

Abstract
While abnormalities related to carbohydrates (glycans) are frequent for patients with rare and undiagnosed diseases as well as in many common diseases, these glycan-related phenotypes (glycophenotypes) are not well represented in knowledge bases (KBs). If glycan-related diseases were more robustly represented and curated with glycophenotypes, these could be used for molecular phenotyping to help to realize the goals of precision medicine. Diagnosis of rare diseases by computational cross-species comparison of genotype–phenotype data has been facilitated by leveraging ontological representations of clinical phenotypes, using Human Phenotype Ontology (HPO), and model organism ontologies such as Mammalian Phenotype Ontology (MP) in the context of the Monarch Initiative. In this article, we discuss the importance and complexity of glycobiology and review the structure of glycan-related content from existing KBs and biological ontologies. We show how semantically structuring knowledge about the annotation of glycophenotypes could enhance disease diagnosis, and propose a solution to integrate glycophenotypes and related diseases into the Unified Phenotype Ontology (uPheno), HPO, Monarch and other KBs. We encourage the community to practice good identifier hygiene for glycans in support of semantic analysis, and clinicians to add glycomics to their diagnostic analyses of rare diseases.
Introduction
From antiquity to present days, clinicians have described diseases with phenotypic features mostly in a free-text representation—from ancient Egyptians using papyrus (1) to today’s disease descriptions in textbooks, publications or medical records. However, with the advance of bioinformatics methods and standards, phenotypes are increasingly being codified in a computable format using ontologies (2). An ontology provides logical classifications of terms in a specified domain and the relationships between them. It also bears textual and logical definitions, synonyms identifiers and cross-references to other ontologies, databases (DB) and knowledge bases (KB) (3). The Open Biological and Biomedical Ontology (OBO) Foundry has developed standards for logically well-formed and interoperable ontologies respectful of the representations of biological reality (4). These ontologies are often used in KBs and DBs to semantically structure information and allow for computational classification and inferencing across data.
Biomedical phenotype and disease ontologies have been used in precision medicine for ‘deep phenotyping’ (5), which is ‘the precise and comprehensive analysis of phenotypic abnormalities in which the individual components of the phenotype are observed and described’ (6). The Human Phenotype Ontology (HPO) (7) is one of the leading biomedical phenotype ontologies and is used by various European and American national rare disease efforts and clinical databases such as 100,000 Genomes Project (8), ClinGen (9), Orphanet (10) and ClinVar (11). The HPO is a source of computable phenotypic descriptions that can support the differential diagnosis process. For example, a set of HPO-encoded phenotypes from a patient with an undiagnosed disease can be compared with the phenotypes of known diseases using semantic similarity algorithms for disease diagnostics (7, 12–15). The HPO is a part of a reconciliation effort to align the logical representation of phenotypes across species (7), which enables their integration into a common, species-independent resource called the Unified Phenotype Ontology (uPheno) (16). These resources provide the basis of semantic similarity algorithms implemented within variant prioritization tools such as the program Exomiser developed by the Monarch Initiative team (14, 17), which uses a protein-interaction network approach to help prioritize variants based on interaction partners (18–20). The Monarch Initiative (monarchinitiative.org) provides ontology-based tools for clinical and translational research applications (12–14). The Monarch platform uses the Mondo Disease Ontology that provides a harmonized and computable foundation for associating phenotypes to diseases (21, 22). Mondo integrates the existing sources of disease definitions, including the Disease Ontology (23), the National Cancer Institute Thesaurus (NCIt) (24), the Online Mendelian Inheritance in Man (OMIM) (25), Systematized Nomenclature of Medicine–Clinical Terms (SNOMED CT) (26), International Classification of Diseases (27), International Classification of Diseases for Oncology (28), OncoTree (29), MedGen (30) and numerous others into a single, coherent merged ontology. Mondo is co-developed with the HPO, to ensure comprehensive representation of diseases and phenotypes.
Use of semantic deep phenotyping approaches has been particularly valuable in cases, where a strictly sequence-based analysis has been insufficient to lead to a diagnosis. This is often the case with patients admitted to national and regional undiagnosed clinics, such as the National Institutes of Health (NIH), Undiagnosed Diseases Program (UDP) and Network (UDN), where only 28% of UDN patients have been diagnosed to date (31). One of the most interesting characteristics of patients in these programs is the high incidence of glycan-related molecular defects, which we refer to here as ‘glycophenotypes’. These include observable abnormalities in the structure, abundance, distribution and activity of glycans, as found in their free or conjugated forms. For example, Gall et al. (32) reported that 50% of patients admitted to the UDP had abnormal glycophenotypes, whether the causal genes were related to glycobiology or not (33). While diseases related to glycobiology have been well-studied (34–36), the integration of glycomics data and glycophenotypes into biological KBs lags behind what we see for genomic, proteomic and metabolomic data (key biological entity types like genes, diseases, pathways, etc.); hence, ‘the need of informatics in glycobiology’ as Campbell et al. state: ‘Databases that provide authoritative information about glycan and glycoconjugate structures, and well-defined standards that allow this information to be exchanged, are required as a foundation for computational tools that give insight into the biological functions and consequences of glycosylation (37)’.
Despite the diagnostic and informatics success of HPO, glycophenotypes are underrepresented in this resource and, thus, limit their value in differential diagnosis. For instance, patients with fucosidosis can have at least five glycophenotypes such as decrease of fucosidase activity, urinary glycosaminoglycan excretion, oligosacchariduria, increase of urinary glycopeptides and accumulation of glycolipids expressing blood group antigens in the liver (38, 39), but only two of these glycophenotypes are in HPO (40, 41). In addition to phenotypes related to glycan-binding protein (GBP) staining, there can be potentially at least six glycophenotypes per glycan-related diseases, hence, 1056 possible HPO glycophenotypes terms for the 176 glycan related diseases (congenital disorder of glycosylation (CDG) and diseases where glycophenotypes are detectable, see online supplementary material for Table S1). Nevertheless, there are only 126 HPO terms related to glycobiology as of time of writing, which are either subclasses of abnormal glucose homeostasis (38/126), abnormal glycosylation (46/126 whether glycolipid metabolism or protein glycosylation), abnormal glycosyltransferases activity (4/126) and abnormal free glycans (38/126), (see online supplementary material for Table S2). In addition, these existing HPO glycophenotypes occupy only 5/20 categories of glycans indicated in Figure 1 (glycosyltransferases and GBP phenotypes excluded). Finally, within Monarch platform, the term ‘abnormal glycosylation’ (HP:0012345) is associated with 31 diseases (42) and the term ‘glycan’ returns only 17 phenotypes (43), yet according to Freeze et al. (34) and Ferreira (44), 130–134 CDG exist (non-including other glycan-related diseases such as diseases related to GBP). Furthermore, Xia et al. (36) reported 54 different urinary glycophenotypes for 10 glycan-related diseases.
Figure 1.
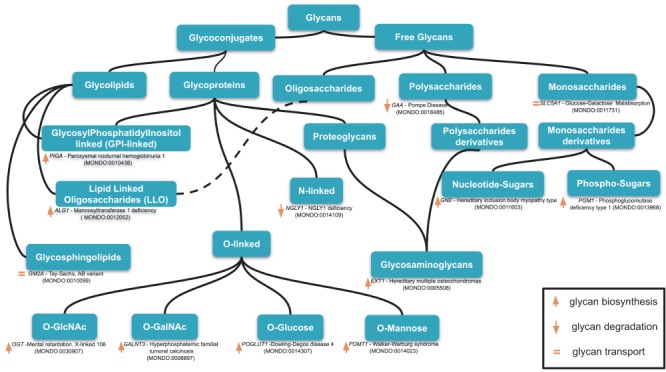
An example of classification of relevant glycans involved in human diseases based on the essential of glycobiology and CHEBI ontology. Glycans can be free or conjugated to macromolecules (protein, lipids). Free glycans can be monosaccharides (n = 1), oligosaccharides (2 < n < 10) or polysaccharides (n > 10), and their derivatives (e.g. acetylated, sulfated) 13 exemplary diseases names along with the mutated genes and MONDO ID (in parentheses) are indicated for 13 classes of glycans. Up, down orange arrows and orange equal signs indicate, respectively, the involvement of the gene products in glycan biosynthesis, degradation or transport. Based on our disease curation, there are 176 glycan-related diseases (CDG and diseases in which glycophenotypes are detectable, see online supplementary material for Table S1).
Here, we provide a short overview of glycobiology, its importance in health and disease and a discussion of some of the technological and informatics challenges that face the use of these data for disease discovery research and diagnostics. We demonstrate the utility in adding glycophenotypes to disease diagnostic pipelines. Finally, we present the results of surveying a selection of ontologies and KBs based on an updated list of glycoscience-related informatics resources from Campbell et al. (37).
Glycans: A Galactic Odyssey
Structures and classification
Glycans, also referred to as carbohydrates and sugars (45), are a fundamental class of biomolecules with the general chemical formula CnH2nOn. They are among the oldest organic molecules found in the Milky Way, and one of the simplest glycans, glycoladehyde, was even discovered on the molecular cloud Sagittarius B2 (46). Glycans, along with amines, may have enriched our solar system to influence life on Earth (47), possibly during meteorite collisions on planet Earth, which led to the formation of more organic molecules (48).
Glycans can be found in bacteria, archaea, eukaryotes and most viruses (49). They are the most abundant biomolecules on Earth with plant-synthesized cellulose (50). In eukaryotic cells, glycans can be found in free forms (monosaccharides, oligosaccharides and polysaccharides) or as bioconjugates, covalently attached to the other major classes of biomolecules such as nucleic acids (sugar-nucleotides), proteins (glycoproteins with N-linked glycans (N-glycans) and O-linked-glycans (O-glycans), glycosylphosphatidylinositol-anchored, proteoglycans) and lipids (lipid-linked oligosaccharides, glycosphingolipids, glycosylphosphatidylinositol-anchored) (Figure 1).
Glycans can be N-acetylated (e.g. N-acetylglucosamine or GlcNAc), phosphorylated (e.g. glucose-6-phosphate), sulfated (e.g. heparan sulfate [HS]), etc. The collection of all glycans in an organism, the glycome, displays an extreme diversity of structures, amounting to up to 104 times more molecules than those found in the proteome (51). The molecular weight of glycans can range from 60 with glycoladehyde (46) to more than 2 × 106 Daltons (Da), with glycan polymers such as hyaluronan (52) making them only partially accessible to metabolomics studies, as metabolomics focuses on the study of molecules below 1,500 Da (53). Glycans, although often regarded at the periphery of metabolomics, proteomics and lipidomics, can play crucial roles in cell biology.
Glycan Roles in Human Biology
Given their ancient evolutionary history, diversity and abundance, it is not surprising that glycans play a pivotal role in human biology. Glycans play many roles that range from structural, modulatory to recognition (49) (Table 1). In terms of structural role, glycans can be a physical barrier, assist protein folding and serve as energy storage. The physical barrier or glycocalyx is a protective coat made of glycoaminoglycans (e.g. HS), glycoproteins (including GPI-anchored) and glycolipids located at the cell surface of eukaryotic cells (54). Glycans at the cell surface and on circulating serum proteins can also provide a shield against proteases and against attachment to certain pathogens (45). Glycans can help protein folding in the endoplasmic reticulum by stabilizing and promoting interaction with GBP (lectins) involved in protein folding such as calnexin and calreticulin (55). Glycans can serve as a structural energy storage with glycogen made of polymer of 55 000 molecules of glucose (56).
Table 1.
Glycan roles, exemplary HPO terms and glycophenotypes associated with six genetic diseases
| Glycan roles | Glycan-related group and pathways | Mutated gene | Disease (Mondo identifier) | Abnormal phenotypes associated with disease | ||
|---|---|---|---|---|---|---|
| Abnormal glycophenotypes | Examplary anatomical, infectious and behavioral phenotypes | |||||
| Structural | Physical barrier | Glycosaminoglycans (HS polymerization) | EXT1 | Hereditary multiple osteochondromas (MONDO:0005508) | Decreased circulating HS level (HP:0410343) | Abnormality of the humerus (HP:003063) Multiple exostoses (HP:0002762) |
| Protein folding | O-glycans synthesis Protein folding | B3GLCT | Peters Plus syndrome (MONDO:0009856) | Shortened O-fucosylated glycan on properdin (HP:0410344) | Anterior chamber synechiae (HP:0007833) Brachycephaly (HP:0000248) | |
| Energy storage | Polysaccharide (glycogen degradation) | GAA | Pompe Disease (MONDO:0009290) | Increase of urinary polyhexose glycans (HP:0410345) | Cardiomegaly (HP:0001640) Cognitive impairment (HP:0100543) | |
| Modulatory | Signaling | O-Glycans synthesis (O-Fucosylation) Notch signaling | LFNG | Spondylocostal dysostosis 3 (MONDO:0012349) | Decreased glycosyltransferase O-fucosylpeptide 3-β-N-acetylglucosaminyltransferase activity (HP:0410349) | Scoliosis (HP:0002650) Slender finger (HP:0001238) |
| Recognition | Intrinsic | O-glycans synthesis (O-mannosylation) laminin-dystroglycan binding | POMT1 | Muscular dystrophy-dystroglycanopathy type A1 (MONDO:0014023) | Hypoglycosylation of alpha-dystroglycan (HP:0030046) | Cataract (HP:0000518) Intellectual disability, severe (HP:0010864) |
| Extrinsic | GBP to pathogen Toll-like receptor signaling Creation of C4 and C2 activators | MBL2 | Mannose-binding lectin (MBL) deficiency (MONDO:0013714) | Decreased mannose-binding protein level (HP:0032305) | Recurrent Klebsiella infections (HP:0002742) Failure to thrive (HP:0001508) | |
The modulatory role of many signaling proteins depends on their own glycosylation, the glycosylation or glycan binding activity of their counter receptors. For instance, human chorionic gonadotropin’s signal transduction depends on its N-linked glycans (57). Similarly, glycans on cell surface proteins are required for the signaling of GBP. For instance, galectin-1 and galectin-8 will signal phosphatidylserine exposure on neutrophils through interaction with polylactosamine containing counter receptors (58, 59). Finally, some receptors can be regulated by signaling glycans such as GM3 glycolipid on the epidermal growth factor receptor (EGFR) (60).
Glycans are involved in intrinsic and extrinsic recognition (45, 49). From the initiation of spermatozoid attachment on sialyl-Lewis(x) on the egg (61) to cell death with glycosylation of Fas/TRAIL death receptor (62), glycans play a role in cell recognition and in the cellular social life, through the interaction of glycan–protein or glycan–glycan interaction. For instance, many antigens are glycan-based, such as the ABO blood group (63). Stem cells growth and differentiation depend on O-fucosylation on Notch (64), and HS is a key regulator of embryonic fate (65). In fact, stem cells’ glycosylation profiles indicate the stage of pluripotency, especially fucosylated glycans (66), and are used for their isolation through specific sets of lectins (67). In cancer biology, glycans are used as markers for many types of cancers (68) and are involved in resistance to cancer in naked mole rats with high molecular weight hyaluronan (69). Glycans are also involved in parasitic infections, during the attachment phase, whether zoonotic (e.g. schistosome (70)), microbial (e.g. Escherichia coli 086, bearing blood group antigen (71)) or viral (e.g. influenza virus H1N1, where H stands for hemagglutinin and N for neuraminidase, respectively, a lectin and a glyco-enzyme (72)).
Glycans in Human Diseases
Alterations in glycan function, such as genetic perturbation in synthesis (involving glycosyltransferases, chaperone of glycosyltransferases, transporter, etc.), degradation or their attachment through GBP, can contribute to the pathophysiology of various diseases. For instance, mutations in the glycosyltransferase EXT1 can lead to the formation of abnormally short HS molecules which accumulate in the Golgi apparatus, and cause abnormal cytoskeleton formation (73) and increase of bone morphogenetic proteins that lead to osteochondromas (74). Glycans also play a role in molecular recognition in innate and acquired immunity. Human milk oligosaccharides contribute to a healthy infant gut microbiome by preventing bacteria and viruses from binding to the intestinal mucosa (75). Bacterial lipopolysaccharide can stimulate innate immune responses (76). Glycosylation of immunoglobulins (Ig) can contribute to many autoimmune diseases such as IgA nephropathy (77). In this disorder, abnormal hypoglycosylated IgA1 displays the glycan epitope (GalNAc) which is recognized as non-self by specific antibodies, forming IgA-immune complexes that are deposited in the renal mesangium and cause glomerular injury (78). In this example, abnormally hypoglycosylated IgA1 is a glycophenotype associated with IgA nephropathy.
Exemplary glycophenotypes measured on biomolecules from cells, tissues and bodily fluids are indicated in Figure 2 and Table 1. For example, assays are performed to quantify and characterize free glycans, glycopeptides, glycoproteins and glycosyltransferase activities in body fluids (urine, blood or serum and cerebrospinal fluid), or in cells, such as fibroblasts. Standard glycomics assays include protein analysis via mass spectrometry, glycosyltransferase activity and glycan binding assays. Glycophenotypes can be described in a structured way as abnormalities of the biomolecule in a given anatomical location, such as abnormal glycopeptide level in the blood.
Figure 2.
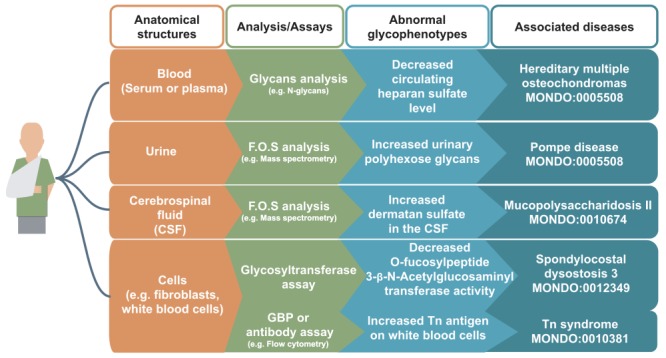
Examples of glycophenotypes that can be captured from various laboratory techniques From a patient’s anatomical structures (indicated in orange boxes, e.g. blood), glycans such as free oligosaccharides (F.O.S.) and glycan-related molecules can be analyzed by standard glycomics assays (indicated in green boxes, e.g. GBP or antibody assay). Patients’ glycophenotypes indicated in the blue boxes can be captured from publications: decreased circulating HS level (82), increased urinary polyhexose glycans (36), increased dermatan sulfate in the CSF (83), decreased O-Fucosylpeptide 3-beta-N-acetylglucosaminyltransferase activity (84) and increased Tn antigen in white blood cells (85). In our preliminary work, we have logically defined design patterns (86) that would generate hundreds of classes, but they are not yet fully integrated in the HPO.
Abnormalities in the structure, abundance, location and biological activity of glycans have been identified in over one hundred genetic disorders, including diseases with abnormalities of glycan degradation, congenital disorders of glycosylation (CDG) and deglycosylation (79). Disorders related to glycosylation often present a multitude of molecular glycophenotypes.
Abnormal glycophenotypes are present in many genetic diseases related to glycobiology as indicated in Table 1, but they have also been described in the ‘fringes’ of our current knowledge of glycan-associated genes (80). For instance, dysfunction in the DNA excision repair enzyme encoded by ERCC6 (excision repair 6, chromatin remodeling factor) can lead to abnormal fucosylated glycans in the urine, which is a marker for Cockayne syndrome type 2 (81).
Glycophenotyping to Improve Human Disease Diagnosis: Fucosidosis
Within the Monarch Initiative platform, whereas hepatomegaly is a common phenotype for 494 diseases (87), oligosacchariduria is a distinctive glycophenotype for nine diseases (40). As unique glycophenotypes can be markers for specific diseases, we hypothesized that expanding the representation of glycophenotypes in the HPO and their use in disease annotations could improve phenotype-based comparisons. As a proof of concept, we performed an analysis with phenotypes associated with the disease fucosidosis (MONDO:0009254). We created 1000 ‘simulated’ patients’ profiles by randomly sampling 10 out the 16 most frequently associated phenotypes with fucosidosis within Monarch Initiative platform (Figure 3A and B). In a second set, we randomly replaced two phenotypes (blue squares, Figure 3B) with two glycophenotypes known to be associated with fucosidosis for each profile (orange squares, Figure 3C). Both sets were compared using the PhenoDigm algorithm (17), which given a list of phenotypes, ranks candidate diseases based on phenotypic similarity. As shown in Figure 3D, the addition of glycophenotypes led to a higher ranking for fucosidosis (rank #1: 82% with glycophenotypes versus 53% without the two glycophenotypes with a Fisher exact P-value = 8.5e-47). The workflow is available in a Jupyter notebook (88) at http://bit.ly/glycop_owlsim_analysis.
Figure 3.
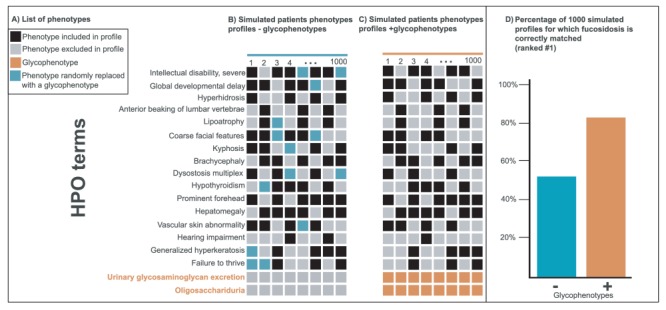
Improvement of disease diagnostic with molecular glycophenotypes for fucosidosis. Panel A lists 18 phenotypes frequently associated with fucosidosis. The columns in Panels B and C illustrate simulated patients phenotypes profiles composed of a random selection of 10 of these 16 phenotypes. The profiles in Panel C include glycophenotypes (bottom in orange), whereas those in Panel B do not. Panel D shows that when these two groups of 1000 simulated profiles are compared for their diagnostic utility, the profiles that contain glycophenotypes (C) significantly outperform those that do not (B) (Fisher exact P-value = 8.5e-47). Moreover, more specific glycophenotypes are more diagnostically useful than more general ones. This underscores the importance of harmonizing glycophenotypes across data resources as well as collecting them from patients.
This demonstrates that the addition of glycophenotypes improves the ranking of relevant glyco-related diseases for candidate diagnoses. Thus, we believe that broadening the representation of molecular phenotypes in phenotype ontologies could help refine rare disease diagnoses.
Landscape Review of Glycobiology and Glyco-Disease Resources: A Need for Improved Glycan Representation and Standards
To the best leverage abnormal glycophenotype data in bioinformatics analyses, as with metabolomics data for the prioritization of pathological variants for whole genome/exome sequencing (WGS/WES) (89), we need comprehensive, standardized and connected representations of glycan terminology. This requirement includes interoperable representation of glycophenotypes in biomedical ontologies, KBs and DBs. While the nomenclature for glycan-related diseases is well established, the nomenclature for glycans can be complex. We face several hurdles to increase the standardized representation of glycophenotypes:
(1) Lack of a unified standard terminology and identifiers for glycans and glycan related entities (e.g. effort such as GlycoCT (90) and the Symbol Nomenclature for Graphical Representations of Glycans (SNFG) (91), respectively, for encoding scheme and pictorial representation of glycans, but they are not mandatory in all journals).
(2) Technical/experimental barriers for interrogating glycophenotypes (standard mass spectrometry cannot differentiate monosaccharide epimers and define linkage between monosaccharides).
(3) Lack of coverage of glycan-related concepts in ontologies (for instance, the Chemical Entities of Biological Interest (CHEBI) ontology (92) contains 264 entries, while more than 100 000 unique cross-species and synthetic glycan structures exist in the glycan repository GlyTouCan (93) and 15 000 unique glycan structures have been estimated for humans (94)).
(4) Lack of associations of glycans and glycan abnormalities with diseases and phenotypes in existing databases such as Monarch (12).
(5) Lack of interoperability across DBs/KBs containing glycan-related data/knowledge (for instance, the semantic barrier between scientific disciplines related to glycobiology as the same molecule can be referred to differently in different subdisciplines of immunology, hematology, biochemistry: CD173, blood group O, H-type 2 antigen (95) or Fuc(α1-2)Gal(β1-4) GlcNAc (96)).
(6) Lack of queryable data store based on glycophenotypes, levels, location, subjects, assays, abnormalities, evaluand, gene, etc. (e.g. an increase/decrease or presence/absence of a particular glycophenotype in a given bodily fluid in a given genetic disease, see Figure 2).
(7) Lack of human and machine-readable formats for diseases, glycans and phenotypes (e.g. International Union of Pure and Applied Chemistry, IUPAC (97)).
(8) Lack of glycophenotype algorithms for disease comparison based on structured rules (e.g. increase of Tn antigen associated with mutation on C1GALT1C1 for cancer (98)), logical structure and relationships between entities (e.g. fuzzy phenotype search (13)).
Hence, there is a need for a standardized vocabulary and identifiers, best practices to facilitate the curation of glycophenotypes related to genetic diseases, especially as the human glycome project aims to define the structures and functions of human glycans which have started (99).
Regardless of this complexity, many glycobiology-related KBs exist with differences in specificity (glycan-related enzymes, diseases, molecules, etc.) that could be used for disease and glycan comparisons in a human readable and computable manner. In Table 2, we provide a review of existing resources and identified gaps and opportunities for additional development in both the HPO and cross-species phenotype ontologies and glyco-KBs and DBs. We focused particularly on diseases and/or glycans by highlighting features, applications, uses and challenges in order to provide potential resources for representing glycophenotypes in a compatible way with the HPO, and applying them toward phenotype-based patient diagnosis and disease-gene discovery.
Table 2.
Description of the KB and Ontologies Overview of relevant knowledge bases and ontologies based on their contents and glycan related data (glycophenotypes, glycan related diseases, genes, GBP, etc.)
| Resources (names and links) | Domains | Descriptions |
|---|---|---|
| Knowledge base | ||
| CAZY (100)http://www.cazy.org/ | Glyco-genes | CAZY has curated data from publications on carbohydrate-active enzymes responsible for the synthesis and breakdown of glycoconjugates, oligosaccharides and polysaccharides. It provides classification of these glyco-enzymes based on their activities (glycoside hydrolases, glycosyltransferases, polysaccharide lyases, carbohydrate esterases and auxiliary activities) and glycan-related genes browser in different species |
| CFG (101)http://www.functionalglycomics.org/ | Glyco-genes GBP glycans diseases | The CFG has generated and collected publicly available data on GBPs (glycan array), glycan profiles in cells and tissue, phenotypic analyses of transgenic mouse lines with knockout glycan related genes (histology, immunology, hematology and metabolism/behavior) |
| GlyConnect (102)https://glyconnect.expasy.org | Glyco-genes GBP Glycans Diseases | GlyConnect integrates of information about protein glycosylation for different species based on taxonomy, protein, tissue, composition disease, glycosylation sites, peptides and references |
| GlycoSciences (103)http://glycosciences.de/ | Glycans diseases | GlycoSciences provides experimental information for glycans such as structure, composition, motifs, biophysical experiments on glycans and curation of comprehensive repository of cluster of differentiation (CD) antigens |
| GlyTouCan (93)https://glytoucan.org/ | Glycans | Glytoucan is a free glycan repository that provides unique accession numbers to any glycan independently of experimental information (n = 110 668). GlyTouCan has made efforts to bridge gaps between experimental glycans and native glycans by creating identifiers from the mass spectrometry data and encourages glycobiologists to use them in their publications |
| JCGGDB (104)https://jcggdb.jp/database_en.html | Glyco-genes GBP glycans diseases | JCGGDB is an integrative database for glycan information and diseases using different resources. It has compiled information related to glycan-related genes or GlycoGene (enzymes, transporter, etc.) and glycan diseases (e.g. CDG-Ia), pathosis, links to other KBs associated gene descriptions (e.g. PMM2) and a genetic glyco-diseases ontology that provides a hierarchical classification of the diseases |
| KEGG (105)https://www.genome.jp/kegg/glycan/ | Glyco-genes GBP glycans diseases | KEGG is a KB that includes a module dedicated to glycobiology (KEGG-glycan) in which glycan identifiers, glycan pathways, genes, and links to other glycan databases. It allows for the search of glycan terms (abbreviation and synonyms) and gives composition, identifiers, reaction, pathways, etc. |
| Monarch (13)monarchinitiative.org | Glyco-genes GBP glycans diseases | Monarch initiative is a platform that provides analytic tools and web services for cross-species comparison of genotype–phenotype associations, disease modeling and precision medicine using semantically integrated data |
| OMIM (25)https://omim.org/ | Glyco-genes GBP diseases | OMIM is a resource containing information about human genes and genetic disorders. It provides information on genetic diseases and associated phenotypes, including disease names and synonyms, unique, phenotype-gene relationships, descriptions of diseases (diagnosis, pathogenesis), clinical and biochemical features, genetic information as well as animal models |
| PubChem (106)https://pubchem.ncbi.nlm.nih.gov/ | Glyco-genes GBP glycans diseases | Pubchem is an open KB from the NIH for chemical structures, identifiers, chemical and physical properties (biological activities, patents, health, safety, toxicity data, etc.). Data can be queried online or downloaded (JSON, XML, ASN.1 files) |
| Reactome (107)https://www.reactome.org/ | Glyco-genes GBP glycans diseases | Reactome is an open-source and peer-reviewed pathway KB that allows search based on biological terms. Reactome has a repertoire of diseases of glycosylation (related to GAG, N-glycans synthesis, O-glycans synthesis and precursors of glycosylation) |
| UniLectin (108)https://www.unilectin.eu/ | GBP glycans | UniLectin is an interactive KB that classifies and curates GBP (or lectin) and their ligands |
| Ontologies | ||
| CHEBI (92)https://www.ebi.ac.uk/chebi/ | Glycans | CHEBI is a dictionary for small molecules developed by the European Bioinformatics Institute using sources from KEGG and developed with an ontology framework. It provides an identifier, name, annotation rating, structure, molecular formula, charge, average mass, ontology, etc. |
| GO (109)http://geneontology.org/ | Glyco-genes GBP glycans | GO consortium is an initiative for the computational representation of genes and their biological functions at the molecular, cellular and histological levels. It provides gene annotations, ontology, mapping and tools such as gene enrichment analysis |
| NCIt (24)https://ncit.nci.nih.gov/ncitbrowser/ | Glyco-genes GBP glycans | NCIt is a thesaurus from the National Cancer Institute Enterprise Vocabulary Services. It provides concepts, terminology, therapies related to cancer and related biomedical topics |
We reviewed a selection of widely used KBs and ontologies with glycan-related content that will help to jump the hurdles we mentioned above. Based on these criteria, we selected relevant KBs/services that could be used to support ontological glycophenotype representations for phenotype-based diagnosis and disease-gene discovery as indicated in Table 2 with CAZY (100), Consortium for Functional Glycomics (CFG) (101), GlyConnect (102), GlycoSciences (103), GlyTouCan (93), Japan Consortium for Glycobiology and Glycotechnology DataBase (JCGGDB) (104), The Kyoto Encyclopedia of Genes and Genomes (KEGG) glycan (105), Monarch (13), OMIM (25), Pubchem (106), Reactome (107), UniLectin (108), CHEBI (92), gene ontology (GO) (109) and NCIt (24).
Following this scope of disease-glycophenotype association, we reviewed key features/applications, use and potential challenges (e.g. using community standards, providing glycan identifiers, links to other resources, etc.) as indicated in Table 3. The majority of the KBs/DBs possess human and machine-readable formats (14/15) and standardized terminologies (15/15) and ontologies (10/15). While all of them have a queryable data store, only two of them possess a phenotype algorithm (Reactome and Monarch) and one has both raw and curated data (CFG). About 8/15 of them have more than 1000 glycans-related terms. Therefore, we propose to build a modular molecular glycophenotype branch that could be integrated into the HPO and other phenotype ontologies as shown in Figure 4.
Table 3.
Review of KB and Ontologies We reviewed relevant knowledge bases and ontologies based on criteria such as presence of human-machine readable, phenotype algorithms, numbers of glycan related terms, etc. Some KBs are richer than other, nevertheless, none of them cover all the criteria
| Resources | Human and machine-readable formats | Queryable data store | Phenotype algorithms | Standardized terminologies and ontologies | Type of data | Glycans-related terms (glycan, sugar, carbohydrate, glycoproteins, glycolipid, glycosyltransferase and lectin) |
|---|---|---|---|---|---|---|
| CAZY | No | Yes | No | Many | Curated | 333 |
| CFG | No | Yes | No | Many | Raw & curated | >1000 |
| Glyconnect | Yes | Yes | No | Many | Curated | >1000 |
| GlycoSciences | Yes | Yes | No | Many | Curated | >1000 |
| Glytoucan | Yes | Yes | No | Many | Curated | >1000 |
| JCGGDB | Yes | Yes | No | Many | Curated | >1000 |
| KEGG glycan | Yes | Yes | No | Many | Curated | >1000 |
| Monarch | Yes | Yes | Yes | Many | Curated | 54 |
| OMIM | Yes | Yes | No | Many | Curated | 227 |
| Pubchem | Yes | Yes | No | Many | Curated | 252 |
| Reactome | Yes | Yes | Yes | Many | Curated | 352 |
| UniLectin | No | Yes | No | Many | Curated | 50 |
| CHEBI | Yes | Yes | No | Many | Curated | >1000 |
| GO | Yes | Yes | No | Many | Curated | >1000 |
| NCIt | Yes | Yes | No | Many | Curated | 364 |
Figure 4.
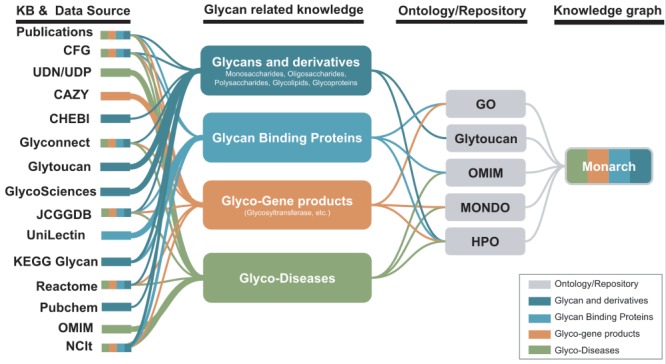
Potential KBs and data sources for the improvement of glycophenotypes representation for HPO and Monarch glycophenotypes related to diseases indicated in publications and KBs could be used to enhance glycan-related knowledge in Monarch.
A Comprehensive Semantic Representation of Glycophenotypes
Integration of glycophenotypes in the HPO and the uPheno framework—prototype of a MGPO
The initial high-level classification of molecular glycophenotypes is now available in HPO, for instance ‘Abnormal protein glycosylation’ (HP:0012346) or ‘Abnormality of glycolipid metabolism’ (HP:0010969). Future efforts will include the integration of subclasses of glycophenotypes in HPO using the resources described above as well as glycan-related terms from clinical measures from the Logical Observation Identifiers Names and Codes (LOINC) within the context of the LOINC2HPO project (110). As this work matures, it will be necessary to create rich educational materials and in-line help for glycobiology curators.
Future effort will also include a developed version of the molecular glycophenotype ontology (MGPO) (86). MGPO phenotypes are logically defined according to the patterns defined by the Unified Phenotype Ontology (uPheno) framework wherever possible (16). Complex patterns specific to glycophenotypes (beyond the scope of uPheno) extend existing uPheno patterns. MGPO prototype includes the following primary characteristics (glycan levels, composition, length, occupancy and binding) and secondary dimensions (glycan type, attachment status, location, residue type and residue position) (https://github.com/monarch-initiative/glyco-phenotype-ontology) (86). The MGPO prototype aimed to inform a more comprehensive ontological representation of glycophenotypes. Considering the biochemical diversity of monosaccharides and possible linkages (e.g. chirality of the molecules, anomeric carbon and 2k stereoisomers, where k is the number of carbon atoms (111)), the diversity of glycan chains quickly becomes exponential. To enable this level of expansion while retaining robust and consistent logical structure, we will use ‘Dead Simple Ontology Design Patterns’ (DOSDP) (112). Use of DOSDPs ensures the interoperability of glycophenotype terms with those from other phenotype ontologies and its future integration into uPheno, currently being developed by the Monarch Initiative and collaborators from the Phenotype Ontology Reconciliation Effort (113).
Challenges with an ontological representation of glycophenotypes
A robust and comprehensive glycophenotype ontological representation would (i) provide synonyms of glycans between disciplines (e.g. Tn antigen or O linked N-acetylgalactosaminyl epitope or O-GalNAc (114)); (ii) gather identifiers (GlyTouCan, Kegg, CHEBI, JCGGDB, IUPAC, etc.); (iii) describe glycophenotypes of genetic disease with higher granularity (e.g. increase of fucosylated glycans in the urine for ERCC6 (81)); (iv) allow comparisons between known and unknown diseases (e.g. answering questions such as ‘what diseases show an increase of fucosylated glycans in the urine?’); and (v) provide phenotype terms for annotations for biocurators. This integration will allow semantic similarity approaches for disease diagnosis based on phenotypes, including glycophenotypes, variant prioritization, patient matchmaking and model system discovery.
However, integrating glycophenotypes in an ontology creates some conceptual challenges that will require community discussion:
#1 Determination of equivalence between native and experimental glycans is challenging and will require the harmonization of nomenclature between IUPAC, CHEBI, GlyTouCan, etc.
#2 Alignment of logical definitions across OBO Foundry ontologies will be difficult due to the different glycobiology modeling that is represented in different contexts, and due to gaps. For example, CHEBI does not include protein; therefore, there is no place for glycoproteins; the protein ontology does not provide information about the glycan portion of a glycoprotein other than the highest level (e.g. N or O glycosylated); the GO represents only biological processes and some glycobiology processes are unknown for some phenotypes (e.g. dysfunctional DNA repair enzyme ERCC6 is associated with increased fucosylated glycan in the urine, yet the biological process is unknown (81)).
#3 Some phenotypes are quantitative and would require conversion to semantic qualitative descriptors, for example glycan array data where there are plots of relative fluorescence unit based on the binding of a protein to an array of hundreds of glycans.
#4 Full granularity of the description of glycophenotypes could be challenging to navigate for curators, for example increase of fucosylation glycans versus the full name of the glycans (e.g. Neu5Ac(α2-3)Gal(β 1-4)GlcNAc(β1-3)Gal(β 1-4)[Fuc(α1-3)]GlcNAc).
The Minimum Requirement for A Glycomics Experiment, Glycomics at Expasy and GlyTouCan have joined effort to address challenge #1 with an automatic attribution of glycan identifiers from the mass spectrometry experiments (115). Nevertheless, a unification with CHEBI will be necessary (challenge #2). Indeed, glycan structures analyzed by mass spectrometry can be ambiguous; for instance, an exact hexose name and linkages can remain undetermined because of the technological limitations. While GlyTouCan tolerates this ambiguity, it differs from CHEBI’s standards. For instance, a urinary trisaccharide, assumed to be three units of glucose, can be a marker for Pompe disease (36). In this case, contrary to CHEBI, GlyTouCan can provide an identifier (G63977XF) regardless of undetermined linkage between the monosaccharides. This will necessarily reveal the gaps and lack of logical interoperability across OBO ontologies (challenge #2), but by working with each of these communities, we will be able to improve glycobiology for all contexts. Close collaboration between glycobiologists and glycoinformaticists will be required to address challenges #3 and #4.
Towards a Glyco-Enriched Knowledge Graph of Disease for Diagnostics and Discovery
A rich set of glycophenotypes will support the integration of disease, pathway, gene function and numerous other biological knowledge (Figure 5). We have begun this integration work within the context of the Monarch knowledge graph (Figure 4) with HPO terms that are semantically associated with Mondo diseases, genes, GO terms, etc. Additionally, we are performing a broader characterization and review of glycophenotypes from the literature: for typical CDG (34), glycan-associated diseases (for instance, disease related to GBP such as Mannose-Binding lectin deficiency), genetic diseases where glycans are markers (e.g. ERCC6 (81)) and across a spectrum of animal models (mouse, zebrafish, rat, fly, etc.). We are focusing on glycan-related knowledge, such as glycans and derivatives, GBP, glycobiology-related genes and diseases. We are also collaborating with GlyGen (https://www.GlyGen.org/) whose aim is to gather glycobiology-related data from multiple resources to provide data mining, sharing and dissemination of glycan-related information, and our curation of glycophenotypes and the Monarch knowledge graph will be made available to the GlyGen platform.
Figure 5.
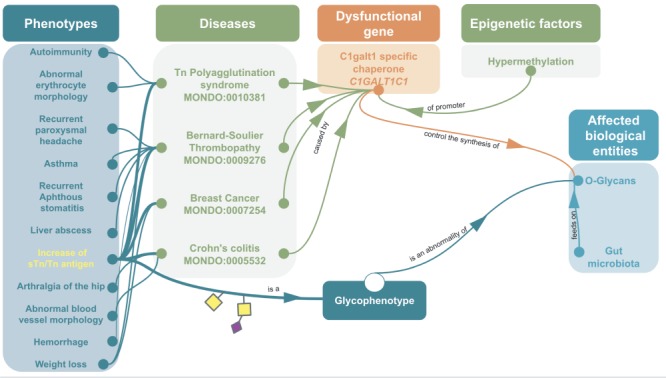
Example of omics integration with ontologies related to glycans: graph representation of the impact of a dysfunctional C1GALT1C1 on health C1GALT1C1 encodes Cosmc, a molecular chaperone for a glycosyltransferase that initiates O-GalNac glycans synthesis (T-synthase) (127). Dysfunctional Cosmc can lead to an improper T-synthase folding, thus abnormal O-glycans with the abnormal glycophenotype: increase of sTn/Tn antigen (SNFG symbols, respectively, yellow square for Tn and a purple diamond/yellow square for sTn) (128). Dysfunctional Cosmc (129) can be due to mutations or epigenetic factors, for instance the hypermethylation of C1GALT1C1’s promoter can lead to increase of sTn/Tn antigen. These two glycophenotypes are also common in many cancers (130). Mouse models have shown that C1GALT1C1 mutation can lead to abnormal O-glycans on platelets, generating bleeding disorders similar to Bernard-Soulier syndrome (MONDO:0009276) (131) Inflammatory bowel disease similar to Crohn’s Colitis (132) and abnormal microbiota (133). In fact, human gut microbiota (HGM) feeds on normal MUC2 glycans (134–136). Hence, the disruption of MUC2 glycosylation due to C1GALT1C1 mutation could potentially lead to microbiota and host physiology issue (137).
Thus far, we have limited our work on glycan representation and integration to human genetic diseases. We plan to extend this work to include data from bacterial and viral glycans and lectins in the context of infectious diseases, as attachment happens through glycans (116). For instance, Norovirus and Parvovirus lectins bind, respectively, to the glycans of ABO and P blood group antigens in host glycans, and also, host glycans and GBPs proteins can bind to pathogens (e.g. Neisseria gonorrhoeae (117) and Neisseria meningitidis (118)); therefore, human genetic variants of glycosyltransferases and lectins may play a role in microbial/viral infection (119). We believe that semantic integration at the molecular level as illustrated in Figure 5 (which shows a graph representation of the potential impact of the dysfunctional gene C1GALT1C1 or COSMC) will support mechanistic discovery and identify interventional targets. A broader integration of glycophenotypes would, therefore, be a valuable part of pathways analysis tools such as Impala (120), Reactome (107) and STITCH (Search Tool for Interacting Chemicals) (121), in support of interconnecting microbiome metabolites. Finally, molecular phenotyping with glycophenotypes and pathway integration could provide better insights toward possible treatments, for instance, dietary supplementation of glycans or glycan-related molecules (122).
Ontology could be a way to integrate glycophenotypes for disease diagnosis; however, another possible approach is to integrate glycomics data to whole genome/exome sequencing (WES/WGS) as recently done by Ashikov et al. (123), similarly to the metabolomics integration in genomics (89, 124, 125). For a more systematic approach, a new bioinformatics pipeline integrating deep molecular glycophenotyping in WES/WGS will be needed.
In conclusion, we have discussed the importance of glycans in health and disease, the technological and informatics challenges for glycan data integration for disease discovery research and diagnostics. We have defined the concept of abnormal glycophenotypes, demonstrated their usefulness in disease diagnostic pipelines with the example of fucosidosis, proposed an integration of selected ontologies/glycoscience KBs and introduced an ontology for glycophenotypes (MGPO). Finally, we urge the community to participate in the advancement of glycophenotype representation and its inclusion in disease research KBs and in clinical diagnostic settings. For instance, glycobiologists should request new abnormal glycophenotypes terms in HPO following the guidelines (126). Similarly, clinicians should report them using the SNFG (91) and GlyTouCan (93) standards. Community coordination and knowledge integration will be critical to overcome the current knowledge gap defined herein.
Glossary
- CAZY
carbohydrate-active enzymes
- CDG
congenital disorders of glycosylation
- C1GALT1C1
core 1 synthase, glycoprotein-N-acetylgalactosamine 3-beta-galactosyltransferase 1 specific chaperone 1
- CFG
consortium for functional glycomics
- CHEBI
chemical entities of biological interest
- ClinGen
central resource that defines the clinical relevance of genes and variants
- ClinVar
public archive of interpretations of clinically relevant variants.
- DB
database
- ERCC6
excision repair 6, chromatin remodeling factor
- Fuc
fucose
- Gal
galactose
- GalNAc
N-acetylgalactosamine
- GBP
glycan-binding protein (lectin)
- Glc
glucose
- GlcNAc
N-acetylglucosamine
- GlyConnect
platform for glycoscience data
- GlycoSciences
KB for glycoscience data
- GlyTouCan
repository for glycans
- GO
gene ontology
- HGNC
Human Genome Organisation Gene Nomenclature Committee
- HMO
human milk oligosaccharides
- HPO
human phenotype ontology
- HS
heparan sulfate
- ID
identifier
- Ig
immunoglobulin
- IUPAC
International Union of Pure and Applied Chemistry
- JCGGDB
Japan Consortium for Glycobiology and Glycotechnology DataBase
- KB
knowledge base
- KEGG
Kyoto Encyclopedia of Genes and Genomes
- LOINC
logical observation identifiers names and codes
- LPS
lipopolysaccharide
- MGPO
Molecular GlycoPhenotype Ontology
- Mondo
Monarch Disease Ontology
- MP
Mammalian Phenotype Ontology
- NCIt
National Center Institute thesaurus
- Neu5Ac
N-acetylneuraminic acid
- NIH
the National Institutes of Health
- OBO
open biological and biomedical ontology
- OMIM
Online Mendelian Inheritance in Man
- Orphanet
KB on rare diseases
- PRO
protein ontology
- Pubchem
NIH’s chemistry KB
- Reactome
pathway KB
- SNFG
symbol nomenclature for glycans
- SNOMED CT
Systematized Nomenclature of Medicine Clinical Terms
- STITCH
search tool for interacting chemicals
- UDP
Undiagnosed Diseases Program
- UDN
Undiagnosed Diseases Network
- UniLectin
KB for glycan-binding protein
- uPheno
unified phenotype ontology
- WES
whole exome sequencing
- WGS
whole genome sequencing
Supplementary Material
Acknowledgement
We would like to thank our colleagues from the Monarch Initiative for comments and suggestions, KidsFirst (U2CHL138346) and Undiagnosed Disease Networks Metabolomics (U01-TR001395-02) for their support.
Funding
National Institutes of Health (5 R24 OD011883).
Conflict of interest. None declared.
References
- 1. Andersen S.R. (1997) The eye and its diseases in ancient Egypt. Acta Ophthalmol. Scand., 75, 338–344. [DOI] [PubMed] [Google Scholar]
- 2. Deans A.R., Lewis S.E., Huala E. et al. (2015) Finding our way through phenotypes. PLoS Biol., 13, e1002033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Arp R., Smith B. and Spear A.D. (2015) Building Ontologies with Basic Formal Ontology, The MIT Press [Google Scholar]
- 4. Smith B., Ashburner M., Rosse C. et al. (2007) The OBO foundry: coordinated evolution of ontologies to support biomedical data integration. Nat. Biotechnol., 25, 1251–1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Haendel M.A., Chute C.G. and Robinson P.N. (2018) Classification, ontology, and precision medicine. N. Engl. J. Med., 379, 1452–1462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Robinson P.N. (2012) Deep phenotyping for precision medicine. Hum. Mutat., 33, 777–780. [DOI] [PubMed] [Google Scholar]
- 7. Köhler S., Carmody L., Vasilevsky N. et al. (2019) Expansion of the human phenotype ontology (HPO) knowledge base and resources. Nucleic Acids Res., 47, D1018-D1027. doi: 10.1093/nar/gky1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Turnbull C., Scott R.H., Thomas E. et al. (2018) The 100 000 genomes project: bringing whole genome sequencing to the NHS. BMJ, k1687, 361. [DOI] [PubMed] [Google Scholar]
- 9. Savatt J.M., Azzariti D.R., Faucett W.A. et al. (2018) ClinGen’s GenomeConnect registry enables patient-centered data sharing. Hum. Mutat., 39, 1668–1676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Orphanet. http://www.orphadata.org/cgi-bin/index.php (30 July 2019, date last accessed).
- 11. Landrum M.J., Lee J.M., Benson M. et al. (2016) ClinVar: public archive of interpretations of clinically relevant variants. Nucleic Acids Res., 44, D862–D868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Monarch Initiative Platform. https://monarchinitiative.org (30 July 2019, date last accessed).
- 13. Mungall C.J., McMurry J.A., Köhler S. et al. (2016) The monarch initiative: an integrative data and analytic platform connecting phenotypes to genotypes across species. Nucleic Acids Res., 45, D712–D722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Smedley D., Jacobsen J.O.B., Jäger M. et al. (2015) Next-generation diagnostics and disease-gene discovery with the exomiser. Nat. Protoc., 10, 2004–2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Köhler S., Schulz M.H., Krawitz P. et al. (2009) Clinical diagnostics in human genetics with semantic similarity searches in ontologies. Am. J. Hum. Genet., 85, 457–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Köhler S., Doelken S.C., Ruef B.J. et al. (2013) Construction and accessibility of a cross-species phenotype ontology along with gene annotations for biomedical research. F1000Res, 2, 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Smedley D., Oellrich A., Köhler S. et al. (2013) PhenoDigm: analyzing curated annotations to associate animal models with human diseases. Database, 2013, bat025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Pengelly R.J., Alom T., Zhang Z. et al. (2017) Evaluating phenotype-driven approaches for genetic diagnoses from exomes in a clinical setting. Sci. Rep., 7, 13509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Robinson P.N., Köhler S., Oellrich A. et al. (2014) Improved exome prioritization of disease genes through cross-species phenotype comparison. Genome Res., 24, 340–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Oellrich A., Koehler S., Washington N. et al. (2014) The influence of disease categories on gene candidate predictions from model organism phenotypes. J. Biomed. Semant., 5, S4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Haendel M.A., McMurry J.A., Relevo R. et al. (2018) A census of disease ontologies. Annu. Rev. Biomed. Data Sci., 1, 305–331. [Google Scholar]
- 22.Monarch Disease Ontology—MONDO. http://www.obofoundry.org/ontology/mondo.html (20 February 2019, date last accessed).
- 23. Schriml L.M., Mitraka E., Munro J. et al. (2019) Human disease ontology 2018 update: classification, content and workflow expansion. Nucleic Acids Res., 47, D955–D962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. de Coronado S., Wright L.W., Fragoso G. et al. (2009) The NCI thesaurus quality assurance life cycle. J. Biomed. Inform., 42, 530–539. [DOI] [PubMed] [Google Scholar]
- 25. Amberger J.S., Bocchini C.A., Schiettecatte F. et al. (2015) OMIM.org: online mendelian inheritance in man (OMIM®), an online catalog of human genes and genetic disorders. Nucleic Acids Res., 43, D789–D798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chu L., Kannan V., Basit M.A. et al. (2019) SNOMED CT concept hierarchies for computable clinical phenotypes from electronic health record data: comparison of Intensional versus extensional value sets. JMIR Med. Inform., e11487, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Office of the Secretary, HHS (2008) HIPAA administrative simplification: modification to medical data code set standards to adopt ICD-10-CM and ICD-10-PCS. Proposed rule. Fed. Regist., 73, 49795–49832. [PubMed] [Google Scholar]
- 28. Fritz A.G. (2013) International Classification of Diseases for Oncology: ICD-O. World Health Organization. [Google Scholar]
- 29.OncoTree. http://oncotree.mskcc.org/#/home (23 July 2019, date last accessed).
- 30.MedGen. https://www.ncbi.nlm.nih.gov/medgen/ (23 July 2019, date last accessed).
- 31. Splinter K., Adams D.R., Bacino C.A. et al. (2018) Effect of genetic diagnosis on patients with previously undiagnosed disease. N. Engl. J. Med., 379, 2131–2139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Gall T., Valkanas E., Bello C. et al. (2017) Defining disease, diagnosis, and translational medicine within a homeostatic perturbation paradigm: the National Institutes of Health undiagnosed diseases program experience. Front. Med., 4, 62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Davids M., Kane M.S., Wolfe L.A. et al. . Glycomics in rare diseases: from diagnosis to mechanism. Transl. Res., 206, 5-17. doi: 10.1016/j.trsl.2018.10.005. [DOI] [PubMed] [Google Scholar]
- 34. Freeze H.H., Schachter H. and Kinoshita T. (2017) In: Varki A, Cummings RD, Esko JD et al. (eds). Essentials of Glycobiology. Cold Spring Harbor Laboratory Press, Cold Spring Harbor (NY). [Google Scholar]
- 35. Xia B., Zhang W., Li X. et al. (2013) Serum N-glycan and O-glycan analysis by mass spectrometry for diagnosis of congenital disorders of glycosylation. Anal. Biochem., 442, 178–185. [DOI] [PubMed] [Google Scholar]
- 36. Xia B., Asif G., Arthur L. et al. (2013) Oligosaccharide analysis in urine by maldi-tof mass spectrometry for the diagnosis of lysosomal storage diseases. Clin. Chem., 59, 1357–1368. [DOI] [PubMed] [Google Scholar]
- 37. Campbell M.P., Aoki-Kinoshita K.F., Lisacek F. et al. (2017) In: Varki A, Cummings RD, Esko JD et al. (eds). Essentials of Glycobiology. Cold Spring Harbor Laboratory Press, Cold Spring Harbor (NY). [Google Scholar]
- 38. Michalski J.-C. and Klein A. (1999) Glycoprotein lysosomal storage disorders: α- and β-mannosidosis, fucosidosis and α-N-acetylgalactosaminidase deficiency. Biochim. Biophys. Acta (BBA) - Mol. Basis Dis., 1455, 69–84. [DOI] [PubMed] [Google Scholar]
- 39. Whitley C.B., Spielmann R.C., Herro G. et al. (2002) Urinary glycosaminoglycan excretion quantified by an automated semimicro method in specimens conveniently transported from around the globe. Mol. Genet. Metab., 75, 56–64. [DOI] [PubMed] [Google Scholar]
- 40.Monarch Oligosacchariduria. http://bit.ly/HP0010471 (4 April 2019, date last accessed).
- 41.Monarch Urinary Glycosaminoglycan Excretion. http://bit.ly/HP0003541 (4 April 2019, date last accessed).
- 42.Monarch Abnormal Glycosylation Diseases. http://bit.ly/Monarch-Glyco (4 Apr 2019, date last accessed).
- 43.Monarch Glycans Related Phenotypes. http://bit.ly/MonarchGlycans (20 July 2019, date last accessed)
- 44. Ferreira C.R., Altassan R., Marques-Da-Silva D. et al. (2018) Recognizable phenotypes in CDG. J. Inherit. Metab. Dis., 41, 541–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Varki A. and Kornfeld S. (2017) In: Varki A, Cummings RD, Esko JD et al. (eds). Essentials of Glycobiology. Cold Spring Harbor Laboratory Press, Cold Spring Harbor (NY). [Google Scholar]
- 46. Hollis J.M., Lovas F.J. and Jewell P.R. (2000) Interstellar glycolaldehyde: the first sugar. ApJ, 540, L107. [Google Scholar]
- 47. Kwok S. (2016) Complex organics in space from solar system to distant galaxies. Astron. Astrophys. Rev., 24, 8. [Google Scholar]
- 48. McCaffrey V.P., Zellner N.E.B., Waun C.M. et al. (2014) Reactivity and survivability of glycolaldehyde in simulated meteorite impact experiments. Orig. Life Evol. Biosph., 44, 29–42. [DOI] [PubMed] [Google Scholar]
- 49. Varki A. (2017) Biological roles of glycans. Glycobiology, 27, 3–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Sun S., Sun S., Cao X. et al. (2016) The role of pretreatment in improving the enzymatic hydrolysis of lignocellulosic materials. Bioresour. Technol., 199, 49–58. [DOI] [PubMed] [Google Scholar]
- 51. Freeze H.H. (2006) Genetic defects in the human glycome. Nat. Rev. Genet., 7, 537–551. [DOI] [PubMed] [Google Scholar]
- 52. Itano N. and Kimata K. (2002) Mammalian hyaluronan synthases. IUBMB Life, 54, 195–199. [DOI] [PubMed] [Google Scholar]
- 53. Viant M.R., Kurland I.J., Jones M.R. et al. (2017) How close are we to complete annotation of metabolomes? Curr. Opin. Chem. Biol., 36, 64–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Tarbell J.M. and Cancel L.M. (2016) The glycocalyx and its significance in human medicine. J. Intern. Med., 280, 97–113. [DOI] [PubMed] [Google Scholar]
- 55. Helenius A. and Aebi M. (2004) Roles of N-linked glycans in the endoplasmic reticulum. Annu. Rev. Biochem., 73, 1019–1049. [DOI] [PubMed] [Google Scholar]
- 56. Zeqiraj E., Tang X., Hunter R.W. et al. (2014) Structural basis for the recruitment of glycogen synthase by glycogenin. Proc. Natl. Acad. Sci. U. S. A., 111, E2831–E2840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Nemansky M., de Leeuw R., Wijnands R.A. et al. (1995) Enzymic remodelling of the N- and O-linked carbohydrate chains of human chorionic gonadotropin. Effects on biological activity and receptor binding. Eur. J. Biochem., 227, 880–888. [DOI] [PubMed] [Google Scholar]
- 58. Karmakar S., Cummings R.D. and McEver R.P. (2005) Contributions of Ca2+ to galectin-1-induced exposure of phosphatidylserine on activated neutrophils. J. Biol. Chem., 280, 28623–28631. [DOI] [PubMed] [Google Scholar]
- 59. Stowell S.R., Arthur C.M., Slanina K.A. et al. (2008) Dimeric galectin-8 induces phosphatidylserine exposure in leukocytes through polylactosamine recognition by the C-terminal domain. J. Biol. Chem., 283, 20547–20559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Coskun Ü., Grzybek M., Drechsel D. et al. (2011) Regulation of human EGF receptor by lipids. Proc. Natl. Acad. Sci. U. S. A., 108, 9044–9048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Pang P.-C., Chiu P.C.N., Lee C.-L. et al. (2011) Human sperm binding is mediated by the sialyl-Lewis(x) oligosaccharide on the zona pellucida. Science, 333, 1761–1764. [DOI] [PubMed] [Google Scholar]
- 62. Lichtenstein R.G. and Rabinovich G.A. (2013) Glycobiology of cell death: when glycans and lectins govern cell fate. Cell Death Differ., 20, 976–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Watkins W.M., Greenwell P., Yates A.D. et al. (1988) Regulation of expression of carbohydrate blood group antigens. Biochimie, 70, 1597–1611. [DOI] [PubMed] [Google Scholar]
- 64. Shi S. and Stanley P. (2003) Protein O-fucosyltransferase 1 is an essential component of notch signaling pathways. Proc. Natl. Acad. Sci. U. S. A., 100, 5234–5239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Kraushaar D.C., Dalton S. and Wang L. (2013) Heparan sulfate: a key regulator of embryonic stem cell fate. Biol. Chem., 394, 741–751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Berger R.P., Dookwah M., Steet R. et al. (2016) Glycosylation and stem cells: regulatory roles and application of iPSCs in the study of glycosylation-related disorders. BioEssays, 38, 1255–1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Wang Y.-C., Nakagawa M., Garitaonandia I. et al. (2011) Specific lectin biomarkers for isolation of human pluripotent stem cells identified through array-based glycomic analysis. Cell Res., 21, 1551–1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Pearce O.M. (2018) Cancer glycan epitopes: biosynthesis, structure, and function. Glycobiology, 28, 670–696. [DOI] [PubMed] [Google Scholar]
- 69. Tian X., Azpurua J., Hine C. et al. (2013) High-molecular-mass hyaluronan mediates the cancer resistance of the naked mole rat. Nature, 499, 346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Mickum M.L., Prasanphanich N.S., Heimburg-Molinaro J. et al. (2014) Deciphering the glycogenome of schistosomes. Front. Genet., 5, 262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Cooling L. (2015) Blood groups in infection and host susceptibility. Clin. Microbiol. Rev., 28, 801–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Types of Influenza Viruses|Seasonal Influenza (Flu)|CDC. https://www.cdc.gov/flu/about/viruses/types.htm (11 Jul 2018, date last accessed).
- 73. Wuyts W., Schmale G.A., Chansky H.A. et al. (2000) In: Adam MP, Ardinger HH, Pagon RA et al. (eds). GeneReviews®. University of Washington, Seattle, Seattle (WA). [Google Scholar]
- 74. Pacifici M. (2018) The pathogenic roles of heparan sulfate deficiency in hereditary multiple exostoses. Matrix Biol., 71-72, 28–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Cacho N.T. and Lawrence R.M. (2017) Innate immunity and breast milk. Front. Immunol., 8, 584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Alexander C. and Rietschel E.T. (2001) Invited review: bacterial lipopolysaccharides and innate immunity. J. Endotoxin Res., 7, 167–202. [PubMed] [Google Scholar]
- 77. Maverakis E., Kim K., Shimoda M. et al. (2015) Glycans in the immune system and the altered glycan theory of autoimmunity: a critical review. J. Autoimmun., 57, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Mestecky J., Tomana M., Moldoveanu Z. et al. (2008) Role of aberrant glycosylation of IgA1 molecules in the pathogenesis of IgA nephropathy. Kidney Blood Press. Res., 31, 29–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Varki A, Cummings RD, Esko JD et al. (eds) (2016) Essentials of Glycobiology. Cold Spring Harbor Laboratory Press, Cold Spring Harbor (NY). [PubMed] [Google Scholar]
- 80. Freeze H.H. (2013) Understanding human glycosylation disorders: biochemistry leads the charge. J. Biol. Chem., 288, 6936–6945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Shehata L., Simeonov D.R., Raams A. et al. (2014) ERCC6 dysfunction presenting as progressive neurological decline with brain hypomyelination. Am. J. Med. Genet. A, 164A, 2892–2900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Anower-E-Khuda M.F., Matsumoto K., Habuchi H. et al. (2013) Glycosaminoglycans in the blood of hereditary multiple exostoses patients: half reduction of heparan sulfate to chondroitin sulfate ratio and the possible diagnostic application. Glycobiology, 23, 865–876. [DOI] [PubMed] [Google Scholar]
- 83. Pan P., Chen M., Zhang Z. et al. (2018) A novel LC-MS/MS assay to quantify dermatan sulfate in cerebrospinal fluid as a biomarker for mucopolysaccharidosis II. Bioanalysis, 10, 825–838. [DOI] [PubMed] [Google Scholar]
- 84. Sparrow D.B., Chapman G., Wouters M.A. et al. (2006) Mutation of the LUNATIC FRINGE gene in humans causes spondylocostal dysostosis with a severe vertebral phenotype. Am. J. Hum. Genet., 78, 28–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Berger E.G. (1999) Tn-syndrome. Biochim. Biophys. Acta, 1455, 255–268. [DOI] [PubMed] [Google Scholar]
- 86. Gourdine J.-P., Metz T., Koeller D., et al. (2016) Building a Molecular Glyco-phenotype Ontology to Decipher Undiagnosed Diseases In ICBO/BioCreative . http://ceur-ws.org/Vol-1747/IP06_ICBO2016.pdf (20 July 2019, date last accessed).
- 87.Monarch Hepatomegaly. http://bit.ly/HP0002240 (4 April 2019, date last accessed).
- 88. Kluyver T. et al. and Jupyter Development Team (2016) In Fernando Loizides B.S. (ed.) Positioning and Power in Academic Publishing: Players, Agents and Agendas, IOS Press, Amsterdam, The Netherlands, pp. 87–90. [Google Scholar]
- 89. Graham E., Lee J., Price M. et al. (2018) Integration of genomics and metabolomics for prioritization of rare disease variants: a 2018 literature review. J. Inherit. Metab. Dis., 41, 435–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Herget S., Ranzinger R., Maass K. et al. (2008) GlycoCT-a unifying sequence format for carbohydrates. Carbohydr. Res., 343, 2162–2171. [DOI] [PubMed] [Google Scholar]
- 91. Neelamegham S., Aoki-Kinoshita K., Bolton E. et al. (2019) Updates to the symbol nomenclature for glycans guidelines. Glycobiology, 29, 620–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Hastings J., Owen G., Dekker A. et al. (2016) ChEBI in 2016: improved services and an expanding collection of metabolites. Nucleic Acids Res., 44, D1214–D1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Tiemeyer M., Aoki K., Paulson J. et al. (2017) GlyTouCan: an accessible glycan structure repository. Glycobiology, 27, 915–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Cummings R.D. and Pierce J.M. (2014) The challenge and promise of glycomics. Chem. Biol., 21, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Cao Y., Merling A., Karsten U. et al. (2001) The fucosylated histo-blood group antigens H type 2 (blood group O, CD173) and Lewis Y (CD174) are expressed on CD34+ hematopoietic progenitors but absent on mature lymphocytes. Glycobiology, 11, 677–683. [DOI] [PubMed] [Google Scholar]
- 96. Stanley P. and Cummings R.D. (2017) In: Varki A, Cummings RD, Esko JD et al. (eds). Essentials of Glycobiology. Cold Spring Harbor Laboratory Press, Cold Spring Harbor (NY). [PubMed] [Google Scholar]
- 97.The International Union of Pure and Applied Chemistry (IUPAC). https://iupac.org/ (10 July 2019, date last accessed).
- 98. Sun X., Ju T. and Cummings R.D. (2018) Differential expression of Cosmc, T-synthase and mucins in Tn-positive colorectal cancers. BMC Cancer, 18, 827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.The Human Glycome Project. https://human-glycome.org/ (10 July 2019, date last accessed).
- 100. Lombard V., Golaconda Ramulu H., Drula E. et al. (2014) The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res., 42, D490–D495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Comelli E.M., Head S.R., Gilmartin T. et al. (2006) A focused microarray approach to functional glycomics: transcriptional regulation of the glycome. Glycobiology, 16, 117–131. [DOI] [PubMed] [Google Scholar]
- 102. Alocci D., Mariethoz J., Gastaldello A. et al. (2019) GlyConnect: glycoproteomics goes visual, interactive, and analytical. J. Proteome Res., 18, 664–677. [DOI] [PubMed] [Google Scholar]
- 103. Böhm M., Bohne-Lang A., Frank M. et al. (2019) Glycosciences.DB: an annotated data collection linking glycomics and proteomics data (2018 update). Nucleic Acids Res., 47, D1195-D1201. doi: 10.1093/nar/gky994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Maeda M., Fujita N., Suzuki Y. et al. (2015) JCGGDB: Japan consortium for glycobiology and glycotechnology database. Methods Mol. Biol., 1273, 161–179. [DOI] [PubMed] [Google Scholar]
- 105. Kanehisa M., Furumichi M., Tanabe M. et al. (2017) KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res., 45, D353–D361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Kim S., Thiessen P.A., Bolton E.E. et al. (2016) PubChem substance and compound databases. Nucleic Acids Res., 44, D1202–D1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Fabregat A., Jupe S., Matthews L. et al. (2018) The reactome pathway knowledgebase. Nucleic Acids Res., 46, D649–D655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Bonnardel F., Mariethoz J., Salentin S. et al. (2019) UniLectin3D, a database of carbohydrate binding proteins with curated information on 3D structures and interacting ligands. Nucleic Acids Res., 27, D1236-D1244. doi: 10.1093/nar/gky832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Ashburner M., Ball C.A., Blake J.A. et al. (2000) Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet., 25, 25–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Zhang X.A., Yates A., Vasilevsky N. et al. (2019) Semantic integration of clinical laboratory tests from electronic health records for deep phenotyping and biomarker discovery. NPJ Digit. Med., 2. pii, 32. doi: 10.1038/s41746-019-0110-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Seeberger P.H. (2017) In: Varki A, Cummings RD, Esko JD et al. (eds). Essentials of Glycobiology. Cold Spring Harbor Laboratory Press, Cold Spring Harbor (NY). [Google Scholar]
- 112. Osumi-Sutherland D., Courtot M., Balhoff J.P. et al. (2017) Dead simple OWL design patterns. J. Biomed. Semant., 8, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Matentzoglu N., Balhoff J.P., Bello S.M. et al. (2018) Phenotype Ontologies Traversing All The Organisms (POTATO) workshop aims to reconcile logical definitions across species; Zenodo, doi: 10.5281/zenodo.2382757. [DOI] [Google Scholar]
- 114. Ju T., Aryal R.P., Kudelka M.R. et al. (2014) The Cosmc connection to the Tn antigen in cancer. Cancer Biomark., 14, 63–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Rojas-Macias M.A., Mariethoz J., Andersson P. et al. (2018) e-Workflow for Recording of Glycomic Mass Spectrometric Data in Compliance with Reporting Guidelines. bioRxiv. doi: 10.1101/401141. [DOI] [Google Scholar]
- 116. Poole J., Day C.J., von Itzstein M. et al. (2018) Glycointeractions in bacterial pathogenesis. Nat. Rev. Microbiol., 16, 440–452. [DOI] [PubMed] [Google Scholar]
- 117. Kahler C.M. (2011) Sticky and sweet: the role of post-translational modifications on neisserial pili. Front. Microbiol., 2, 87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Mubaiwa T.D., Hartley-Tassell L.E., Semchenko E.A. et al. (2017) The glycointeractome of serogroup B Neisseria meningitidis strain MC58. Sci. Rep., 7, 5693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Kenney A.D., Dowdle J.A., Bozzacco L. et al. (2017) Human genetic determinants of viral diseases. Annu. Rev. Genet., 51, 241–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Kamburov A., Cavill R., Ebbels T.M.D. et al. (2011) Integrated pathway-level analysis of transcriptomics and metabolomics data with IMPaLA. Bioinformatics, 27, 2917–2918. [DOI] [PubMed] [Google Scholar]
- 121. Szklarczyk D., Santos A., von Mering C. et al. (2016) STITCH 5: augmenting protein-chemical interaction networks with tissue and affinity data. Nucleic Acids Res., 44, D380–D384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Dalziel M., Crispin M., Scanlan C.N. et al. (2014) Emerging principles for the therapeutic exploitation of glycosylation. Science, 343, 1235681. [DOI] [PubMed] [Google Scholar]
- 123. Ashikov A., Abu Bakar N., Wen X.-Y. et al. (2018) Integrating glycomics and genomics uncovers SLC10A7 as essential factor for bone mineralization by regulating post-Golgi protein transport and glycosylation. Hum. Mol. Genet., 27, 3029-3045. doi: 10.1093/hmg/ddy213. [DOI] [PubMed] [Google Scholar]
- 124. Tarailo-Graovac M., Shyr C., Ross C.J. et al. (2016) Exome sequencing and the Management of Neurometabolic Disorders. N. Engl. J. Med., 374, 2246–2255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Shin S.-Y., Fauman E.B., Petersen A.-K. et al. (2014) An atlas of genetic influences on human blood metabolites. Nat. Genet., 46, 543–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Guideline for HPO Term Request. https://github.com/obophenotype/human-phenotype-ontology/wiki/How-to-make-a-good-term-request (23 July 2019, date last accessed).
- 127. Ju T. and Cummings R.D. (2002) A unique molecular chaperone Cosmc required for activity of the mammalian core 1 beta 3-galactosyltransferase. Proc. Natl. Acad. Sci. U. S. A., 99, 16613–16618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Wang Y., Ju T., Ding X. et al. (2010) Cosmc is an essential chaperone for correct protein O-glycosylation. Proc. Natl. Acad. Sci. U. S. A., 107, 9228–9233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Mi R., Song L., Wang Y. et al. (2012) Epigenetic silencing of the chaperone Cosmc in human leukocytes expressing Tn antigen. J. Biol. Chem., 287, 41523–41533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Fu C., Zhao H., Wang Y. et al. (2016) Tumor-associated antigens: Tn antigen, sTn antigen, and T antigen. Hladnikia, 88, 275–286. [DOI] [PubMed] [Google Scholar]
- 131. Wang Y., Jobe S.M., Ding X. et al. (2012) Platelet biogenesis and functions require correct protein O-glycosylation. Proc. Natl. Acad. Sci. U. S. A., 109, 16143–16148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Theodoratou E., Campbell H., Ventham N.T. et al. (2014) The role of glycosylation in IBD. Nat. Rev. Gastroenterol. Hepatol., 11, 588–600. [DOI] [PubMed] [Google Scholar]
- 133. Kudelka M.R., Hinrichs B.H., Darby T. et al. (2016) Cosmc is an X-linked inflammatory bowel disease risk gene that spatially regulates gut microbiota and contributes to sex-specific risk. Proc. Natl. Acad. Sci. U. S. A., 113, 14787–14792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Johansson M.E.V., Larsson J.M.H. and Hansson G.C. (2011) The two mucus layers of colon are organized by the MUC2 mucin, whereas the outer layer is a legislator of host-microbial interactions. Proc. Natl. Acad. Sci. U. S. A., 108, 4659–4665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Arike L. and Hansson G.C. (2016) The densely O-glycosylated MUC2 mucin protects the intestine and provides food for the commensal bacteria. J. Mol. Biol., 428, 3221–3229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Tadesse S., Corner G., Dhima E. et al. (2017) MUC2 mucin deficiency alters inflammatory and metabolic pathways in the mouse intestinal mucosa. Oncotarget, 8, 71456–71470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Cohen L.J., Esterhazy D., Kim S.-H. et al. (2017) Commensal bacteria make GPCR ligands that mimic human signalling molecules. Nature, 549, 48–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.