of novel high affinity opiate receptor binding in rat brain. Nature, 253, 563–565. [DOI] [PubMed] [Google Scholar]
- Pearson KA, Stephen A, Beck SG, & Valentino RJ (2006). Identifying genes in monoamine nuclei that may determine stress vulnerability and depressive behavior in Wistar–Kyoto rats. Neuropsychopharmacology, 31, 2449–2461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pecina M, Karp JF, Mathew S, Todtenkopf MS, Ehrich EW, & Zubieta JK (2018). Endogenous opioid system dysregulation in depression: implications for new therapeutic approaches. Mol Psychiatry [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peckys D, & Hurd YL (2001). Prodynorphin and kappa opioid receptor mRNA expression in the cingulate and prefrontal cortices of subjects diagnosed with schizophrenia or affective disorders. Brain Res Bull, 55, 619–624. [DOI] [PubMed] [Google Scholar]
- Pennock RL, & Hentges ST (2016). Desensitization–resistant and –sensitive GPCR–mediated inhibition of GABA release occurs by Ca2+–dependent and –independent mechanisms at a hypothalamic synapse. J Neurophysiol, 115, 2376–2388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrine SA, Sheikh IS, Nwaneshiudu CA, Schroeder JA, & Unterwald EM (2008). Withdrawal from chronic administration of cocaine decreases delta opioid receptor signaling and increases anxiety–and depression–like behaviors in the rat. Neuropharmacology, 54, 355–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeiffer A, Brantl V, Herz A, & Emrich HM (1986). Psychotomimesis mediated by kappa opiate receptors. Science, 233, 774–776. [DOI] [PubMed] [Google Scholar]
- Pietrzak RH, Naganawa M, Huang Y, Corsi–Travali S, Zheng MQ, Stein MB, et al. (2014). Association of in vivo kappa–opioid receptor availability and the transdiagnostic dimensional expression of trauma–related psychopathology. JAMA Psychiatry, 71, 1262–1270. [DOI] [PubMed] [Google Scholar]
- Pizzagalli DA, & Carlezon WA (2017). Error Processing in Depressive States: A Translational Opportunity? Neuropsychopharmacology, 42, 372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pliakas AM, Carlson RR, Neve RL, Konradi C, Nestler EJ, & Carlezon WA Jr. (2001). Altered responsiveness to cocaine and increased immobility in the forced swim test associated with elevated cAMP response element–binding protein expression in nucleus accumbens. J Neurosci, 21, 7397–7403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pohorecky LA, Skiandos A, Zhang X, Rice KC, & Benjamin D (1999). Effect of chronic social stress on delta–opioid receptor function in the rat. J Pharmacol Exp Ther, 290, 196–206. [PubMed] [Google Scholar]
- Polter AM, Barcomb K, Chen RW, Dingess PM, Graziane NM, Brown TE, et al. (2017). Constitutive activation of kappa opioid receptors at ventral tegmental area inhibitory synapses following acute stress. Elife, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polter AM, Bishop RA, Briand LA, Graziane NM, Pierce RC, & Kauer JA (2014). Poststress block of kappa opioid receptors rescues long–term potentiation of inhibitory synapses and prevents reinstatement of cocaine seeking. Biol Psychiatry, 76, 785–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Post A, Smart TS, Jackson K, Mann J, Mohs R, Rorick–Kehn L, et al. (2016a). Proof–of–Concept Study to Assess the Nociceptin Receptor Antagonist LY2940094 as a New Treatment for Alcohol Dependence. Alcohol Clin Exp Res, 40, 1935–1944. [DOI] [PubMed] [Google Scholar]
- Post A, Smart TS, Krikke–Workel J, Dawson GR, Harmer CJ, Browning M, et al. (2016b). A Selective Nociceptin Receptor Antagonist to Treat Depression: Evidence from Preclinical and Clinical Studies. Neuropsychopharmacology, 41, 1803–1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potter DN, Damez–Werno D, Carlezon WA Jr., Cohen BM, & Chartoff EH (2011). Repeated exposure to the kappa–opioid receptor agonist salvinorin A modulates extracellular signal–regulated kinase and reward sensitivity. Biol Psychiatry, 70, 744–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poulin JF, Berube P, Laforest S, & Drolet G (2013). Enkephalin knockdown in the central amygdala nucleus reduces unconditioned fear and anxiety. Eur J Neurosci, 37, 1357–1367. [DOI] [PubMed] [Google Scholar]
- Poulin JF, Laforest S, & Drolet G (2014). Enkephalin downregulation in the nucleus accumbens underlies chronic stress–induced anhedonia. Stress, 17, 88–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pucci M, Micioni Di Bonaventura MV, Giusepponi ME, Romano A, Filaferro M, Maccarrone M, et al. (2016). Epigenetic regulation of nociceptin/orphanin FQ and corticotropin–releasing factor system genes in frustration stress–induced binge–like palatable food consumption. Addict Biol, 21, 1168–1185. [DOI] [PubMed] [Google Scholar]
- Quednow BB, Csomor PA, Chmiel J, Beck T, & Vollenweider FX (2008). Sensorimotor gating and attentional set–shifting are improved by the mu–opioid receptor agonist morphine in healthy human volunteers. Int J Neuropsychopharmacol, 11, 655–669. [DOI] [PubMed] [Google Scholar]
- Quock RM, Burkey TH, Varga E, Hosohata Y, Hosohata K, Cowell SM, et al. (1999). The delta–opioid receptor: molecular pharmacology, signal transduction, and the determination of drug efficacy. Pharmacol Rev, 51, 503–532. [PubMed] [Google Scholar]
- Raddad E, Chappell A, Meyer J, Wilson A, Ruegg CE, Tauscher J, et al. (2016). Occupancy of Nociceptin/Orphanin FQ Peptide Receptors by the Antagonist LY2940094 in Rats and Healthy Human Subjects. Drug Metab Dispos, 44, 1536–1542. [DOI] [PubMed] [Google Scholar]
- Raehal KM, & Bohn LM (2011). The role of beta–arrestin2 in the severity of antinociceptive tolerance and physical dependence induced by different opioid pain therapeutics. Neuropharmacology, 60, 58–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramaker MJ, & Dulawa SC (2017). Identifying fast–onset antidepressants using rodent models. Mol Psychiatry, 22, 656–665. [DOI] [PubMed] [Google Scholar]
- Randall–Thompson JF, Pescatore KA, & Unterwald EM (2010). A role for delta opioid receptors in the central nucleus of the amygdala in anxiety–like behaviors. Psychopharmacology (Berl), 212, 585–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranganathan M, Schnakenberg A, Skosnik PD, Cohen BM, Pittman B, Sewell RA, et al. (2012). Dose–related behavioral, subjective, endocrine, and psychophysiological effects of the kappa opioid agonist Salvinorin A in humans. Biol Psychiatry, 72, 871–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rawls SM, & McGinty JF (2000). Delta opioid receptors regulate calcium–dependent, amphetamine–evoked glutamate levels in the rat striatum: an in vivo microdialysis study. Brain Res, 861, 296–304. [DOI] [PubMed] [Google Scholar]
- Ray R, Jepson C, Patterson F, Strasser A, Rukstalis M, Perkins K, et al. (2006). Association of OPRM1 A118G variant with the relative reinforcing value of nicotine. Psychopharmacology (Berl), 188, 355–363. [DOI] [PubMed] [Google Scholar]
- Razzoli M, Andreoli M, Michielin F, Quarta D, & Sokal DM (2011). Increased phasic activity of VTA dopamine neurons in mice 3 weeks after repeated social defeat. Behav Brain Res, 218, 253–257. [DOI] [PubMed] [Google Scholar]
- Reed B, Butelman ER, Fry RS, Kimani R, & Kreek MJ (2018). Repeated Administration of Opra Kappa (LY2456302), a Novel, Short–Acting, Selective KOP–r Antagonist, in Persons with and without Cocaine Dependence. Neuropsychopharmacology, 43, 739–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed B, Fang N, Mayer–Blackwell B, Chen S, Yuferov V, Zhou Y, et al. (2012). Chromatin alterations in response to forced swimming underlie increased prodynorphin transcription. Neuroscience, 220, 109–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinoso–Barbero F, & de Andres I (1995). Effects of opioid microinjections in the nucleus of the solitary tract on the sleep–wakefulness cycle states in cats. Anesthesiology, 82, 144–152. [DOI] [PubMed] [Google Scholar]
- Reinscheid RK, Nothacker HP, Bourson A, Ardati A, Henningsen RA, Bunzow JR, et al. (1995). Orphanin FQ: a neuropeptide that activates an opioidlike G protein–coupled receptor. Science, 270, 792–794. [DOI] [PubMed] [Google Scholar]
- Ren Z, Yan P, Zhu L, Yang H, Zhao Y, Kirby BP, et al. (2018). Dihydromyricetin exerts a rapid antidepressant–like effect in association with enhancement of BDNF expression and inhibition of neuroinflammation. Psychopharmacology (Berl), 235, 233–244. [DOI] [PubMed] [Google Scholar]
- Resnick RB, Resnick E, & Galanter M (1991). Buprenorphine responders: a diagnostic subgroup of heroin addicts? Prog Neuropsychopharmacol Biol Psychiatry, 15, 531–538. [DOI] [PubMed] [Google Scholar]
- Reyes BA, Chavkin C, & Van Bockstaele EJ (2010). Agonist–induced internalization of kappa–opioid receptors in noradrenergic neurons of the rat locus coeruleus. J Chem Neuroanat, 40, 301–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes BA, Kravets JL, Connelly KL, Unterwald EM, & Van Bockstaele EJ (2017). Localization of the delta opioid receptor and corticotropin–releasing factor in the amygdalar complex: role in anxiety. Brain Struct Funct, 222, 1007–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes BA, Zitnik G, Foster C, Van Bockstaele EJ, & Valentino RJ (2015). Social Stress Engages Neurochemically–Distinct Afferents to the Rat Locus Coeruleus Depending on Coping Strategy. eNeuro, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards EM, Mathews DC, Luckenbaugh DA, Ionescu DF, Machado–Vieira R, Niciu MJ, et al. (2016). A randomized, placebo–controlled pilot trial of the delta opioid receptor agonist AZD2327 in anxious depression. Psychopharmacology (Berl), 233, 1119–1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson SA, Erickson RL, Browne CA, & Lucki I (2017). A role for the mu opioid receptor in the antidepressant effects of buprenorphine. Behav Brain Res, 319, 96–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robles CF, McMackin MZ, Campi KL, Doig IE, Takahashi EY, Pride MC, et al. (2014). Effects of kappa opioid receptors on conditioned place aversion and social interaction in males and females. Behav Brain Res, 262, 84–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogala B, Li Y, Li S, Chen X, & Kirouac GJ (2012). Effects of a post–shock injection of the kappa opioid receptor antagonist norbinaltorphimine (norBNI) on fear and anxiety in rats. PLoS One, 7, e49669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronken E, Mulder AH, & Schoffelmeer AN (1993a). Interacting presynaptic kappa–opioid and GABAA receptors modulate dopamine release from rat striatal synaptosomes. J Neurochem, 61, 1634–1639. [DOI] [PubMed] [Google Scholar]
- Ronken E, Van Muiswinkel FL, Mulder AH, & Schoffelmeer AN (1993b). Opioid receptor–mediated inhibition of evoked catecholamine release from cultured neurons of rat ventral mesencephalon and locus coeruleus. Eur J Pharmacol, 230, 349–355. [DOI] [PubMed] [Google Scholar]
- Rorick–Kehn LM, Ciccocioppo R, Wong CJ, Witkin JM, Martinez–Grau MA, Stopponi S, et al. (2016). A Novel, Orally Bioavailable Nociceptin Receptor Antagonist, LY2940094, Reduces Ethanol Self–Administration and Ethanol Seeking in Animal Models. Alcohol Clin Exp Res, 40, 945–954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rorick–Kehn LM, Witcher JW, Lowe SL, Gonzales CR, Weller MA, Bell RL, et al. (2014a). Determining pharmacological selectivity of the kappa opioid receptor antagonist LY2456302 using pupillometry as a translational biomarker in rat and human. Int J Neuropsychopharmacol, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rorick–Kehn LM, Witkin JM, Statnick MA, Eberle EL, McKinzie JH, Kahl SD, et al. (2014b). LY2456302 is a novel, potent, orally–bioavailable small molecule kappa–selective antagonist with activity in animal models predictive of efficacy in mood and addictive disorders. Neuropharmacology, 77, 131–144. [DOI] [PubMed] [Google Scholar]
- Rossier J, Vargo TM, Minick S, Ling N, Bloom FE, & Guillemin R (1977). Regional dissociation of beta–endorphin and enkephalin contents in rat brain and pituitary. Proc Natl Acad Sci U S A, 74, 5162–5165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothman RB, Mahboubi A, Bykov V, Kim CH, de Costa BR, Jacobson AE, et al. (1992). Probing the opioid receptor complex with (+)–trans–SUPERFIT. II. Evidence that mu ligands are noncompetitive inhibitors of the delta cx opioid peptide binding site. Peptides, 13, 1137–1143. [DOI] [PubMed] [Google Scholar]
- Rothman RB, Mahboubi A, Bykov V, Kim CH, Jacobson AE, & Rice KC (1991). Probing the opioid receptor complex with (+)–trans–superfit. I. Evidence that [D–Pen2,D–Pen5]enkephalin interacts with high affinity at the delta cx binding site. Peptides, 12, 359–364. [DOI] [PubMed] [Google Scholar]
- Rozenfeld R, & Devi LA (2011). Exploring a role for heteromerization in GPCR signalling specificity. Biochem J, 433, 11–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell SE, Rachlin AB, Smith KL, Muschamp J, Berry L, Zhao Z, et al. (2014). Sex differences in sensitivity to the depressive–like effects of the kappa opioid receptor agonist U–50488 in rats. Biol Psychiatry, 76, 213–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutz S, Riegert C, Rothmaier AK, & Jackisch R (2007). Presynaptic modulation of 5–HT release in the rat septal region. Neuroscience, 146, 643–658. [DOI] [PubMed] [Google Scholar]
- Saito M, Ehringer MA, Toth R, Oros M, Szakall I, Sikela JM, et al. (2003). Variants of kappa–opioid receptor gene and mRNA in alcohol–preferring and alcohol–avoiding mice. Alcohol, 29, 39–49. [DOI] [PubMed] [Google Scholar]
- Saitoh A, Kimura Y, Suzuki T, Kawai K, Nagase H, & Kamei J (2004). Potential anxiolytic and antidepressant–like activities of SNC80, a selective delta–opioid agonist, in behavioral models in rodents. J Pharmacol Sci, 95, 374–380. [DOI] [PubMed] [Google Scholar]
- Saitoh A, & Nagase H (2018). Delta Opioid Receptor (DOR) Ligands and Pharmacology: Development of Indolo–and Quinolinomorphinan Derivatives Based on the Message–Address Concept. Handb Exp Pharmacol, 247, 3–19. [DOI] [PubMed] [Google Scholar]
- Saitoh A, Sugiyama A, Nemoto T, Fujii H, Wada K, Oka J, et al. (2011). The novel delta opioid receptor agonist KNT–127 produces antidepressant–like and antinociceptive effects in mice without producing convulsions. Behav Brain Res, 223, 271–279. [DOI] [PubMed] [Google Scholar]
- Saitoh A, Yoshikawa Y, Onodera K, & Kamei J (2005). Role of delta–opioid receptor subtypes in anxiety–related behaviors in the elevated plus–maze in rats. Psychopharmacology (Berl), 182, 327–334. [DOI] [PubMed] [Google Scholar]
- Samaco RC, Mandel–Brehm C, McGraw CM, Shaw CA, McGill BE, & Zoghbi HY (2012). Crh and Oprm1 mediate anxiety–related behavior and social approach in a mouse model of MECP2 duplication syndrome. Nat Genet, 44, 206–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders MJ, Kieffer BL, & Fanselow MS (2005). Deletion of the mu opioid receptor results in impaired acquisition of Pavlovian context fear. Neurobiol Learn Mem, 84, 33–41. [DOI] [PubMed] [Google Scholar]
- Scherrer G, Tryoen–Toth P, Filliol D, Matifas A, Laustriat D, Cao YQ, et al. (2006). Knockin mice expressing fluorescent delta–opioid receptors uncover G protein–coupled receptor dynamics in vivo. Proc Natl Acad Sci U S A, 103, 9691–9696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid CL, & Bohn LM (2009). Physiological and pharmacological implications of beta–arrestin regulation. Pharmacol Ther, 121, 285–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt HD, & Duman RS (2010). Peripheral BDNF produces antidepressant–like effects in cellular and behavioral models. Neuropsychopharmacology, 35, 2378–2391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serrano–Gomez A, Thompson JP, & Lambert DG (2011). Nociceptin/orphanin FQ in inflammation and sepsis. Br J Anaesth, 106, 6–12. [DOI] [PubMed] [Google Scholar]
- Sharma S, & Fulton S (2013). Diet–induced obesity promotes depressive–like behaviour that is associated with neural adaptations in brain reward circuitry. Int J Obes (Lond), 37, 382–389. [DOI] [PubMed] [Google Scholar]
- Shippenberg TS, & Herz A (1986). Differential effects of mu and kappa opioid systems on motivational processes. NIDA Res Monogr, 75, 563–566. [PubMed] [Google Scholar]
- Shirayama Y, Ishida H, Iwata M, Hazama GI, Kawahara R, & Duman RS (2004). Stress increases dynorphin immunoreactivity in limbic brain regions and dynorphin antagonism produces antidepressant–like effects. J Neurochem, 90, 1258–1268. [DOI] [PubMed] [Google Scholar]
- Sia AT, Lim Y, Lim EC, Goh RW, Law HY, Landau R, et al. (2008). A118G single nucleotide polymorphism of human mu–opioid receptor gene influences pain perception and patient–controlled intravenous morphine consumption after intrathecal morphine for postcesarean analgesia. Anesthesiology, 109, 520–526. [DOI] [PubMed] [Google Scholar]
- Simonin F, Befort K, Gaveriaux–Ruff C, Matthes H, Nappey V, Lannes B, et al. (1994). The human delta–opioid receptor: genomic organization, cDNA cloning, functional expression, and distribution in human brain. Mol Pharmacol, 46, 1015–1021. [PubMed] [Google Scholar]
- Simonin F, Gaveriaux–Ruff C, Befort K, Matthes H, Lannes B, Micheletti G, et al. (1995). kappa–Opioid receptor in humans: cDNA and genomic cloning, chromosomal assignment, functional expression, pharmacology, and expression pattern in the central nervous system. Proc Natl Acad Sci U S A, 92, 7006–7010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siniscalchi A, Rodi D, Morari M, Marti M, Cavallini S, Marino S, et al. (2002). Direct and indirect inhibition by nociceptin/orphanin FQ on noradrenaline release from rodent cerebral cortex in vitro. Br J Pharmacol, 136, 1178–1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith JS, Zubieta JK, Price JC, Flesher JE, Madar I, Lever JR, et al. (1999). Quantification of delta–opioid receptors in human brain with N1’–([11C]methyl) naltrindole and positron emission tomography. J Cereb Blood Flow Metab, 19, 956–966. [DOI] [PubMed] [Google Scholar]
- Sofuoglu M, Portoghese PS, & Takemori AE (1991). Differential antagonism of delta opioid agonists by naltrindole and its benzofuran analog (NTB) in mice: evidence for delta opioid receptor subtypes. J Pharmacol Exp Ther, 257, 676–680. [PubMed] [Google Scholar]
- Sofuoglu M, Portoghese PS, & Takemori AE (1992). delta–Opioid receptor binding in mouse brain: evidence for heterogeneous binding sites. Eur J Pharmacol, 216, 273–277. [DOI] [PubMed] [Google Scholar]
- Statnick MA, Chen Y, Ansonoff M, Witkin JM, Rorick–Kehn L, Suter TM, et al. (2016). A Novel Nociceptin Receptor Antagonist LY2940094 Inhibits Excessive Feeding Behavior in Rodents: A Possible Mechanism for the Treatment of Binge Eating Disorder. J Pharmacol Exp Ther, 356, 493–502. [DOI] [PubMed] [Google Scholar]
- Striebel JM, & Kalapatapu RK (2014). The anti–suicidal potential of buprenorphine: a case report. Int J Psychiatry Med, 47, 169–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stubbs B, Vancampfort D, Veronese N, Thompson T, Fornaro M, Schofield P, et al. (2017). Depression and pain: primary data and meta–analysis among 237 952 people across 47 low–and middle–income countries. Psychol Med, 47, 2906–2917. [DOI] [PubMed] [Google Scholar]
- Sugiyama A, Nagase H, Oka J, Yamada M, & Saitoh A (2014). DOR(2)–selective but not DOR(1)–selective antagonist abolishes anxiolytic–like effects of the delta opioid receptor agonist KNT–127. Neuropharmacology, 79, 314–320. [DOI] [PubMed] [Google Scholar]
- Sundaramurthy S, Annamalai B, Samuvel DJ, Shippenberg TS, Jayanthi LD, & Ramamoorthy S (2017). Modulation of serotonin transporter function by kappa–opioid receptor ligands. Neuropharmacology, 113, 281–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szczytkowski–Thomson JL, Lebonville CL, & Lysle DT (2013). Morphine prevents the development of stress–enhanced fear learning. Pharmacol Biochem Behav, 103, 672–677. [DOI] [PubMed] [Google Scholar]
- Szklarczyk K, Korostynski M, Cieslak PE, Wawrzczak–Bargiela A, & Przewlocki R (2015). Opioid–dependent regulation of high and low fear responses in two inbred mouse strains. Behav Brain Res, 292, 95–101. [DOI] [PubMed] [Google Scholar]
- Takahashi K, Nakagawasai O, Sugawara M, Sato A, Nemoto W, Tadano T, et al. (2018). Kappa Opioid Receptor Agonist Administration in Olfactory Bulbectomized Mice Restores Cognitive Impairment through Cholinergic Neuron Activation. Biol Pharm Bull, 41, 957–960. [DOI] [PubMed] [Google Scholar]
- Tang MM, Lin WJ, Zhang JT, Zhao YW, & Li YC (2017). Exogenous FGF2 reverses depressive–like behaviors and restores the suppressed FGF2–ERK½ signaling and the impaired hippocampal neurogenesis induced by neuroinflammation. Brain Behav Immun, 66, 322–331. [DOI] [PubMed] [Google Scholar]
- Tejeda HA, Counotte DS, Oh E, Ramamoorthy S, Schultz–Kuszak KN, Backman CM, et al. (2013). Prefrontal cortical kappa–opioid receptor modulation of local neurotransmission and conditioned place aversion. Neuropsychopharmacology, 38, 1770–1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tejeda HA, Hanks AN, Scott L, Mejias–Aponte C, Hughes ZA, & O’Donnell P (2015). Prefrontal Cortical Kappa Opioid Receptors Attenuate Responses to Amygdala Inputs. Neuropsychopharmacology, 40, 2856–2864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tejeda HA, Wu J, Kornspun AR, Pignatelli M, Kashtelyan V, Krashes MJ, et al. (2017). Pathway–and Cell–Specific Kappa–Opioid Receptor Modulation of Excitation–Inhibition Balance Differentially Gates D1 and D2 Accumbens Neuron Activity. Neuron, 93, 147–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorat SN, & Hammond DL (1997). Modulation of nociception by microinjection of delta–1 and delta–2 opioid receptor ligands in the ventromedial medulla of the rat. J Pharmacol Exp Ther, 283, 1185–1192. [PubMed] [Google Scholar]
- Tjoumakaris SI, Rudoy C, Peoples J, Valentino RJ, & Van Bockstaele EJ (2003). Cellular interactions between axon terminals containing endogenous opioid peptides or corticotropin–releasing factor in the rat locus coeruleus and surrounding dorsal pontine tegmentum. J Comp Neurol, 466, 445–456. [DOI] [PubMed] [Google Scholar]
- Tollefson S, Himes M, & Narendran R (2017). Imaging corticotropin–releasing–factor and nociceptin in addiction and PTSD models. Int Rev Psychiatry, 29, 567–579. [DOI] [PubMed] [Google Scholar]
- Torregrossa MM, Jutkiewicz EM, Mosberg HI, Balboni G, Watson SJ, & Woods JH (2006). Peptidic delta opioid receptor agonists produce antidepressant–like effects in the forced swim test and regulate BDNF mRNA expression in rats. Brain Res, 1069, 172–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapaidze N, Gomes I, Cvejic S, Bansinath M, & Devi LA (2000). Opioid receptor endocytosis and activation of MAP kinase pathway. Brain Res Mol Brain Res, 76, 220–228. [DOI] [PubMed] [Google Scholar]
- Trentani A, Kuipers SD, Ter Horst GJ, & Den Boer JA (2002). Selective chronic stress–induced in vivo ERK½ hyperphosphorylation in medial prefrontocortical dendrites: implications for stress–related cortical pathology? Eur J Neurosci, 15, 1681–1691. [DOI] [PubMed] [Google Scholar]
- Trivedi MH (2006). Major depressive disorder: remission of associated symptoms. J Clin Psychiatry, 67 Suppl 6, 27–32. [PubMed] [Google Scholar]
- Tsui JI, Burt R, Thiede H, & Glick SN (2018). Utilization of buprenorphine and methadone among opioid users who inject drugs. Subst Abus, 39, 83–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valentino RJ, & Bangasser DA (2016). Sex–biased cellular signaling: molecular basis for sex differences in neuropsychiatric diseases. Dialogues Clin Neurosci, 18, 385–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valenza M, Butelman ER, & Kreek MJ (2017). “Effects of the novel relatively short–acting kappa opioid receptor antagonist LY2444296 in behaviors observed after chronic extended–access cocaine self–administration in rats”. Psychopharmacology (Berl), 234, 2219–2231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van’t Veer A, Bechtholt AJ, Onvani S, Potter D, Wang Y, Liu–Chen LY, et al. (2013). Ablation of kappa–opioid receptors from brain dopamine neurons has anxiolytic–like effects and enhances cocaine–induced plasticity. Neuropsychopharmacology, 38, 1585–1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Bronswijk S, Moopen N, Beijers L, Ruhe HG, & Peeters F (2019). Effectiveness of psychotherapy for treatment–resistant depression: a meta–analysis and meta–regression. Psychol Med, 49, 366–379. [DOI] [PubMed] [Google Scholar]
- van Rijn RM, Brissett DI, & Whistler JL (2010). Dual efficacy of delta opioid receptor–selective ligands for ethanol drinking and anxiety. J Pharmacol Exp Ther, 335, 133–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Rijn RM, Defriel JN, & Whistler JL (2013). Pharmacological traits of delta opioid receptors: pitfalls or opportunities? Psychopharmacology (Berl), 228, 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanz F, Bicca MA, Linartevichi VF, Giachero M, Bertoglio LJ, & Monteiro de Lima TC (2018). Role of dorsal hippocampus kappa opioid receptors in contextual aversive memory consolidation in rats. Neuropharmacology, 135, 253–267. [DOI] [PubMed] [Google Scholar]
- Varona A, Gil J, Saracibar G, Maza JL, Echevarria E, & Irazusta J (2003). Effects of imipramine treatment on delta–opioid receptors of the rat brain cortex and striatum. Arzneimittelforschung, 53, 21–25. [DOI] [PubMed] [Google Scholar]
- Vaughan CW, & Christie MJ (1996). Increase by the ORL1 receptor (opioid receptor–like1) ligand, nociceptin, of inwardly rectifying K conductance in dorsal raphe nucleus neurones. Br J Pharmacol, 117, 1609–1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vijay A, Wang S, Worhunsky P, Zheng MQ, Nabulsi N, Ropchan J, et al. (2016). PET imaging reveals sex differences in kappa opioid receptor availability in humans, in vivo. Am J Nucl Med Mol Imaging, 6, 205–214. [PMC free article] [PubMed] [Google Scholar]
- Vitale G, Arletti R, Ruggieri V, Cifani C, & Massi M (2006). Anxiolytic–like effects of nociceptin/orphanin FQ in the elevated plus maze and in the conditioned defensive burying test in rats. Peptides, 27, 2193–2200. [DOI] [PubMed] [Google Scholar]
- Vitale G, Ruggieri V, Filaferro M, Frigeri C, Alboni S, Tascedda F, et al. (2009). Chronic treatment with the selective NOP receptor antagonist [Nphe 1, Arg 14, Lys 15]N/OFQ–NH 2 (UFP–101) reverses the behavioural and biochemical effects of unpredictable chronic mild stress in rats. Psychopharmacology (Berl), 207, 173–189. [DOI] [PubMed] [Google Scholar]
- Vorspan F, Mehtelli W, Dupuy G, Bloch V, & Lepine JP (2015). Anxiety and substance use disorders: co–occurrence and clinical issues. Curr Psychiatry Rep, 17, 4. [DOI] [PubMed] [Google Scholar]
- Wagner JJ, Etemad LR, & Thompson AM (2001). Opioid–mediated facilitation of long–term depression in rat hippocampus. J Pharmacol Exp Ther, 296, 776–781. [PubMed] [Google Scholar]
- Walker BM, & Koob GF (2008). Pharmacological evidence for a motivational role of kappa–opioid systems in ethanol dependence. Neuropsychopharmacology, 33, 643–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker EA, Zernig G, & Woods JH (1995). Buprenorphine antagonism of mu opioids in the rhesus monkey tail–withdrawal procedure. J Pharmacol Exp Ther, 273, 1345–1352. [PubMed] [Google Scholar]
- Wang J, Charboneau R, Barke RA, Loh HH, & Roy S (2002). Mu–opioid receptor mediates chronic restraint stress–induced lymphocyte apoptosis. J Immunol, 169, 3630–3636. [DOI] [PubMed] [Google Scholar]
- Wang J, Song Q, Xu A, Bao Y, Xu Y, & Zhu Q (2017). Design, synthesis and biological evaluation of aminobenzyloxyarylamide derivatives as selective kappa opioid receptor antagonists. Eur J Med Chem, 130, 15–25. [DOI] [PubMed] [Google Scholar]
- Wang JB, Johnson PS, Persico AM, Hawkins AL, Griffin CA, & Uhl GR (1994). Human mu opiate receptor. cDNA and genomic clones, pharmacologic characterization and chromosomal assignment. FEBS Lett, 338, 217–222. [DOI] [PubMed] [Google Scholar]
- Wang Q, Yue XF, Qu WM, Tan R, Zheng P, Urade Y, et al. (2013). Morphine inhibits sleep–promoting neurons in the ventrolateral preoptic area via mu receptors and induces wakefulness in rats. Neuropsychopharmacology, 38, 791–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber MM, & Emrich HM (1988). Current and historical concepts of opiate treatment in psychiatric disorders. Int Clin Psychopharmacol, 3, 255–266. [DOI] [PubMed] [Google Scholar]
- Weerts EM, Wand GS, Kuwabara H, Munro CA, Dannals RF, Hilton J, et al. (2011). Positron emission tomography imaging of mu–and delta–opioid receptor binding in alcohol–dependent and healthy control subjects. Alcohol Clin Exp Res, 35, 2162–2173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei LN, Hu X, Bi J, & Loh H (2000). Post–transcriptional regulation of mouse kappa–opioid receptor expression. Mol Pharmacol, 57, 401–408. [PubMed] [Google Scholar]
- Weiss N, Tadmouri A, Mikati M, Ronjat M, & De Waard M (2007). Importance of voltage–dependent inactivation in N–type calcium channel regulation by G–proteins. Pflugers Arch, 454, 115–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss RD, Potter JS, Fiellin DA, Byrne M, Connery HS, Dickinson W, et al. (2011). Adjunctive counseling during brief and extended buprenorphine–naloxone treatment for prescription opioid dependence: a 2–phase randomized controlled trial. Arch Gen Psychiatry, 68, 1238–1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wells AM, Ridener E, Bourbonais CA, Kim W, Pantazopoulos H, Carroll FI, et al. (2017). Effects of Chronic Social Defeat Stress on Sleep and Circadian Rhythms Are Mitigated by Kappa–Opioid Receptor Antagonism. J Neurosci, 37, 7656–7668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wentland MP, Lou R, Lu Q, Bu Y, Denhardt C, Jin J, et al. (2009a). Syntheses of novel high affinity ligands for opioid receptors. Bioorg Med Chem Lett, 19, 2289–2294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wentland MP, Lou R, Lu Q, Bu Y, VanAlstine MA, Cohen DJ, et al. (2009b). Syntheses and opioid receptor binding properties of carboxamido–substituted opioids. Bioorg Med Chem Lett, 19, 203–208. [DOI] [PubMed] [Google Scholar]
- Wentland MP, Lu Q, Lou R, Bu Y, Knapp BI, & Bidlack JM (2005). Synthesis and opioid receptor binding properties of a highly potent 4–hydroxy analogue of naltrexone. Bioorg Med Chem Lett, 15, 2107–2110. [DOI] [PubMed] [Google Scholar]
- Westbrook RF, Good AJ, & Kiernan MJ (1997). Microinjection of morphine into the nucleus accumbens impairs contextual learning in rats. Behav Neurosci, 111, 996–1013. [DOI] [PubMed] [Google Scholar]
- Westbrook RF, Greeley JD, Nabke CP, & Swinbourne AL (1991). Aversive conditioning in the rat: effects of a benzodiazepine and of an opioid agonist and antagonist on conditioned hypoalgesia and fear. J Exp Psychol Anim Behav Process, 17, 219–230. [DOI] [PubMed] [Google Scholar]
- WHO. (2017). Depression and Other Common Mental Disorders: Global Health Estimates . Licence: CC BY–NC–SA 3.0 IGO. [Google Scholar]
- Williams AV, Laman–Maharg A, Armstrong CV, Ramos–Maciel S, Minie VA, & Trainor BC (2018). Acute inhibition of kappa opioid receptors before stress blocks depression–like behaviors in California mice. Prog Neuropsychopharmacol Biol Psychiatry, 86, 166–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams TJ, Akama KT, Knudsen MG, McEwen BS, & Milner TA (2011). Ovarian hormones influence corticotropin releasing factor receptor colocalization with delta opioid receptors in CA1 pyramidal cell dendrites. Exp Neurol, 230, 186–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson MA, & Junor L (2008). The role of amygdalar mu–opioid receptors in anxiety–related responses in two rat models. Neuropsychopharmacology, 33, 2957–2968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winters BL, Gregoriou GC, Kissiwaa SA, Wells OA, Medagoda DI, Hermes SM, et al. (2017). Endogenous opioids regulate moment–to–moment neuronal communication and excitability. Nat Commun, 8, 14611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witkin JM, Rorick–Kehn LM, Benvenga MJ, Adams BL, Gleason SD, Knitowski KM, et al. (2016). Preclinical findings predicting efficacy and side–effect profile of LY2940094, an antagonist of nociceptin receptors. Pharmacol Res Perspect, 4, e00275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witkin JM, Statnick MA, Rorick–Kehn LM, Pintar JE, Ansonoff M, Chen Y, et al. (2014). The biology of Nociceptin/Orphanin FQ (N/OFQ) related to obesity, stress, anxiety, mood, and drug dependence. Pharmacol Ther, 141, 283–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolozin BL, & Pasternak GW (1981). Classification of multiple morphine and enkephalin binding sites in the central nervous system. Proc Natl Acad Sci U S A, 78, 6181–6185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood SK, Walker HE, Valentino RJ, & Bhatnagar S (2010). Individual differences in reactivity to social stress predict susceptibility and resilience to a depressive phenotype: role of corticotropin–releasing factor. Endocrinology, 151, 1795–1805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wook Koo J, Labonte B, Engmann O, Calipari ES, Juarez B, Lorsch Z, et al. (2016). Essential Role of Mesolimbic Brain–Derived Neurotrophic Factor in Chronic Social Stress–Induced Depressive Behaviors. Biol Psychiatry, 80, 469–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yovell Y, Bar G, Mashiah M, Baruch Y, Briskman I, Asherov J, et al. (2016). Ultra–Low–Dose Buprenorphine as a Time–Limited Treatment for Severe Suicidal Ideation: A Randomized Controlled Trial. Am J Psychiatry, 173, 491–498. [DOI] [PubMed] [Google Scholar]
- Yu TP, Fein J, Phan T, Evans CJ, & Xie CW (1997). Orphanin FQ inhibits synaptic transmission and long–term potentiation in rat hippocampus. Hippocampus, 7, 88–94. [DOI] [PubMed] [Google Scholar]
- Zaveri NT (2016). Nociceptin Opioid Receptor (NOP) as a Therapeutic Target: Progress in Translation from Preclinical Research to Clinical Utility. J Med Chem, 59, 7011–7028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Torregrossa MM, Jutkiewicz EM, Shi YG, Rice KC, Woods JH, et al. (2006). Endogenous opioids upregulate brain–derived neurotrophic factor mRNA through delta–and micro–opioid receptors independent of antidepressant–like effects. Eur J Neurosci, 23, 984–994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Butelman ER, Schlussman SD, Ho A, & Kreek MJ (2005a). Effects of the plant–derived hallucinogen salvinorin A on basal dopamine levels in the caudate putamen and in a conditioned place aversion assay in mice: agonist actions at kappa opioid receptors. Psychopharmacology (Berl), 179, 551–558. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Wang D, Johnson AD, Papp AC, & Sadee W (2005b). Allelic expression imbalance of human mu opioid receptor (OPRM1) caused by variant A118G. J Biol Chem, 280, 32618–32624. [DOI] [PubMed] [Google Scholar]
- Zheng MQ, Kim SJ, Holden D, Lin SF, Need A, Rash K, et al. (2014). An Improved Antagonist Radiotracer for the kappa–Opioid Receptor: Synthesis and Characterization of (11)C–LY2459989. J Nucl Med, 55, 1185–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng MQ, Nabulsi N, Kim SJ, Tomasi G, Lin SF, Mitch C, et al. (2013). Synthesis and evaluation of 11C–LY2795050 as a kappa–opioid receptor antagonist radiotracer for PET imaging. J Nucl Med, 54, 455–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Hsu MS, & Pintar JE (1998). Developmental expression of the mu, kappa, and delta opioid receptor mRNAs in mouse. J Neurosci, 18, 2538–2549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zubieta JK, Dannals RF, & Frost JJ (1999). Gender and age influences on human brain mu–opioid receptor binding measured by PET. Am J Psychiatry, 156, 842–848. [DOI] [PubMed] [Google Scholar]
- Zubieta JK, Smith YR, Bueller JA, Xu Y, Kilbourn MR, Jewett DM, et al. (2001). Regional mu opioid receptor regulation of sensory and affective dimensions of pain. Science, 293, 311–315. [DOI] [PubMed] [Google Scholar]