

Abstract
High‐grade neuroepithelial tumor with BCOR exon 15 internal tandem duplication (HGNET BCOR ex15 ITD) is a recently proposed tumor entity of the central nervous system (CNS) with a distinct methylation profile and characteristic genetic alteration. The complete spectrum of histologic features, accompanying genetic alterations, clinical outcomes, and optimal treatment for this new tumor entity are largely unknown. Here, we performed a comprehensive assessment of 10 new cases of HGNET BCOR ex15 ITD. The tumors mostly occurred in young children and were located in the cerebral or cerebellar hemispheres. On imaging all tumors were large, well‐circumscribed, heterogeneous masses with variable enhancement and reduced diffusion. They were histologically characterized by predominantly solid growth, glioma‐like fibrillarity, perivascular pseudorosettes, and palisading necrosis, but absence of microvascular proliferation. They demonstrated sparse to absent GFAP expression, no synaptophysin expression, variable OLIG2 and NeuN positivity, and diffuse strong BCOR nuclear positivity. While BCOR exon 15 internal tandem duplication was the solitary pathogenic alteration identified in six cases, four cases contained additional alterations including CDKN2A/B homozygous deletion, TERT amplification or promoter hotspot mutation, and damaging mutations in TP53, BCORL1, EP300, SMARCA2 and STAG2. While the limited clinical follow‐up in prior reports had indicated a uniformly dismal prognosis for this tumor entity, this cohort includes multiple long‐term survivors. Our study further supports inclusion of HGNET BCOR ex15 ITD as a distinct CNS tumor entity and expands the known clinicopathologic, radiographic, and genetic features.
Keywords: BCOR exon 15 internal tandem duplication, brain tumor, HGNET, high‐grade neuroepithelial tumor, molecular neuro‐oncology, molecular neuropathology
Introduction
A recent genomic profiling study of tumors previously diagnosed as primitive neuroectodermal tumor of the central nervous system (CNS‐PNET) identified a new subtype of high‐grade neuroepithelial tumor unified by a recurrent internal tandem duplication within exon 15 of the BCOR transcriptional co‐repressor gene and a distinct genome‐wide methylation profile compared to all other CNS tumor entities assessed to date 26. These tumors (hereafter abbreviated HGNET BCOR ex15 ITD) predominantly arose in the cerebral or cerebellar hemispheres of young children, had an approximately equal male to female distribution, were histologically characterized by perivascular pseudorosettes and glioma‐like fibrillarity, and had poor outcomes in the small number of cases with available clinical follow‐up.
The protein product of the BCOR gene was initially identified in 2000 as a novel binding partner of BCL6, which is a POZ/zinc finger domain‐containing transcriptional repressor protein 9. BCOR was demonstrated to function as a transcriptional co‐repressor when tethered to DNA that potentiated BCL6 mediated repression, specifically through its association with class I and II histone deacetylases 9. Inherited/constitutional mutations in the BCOR gene were identified in 2004 as the cause of an X‐linked oculofaciocardiodental syndrome (Online Mendelian Inheritance in Man #300166) characterized by microphthalmia, congenital cataracts, long narrow face, dental radiculomegaly with persistent primary teeth, and cardiac septal defects 15. Studies in osteodentinogenic mesenchymal stem cells from a patient with oculofaciocardiodental syndrome found that BCOR mutation disrupted homeostasis by resulting in increased methylation of lysine 4 and lysine 36 on the tail of histone H3, thereby reactivating transcription of silenced target genes 6. Thus, BCOR appears to be a critical epigenetic regulatory gene whose constitutional disruption results in a severe developmental syndrome affecting multiple organ systems.
Whereas constitutional mutations in the BCOR gene perturb organogenesis during development, somatic alterations in BCOR have now been identified as recurrent genetic drivers in a wide spectrum of human tumor types. A recurrent internal tandem duplication within exon 15 of BCOR has been identified as the defining genetic alteration in clear cell sarcoma of the kidney, primitive myxoid mesenchymal tumor of infancy, and a subset of CNS high‐grade neuroepithelial tumors 2, 10, 23, 24, 26, 28. Distinct from exon 15 internal tandem duplication, in‐frame gene fusions involving the BCOR gene are present in a subset of endometrial stromal sarcomas, pediatric low‐grade gliomas, and undifferentiated round cell sarcomas of bone and soft tissue, most often with the ZC3H7B gene in endometrial stromal sarcomas, the EP300 gene in pediatric low‐grade gliomas, and the CCNB3 gene in bone and soft tissue sarcomas 13, 18, 20, 21, 25, 27. Lastly, somatic truncating mutations or homozygous deletions of BCOR have been recurrently found in acute myeloid leukemia, retinoblastoma, medulloblastoma, and diffuse gliomas 5, 8, 12, 14, 16, 22, 29. Thus, the BCOR gene appears to be an important oncogenic driver in a broad spectrum of human tumor types, with distinct genetic alterations specific to different tumor entities.
Only a few additional patients with HGNET BCOR ex15 ITD have been reported since the initial description of this tumor entity by Sturm et al. 1, 19, 26, 30. As such, the full spectrum of histologic features, accompanying genetic alterations, clinical outcomes, and optimal treatment for this new tumor entity remain largely undefined. Here, we report our experience with the clinical, radiographic, histologic, ultrastructural, and genetic features of 10 new cases of HGNET BCOR ex15 ITD.
Methods
Patient cohort
Ten children diagnosed with high‐grade neuroepithelial tumors found to harbor BCOR exon 15 internal tandem duplication by targeted next‐generation sequencing analysis at UCSF Medical Center were included in this study. Patient SF‐BCOR‐2 has been previously reported in part 11. Preoperative imaging studies were reviewed for each patient by two expert neuroradiologists (M.A. and S.C.). This study was approved by the Committee on Human Research of the University of California, San Francisco, with a waiver of patient consent.
Tumor samples and histology review
All tumor specimens were fixed in 10% neutral‐buffered formalin and embedded in paraffin. Pathologic review of all tumors was conducted by a group of expert neuropathologists (S.P.F., M.P., A.W.B., T.T., A.P., and D.A.S.).
Immunohistochemistry
Immunohistochemistry was performed on whole formalin‐fixed, paraffin‐embedded tissue sections using the following antibodies: glial fibrillary acidic protein (GFAP, Dako, cat# GA524, polyclonal, 1:3000 dilution, 15‐minute incubation); oligodendrocyte transcription factor 2 (OLIG2, Immuno Bio Labs, polyclonal, 1:200 dilution, 30‐minute incubation); NeuN (Chemicon, cat# MAB377, clone A60, 1:4000 dilution, 15‐minute incubation); synaptophysin (Cell Marque, cat# 336A, polyclonal, 1:100 dilution, 30‐minute incubation); neurofilament (Cell Marque, cat# 302M, clone 2F11, undiluted, 30‐minute incubation); epithelial membrane antigen (EMA, Leica, cat# PA0035, clone GP1.4, undiluted, 15‐minute incubation); BCOR (Santa Cruz Biotechnology, cat# sc‐514576, clone C‐10, 1:200 dilution, 30‐minute incubation); p53 (Leica, cat# PA0057, clone DO‐7, undiluted, 15‐minute incubation); Ki67 (Dako, cat# GA626, clone MIB1, 1:50 dilution, 30‐minute incubation). All immunostaining was performed on a Leica Bond‐III automated stainer. ER1 antigen retrieval was used for OLIG2, neurofilament, NeuN and EMA antibodies. ER2 antigen retrieval was used for synaptophysin, BCOR, p53 and Ki67 antibodies. No antigen retrieval was performed for GFAP. Diaminobenzidine was used as the chromogen, followed by hematoxylin counterstain.
Electron microscopy
Ultrathin (80 nm) sections of glutaraldehyde‐fixed, Epon‐embedded tissue were stained with 2% uranyl acetate at the UCSF Electron Microscopy Core Lab. Sections were subsequently examined in a JEOL 1400 transmission electron microscope at 120 kV. Images were recorded with a Gatan SC1000 CCDE camera.
Targeted next‐generation sequencing
Genomic DNA was extracted from formalin‐fixed, paraffin‐embedded blocks of tumor tissue from the 10 tumors using the QIAamp DNA FFPE Tissue Kit (Qiagen). In nine cases, tumor tissue from the initial resection was used for sequencing analysis. The tumor tissue analyzed for patient SF‐BCOR‐8 was from the recurrent tumor following initial gross total resection, 60 Gy cranial radiation, and adjuvant chemotherapy with temozolomide and bevacizumab. Genomic DNA was also extracted from a peripheral blood sample for four patients (SF‐BCOR‐1, SF‐BCOR‐2, SF‐BCOR‐5 and SF‐BCOR‐7) using the QIAamp DNA Blood Midi Kit (Qiagen). Capture‐based next‐generation DNA sequencing was performed using an assay that targets all coding exons of 479 cancer‐related genes, select introns and upstream regulatory regions of 47 genes to enable detection of structural variants including gene fusions, and DNA segments at regular intervals along each chromosome to enable genome‐wide copy number and zygosity analysis, with a total sequencing footprint of 2.8 Mb (UCSF500 Cancer Panel; Supplementary Table 1; reference 11). Multiplex library preparation was performed using the KAPA Hyper Prep Kit (Roche) according to the manufacturer’s specifications using 250 ng of sample DNA. Hybrid capture of pooled libraries was performed using a custom oligonucleotide library (Nimblegen SeqCap EZ Choice). Captured libraries were sequenced as paired‐end 100 bp reads on an Illumina HiSeq 2500 instrument. Sequence reads were mapped to the reference human genome build GRCh37 (hg19) using the Burrows–Wheeler aligner (BWA). Recalibration and deduplication of reads was performed using the Genome Analysis Toolkit (GATK). Coverage and sequencing statistics were determined using Picard CalculateHsMetrics and Picard CollectInsertSizeMetrics. Single nucleotide variant and insertion/deletion mutation calling was performed with FreeBayes and PinDel. Structural variant calling was performed with Delly. Variant annotation was performed with Annovar. Single nucleotide variants and insertions/deletions were visualized and verified using Integrated Genome Viewer. Genome‐wide copy number analysis based on on‐target and off‐target reads was performed by CNVkit and visualized using Nexus Copy Number (Biodiscovery).
Clinical summary and Kaplan–Meier survival analysis
Kaplan–Meier survival analysis was performed using GraphPad Prism software. In addition to the 10 patients from this cohort, all previously reported cases of high‐grade neuroepithelial tumors with confirmed BCOR exon 15 internal tandem duplication by targeted Sanger or next‐generation sequencing were included in the clinical summary and survival analysis in Figure 10. These included 15 cases from Sturm et al., 6 cases from Yoshida et al., 3 cases from Appay et al., and 1 case from Paret et al. 1, 19, 26, 30. The clinical features and data source of these previously reported 25 patients are shown in Supplementary Table 9. This analysis excluded the 19 cases from Sturm et al. that clustered by methylation profiling with “CNS high‐grade neuroepithelial tumor with BCOR alteration” but did not have genetic analysis confirming BCOR exon 15 internal tandem duplication 26.
Figure 10.
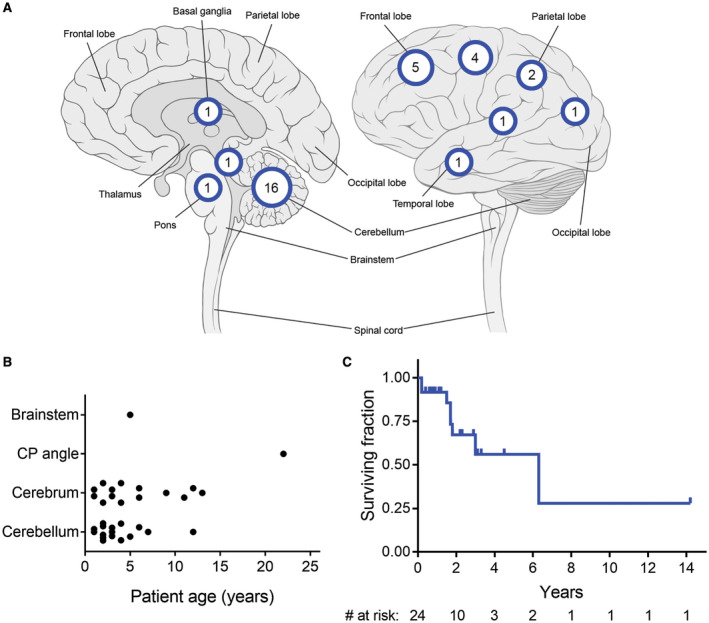
Clinical features of CNS high‐grade neuroepithelial tumor with BCOR exon 15 internal tandem duplication. Clinical data from the 10 patients in this cohort (Supplementary Table 2), as well as all previously reported cases of this tumor entity with confirmed BCOR exon 15 internal tandem duplication (Supplementary Table 9), were aggregated for analysis. A. Location of the 33 tumors with specified anatomic site in the central nervous system. B. Age at initial diagnosis stratified by location for the 32 tumors with specified age and anatomic site. C. Kaplan–Meier survival analysis for the 24 patients with available clinical outcome data.
Results
Clinical features
The three male and seven female patients ranged from 1 to 13 years old (median 3 years) at time of initial diagnosis (Table 1 and Supplementary Table 2). Seven tumors were in young children less than 5 years old, while three tumors were in older children. Presenting symptoms were variable ranging from headaches to seizures to focal neurologic deficits. Tumors were located in the cerebral hemispheres in five patients, in the cerebellar hemispheres in four patients, and in the basal ganglia in one patient. The cerebellar tumors were exclusively present in young children less than 5 years old, while the supratentorial tumors were present in both young and older children.
Table 1.
Clinical features of the 10 patients with CNS high‐grade neuroepithelial tumors with BCOR exon 15 internal tandem duplication.
| Patient ID | Age (yrs) | Sex | Tumor location | Extent of resection | Radiation therapy | Initial chemotherapy | Recurrence | Follow‐up (yrs) | Status at last follow‐up |
|---|---|---|---|---|---|---|---|---|---|
| SF‐BCOR‐1 | 1 | F | Frontoparietal lobe | GTR | None | Multiagent, platinum based | 14 months | 14.2 | Alive, NED |
| SF‐BCOR‐2 | 4 | F | Cerebellum | GTR | Craniospinal | Multiagent, platinum based | None | 1.8 | Alive, NED |
| SF‐BCOR‐3 | 3 | F | Frontal lobe | GTR | Cranial | Multiagent, platinum based | None | 0.4 | Alive, NED |
| SF‐BCOR‐4 | 3 | M | Cerebellum | GTR | None | None | 4 months | 2.3 | Alive w/ disseminated disease |
| SF‐BCOR‐5 | 2 | F | Cerebellum | GTR | None | Multiagent, platinum based | None | 0.7 | Alive, NED |
| SF‐BCOR‐6 | 2 | F | Frontoparietal lobe | GTR | None | Multiagent, platinum based | None | 0.8 | Alive, NED |
| SF‐BCOR‐7 | 9 | F | Basal ganglia | GTR | Cranial | Temozolomide & bevacizumab | None | 2.2 | Alive, NED |
| SF‐BCOR‐8 | 13 | M | Frontal lobe | GTR | Cranial | Temozolomide & bevacizumab | 49 months | 4.5 | Alive, NED |
| SF‐BCOR‐9 | 2 | M | Cerebellum | GTR | Cranial | Temozolomide & bevacizumab | 31 months | 2.9 | Alive, NED |
| SF‐BCOR‐10 | 12 | F | Frontoparietal lobe | GTR | Craniospinal | Multiagent, platinum based | None | 1.1 | Alive, NED |
Imaging features
Preoperative magnetic resonance imaging revealed solid, well‐circumscribed masses in each of the 10 patients (Figure 1 and Supplementary Table 3). The tumors were all large with associated mass effect. Maximal dimension ranged from 3.8 to 10.2 cm. Many of the tumors demonstrated central areas of necrosis or blood products. Contrast enhancement was variable but never showed the ring‐enhancing pattern characteristic of most glioblastomas. Diffusion‐weighted imaging often showed reduced diffusion suggestive of high cellularity neoplasms. Most tumors abutted the overlying dura without definite invasion. No cerebrospinal dissemination was seen at time of diagnosis in any of the patients.
Figure 1.
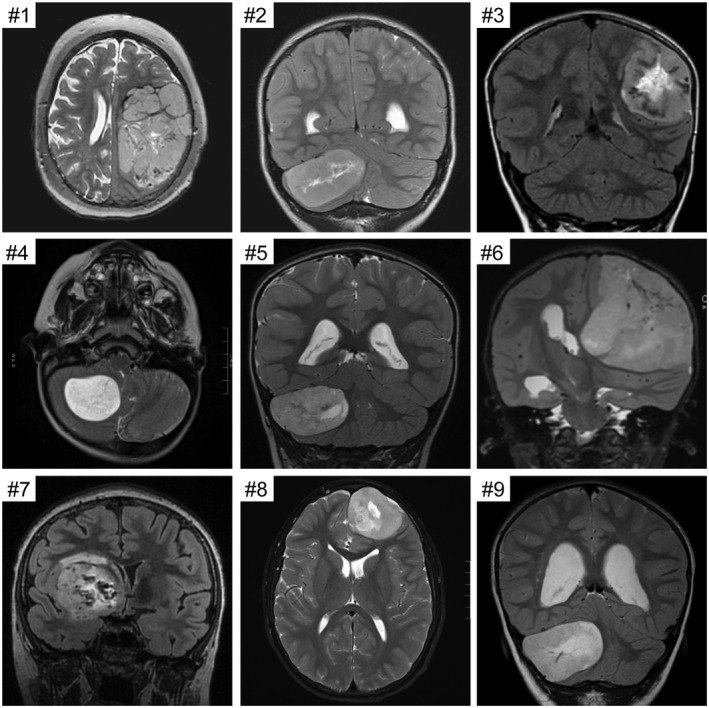
Imaging features of the CNS high‐grade neuroepithelial tumors with BCOR exon 15 internal tandem duplication. Shown are preoperative magnetic resonance images for cases #1–9. All tumors were large, well‐circumscribed, heterogeneous masses with variable enhancement and reduced diffusion. Many of the tumors demonstrated central areas of necrosis or blood products.
Histologic features
The 10 tumors all demonstrated a predominantly solid growth pattern with a sharp border with adjacent brain parenchyma, although a couple of tumors showed infiltration at their interface with adjacent brain (Figures 2, 3, 4, Table 2, and Supplementary Table 4). A prominent feature uniformly seen in all tumors was ependymoma‐like perivascular pseudorosettes with tumor cells aggregated around blood vessels with an intervening anuclear zone; however, in contrast to ependymoma, the perivascular processes were negative for GFAP (see below). The tumor cells were characterized by round to oval nuclei with fine chromatin. Most examples demonstrated glioma‐like fibrillarity. Necrosis was observed in all tumors, almost always with palisading of the tumor cells at the periphery. All tumors were highly vascular with a rich branching capillary network. However, well‐developed microvascular proliferation was not identified in any of the 10 cases. Cellularity and mitotic activity was variable, ranging from areas with low cellularity and scant mitoses to densely cellular areas with numerous mitoses. Some of the tumors had a myxoid and microcystic background, while others had marked stromal and perivascular hyalinization reminiscent of astroblastoma. Microcalcifications were seen in a minority of cases. Rosenthal fibers and eosinophilic granular bodies were not observed in any of the tumors. Three cases featured distinctive Homer Wright‐like rosettes, with tumor cells rosetted around central areas of eosinophilic fibrillar processes; this often raised a differential diagnostic consideration of medulloblastoma or other embryonal neoplasms. However, both the tumor cells and central cores of these rosettes lacked synaptophysin expression (see below), differentiating them from true Homer Wright (neuroblastic) rosettes.
Figure 2.
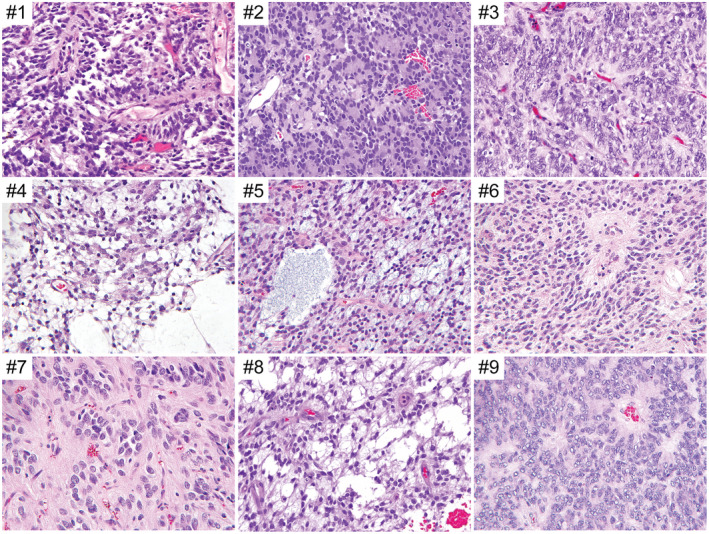
Histologic features of the CNS high‐grade neuroepithelial tumors with BCOR exon 15 internal tandem duplication. Shown are representative hematoxylin and eosin (H&E)‐stained sections of cases #1–9.
Figure 3.
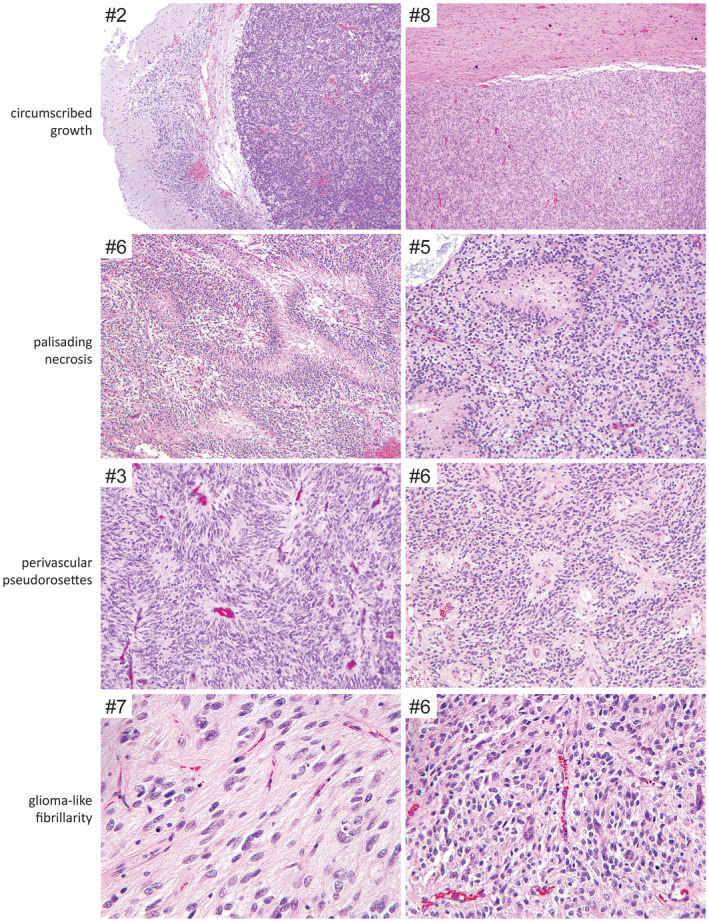
Recurrent histologic features observed in CNS high‐grade neuroepithelial tumor with BCOR exon 15 internal tandem duplication. Shown are H&E‐stained sections demonstrating the circumscribed growth, palisading necrosis, perivascular pseudorosettes, and glioma‐like fibrillarity frequently observed in this tumor entity.
Figure 4.
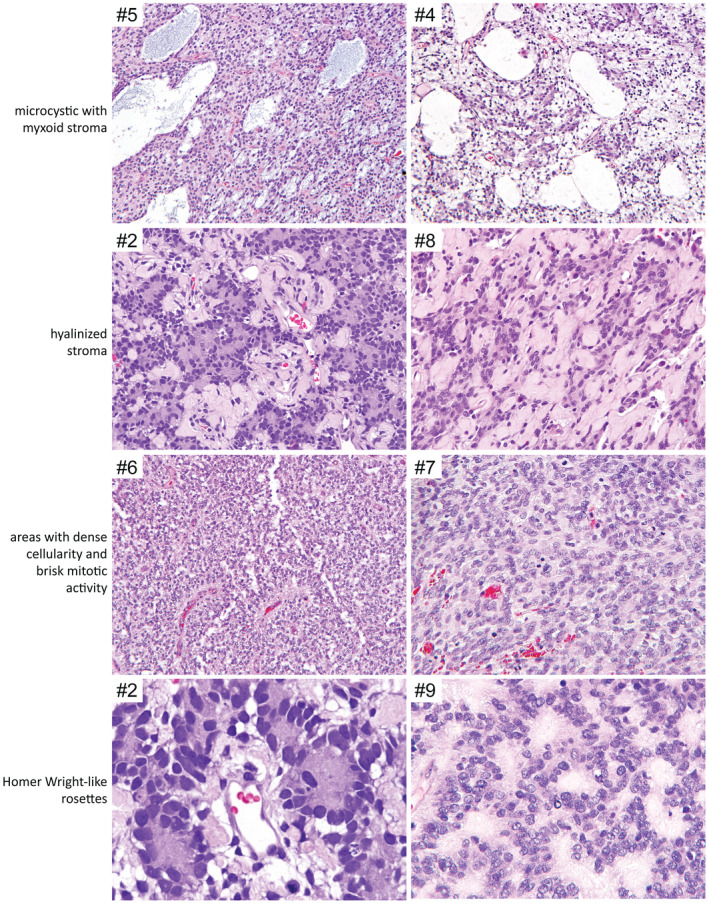
Additional recurrent histologic features observed in a subset of CNS high‐grade neuroepithelial tumor with BCOR exon 15 internal tandem duplication. Shown are H&E‐stained sections demonstrating the microcystic/myxoid background, hyalinized stroma, areas with dense cellularity and brisk mitotic activity, and Homer Wright‐like rosettes observed in a subset of the cases.
Table 2.
Histologic features of the 10 CNS high‐grade neuroepithelial tumors with BCOR exon 15 internal tandem duplication.
| Tumor ID | Growth pattern | Perivascular pseudorosettes | Homer Wright‐like rosettes | Collagenous stroma | Myxoid/microcystic areas | Necrosis | Microvascular proliferation |
|---|---|---|---|---|---|---|---|
| SF‐BCOR‐1 | Mostly solid | Present | Absent | Absent | Absent | Palisading | Absent |
| SF‐BCOR‐2 | Solid | Present | Present | Present | Absent | Palisading | Absent |
| SF‐BCOR‐3 | Solid | Present | Absent | Absent | Absent | Palisading | Absent |
| SF‐BCOR‐4 | Solid | Present | Present | Absent | Present | Palisading | Absent |
| SF‐BCOR‐5 | Mostly solid | Present | Absent | Absent | Present | Palisading | Absent |
| SF‐BCOR‐6 | Solid | Present | Absent | Present | Present | Palisading | Absent |
| SF‐BCOR‐7 | Solid | Present | Absent | Absent | Absent | Non‐palisading | Absent |
| SF‐BCOR‐8 | Solid and infiltrative | Present | Absent | Present | Present | Palisading | Absent |
| SF‐BCOR‐9 | Solid | Present | Present | Absent | Present | Palisading | Absent |
| SF‐BCOR‐10 | Solid and infiltrative | Present | Absent | Present | Absent | Palisading | Absent |
Immunohistochemical features
Immunostaining for GFAP was negative in all or the vast majority of tumor cells in the nine evaluated cases (Figure 5 and Table 3). However, OLIG2 positivity was observed in most tumors, with variable labeling ranging from 10% to 40% of tumor cells. NeuN positivity was also observed in most tumors, with variable labeling of tumor nuclei ranging from 10% to 80%. Synaptophysin labeling was uniformly negative in the nine evaluated tumors. Immunohistochemistry for neurofilament protein often showed scattered cells with cytoplasmic staining. Immunostaining for epithelial membrane antigen (EMA) was typically negative or showed faint granular cytoplasmic staining, distinct from the dot‐like or ring‐like staining pattern typically seen in ependymomas. Diffuse strong nuclear staining for BCOR protein was observed in the eight evaluated cases. The Ki‐67 labeling index was variable ranging from 15% to 60% in the highest areas.
Figure 5.
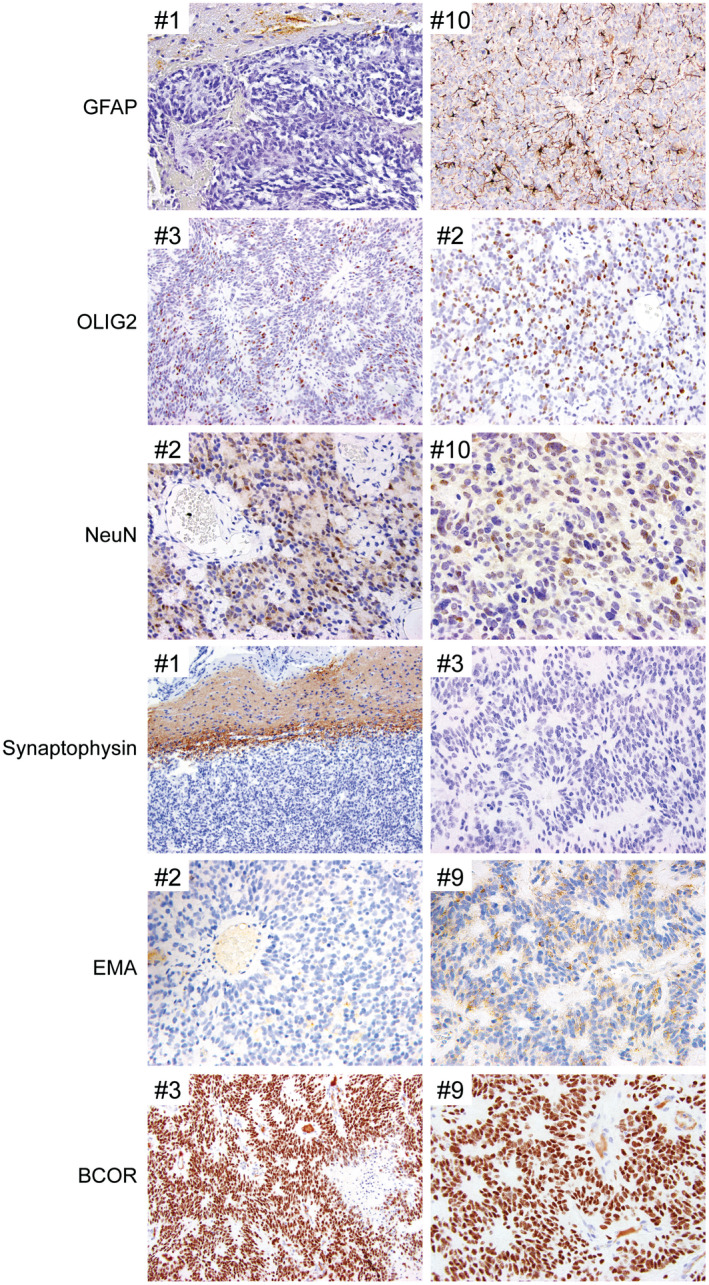
Immunohistochemical features of CNS high‐grade neuroepithelial tumor with BCOR exon 15 internal tandem duplication. Shown are representative immunohistochemical stains demonstrating the sparse to absent GFAP positivity, variable OLIG2 positivity, consistent NeuN positivity, synaptophysin negativity, granular cytoplasmic EMA staining with absence of paranuclear dot‐like positivity, and diffuse strong nuclear BCOR expression.
Table 3.
Immunohistochemical features of the 10 CNS high‐grade neuroepithelial tumors with BCOR exon 15 internal tandem duplication.
| Tumor ID | GFAP | OLIG2 | NeuN | Synaptophysin | Neurofilament | EMA | p53 | Ki‐67 | BCOR |
|---|---|---|---|---|---|---|---|---|---|
| SF‐BCOR‐1 | Negative | – | – | Negative | Positive (10%) | – | 5% | 20% | Strongly positive |
| SF‐BCOR‐2 | Negative | Positive (20%) | Positive (10%) | Negative | Positive (10%) | Granular cytoplasmic positivity | 30% | – | – |
| SF‐BCOR‐3 | Negative | Positive (20%) | Negative | Negative | Positive (5%) | Granular cytoplasmic positivity | – | 40% | Strongly positive |
| SF‐BCOR‐4 | Negative | Positive (10%) | Positive | Negative | – | Negative | 15% | 20% | – |
| SF‐BCOR‐5 | Focally positive | – | Positive | Negative | Negative | Negative | 10% | 15% | Strongly positive |
| SF‐BCOR‐6 | Negative | – | Positive (40%) | Negative | Positive (1%) | Negative | 10% | 60% | Strongly positive |
| SF‐BCOR‐7 | Negative | Positive (30%) | Positive (80%) | – | Negative | Negative | – | – | Strongly positive |
| SF‐BCOR‐8 | – | Positive (40%) | Positive (70%) | Negative | Negative | – | – | – | Strongly positive |
| SF‐BCOR‐9 | Negative | Negative | Positive (20%) | Negative | Positive (10%) | Granular cytoplasmic positivity | – | – | Strongly positive |
| SF‐BCOR‐10 (LG) | Negative | Positive (20%) | Positive (80%) | Negative | Negative | Granular cytoplasmic positivity | 0% | 5% | Strongly positive |
| SF‐BCOR‐10 (HG) | Focally positive | Negative | Positive (20%) | Negative | Positive (1%) | Granular cytoplasmic positivity | 90% | 60% | Strongly positive |
HG = high‐grade/anaplastic component; LG = low‐grade appearing component.
Ultrastructural features
Transmission electron microscopy was performed on two of the tumors (SF‐BCOR‐1 and SF‐BCOR‐7). This analysis showed primitive cells with abundant rough endoplasmic reticulum and limited intermediate cytoskeletal filaments (Figure 6). No tight junctions, cilia, or microvilli characteristic of ependymoma were seen. Additionally, no neurosecretory granules or synaptic vesicles were seen.
Figure 6.
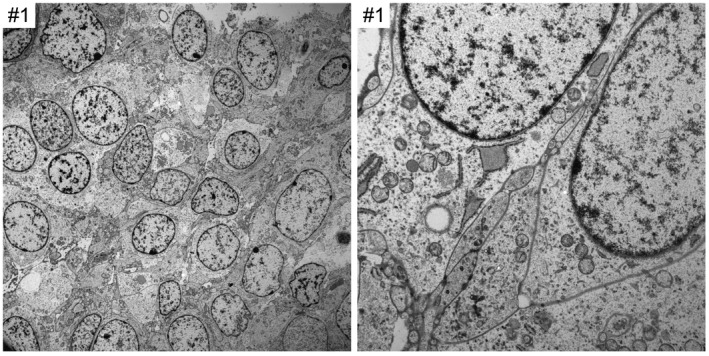
Ultrastructural features of CNS high‐grade neuroepithelial tumor with BCOR exon 15 internal tandem duplication. Shown are electron microscopy images demonstrating primitive cells with abundant rough endoplasmic reticulum. No tight junctions, cilia or microvilli characteristic of ependymal differentiation are seen. Additionally, no neurosecretory granules or synaptic vesicles are observed.
Targeted next‐generation sequencing results
Targeted next‐generation sequencing of approximately 500 cancer‐associated genes and genome‐wide copy number analysis was performed on the 10 tumors as described in the Methods. A tandem duplication within exon 15 of the BCOR gene was identified in all 10 cases (Figures 7 and 8, Supplementary Table 5). The minimally duplicated codons across all 10 tumors were p.L1713_G1738 (RefSeq transcript NM_001123385).
Figure 7.
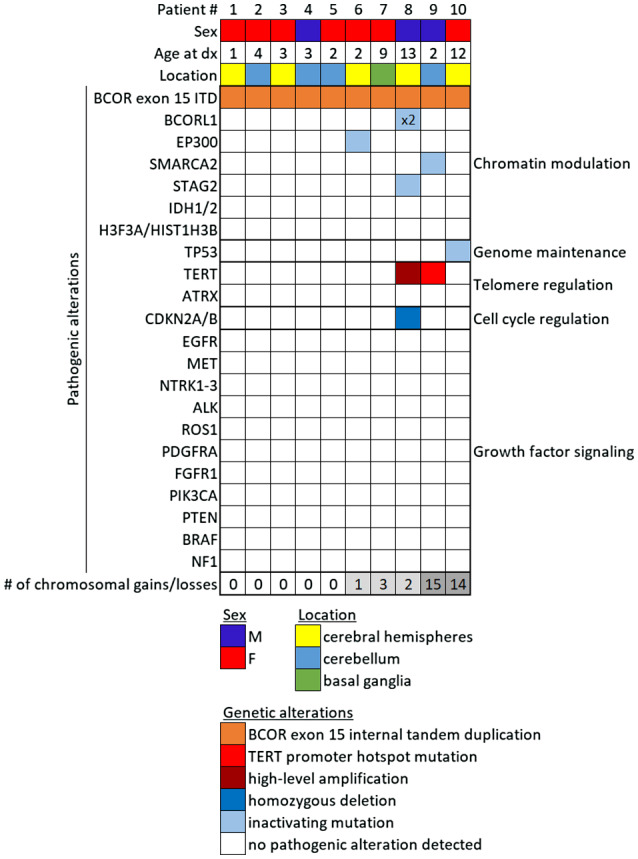
Genetic landscape of CNS high‐grade neuroepithelial tumor with BCOR exon 15 internal tandem duplication. Oncoprint table of the clinical features, likely pathogenic genetic alterations, and quantity of chromosomal copy number alterations in the 10 cases.
Figure 8.
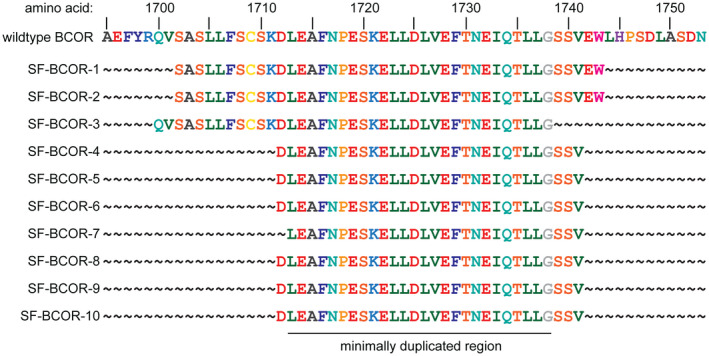
Diagram of the amino acid sequence at the C‐terminus of the BCOR protein showing the duplicated amino acids within exon 15 for the 10 CNS high‐grade neuroepithelial tumors with BCOR exon 15 internal tandem duplication.
In six cases, the BCOR exon 15 internal tandem duplication was the solitary pathogenic alteration identified. Four cases contained additional genetic alterations considered likely to be contributing to tumor pathogenesis (Supplementary Tables 6 and 7). These included SF‐BCOR‐9 with TERT promoter hotspot mutation and a splice site mutation in the SMARCA2 chromatin remodeling gene. SF‐BCOR‐6 contained an additional truncating frameshift mutation in the CREBBP histone acetyltransferase gene, while SF‐BCOR‐10 contained a damaging missense mutation in the TP53 tumor suppressor gene. The genomic profiling that was performed for patient SF‐BCOR‐8 was on the recurrent tumor following initial gross total resection, 60 Gy cranial radiation, and adjuvant chemotherapy with temozolomide and bevacizumab. This recurrent tumor SF‐BCOR‐8 harbored BCOR exon 15 internal tandem duplication along with additional CDKN2A/B homozygous deletion, TERT promoter hotspot mutation (c.‐124C>T), two truncating frameshift mutations in the BCORL1 gene, and a splice site mutation in the STAG2 gene. Whether any of these alterations were present in the initial tumor vs. acquired during disease progression after therapy is unknown (Table 4).
Table 4.
Genetic alterations in the 10 CNS high‐grade neuroepithelial tumors with BCOR exon 15 internal tandem duplication.
| Tumor ID | BCOR exon 15 duplicated amino acids | Additional likely pathogenic alterations | Cytogenetic alterations |
|---|---|---|---|
| SF‐BCOR‐1 | p.S1702_W1743 | None | None |
| SF‐BCOR‐2 | p.S1702_W1743 | None | None |
| SF‐BCOR‐3 | p.Q1700_G1738 | None | None |
| SF‐BCOR‐4 | p.D1712_V1741 | None | None |
| SF‐BCOR‐5 | p.D1712_V1741 | None | None |
| SF‐BCOR‐6 | p.D1712_V1741 | EP300 frameshift mutation | −18 |
| SF‐BCOR‐7 | p.L1713_V1741 | none | +2p, −2q, −10 |
| SF‐BCOR‐8 | p.D1712_V1741 | BCORL1 frameshift mutation x2, STAG2 splice site mutation, TERT promoter hotspot mutation, CDKN2A/B homozygous deletion | −9, −interstitial 10q |
| SF‐BCOR‐9 | p.D1712_V1741 | SMARCA2 splice site mutation, TERT amplification | −distal 1p, +interstitial 1p, +1q, +2q, +interstitial 3q, +5p, +proximal 5q, +7, −9p, +12, −distal 14q, −proximal 15q, +20 |
| SF‐BCOR‐10 | p.D1712_V1741 | TP53 missense mutation | +1p, +1q, +2, +6, +7, +12, +14q, +15q, +17, +18, +19, +21q, +22q |
The somatic mutation burden was uniformly very low (less than 2 somatic mutations per Mb within the 2.8 Mb of the tumor genome that was interrogated by the sequencing assay). Among the four patients in which a normal sample was also sequenced, no pathogenic germline alterations associated with increased cancer risk were identified.
Five of the tumors demonstrated a balanced diploid genome without chromosomal gains or losses (Supplementary Table 8). Three of the tumors demonstrated a paucity of chromosomal gains/losses (fewer than 4). Two of the tumors (SF‐BCOR‐9 and SF‐BCOR‐10) demonstrated markedly aneuploid genomes with numerous chromosomal gains and losses, both at time of initial resection in the absence of prior therapy. No recurrent chromosomal gains or losses in more than 2 of the 10 tumors were observed.
Anaplastic features in HGNET BCOR exon 15 ITD
Case SF‐BCOR‐10 demonstrated two distinct histologic components (Figure 9). One was a lower grade appearing component with moderate cellularity, abundant fibrillarity, numerous calcifications, and scant mitoses. This was apposed with an overtly anaplastic component featuring dense cellularity, increased nuclear pleomorphism, and brisk mitotic activity. The anaplastic component was sharply demarcated from the lower grade component enabling genomic profiling to be performed separately on the two regions. Both components contained the identical BCOR exon 15 internal tandem duplication and a damaging missense mutation in the TP53 tumor suppressor gene. The lower grade component had monosomy 13q as the solitary chromosomal copy number alteration, whereas the anaplastic component harbored numerous chromosomal gains and losses (+1p, +1q [4N], +2, +6, +7, +12, +14q, +15q, +17, +18 [4N], +19, +21q [4N], and +22q).
Figure 9.
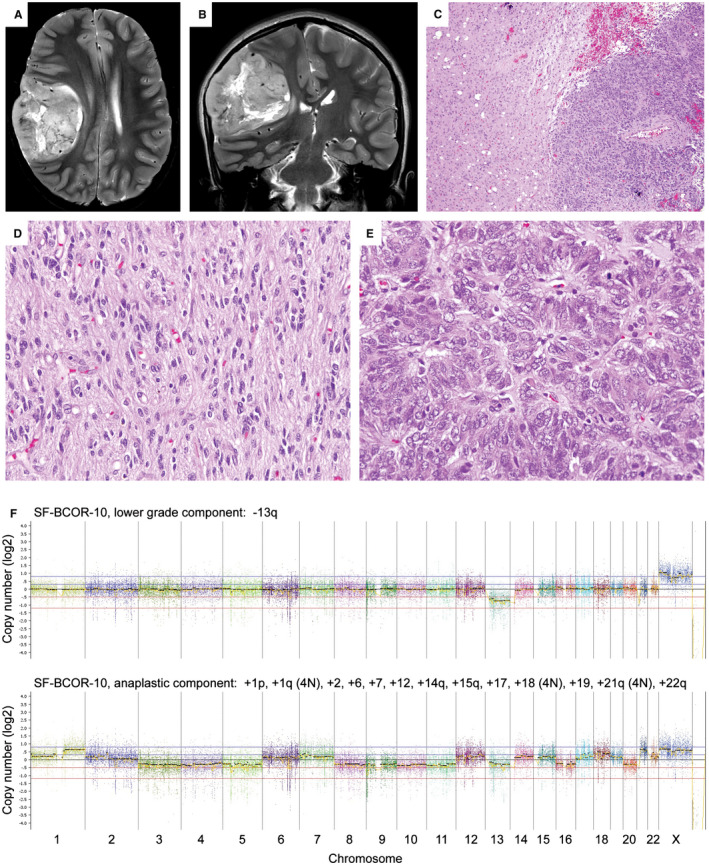
Anaplastic features in CNS HGNET BCOR ex15 ITD (case #10) in association with TP53 inactivation and marked aneuploidy. A,B. Preoperative axial and coronal T2‐weighted MR images showing a circumscribed mass in the right cerebral hemisphere. C–E. H&E‐stained sections showing a biphasic tumor composed of a lower grade appearing component with abundant fibrillar processes (C left, D), and an anaplastic component with dense cellularity, severe nuclear pleomorphism and brisk mitotic activity (C right, E). F. Genome‐wide copy number plots for the lower grade appearing component showing monosomy 13q as the solitary copy number alteration (top), and for the anaplastic component showing numerous chromosomal gains and losses (bottom).
Clinical outcomes
The complete clinical data including extent of resection, treatment regimen, and outcome data from the 10 patients are presented in Table 1 and Supplementary Table 2. All 10 patients initially underwent gross total resection. Four children were subsequently treated with cranial radiation, two children with craniospinal radiation with a boost to the tumor bed and four children did not receive radiation as part of their immediate post‐resection therapy. The initial chemotherapy regimen was temozolomide and bevacizumab following a high‐grade glioma therapy protocol for three children, while six children were initially treated with an intensive multiagent chemotherapy regimen following an embryonal tumor therapy protocol. One child (SF‐BCOR‐4) did not receive any adjuvant radiation or chemotherapy following initial resection.
Clinical follow‐up for this cohort of 10 children ranged from 0.4 to 14.2 years (median 2.0 years). Four children experienced tumor recurrence at 4, 14, 31 and 49 months after initial resection. The earliest recurrence at 4 months was in the child who did not receive any adjuvant therapy (SF‐BCOR‐4), who later developed disseminated disease along the spinal cord at 20 months after initial diagnosis. Two of the other children who experienced recurrence at 31 and 49 months had been treated with cranial radiation and chemotherapy following a high‐grade glioma therapy protocol with temozolomide and bevacizumab (SF‐BCOR‐8 and SF‐BCOR‐9). The fourth child who experienced recurrence at 14 months (SF‐BCOR‐1) had not received radiation therapy but was treated with platinum‐based multiagent chemotherapy following an embryonal tumor therapy protocol. These four children all underwent a second resection confirming tumor recurrence, followed by additional radiation and/or chemotherapy. The recurrent disease in these four children was localized (adjacent to the prior resection cavity), with only one patient in this cohort later experiencing cerebrospinal dissemination (SF‐BCOR‐4). All of the 10 children in this cohort were alive at last clinical follow‐up, including two long‐term survivors at 4.5 years (SF‐BCOR‐8) and 14.2 years (SF‐BCOR‐1) after initial diagnosis.
We next curated clinical data from all reported cases of CNS high‐grade neuroepithelial tumor with confirmed BCOR exon 15 internal tandem duplication (Supplementary Table 9). Patient age, sex, tumor location and survival data were analyzed from the 10 patients in our cohort together with these 25 previously reported patients (Figure 10). The median patient age was 3.5 years (range 0–22 years) at time of initial diagnosis. These 35 patients included 16 males and 19 females. Tumors were located in the cerebellar hemispheres (n = 16), cerebral hemispheres (n = 14), basal ganglia (n = 1), brainstem (n = 1) and cerebellopontine angle (n = 1) (Figure 10A). No significant association of tumor location with patient age at diagnosis was apparent (Figure 10B). Kaplan–Meier analysis of overall survival in the 24 patients with available data revealed a poor prognosis in general, although the number of cases with adequate follow‐up remains limited (Figure 10C).
Discussion
HGNET BCOR ex15 ITD is a recently proposed tumor entity of the central nervous system for which the clinicopathologic features have yet to be fully defined. Here, we have performed comprehensive clinicopathologic, radiographic, and genomic studies on a cohort of 10 new cases. Together with the previously reported 25 cases in the scientific literature to date, our study better defines the distinctive radiographic and pathologic features that characterize this tumor entity, as well as providing detailed outcome data for children treated following either high‐grade glioma or CNS embryonal tumor therapy protocols.
HGNET BCOR ex15 ITD usually presents as a large, well‐circumscribed, heterogeneous mass with reduced diffusion and variable enhancement in the cerebral or cerebellar hemispheres. The majority arise in children younger than 5 years of age, but multiple cases in teenagers or young adults have now been observed. No sex predilection is apparent for this tumor entity, unlike other brain tumor entities such as astroblastoma‐like neuroepithelial tumors with MN1 alteration that demonstrate a significant female predominance 26.
HGNET BCOR ex15 ITD can demonstrate a wide morphologic spectrum, but usually feature a distinctive set of histologic and immunohistochemical features that provide clues to the diagnosis prior to molecular testing. Common features are a mostly solid growth pattern, GFAP‐negative perivascular pseudorosettes, and monotonous round to ovoid nuclei with fine chromatin. Also, the characteristic combination of palisading necrosis without microvascular proliferation is helpful to differentiate these tumors from glioblastoma. The tumors can resemble anaplastic ependymomas due to perivascular pseudorosettes, astroblastomas due to perivascular pseudorosettes and hyalinized/collagenous stroma, or diffuse gliomas due to glial‐like fibrillarity and infiltrative areas at their periphery. Those that contain structures resembling Homer Wright rosettes may also mimic medulloblastoma or CNS neuroblastoma, but differ from these entities based on their lack of synaptophysin expression. These are some of the most likely diagnoses that HGNET BCOR ex15 ITD may have received in the past. However, HGNET BCOR ex15 ITD have an unusual immunohistochemical profile with dual OLIG2 and NeuN positivity, along with sparse to absent GFAP expression and no synaptophysin expression, that can be helpful in distinguishing these tumors from potential histologic mimics. For example, this pattern is distinct from anaplastic ependymomas (usually OLIG2 negative, GFAP positive, and EMA positive with paranuclear dot‐like staining), astroblastomas (usually GFAP positive), and diffuse gliomas (usually GFAP positive and NeuN negative). Additionally, the strong nuclear positivity for BCOR in virtually all tumor cells may be another helpful clue. However, the specificity of diffuse strong nuclear BCOR expression for this tumor entity needs to be further evaluated. For example, the pediatric low‐grade gliomas with EP300‐BCOR fusion can also demonstrate diffuse strong nuclear BCOR expression 27, and we have observed diffuse strong nuclear BCOR expression in an astroblastoma‐like neuroepithelial tumor with MN1 rearrangement that lacked BCOR exon 15 ITD (data not shown).
We believe these HGNET BCOR ex15 ITD are almost certainly of neuroepithelial origin (and therefore not sarcomas), based on the combination of their intraparenchymal location within the brain, glioma‐like fibrillarity, expression of OLIG2 and NeuN proteins, and absence of appreciable intercellular basement membrane deposition in most cases. While the identical BCOR exon 15 ITD is also present in two sarcoma entities, this most likely reflects a common molecular pathogenesis arising in distinct cells of origin: neural progenitor cell for HGNET BCOR ex15 ITD vs. a mesenchymal progenitor cell for clear cell sarcoma of the kidney and primitive myxoid mesenchymal tumor of infancy. Future comparison of the genome‐wide methylation and transcriptome profiles between the different tumor entities that all share the identical BCOR exon 15 ITD is likely to be informative in this regard.
While optimal treatment strategies remain uncertain for this tumor entity, the clinical data from this cohort do provide some new valuable insight. For instance, the one patient (SF‐BCOR‐4) in this cohort who did not receive any adjuvant radiation or chemotherapy after gross total resection experienced rapid local recurrence and also subsequently cerebrospinal dissemination. In combination with the poor outcomes observed for most patients to date, we believe that additional adjuvant therapy beyond the maximal safest resection possible should be strongly considered in all patients. Among the three children in our cohort that were treated following a high‐grade glioma therapy protocol with cranial radiation and adjuvant temozolomide plus bevacizumab, two experienced local recurrence within 4 years after initial resection, whereas the third patient remains recurrence free at approximately 2 years after initial resection. Among the six children in our cohort that were treated following a CNS embryonal tumor therapy protocol with intensive platinum‐based chemotherapy regimens (three without radiation due to young patient age and three with cranial or craniospinal radiation), only one child experienced local recurrence at 14 months but is a long‐term survivor who is currently alive without evidence of disease at 14 years after initial diagnosis. However, the follow‐up interval is less than 2 years for the other five children, making the efficacy of this CNS embryonal tumor therapy protocol inconclusive at this point.
The high‐grade neuroepithelial tumors in this cohort are all unified by the presence of an internal tandem duplication within exon 15 of the BCOR gene. This recurrent internal tandem duplication that is heterozygous (ie, without loss of the remaining wild‐type allele) and localizes within exon 15 that encodes the BCORL‐PCGF1‐binding domain is very likely to function as an activating, gain‐of‐function event. However, the specific mechanism by which this recurrent internal tandem duplication event in BCOR drives tumor development remains unknown, as are methods to therapeutically intervene using a precision medicine approach for these aggressive malignancies of childhood driven by BCOR exon 15 ITD.
Recent genomic investigation has revealed that distinct alterations in the BCOR gene are selected for in different brain tumor entities. Unlike the high‐grade neuroepithelial tumors in this cohort defined by BCOR exon 15 ITD, a group of children with low‐grade gliomas harboring in‐frame EP300–BCOR gene fusions were recently reported that had divergent histologic features and a distinct genome‐wide methylation profile compared to HGNET BCOR ex15 ITD 27. These gliomas with EP300–BCOR fusions had histologic features somewhat resembling either pilocytic astrocytoma or dysembryoplastic neuroepithelial tumor and lacked the perivascular pseudorosettes and palisading necrosis that characterize HGNET BCOR ex15 ITD. Additionally, truncating mutations or homozygous deletions of BCOR or its homolog BCORL1 have been recurrently found in retinoblastoma, medulloblastoma, and diffuse gliomas 12, 14, 16, 22, 29. Among diffuse gliomas, truncating mutations or homozygous deletions in the BCOR or BCORL1 genes are present in a significant fraction of H3 K27M‐mutant diffuse midline gliomas, as well as high‐grade gliomas in the cerebral hemispheres of children 14, 29. In contrast to the exon 15 internal tandem duplication and in‐frame fusion with EP300 that are likely activating gain‐of‐function events, these recurrent nonsense or frameshift mutations as well as homozygous deletions in diffuse gliomas, medulloblastomas, and retinoblastomas are almost certainly functionally inactivating events. Thus, the oncogenic mechanisms by which BCOR alterations promote tumorigenesis are likely to be divergent dependent on the specific genetic alteration present. While Sturm et al. initially proposed the terminology “CNS high‐grade neuroepithelial tumor with BCOR alteration,” it is now clear that the described entity was limited to those neuroepithelial tumors with exon 15 ITD and not merely any BCOR alteration 26. We thus recommend the more precise terminology of “CNS high‐grade neuroepithelial tumor with BCOR exon 15 internal tandem duplication” for this tumor entity moving forward.
While the majority of cases in this patient cohort harbored BCOR exon 15 internal tandem duplication as the solitary pathogenic alteration, a subset harbored additional genetic alterations likely contributing to tumor pathogenesis. These were most frequently inactivating mutations within other transcriptional or epigenetic regulatory genes, including EP300, SMARCA2, STAG2, and BCORL1. Why these tumors selected for additional genetic alterations predicted to disrupt gene expression profiles beyond the BCOR exon 15 ITD is uncertain. Additionally, two of the cases contained TERT alterations, one with gene amplification and one with promoter hotspot mutation, indicating that telomere maintenance in a subset of HGNET BCOR ex15 ITD is accomplished by TERT activation. However, none of the cases harbored ATRX mutation or deletion, indicating that the alternative lengthening of telomeres typical of IDH‐mutant and histone H3‐mutant diffuse gliomas is not common in HGNET BCOR ex15 ITD.
Notably, none of the cases contained IDH1 p.R132 or IDH2 p.R172 mutations that define diffuse lower‐grade gliomas in the cerebral hemispheres of adults 4. None of the cases contained H3F3A or HIST1H3B p.K27M mutation that define the majority of diffuse gliomas within midline structures of the CNS 14, 29. H3F3A p.G34 mutation or SETD2 truncating mutation that define a subset of high‐grade gliomas in the cerebral hemispheres of teenagers and young adults were not present in any of the cases 7, 14. No cases contained amplification, mutation, or rearrangement of receptor tyrosine kinase genes such as EGFR, PDGFRA, MET, FGFR1‐3, NTRK1‐3, ALK, or ROS1 that are common in high‐grade gliomas in children and adults 3, 14, 29. None of the cases contained BRAF mutation or rearrangement, nor any other alteration in components of the Ras‐Raf‐MAP kinase signaling pathway that are common in pediatric low‐grade gliomas 31. None of the cases contained alterations in components of the PI3‐kinase‐Akt‐mTOR signaling pathway including the PTEN, TSC1, TSC2, PIK3CA, or PIK3R1 genes that are common in multiple glioma subtypes 3, 14, 31. MYB or MYBL1 rearrangements that are common in pediatric low‐grade gliomas were not found in any of the cases 31. None of the cases contained MYC or MYCN amplification that are common in Groups 3 and 4 medulloblastomas, as well as a subset of pediatric glioblastomas 14, 16. Additionally, none of the cases contained RELA or YAP1 fusions or NF2 mutation that are common in ependymomas 17. None of the cases contained SMARCB1 or SMARCA4 biallelic inactivation that defines atypical teratoid/rhabdoid tumor, although one tumor did harbor a heterozygous truncating mutation in the related SMARCA2 chromatin remodeling gene. Thus, HGNET BCOR ex15 ITD appear to be genetically distinct from all other CNS tumor entities that have been molecularly defined to date.
In summary, we have comprehensively characterized the new tumor entity “High‐grade neuroepithelial tumor with BCOR exon 15 internal tandem duplication.” While the BCOR exon 15 ITD appears to be the solitary genetic driver in most cases, a subset also acquires additional genetic alterations that include TERT activation, CDKN2A homozygous deletion, and inactivating mutations in other transcriptional and epigenetic regulatory genes including EP300, SMARCA2, STAG2, and BCORL1. Rare examples may also acquire TP53 mutational inactivation along with numerous chromosomal gains/losses that corresponds with histologic anaplasia. Future studies are warranted to identify the cellular mechanisms by which BCOR exon 15 ITD drives tumorigenesis and determine the optimal treatment strategies for affected children.
Conflict of interest
None of the authors have any conflicts of interest to disclose.
Supporting information
Table S1. List of the 479 genes targeted for sequencing on the UCSF500 Cancer Panel. All coding exons were captured for sequencing from each of these genes, with those highlighted genes also having select intronic or upstream regulatory regions that were captured for sequencing to enable detection of structural variants.
Table S2. Clinical features of the 10 patients with CNS high‐grade neuroepithelial tumor with BCOR exon 15 internal tandem duplication.
Table S3. Imaging features at time of presentation for the 10 cases of CNS high‐grade neuroepithelial tumor with BCOR exon 15 internal tandem duplication.
Table S4. Histologic features of the 10 CNS high‐grade neuroepithelial tumors with BCOR exon 15 internal tandem duplication.
Table S5. BCOR exon 15 internal tandem duplications present in the 10 high‐grade neuroepithelial tumors.
Table S6. Likely pathogenic single nucleotide variants and indels identified in the six cases with tumor‐only sequencing analysis.
Table S7. Complete list of somatic nonsynonymous single nucleotide variants and indels identified in the four cases with paired tumor‐normal sequencing analysis.
Table S8. Chromosomal copy number alterations identified in the 10 CNS high‐grade neuroepithelial tumors with BCOR exon 15 internal tandem duplication.
Table S9. Clinical features of the 35 reported cases of CNS high‐grade neuroepithelial tumor with confirmed BCOR exon 15 internal tandem duplication.
Acknowledgements
This study was supported by the NIH Director’s Early Independence Award (DP5 OD021403) to D.A.S. We thank the staff of the UCSF Clinical Cancer Genomics Laboratory for assistance with genetic profiling. We thank the staff of the UCSF Electron Microscopy Core Lab (Cosima Carnahan and Sherry Kamiya) for assistance with ultrastructural analysis. We thank Sarah Bowman and Torrick Taylor for assistance with digital slide scanning.
Data Availability Statement
Scanned image files of H&E‐stained sections from the 10 tumors from which representative images are presented are available for downloading and viewing at the following link: https://figshare.com/projects/High-grade_neuroepithelial_tumor_with_BCOR_exon_15_internal_tandem_duplication/61691. Sequencing data files are available from the authors upon request.
References
- 1. Appay R, Macagno N, Padovani L, Korshunov A, Kool M, Andre N et al (2017) HGNET‐BCOR tumors of the cerebellum: clinicopathologic and molecular characterization of 3 cases. Am J Surg Pathol 41:1254–1260. [DOI] [PubMed] [Google Scholar]
- 2. Astolfi A, Melchionda F, Perotti D, Fois M, Indio V, Urbini M et al (2015) Whole transcriptome sequencing identifies BCOR internal tandem duplication as a common feature of clear cell sarcoma of the kidney. Oncotarget 6:40934–40939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Brennan CW, Verhaak RG, McKenna A, Campos B, Noushmehr H, Salama SR et al (2013) The somatic genomic landscape of glioblastoma. Cell 155:462–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Cancer Genome Atlas Research Network , Brat DJ, Verhaak RG, Aldape KD, Yung WK, Salama SR et al (2015) Comprehensive, integrative genomic analysis of diffuse lower‐grade gliomas. N Engl J Med 372:2481–2498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Damm F, Chesnais V, Nagata Y, Yoshida K, Scourzic L, Okuno Y et al (2013) BCOR and BCORL1 mutations in myelodysplastic syndromes and related disorders. Blood 122:3169–3177. [DOI] [PubMed] [Google Scholar]
- 6. Fan Z, Yamaza T, Lee JS, Yu J, Wang S, Fan G et al (2009) BCOR regulates mesenchymal stem cell function by epigenetic mechanisms. Nat Cell Biol 11:1002–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Fontebasso AM, Schwartzentruber J, Khuong‐Quang DA, Liu XY, Sturm D, Korshunov A et al (2013) Mutations in SETD2 and genes affecting histone H3K36 methylation target hemispheric high‐grade gliomas. Acta Neuropathol 125:659–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Grossmann V, Tiacci E, Holmes AB, Kohlmann A, Martelli MP, Kern W et al (2011) Whole‐exome sequencing identifies somatic mutations of BCOR in acute myeloid leukemia with normal karyotype. Blood 118:6153–6163. [DOI] [PubMed] [Google Scholar]
- 9. Huynh KD, Fischle W, Verdin E, Bardwell VJ (2000) BCoR, a novel corepressor involved in BCL‐6 repression. Genes Dev 14:1810–1823. [PMC free article] [PubMed] [Google Scholar]
- 10. Kao YC, Sung YS, Zhang L, Huang SC, Argani P, Chung CT et al (2016) Recurrent BCOR internal tandem duplication and YWHAE‐NUTM2B fusions in soft tissue undifferentiated round cell sarcoma of infancy: overlapping genetic features with clear cell sarcoma of kidney. Am J Surg Pathol 40:1009–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kline CN, Joseph NM, Grenert JP, van Ziffle J, Talevich E, Onodera C et al (2017) Targeted next‐generation sequencing of pediatric neuro‐oncology patients improves diagnosis, identifies pathogenic germline mutations, and directs targeted therapy. Neuro‐oncology 19:699–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kooi IE, Mol BM, Massink MP, Ameziane N, Meijers‐Heijboer H, Dommering CJ et al (2016) Somatic genomic alterations in retinoblastoma beyond RB1 are rare and limited to copy number changes. Sci Rep 6:25264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lewis N, Soslow RA, Delair DF, Park KJ, Murali R, Hollmann TJ et al (2018) ZC3H7B‐BCOR high‐grade endometrial stromal sarcomas: a report of 17 cases of a newly defined entity. Mod Pathol 31:674–684. [DOI] [PubMed] [Google Scholar]
- 14. Mackay A, Burford A, Carvalho D, Izquierdo E, Fazal‐Salom J, Taylor KR et al (2017) Integrated molecular meta‐analysis of 1,000 pediatric high‐grade and diffuse intrinsic pontine glioma. Cancer Cell 32:520–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ng D, Thakker N, Corcoran CM, Donnai D, Perveen R, Schneider A et al (2004) Oculofaciocardiodental and Lenz microphthalmia syndromes result from distinct classes of mutations in BCOR. Nat Genet 36:411–416. [DOI] [PubMed] [Google Scholar]
- 16. Northcott PA, Buchhalter I, Morrissy AS, Hovestadt V, Weischenfeldt J, Ehrenberger T et al (2017) The whole‐genome landscape of medulloblastoma subtypes. Nature 547:311–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Pajtler KW, Witt H, Sill M, Jones DT, Hovestadt V, Kratochwil F et al (2015) Molecular classification of ependymal tumors across all CNS compartments, histopathological grades, and age groups. Cancer Cell 27:728–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Panagopoulos I, Thorsen J, Gorunova L, Haugom L, Bjerkehagen B, Davidson B et al (2013) Fusion of the ZC3H7B and BCOR genes in endometrial stromal sarcomas carrying an X;22‐translocation. Genes Chromosomes Cancer 52:610–618. [DOI] [PubMed] [Google Scholar]
- 19. Paret C, Theruvath J, Russo A, Kron B, El Malki K, Lehmann N et al (2016) Activation of the basal cell carcinoma pathway in a patient with CNS HGNET‐BCOR diagnosis: consequences for personalized targeted therapy. Oncotarget 7:83378–83391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Peters TL, Kumar V, Polikepahad S, Lin FY, Sarabia SF, Liang Y et al (2015) BCOR‐CCNB3 fusions are frequent in undifferentiated sarcomas of male children. Mod Pathol 28:575–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Pierron G, Tirode F, Lucchesi C, Reynaud S, Ballet S, Cohen‐Gogo S et al (2012) A new subtype of bone sarcoma defined by BCOR‐CCNB3 gene fusion. Nat Genet 44:461–466. [DOI] [PubMed] [Google Scholar]
- 22. Pugh TJ, Weeraratne SD, Archer TC, Pomeranz Krummel DA, Auclair D, Bochicchio J et al (2012) Medulloblastoma exome sequencing uncovers subtype‐specific somatic mutations. Nature 488:106–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Roy A, Kumar V, Zorman B, Fang E, Haines KM, Doddapaneni H et al (2015) Recurrent internal tandem duplications of BCOR in clear cell sarcoma of the kidney. Nat Commun 6:8891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Santiago T, Clay MR, Allen SJ, Orr BA (2017) Recurrent BCOR internal tandem duplication and BCOR or BCL6 expression distinguish primitive myxoid mesenchymal tumor of infancy from congenital infantile fibrosarcoma. Mod Pathol 30:884–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Specht K, Zhang L, Sung YS, Nucci M, Dry S, Vaiyapuri S et al (2016) Novel BCOR‐MAML3 and ZC3H7B‐BCOR gene fusions in undifferentiated small blue round cell sarcomas. Am J Surg Pathol 40:433–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sturm D, Orr BA, Toprak UH, Hovestadt V, Jones DTW, Capper D et al (2016) New brain tumor entities emerge from molecular classification of CNS‐PNETs. Cell 164:1060–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Torre M, Meredith DM, Dubuc A, Solomon DA, Perry A, Vasudevaraja V et al (2019) Recurrent EP300‐BCOR fusions in pediatric gliomas with distinct clinicopathologic features. J Neuropathol Exp Neurol 78:305–314. [DOI] [PubMed] [Google Scholar]
- 28. Ueno‐Yokohata H, Okita H, Nakasato K, Akimoto S, Hata J, Koshinaga T et al (2015) Consistent in‐frame internal tandem duplications of BCOR characterize clear cell sarcoma of the kidney. Nat Genet 47:861–863. [DOI] [PubMed] [Google Scholar]
- 29. Wu G, Diaz AK, Paugh BS, Rankin SL, Ju B, Li Y et al (2014) The genomic landscape of diffuse intrinsic pontine glioma and pediatric non‐brainstem high‐grade glioma. Nat Genet 46:444–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yoshida Y, Nobusawa S, Nakata S, Nakada M, Arakawa Y, Mineharu Y et al (2018) CNS high‐grade neuroepithelial tumor with BCOR internal tandem duplication: a comparison with its counterparts in the kidney and soft tissue. Brain Pathol 28:710–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zhang J, Wu G, Miller CP, Tatevossian RG, Dalton JD, Tang B et al (2013) Whole‐genome sequencing identifies genetic alterations in pediatric low‐grade gliomas. Nat Genet 45:602–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. List of the 479 genes targeted for sequencing on the UCSF500 Cancer Panel. All coding exons were captured for sequencing from each of these genes, with those highlighted genes also having select intronic or upstream regulatory regions that were captured for sequencing to enable detection of structural variants.
Table S2. Clinical features of the 10 patients with CNS high‐grade neuroepithelial tumor with BCOR exon 15 internal tandem duplication.
Table S3. Imaging features at time of presentation for the 10 cases of CNS high‐grade neuroepithelial tumor with BCOR exon 15 internal tandem duplication.
Table S4. Histologic features of the 10 CNS high‐grade neuroepithelial tumors with BCOR exon 15 internal tandem duplication.
Table S5. BCOR exon 15 internal tandem duplications present in the 10 high‐grade neuroepithelial tumors.
Table S6. Likely pathogenic single nucleotide variants and indels identified in the six cases with tumor‐only sequencing analysis.
Table S7. Complete list of somatic nonsynonymous single nucleotide variants and indels identified in the four cases with paired tumor‐normal sequencing analysis.
Table S8. Chromosomal copy number alterations identified in the 10 CNS high‐grade neuroepithelial tumors with BCOR exon 15 internal tandem duplication.
Table S9. Clinical features of the 35 reported cases of CNS high‐grade neuroepithelial tumor with confirmed BCOR exon 15 internal tandem duplication.
Data Availability Statement
Scanned image files of H&E‐stained sections from the 10 tumors from which representative images are presented are available for downloading and viewing at the following link: https://figshare.com/projects/High-grade_neuroepithelial_tumor_with_BCOR_exon_15_internal_tandem_duplication/61691. Sequencing data files are available from the authors upon request.