

Abstract
Becker muscular dystrophy (BMD) has onset usually in childhood, frequently by 11 years. BMD can present in several ways such as waddling gait, exercise related cramps with or without myoglobinuria. Rarely cardiomyopathy might be the presenting feature. The evolution is variable. BMD is caused by dystrophin deficiency due to inframe deletions, mutations or duplications in dystrophin gene (Xp21.2) We review here the evolution and current therapy presenting a personal series of cases followed for over two decades, with multifactorial treatment regimen. Early treatment includes steroid treatment that has been analized and personalized for each case. Early treatment of cardiomyopathy with ACE inhibitors is recommended and referral for cardiac transplantation is appropriate in severe cases. Management includes multidisciplinary care with physiotherapy to reduce joint contractures and prolong walking. BMD is slowly progressive with phenotypic variability. Despite childhood onset, independent walking is never lost before the third decade. Personalized medicine is required to tailor treatment to individual cases.
Key words: Becker muscular dystrophy, BMD, steroids
Introduction
After the description by Becker and Kiener in 1955 in affected families, the discovery by Monaco et al. (1) of the dystrophin gene (DMD) and the diagnostic use of dystrophin protein testing led to a drastic re-consideration of clinical phenotypes associated with deletion or duplication of the dystrophin gene: several different clinical entities were described associated with different prognosis according to the localization of mutation and residual amount of dystrophin protein (2, 3). Among these phenotypes, there are patients with cramps and myalgia, myoglobinuria, mild myopathy, quadriceps myopathy, late-onset myopathy, X-linked dilated cardiomyopathy (2, 3). It is not uncommon to find in diagnostic procedures patients with a deletion in the dystrophin gene that have normal muscle strength and endurance, but present high CK, and so far their follow-up and treatment recommendations are still a matter of debate. Patients with early cardiomyopathy are also a possible variant of BMD (4, 5) and may be susceptible either to specific drug therapy and/or to cardiac transplantation (6-8). Here we cover emerging therapies considering follow-up, and exemplifying some phenotypes and treatments by a few study cases.
Pathophysiology and rationale of therapeutic targets
All patients with dystrophin of abnormal quantity or size have clinical manifestations compatible with BMD range of phenotypes and their diagnostic traits have been identified (9). In addition to classical BMD cases, on the basis of DMD gene mutations or dystrophin protein abnormality it is possible to diagnose a number of “preclinical” or “asymptomatic” cases.
Following them for decades as atypical cases, most “asymptomatic” BMD patients show less ability in term to run or perform Gowers’ maneuver, episodes of myoglobinuria or fatigability. This progression is mainly to be attributed to two patho-mechanism factors:
inflammatory cytokines and TNF-alpha-induced microRNAs control dystrophin expression (10). Inflammatory cells could play a role in microRNAs induction and cross talk, and likely the microRNA pattern could not only be a signaling of NF-kB pathway but also play a role in promoting myogenesis, differentiation and muscle regeneration (11);
oxidative stress refers to an imbalance between the generation of free radicals- chemical species with a high reactivity and instability of oxygen (Reactive Oxygen Species, ROS) and nitrogen (Reactive Nitrogen Species, RNS), and the activity of the antioxidant defense systems (12). Among the ROS that are mainly generated during the mitochondrial electron transport chain, the superoxide anion, the hydrogen peroxide, and the hydroxyl radicals are the most studied.
The nitric oxide (NO) and the peroxynitrite are the most known among the RNS. The NO, a low reactive molecule that can become toxic forming peroxynitrite in the presence of superoxide anion, is synthesized by the enzyme NO synthase (NOS), among which isoforms there is the neuronal NOS (nNOS), localized in the sarcolemma of muscle fibers and deemed to be the main producer of RNS (12). Increasing levels of both ROS and RNS can damage different intracellular macromolecules, such as lipids, proteins, and nucleic acids (13). In particular, lipids of the sarcolemma are frequently attacked in a process called lipid peroxidation (14) and, for this reason, the products of lipid peroxidation are often used as biomarkers of oxidative stress (12).
Among therapeutic targets of metabolic pathways involved in muscle plasticity, there are increasing utrophin (15), NO (16), and inhibiting ROS and RNS (17, 18).
Case reports (Tab. 1)
Table 1.
Clinical and molecular features in the present BMD patient series.
| Clinical feature | Case 1 | Case 2 | Case 3 | Case 4 | Case 5 | Case 6 | Case 7 |
|---|---|---|---|---|---|---|---|
| Dystrophin protein | 427 kDa, 10% | 600 kDa, 100% | n.d. | n.d. | 427 kDa, 5% | 370 kDa, 30% | 380 kDa, 60% |
| Dystrophin gene mutation | Missense p.T160P | Dupl. ex.14-42 | Del. ex.48-49 | Del. ex.47-49 | Point 5’-UTR | Del. ex.45-47 | del. ex.45-49 |
| Age at onset (yrs) | 15 | 4 | 9 | Childhood | 16 | 5 | 17 |
| Cramps/myalgia | No | Yes | No | No | Yes | No | No |
| Myoglobinuria | Yes | No | No | No | Yes | No | No |
| Fatigability | Yes | No | Yes | No | No | Yes | No |
| Calf hypertrophy | Yes | Yes | No | Yes | No | Yes | Yes |
| Pes cavus | Yes | No | No | No | No | No | Yes |
| Joint contractures | No | No | Yes | No | No | Yes | Yes |
| Cardiac involvement | +++ | +/- | + | +/++ | +++ | ++/+ | + |
| Left ventricular ejection fraction | n.r. | n.r. | 54-55% | 50-51% | 34% | 45-55% | 55% |
| Cardiac transplantation | At 25 yrs | No | No | No | at 32 yrs | No | No |
| CK levels (U/L) | 2930-3479 | 1400-8630 | 1003-3600 | 656-919 | 488-1700 | 1106-2794 | 459 |
| Forced vital capacity | n.d. | n.d. | 89% | n.d. | n.d. | Normal | 76% |
| Mental/behavioral changes | Yes | No | Yes | No | No | No | Yes |
| Steroid treatment | Prednisone | Prednisone | No | No | No | Deflazacort | Deflazacort |
| Duration of steroid treatment | 10 years | 5 months | - | - | - | 26 years | 20 years |
| Effects of steroid treatment | Initial benefit | No benefit | - | - | - | Functional stabilization on long-term | Initial benefit, functional stabilization on long-term |
N.d.: not determined; N.r.: not reported
Case 1
This 15-year-old boy presented with muscle weakness, high CK levels, recurrent myoglobinuric episodes, scoliosis, calf hypertrophy, waddling gait, pes cavus, and thigh hypertrophy. He was found to carry a novel missense variant p.Thr160Pro in exon 6 of DMD gene, dystrophin protein was of normal molecular weight but reduced to 10% of controls. CK levels ranged between 2930-3479 U/L. He developed an early and severe form of dilated cardiomyopathy, which required cardiac transplantation at age 25 years (Fig. 1A). He was then closely followed both by cardiologist and neurologist. Since the heart transplant, he has been treated with 160 mg cyclosporin, 75 mg azathioprine, 20 mg prednisone daily, and has been followed once a year. He first presented some gait improvement and followed aerobic rehabilitation at home, as well as in a rehabilitative hospital. At age 35 he was able to perform a 6-meter walking test (6MWT): at two minutes he walked 50 m, and at 6 minutes 160 m. He had some behavioral problems, but was still able to work as a telephone operator. At last examination, he could stand, but walked only few steps; he was referred by family relatives to present rage tempers, but then he started coping with disease limitations and required drugs.
Figure 1.
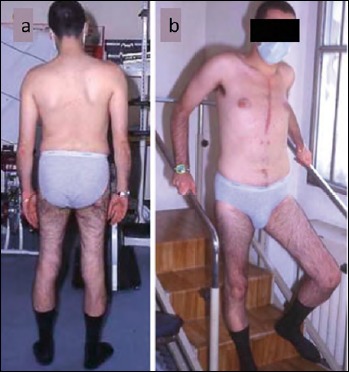
The patient (Case 1) after cardiac transplantation uses the handrail in descending stairs. Note hypotrophy of quadriceps muscle.
Case 2
At 4 years of age this child had onset of calf myalgia with cramps and CK levels ranging between 1400-8630 U/L. He was treated for 5 months with 50 mg prednisone without a clear benefit during his early teens. A muscle biopsy performed at 10 years of age showed active degeneration and regeneration foci, and a few inflammatory cells. By immunohistochemistry, there were many fetal myosin positive fibers (regenerating fibers). Immunoblot analysis showed normal quantity of an abnormally large dystrophin (600 kDa) as compared to normal size (427 kDa), originated by a duplication in the DMD gene involving exons 14-42(19). Clinical severity was mild. In the subsequent follow-ups, he discontinued prednisone treatment, was able to walk, but presented a definite deterioration, was apparently weak with persistent cramps and walking difficulty. He presented also ECG changes, such as right bundle branch block, increased R/S ratio and T-wave abnormalities. This case is interesting because it demonstrates that an enlarged dystrophin molecule is only partially functional and causes progressive weakness.
Case 3
This patient was an hyperactive child. He started walking at 14 months of age. He suffered from stuttering and headache and was seen as “lazy“ respect to peers. A muscle biopsy showed abnormal dystrophin, due to a deletion of exons 48-49 in the DMD gene. He then presented restrictive ventilatory dysfunction and elevated CK (3600 U/L), and had to leave elementary school for his poor performance. At 9 years he was able to raise from floor with one hand. At age 19 he was still reported as “nervous” and presented joint contractures, normal limb strength and grip, but weakness of extrarotator upper limb muscles and curved shoulders. On spirometry, forced vital capacity was 89%. He presented easy fatigability during cycling and elevated CK levels (1003 U/L). On echocardiography, left ventricular ejection fraction was 54-55%. This pauci-symptomatic patient did not follow any drug treatment.
Case 4
This child had high CK but presented normal muscle strength, and had only slight scapular winging and calf hypertrophy. A muscle biopsy showed few hyper-contracted fibers. Genetic analysis showed a deletion of exons 47-49 in DMD gene. At last examination, CK levels were moderately increased (656-919 U/L), and he could perform normal sporting ability and occasionally mountain climbing. Echocardiography showed altered diastolic relaxation and left ventricular ejection fraction was 50-51%.
Case 5
This 16-years-old boy (with a brother affected with severe cardiomyopathy who had died two years after cardiac transplantation) suffered from myalgia, recurrent myoglobinuric episodes, CK = 1700 U/L, and dilated cardiomyopathy. At age 32 years he had no muscle weakness and muscle biopsy showed dystrophin of normal molecular weight but severely reduced amount (5% of controls) caused by a splicing mutation in 5’-UTR region of DMD gene (20). The severity of cardiomyopathy progressed and he required a cardiac transplantation. This is a rare case of X-linked dilated cardiomyopathy caused by a mutation in the 5’-end of the DMD gene, leading to the absence of the muscle-promoter and the first exon of DMD gene, resulting in no dystrophin transcript in the cardiac muscle alone, whereas two alternative promoters were active in skeletal muscle (brain and Purkinje-cells promoters) and expressed sufficient dystrophin to prevent muscle symptoms (21).
Case 6
Since age 5 years this boy presented difficulty running and at age 11 years he had difficulty walking. Spirometry and echocardiography were normal. At age 14 he had a waddling gait, proximal lower limbs hypotrophy, Achilles tendon retractions, and calf pseudo-hypertrophy. CK level was 1106 U/L (n.v. 10-80), EMG was myogenic. Muscle biopsy showed dystrophin protein of 370 kDa, 30% of controls, due to a deletion in exons 45-47 of DMD gene.
Since age 16 years he presented difficulty climbing stairs and since age 18 years he had difficulty performing aerobic exercises. He was then started on deflazacort 30 mg/10 days/month, that at age 28 was increased to 60 mg/10 days/month and associated to diphosphonate for bone osteoporosis.
At 28 years of age he was found to have a left ventricular enlargement, with ejection fraction 45%. He was started on ACE-inhibitors (2.5 mg Triatec) and β-blockers (Congestor 1.25 mg): after 15 years of this drug regimen, his left ventricular ejection fraction improved to 55%, and CK was 2794 U/L. At age 39 years, echocardiography showed left ventricle hypokinesia with dyastolic dysfunction (altered relaxation).
At age 44 years (after 26 years of steroid treatment), he presented calf pseudo-hypertrophy, was able to rise from floor putting hands on the table, he had proximal muscle weakness in iliopsoas, semitendinosus, semimembranosus 3/5, quadriceps 4/5, triceps 4/5, biceps 3+/5 (MRC), his overall muscle functions were stable, walking ability was preserved but waddling. Muscle MRI demonstrated fibro-fatty replacement of posterior thigh and calf muscles.
Case 7
At 17 years of age he started having difficulty walking, had pes cavus and was first mis-diagnosed as affected with spinal muscular atrophy (Kugelberg-Welander syndrome). A muscle biopsy showed myopathic changes, dystrophin protein of 380 kDa, 60% of controls, due to a deletion in exons 45-49 of DMD gene. Since age 18 he was put on deflazacort (60 mg/alternate day) with improvement for about 7 years. Since age 25 he has difficulty climbing stairs requiring the use of handrail. At age 28 years he underwent cataract surgery. At age 24 echocardiography showed hypokinesia of left ventricle, with ejection fraction 55%.
Spirometry showed respiratory insufficiency, with FVC = 76%, compatible with slight restrictive ventilatory dysfunction. At age 33 years he had waddling gait, calf hypertrophy, weakness in ileopsoas, quadriceps and extrarotator muscles. CK was 459 U/L.
At age 37 he complained of limb pain: a spine MRI evidenced disc compression at L3-L4 and L4-L5 roots. The patient had also panic attacks and was treated with benzodiazepines, but continued alternate-day deflazacort with strength stabilization.
At age 38 years (after 20 years steroid treatment) he had waddling gait with lordotic posture, was able to raise from a chair using one hand, was able to raise from floor in 5-8 secs, muscle strength of ileopsoas and quadriceps was 4/5.
Genotype-phenotype correlations and prognostic features
The cases above reported show that there is a wide clinical variability for BMD, ranging from asymptomatic hyperCKemia with cramps/myalgia syndrome and myoglobinuria to proximal myopathy with cardiomyopathy.
Mutations in the DMD gene that do not alter the reading-frame (in-frame deletions/duplications) are usually associated with BMD phenotypes (22). In general, mutations in the proximal region of the dystrophin gene (exons 2-10) are associated with early-onset and severe phenotype, while those in the proximal region of central rod domain (exons 11-43) are associated with mild or asymptomatic phenotype, and those in the distal region of central rod domain (exons 44-55) are associated with the classical phenotype (23); we also observed a possible relationship between the presence of dilated cardiomyopathy and mutations in particular regions (exons 48-49, 5ʹ-region) (4-6, 20). Dystrophin immunoblot is the most important biochemical tool to diagnose patients even in the preclinical stage of the disease; this analysis is able to demonstrate the abnormality of dystrophin quantity and molecular weight, usually due to in-frame deletion/duplications in the DMD gene. For example, in young patients with high CK, a good prognosis can be entertained by dystrophin protein quantity over 70%. The clinician should not restrict the research to DNA genetic analysis, but prognostic assessment might need a muscle biopsy. Severe cases usually have less than 20% dystrophin protein amount, while mild cases with over 70% dystrophin would be late-onset ambulatory or even asymptomatic (2, 3).
Management of clinical manifestations
Cardiac involvement
Heart failure from dilated cardiomyopathy is a common cause of morbidity and the most common cause of death in BMD patients. Early diagnosis and treatment of cardiomyopathy is important for improving quality of life and maximizing patients’ survival.
ENMC (European Neuro Muscular Center) recommendations include a yearly monitoring by ECG and echocardiography starting soon after diagnosis. The importance of a frequent cardiac surveillance also in BMD patients without or with minor muscle symptoms has been highlighted by the observation that a subclinical cardiac involvement is very frequent in such patients (5, 6), who may later develop an overt dilated cardiomyopathy requiring proper treatment.
The frequency of cardiac monitoring should be increased as directed by the cardiologist, with the onset of heart failure symptoms. Treatment of cardiac involvement in dystrophinopathy patients was pioneered by Nigro (24, 25) and described by Melacini (4).
The consensus guidelines recommend initiation of angiotensin converting enzyme (ACE) inhibitors therapy with or without beta-blockers. Most cardiologists start treatment when the left ventricular ejection fraction drops below 55%. The possible preventive efficacy of ACE inhibitors was evaluated in a randomized trial of 57 children with BMD (mean age 10.7 years) who had a mean left ventricular ejection fraction of 65% (26): the children were assigned to perindopril or placebo and treated up others. Survival for the perindopril and placebo groups was 93 and 66%, respectively.
The use of eplerenone in a combined cardioprotective therapy, was able to attenuate the progressive decline of left ventricular systolic function in Duchenne patients (27), and, due to the absence of important contraindications, it has been suggested as a new drug to be used in future trials also in BMD patients, aimed at investigate its possible benefit as a cardiomyopathy treatment (28, 29).
When dilated cardiomyopathy evolves towards the stage of heart failure, this rapidly becomes intractable and cardiac transplantation is the only life-saving option. Heart transplant has so far only been reserved to BMD patients with both severe dilated cardiomyopathy and limited evidence of skeletal muscle disease (7). Long-term follow-up of transplanted patients suggested that this procedure is able to consistently prolong life expectancy (8), provided that the exact modulation of immunosuppressive dosage to avoid worsening of myopathy and the adoption of proper respiratory and muscle functional rehabilitation are pursued.
In the present series of cases, cardiac involvement was present in Cases 1, 3, 4, 5, 6 and it appeared as a prominent clinical feature in Cases 1 and 5, who successfully underwent cardiac transplantation.
Intellectual disability
Mental retardation (IQ < 75) has been rarely reported in BMD patients (30), possibly related to deletion mutations removing the Dp140 regulatory region of the DMD gene. However, some patients may present behavioral problems, panic attacks, language problem, hyperactivity and sometimes difficulty on fining adequate jobs (as in the present Cases 1, 3 and 7). Some of these difficulties might be attributed to social problems (31), but the study of central nervous system involvement in BMD would need a large cooperative study.
Rehabilitative therapy
A relevant aspect to consider when advising physical exercise in BMD patients is related their muscle metabolism. As known, the muscle fibers can be distinguished into two main categories (17): type I fibers (slow), which predominantly use an oxidative metabolism and are predisposed for an aerobic work; and type II fibers (fast), which have a glycolytic metabolism and therefore are predisposed for an anaerobic work. There are also the type IIa fibers, that have intermediate characteristics between the oxidative fibers and the glycolytic fibers.
Furthermore, a characteristic of the muscle fiber is that it is endowed with plasticity, i.e. the ability to modify metabolism and physiological properties depending on external stimuli, especially exercise and drugs (18). The transition from the fast fibers to the slow fibers of healthy muscles is not complete, and intermediate fibers characteristics predominate.
This transition may have crucial implications from a therapeutic perspective, since it has been demonstrated that the slow muscle fibers are more resistant to necrosis and, therefore, seem to be more spared in muscular dystrophies (17).
Several researches are underway to identify which properties make the slow fibers more resistant to the dystrophic process. To date, it is known that these fibers: a) are characterized by an increase in the expression of utrophin, a protein with a structure similar to dystrophin and that can partially compensate for its deficiency (15); b) releases less ROS; c) are more resistant to fatigue and, consequently, can be more resistant to the injury caused by extreme contractions; d) have a higher expression of the adenosine monophosphate activated protein kinase (AMPK), which protects the cells through different mechanisms, including the reduction of fibrosis by increasing the number of mitochondria. Overall, a better understanding of how the transition of fast fibers to slow fibers changes the metabolism and physiological properties of muscle will play an important role in the development of new treatment options for muscular dystrophies.
A better understanding of the disease process and progression, including comorbidities and complications, is mandatory for a comprehensive and individually-tailored planning and monitoring of rehabilitative activities. This will also shed lights on the intrinsic properties of the dystrophic muscle to respond to different interventions, possibly involving the enhancement of the residual motor activities and adaptive plastic changes in response to fiber loss. Interestingly, multimodal integrated interventions targeting both perception abilities and motor skills have been shown to be effective also in other clinical contexts (32). In particular, it has been demonstrated that motor learning and memory mediated mechanisms of plasticity might underlie the improvement of hand motor coordination, speed-accuracy, and fine motor performance in some neurological disorders.
The loss of nNOS may cause functional ischemia contributing to skeletal and cardiac muscle cell injury. The effects of NO is augmented (by inhibiting degradation of cGMD) by the use of sildenafil and tadanafil. Although promising results have been observed using these drugs in dystrophinopathy animal models, the effects in Duchenne and Becker patients have been disappointing, with minor effects on upper limb performance and none on ambulation (16).
Endurance training has been demonstrated to be a safe method to increase exercise performance and daily function in BMD patients (33), suggesting the usefulness of an active approach to rehabilitation (34, 35).
Prevention of other secondary complications
To avoid respiratory complications, pneumococcal and influenza annual immunization, as well as annual spirometry is recommended. In patients presenting a decline of forced vital capacity leading to respiratory insufficiency, supportive respiratory aids should be offered when necessary.
Other complications in BMD are joint contractures (pes cavus was reported in 15-70% of cases, and it was present in Cases 1 and 7), which can be prevented by physical therapy to promote mobility and reduce cramps and muscle fatigue.
A proper control of dietary intake is necessary to avoid obesity and reduce the risk of bone fractures (diet rich in vitamin D and calcium).
Current and future therapies
Medical treatments
Anti-inflammatory steroid therapy have been so far adopted in BMD patients, since steroids improve muscle strength and function. Steroids are the most relevant available treatment, and we present our experience in this patients series.
Long-term treatment with deflazacort on intermittent schedule has been used in two patients in the present series (Cases 6 and 7), in whom we have been able to obtain an initial improvement, followed by stabilization of the patient condition, with preservation of muscle function and prolongation of walking ability. Furthermore, this therapy is expected to maintain upper limb muscle strength, and delay the decline of respiratory and cardiac function, although this field needs further investigations. Prednisone was less successful, since both patients (Cases 1 and 2) had muscle strength deterioration, although other comorbidities such as cardiomyopathy were relevant clinical features. The two drugs had different cardiological and side effects. Steroid treatment may be continued on a long-term basis if side effects (weight gain, increased risk of bone fractures, cataract, behavioral changes) are not severe, and the schedule and dosage of treatment should be changed and personalized if necessary.
Givinostat, anti-fibrotic agent, is under trial with a morphological outcome.
Emerging molecular therapies: personalized medicine and future directions
The field of dystrophinopathies has seen an unprecedented advancement in the field of new molecular therapies in the last two decades. Most of them aim to the molecular correction of dystrophin deficiency in selected series of patients with specific mutations. A series of antisense oligonucleotides (AON), such as eteplirsen, have been demonstrated to be able to induce specific exon skipping during splicing, to correct the open-reading-frame, and to restore dystrophin production (36). This potential of this therapy has been so far investigated in Duchenne muscular dystrophy patients, showing beneficial results (37), but not in BMD patients. Unfortunately, most of the molecular defects in BMD concern the quantity and quality of dystrophin and are not amenable to correction.
The CRISP/Cas9 technique, which has been shown to partially restore dystrophin expression in cardiac and skeletal muscle in dystrophinopathy mouse model by removing the noncoding introns that flank the mutation-containing exon (38), could be applied in BMD patients, especially in those bearing a gene duplication.
Conclusions
The clinical heterogeneity of BMD is extreme, with patients presenting only with cramps/myalgia syndrome and high CK, other recurrent myoglobinuric episodes, while loss of strength is usually seen in the typical cases. A good term to define this heterogeneous group of disorders/phenotypes is mild dystrophinopathy.
Since some cases have a predominant heart phenotype, their cardiac prognosis is crucially determined by an adequate treatment aimed to prevent cardiac failure and death. In selected patients, cardiac transplantation may be offered as a life-saving therapy, which is able to considerably prolong life expectancy.
Although new molecular therapies are emerging as a future way to systemically treat dystrophin deficiency, at the moment no curative treatments are available, and we can only treat symptoms, such as muscle functional impairment with steroids, heart failure with specific drugs and transplantation, joint contractures with physical therapy.
However, to obtain a long survival, it is of crucial importance that patients with BMD stay in shape and continue to use their muscles. This can include physical therapy treatment and aerobic exercise, which are directed to maximize muscle function.
Figures and tables
Footnotes
Conflict of interest
The Authors declare to have no conflict of interest.
References
- 1.Monaco AP, Bertelson CJ, Middlesworth W, et al. Detection of deletions spanning the Duchenne muscular dystrophy locus using a tightly linked DNA segment. Nature 1985;316:842-5. [DOI] [PubMed] [Google Scholar]
- 2.Angelini C, Fanin M, Pegoraro E, et al. Clinical-molecular correlation in 104 mild X-linked muscular dystrophy patients: characterization of subclinical phenotypes. Neuromusc Disord 1994;4:349-58. [DOI] [PubMed] [Google Scholar]
- 3.Angelini C, Fanin M, Freda MP, et al. Prognostic factors in mild dystrophinopathies. J Neurol Sci 1996;142:70-8. [DOI] [PubMed] [Google Scholar]
- 4.Melacini P, Fanin M, Danieli GA, et al. Cardiac involvement in Becker muscular dystrophy. J Am Coll Cardiol 1993;22:1927-34. [DOI] [PubMed] [Google Scholar]
- 5.Melacini P, Fanin M, Danieli GA, et al. Myocardial involvement is very frequent among patients affected with subclinical Becker muscular dystrophy. Circulation 1996;94:3168-75. [DOI] [PubMed] [Google Scholar]
- 6.Nigro G, Comi LI, Politano L, et al. Evaluation of the cardiomyopathy in Becker muscular dystrophy. Muscle Nerve 1995;18:283-91. [DOI] [PubMed] [Google Scholar]
- 7.Melacini P, Gambino A, Caforio A, et al. Heart transplantation in patients with inherited myopathies associated with end-stage cardiomyopathy: molecular and biochemical defects on cardiac and skeletal muscle. Transplant Proc 2001;33:1596-9. [DOI] [PubMed] [Google Scholar]
- 8.Papa AA, D’Ambrosio P, Petillo R, et al. Heart transplantation in patients with dystrophinopathic cardiomyopathy: review of the literature and personal series. Intract Rare Dis Res 2017;6:95-101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hoffman EP, Kunkel LM, Angelini C, et al. Improved diagnosis of Becker muscular dystrophy by dystrophin testing. Neurology 1989;39:1011-7. [DOI] [PubMed] [Google Scholar]
- 10.Fiorillo AA, Heier CR, Novak JS, et al. TNF-α-Induced microRNAs control dystrophin expression in Becker muscular dystrophy. Cell Rep 2015;12:1-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Roberts TC, Godfrey C, McClorey G, et al. Extracellular microRNAs are dynamic non-vesicular biomarkers of muscle turnover. Nucleic Acids Res 2013;41:9500-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chico L, Ricci G, Cosci O, et al. Physical exercise and oxidative stress in muscular dystrophies: is there a good balance? Arch Ital Biol 2017;155:11-24. [DOI] [PubMed] [Google Scholar]
- 13.Phaniendra A, Jestadi DB, Periyasamy L. Free radicals: properties, sources, targets, and their implication in various diseases. Indian J Clin Biochem 2015;30:11-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Markert CD, Ambrosio F, Call JA, et al. Exercise and Duchenne muscular dystrophy: toward evidence-based exercise prescription. Muscle Nerve 2011;43:463-78. [DOI] [PubMed] [Google Scholar]
- 15.Blake DJ, Tinsley JM, Davies KE. Utrophin: a structural and functional comparison to dystrophin. Brain Pathol 1996;6:37-47. [DOI] [PubMed] [Google Scholar]
- 16.Dombernowsky NW, Olmestig JNE, Witting N, et al. Role of neuronal nitric oxide synthase (nNOS) in Duchenne and Becker muscular dystrophies. Still a possible treatment modality? Neuromusc Disord 2018;28:914-26. [DOI] [PubMed] [Google Scholar]
- 17.Heydemann A. Skeletal muscle metabolism in Duchenne and Becker muscular dystrophy – implications for therapies. Nutrients 2018;10 pii: E796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ferraro E, Giammarioli AM, Chiandotto S, et al. Exercise-induced skeletal muscle remodeling and metabolic adaptation: redox signaling and role of autophagy. Antioxid Redox Signal 2014;21:154-76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Angelini C, Beggs AH, Hoffman EP, et al. Enormous dystrophin in a patient with Becker muscular dystrophy. Neurology 1990;40:808-12. [DOI] [PubMed] [Google Scholar]
- 20.Milasin J, Muntoni F, Severini GM, et al. A point mutation in the 5’ splice site of the dystrophin gene first intron responsible for X-linked dilated cardiomyopathy. Hum Molec Genet 1996;5:73-9. [DOI] [PubMed] [Google Scholar]
- 21.Towbin JA, Hejtmancik JF, Brink P, et al. X-linked dilated cardiomyopathy. Molecular genetic evidence of linkage to the Duchenne muscular dystrophy (dystrophin) gene at the Xp21 locus. Circulation 1993;87:1854-65. [DOI] [PubMed] [Google Scholar]
- 22.Monaco AP, Bertelson CJ, Liechti-Gallati S, et al. An explanation for the phenotypic differences between patients bearing partial deletions of the DMD locus. Genomics 1988;2:90-5. [DOI] [PubMed] [Google Scholar]
- 23.Bello L, Campadello P, Barp A, et al. Functional changes in Becker muscular dystrophy: implications for clinical trials in dystrophinopathies. Scient Rep 2016;6:32439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nigro G, Comi L, Politano L, et al. The incidence and evolution of cardiomyopathy in Duchenne muscular dystrophy. Int J Cardiol 1990;26:271-7. [DOI] [PubMed] [Google Scholar]
- 25.Politano L, Nigro G. Treatment of dystrophinopathic cardiomyopathy: review of the literature and personal results. Acta Myol 2012;31:24-30. [PMC free article] [PubMed] [Google Scholar]
- 26.Duboc D, Meune C, Lerebours G, et al. Effect of perindopril on the onset and progression of left ventricular dysfunction in Duchenne muscular dystrophy. J Am Coll Cardiol 2005;45:855-7. [DOI] [PubMed] [Google Scholar]
- 27.Raman SV, Hor KN, Mazur W, et al. Eplerenone for early cardiomyopathy in Duchenne muscular dystrophy: a randomized, double-blind, placebo-controlled trial. Lancet Neurol 2015;14:153-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Breitenbach S, Lehmann-Horn F, Jurkat-Rott K. Eplerenone repolarizes muscle membrane through Na,K-ATPase activation by Tyr10 dephosphorylation. Acta Myol 2016;35:86-9. [PMC free article] [PubMed] [Google Scholar]
- 29.Angelini C. Prevention of cardiomyopathy in Duchenne muscular dystrophy. Lancet Neurol 2015;14:127-8. [DOI] [PubMed] [Google Scholar]
- 30.Bardoni A, Sironi M, Felisari G, et al. Absence of brain Dp140 isoform and cognitive impairment in Becker muscular dystrophy. Lancet 1999;353:897-8. [DOI] [PubMed] [Google Scholar]
- 31.Magliano L, Scutifero M, Patalano M, et al. Integrated care of muscular dystrophies in Italy. Part 2. Psychological treatments, social and welfare support, and financial costs. Acta Myol 2017;36:41-5. [PMC free article] [PubMed] [Google Scholar]
- 32.Cantone M, Catalano MA, Lanza G, et al. Motor and perceptual recovery in adult patients with mild intellectual disability. Neural Plast 2018;2018:3273246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sveen ML, Jeppesen TD, Hauerslev S, et al. Endurance training improves fitness and strength in patients with Becker muscular dystrophy. Brain 2008;131:2824-31. [DOI] [PubMed] [Google Scholar]
- 34.Sveen ML, Andersen SP, Ingelsrud LH, et al. Resistance training in patients with limb-girdle and Becker muscular dystrophies. Muscle Nerve 2013;47:163-9. [DOI] [PubMed] [Google Scholar]
- 35.Jensen BR, Berthelsen MP, Husu E, et al. Body weight-supported training in Becker and limb girdle 2I muscular dystrophy. Muscle Nerve 2016;54:239-43. [DOI] [PubMed] [Google Scholar]
- 36.Goemans NM, Tulinius M, Van den Akker JT, et al. Systemic administration of PRO051 in Duchenne’s muscular dystrophy. N Engl J Med 2011;364:1513-22. [DOI] [PubMed] [Google Scholar]
- 37.Mendell JR, Goemans N, Lowes LP, et al. Longitudinal effect of eteplirsen versus historical control on ambulation in Duchenne muscular dystrophy. Ann Neurol 2016;79:257-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Long C, Amoasii L, Mireault AA, et al. Postnatal genome editing partially restores dystrophin expression in a mouse model of muscular dystrophy. Science 2016;351:400-3. [DOI] [PMC free article] [PubMed] [Google Scholar]