

Abstract
Lower lobe predominant pulmonary cysts occur in up to 90% of patients with Birt–Hogg–Dubé (BHD) syndrome, but the key pathologic cell type and signaling events driving this distinct phenotype remain elusive. Through examination of the LungMAP database, we found that folliculin (FLCN) is highly expressed in neonatal lung mesenchymal cells. Using RNA-Seq, we found that inactivation of Flcn in mouse embryonic fibroblasts leads to changes in multiple Wnt ligands, including a 2.8-fold decrease in Wnt2. This was associated with decreased TCF/LEF activity, a readout of canonical WNT activity, after treatment with a GSK3-α/β inhibitor. Similarly, FLCN deficiency in HEK293T cells decreased WNT pathway activity by 76% post-GSK3-α/β inhibition. Inactivation of FLCN in human fetal lung fibroblasts (MRC-5) led to ~ 100-fold decrease in Wnt2 expression and a 33-fold decrease in Wnt7b expression—two ligands known to be necessary for lung development. Furthermore, canonical WNT activity was decreased by 60%. Classic WNT targets such as AXIN2 and BMP4, and WNT enhanceosome members including TCF4, LEF1 and BCL9 were also decreased after GSK3-α/β inhibition. FLCN-deficient MRC-5 cells failed to upregulate LEF1 in response to GSK3-α/β inhibition. Finally, we found that a constitutively active β-catenin could only partially rescue the decreased WNT activity phenotype seen in FLCN-deficient cells, whereas silencing the transcription factor TFE3 completely reversed this phenotype. In summary, our data establish FLCN as a critical regulator of the WNT pathway via TFE3 and suggest that FLCN-dependent defects in WNT pathway developmental cues may contribute to lung cyst pathogenesis in BHD.
Introduction
Inactivating germline mutations in Folliculin (FLCN) lead to Birt–Hogg–Dubé (BHD, OMIM #135150) syndrome, an autosomal disorder characterized by facial fibrofolliculomas, renal cell carcinomas (RCCs) and lower lobe predominant pulmonary cysts (1,2). RCCs occur in up to one-third of BHD patients, and pulmonary cysts in up to 90%. Pneumothorax occurs in ~ 30% of BHD patients, with a recurrence rate of around 85% (3–5). In ~12% of all primary spontaneous pneumothorax cases a family history of pneumothorax can be identified, and of those familial spontaneous pneumothorax cases, germline FLCN mutations have been found in up to 50% of patients (6,7).
FLCN has diverse functions in the cell that include regulation of mechanistic target of rapamycin complex 1 (mTORC1) (8–14), TGF-β signaling (9,15), mitochondrial biogenesis through PGC1-α (16), autophagy (17), AMPK (12,18,19), cytoplasmic-nuclear localization of the transcription factor TFE3 (20–22), cell–cell adhesion and Rho A signaling (23,24), formation of primary cilia (25) and EGFR recycling (26). The DENN (differentially expressed in normal and neoplastic cells) domain within FLCN, which is near the C-terminus, is believed to regulate small GTPases (27). FLCN is now known to localize to the lysosome where it functions as a GAP (GTPase-activating protein) for RagC/D (10), interacts with RagA/B (11) and is required for mTOR activation by amino acids downstream of GATOR1 complex (28). Lysosome-dependent positive regulation of mTOR activity appears to be one of the most fundamental and evolutionarily conserved functions of FLCN, with similar mechanisms present in yeast (29).
The mechanisms of lung-cyst development and the lower lobe predominance of lung cysts in BHD are not well understood. Pulmonary cysts in BHD patients typically develop below the level of the carina, in a sub-pleural or paramediastinal distribution, with up to 88% of these abutting the interlobular septae on histologic examination (30,31). The timing of the onset of cyst formation in BHD is also not fully understood. Pneumothorax most frequently occurs in the 3rd, 4th or 5th decade of life in BHD, but cysts may arise much earlier, with some case reports of cystic disease in pediatric and prenatal BHD patients (3,32,33), suggesting a developmental origin. To date, FLCN’s functions have been studied primarily in epithelial and hematopoietic lineages (17,18,25,34–36). Given the key role of embryonic mesenchyme in lung development, we chose to focus here on the role of FLCN in the mesenchyme. We found that canonical Wnt signaling is decreased in FLCN-deficient mesenchymal cells from both mice and humans and associated with decreased levels of multiple components of the Wnt enhanceosome. This phenotype can be rescued by silencing TFE3. These findings lead to the hypothesis that lung cysts in BHD may arise through defective mesenchymal signaling via Wnt, beginning as early as anterior foregut endoderm specification.
Results
Loss of Flcn in mouse embryonic fibroblasts leads to altered expression of Wnt ligands
Publicly available RNA-Seq data on the LungMAP platform shows that in children, FLCN is expressed in multiple cell types including pulmonary mesenchyme and pulmonary epithelium (Fig. 1A). To assess the role of Flcn in mesenchyme, mouse embryonic fibroblasts (MEFs) were derived from a Flcnfl/fl: Rosa26Cre-ERT2 mouse and treated with 4-hydroxy-tamoxifen (2 μM) for 3 days, resulting in loss of Flcn at the protein (Fig. 1B) and mRNA (Fig. 1D) levels in these cells, referred to as Flcn knock-out (KO) MEFs, compared with ethanol-treated controls. RNA sequencing was performed on these Flcn KO and control MEFs (Fig. 1C). Positive regulation of WNT signaling was the third most down regulated Gene Ontology (GO) term identified (Supplementary Material, Figure S1). Seventy-two transcripts were upregulated in Flcn KO versus control cells with a Log2 fold change of >1, and 22 transcripts were downregulated with a Log2 fold change of < 1 (p adj < 0.01). The most upregulated transcript from the RNA-Seq analysis was Wnt7a (2.3 fold, P adj = 7.55 ×10−38), and the most downregulated transcript was Wnt2 (−1.5-fold, P adj = 4.70 ×10−18, Fig. 1C). Several other transcripts that have been implicated in Wnt signaling were also differentially regulated including SFRP2 and DKK2, decreased by 53% and 44%, respectively.
Figure 1.
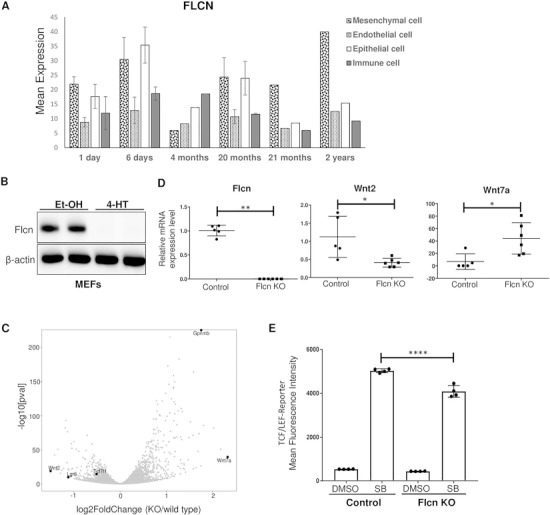
Expression of FLCN in human lungs and expression of Wnt ligands in Flcn-deficient cells. (A) RNA-Seq data from the LungMAP database showing FLCN expression patterns by cell type in children from the age of 1 day to 2 years. (B) Western blot of Flcn fl/fl Rosa26Cre-ERT2 MEFs after 3 days of treatment with ethanol (Et-OH) or 2 μM 4-hydroxy-tamoxifen (4-HT). β-actin is a loading control. The data are representative of three independent experiments. (C) Volcano plot generated from RNA-Seq differential expression analysis using MEF-derived RNA. Scale is Log2 fold change; Wnt2 = −1.5, Wnt7a = +2.3. (D) qRT-PCR of selected target transcripts (Flcn, Wnt2 and Wnt7a) in multiple MEF clones (n = 5 for control and n = 6 for Flcn KO MEFs). (E) Mean fluorescence intensity of control or Flcn-deficient MEFs infected with lentivirus containing the TCF/LEF-GFP reporter construct and treated with DMSO or SB216763 (1 μM) for 24 h. Data are representative of three independent experiments and presented as mean ± SD, *P < 0.05, **P < 0.01, Student’s t-test, ****P < 0.0001 one-way ANOVA.
Single-cell clones of Flcn knock-out MEFs were generated from the parent populations. Quantitative PCR was performed on five control clones and six KO clones. These single-cell clones confirmed the increased Wnt7a (~6.5-fold increase, P = 0.015) and decreased Wnt2 mRNA levels (~3-fold decrease, P = 0.014) in the KO cells (Fig. 1D). Glycoprotein non-metastatic B (Gpnmb), a cell surface protein and TFE3 target that has been previously shown to be overexpressed in BHD-associated RCC when compared with sporadic RCC (37), was also upregulated in the KO MEFs (Fig. 1C), providing an internal control for the data set.
Loss of Flcn in MEFs leads to decreased canonical Wnt signaling after GSK3-α/β inhibition
To determine how Flcn-deficiency impacts Wnt signaling, control and KO MEFs were infected with a lentiviral TCF/LEF-GFP reporter plasmid. Canonical Wnt activity was assessed by measuring the mean fluorescence intensity of the GFP reporter by flow cytometry, with or without treatment with the GSK3-α/β inhibitor SB216763 (SB). As expected, GSK3-α/β inhibition stimulated Wnt activity in both the KO and control cells. SB-stimulated Wnt activity was decreased by 19% (P ≤ 0.0001) in the Flcn KO MEFs when compared to the control MEFs (Fig. 1E). Canonical Wnt signaling activity was comparable between Flcn KO and control MEFs in the absence of SB stimulation.
Canonical WNT signaling activity is decreased in FLCN-deficient HEK293T cells
To determine whether FLCN regulates WNT activity in human cells, we generated a CRISPR/Cas-9 plasmid targeting the ATG of FLCN and generated seven complete KO HEK293T cell lines (Fig. 2A). We infected one of the KO clones (clone 7) and the parental cells with the lentiviral TCF/LEF-GFP reporter construct to generate stable reporter cell lines. TCF/LEF-GFP reporter activity was decreased by 76% (P = 0.03, one-way ANOVA) in FLCN KO HEK293T cells after stimulation with the GSK3-α/β inhibitor CHIR99021 (CHIR) (Fig. 2B). CHIR is widely used to stimulate canonical WNT signaling, but is also known to have off-target effects. To more precisely probe the canonical WNT pathway, we utilized the compound BRD0320, a recently developed and more specific GSK3-α/β inhibitor (38). WNT activity was decreased by 60% (P ≤ 0.0001, one-way ANOVA) in FLCN KO HEK293T cells after stimulation with BRD0320 (Fig. 2C). Next, we used Wnt3a, a canonical WNT signaling ligand, to stimulate WNT signaling in FLCN KO cells through the endogenous cell surface receptor machinery. Stimulation with Wnt3a for 48 h lead to a 31% decrease in WNT reporter activity in comparison with control cells (P ≤ 0.0001, one-way ANOVA) (Fig. 2D). This phenotype is not associated with differential β-catenin translocation to the nucleus in these cells after CHIR stimulation (Supplementary Material, Figure S1).
Figure 2.
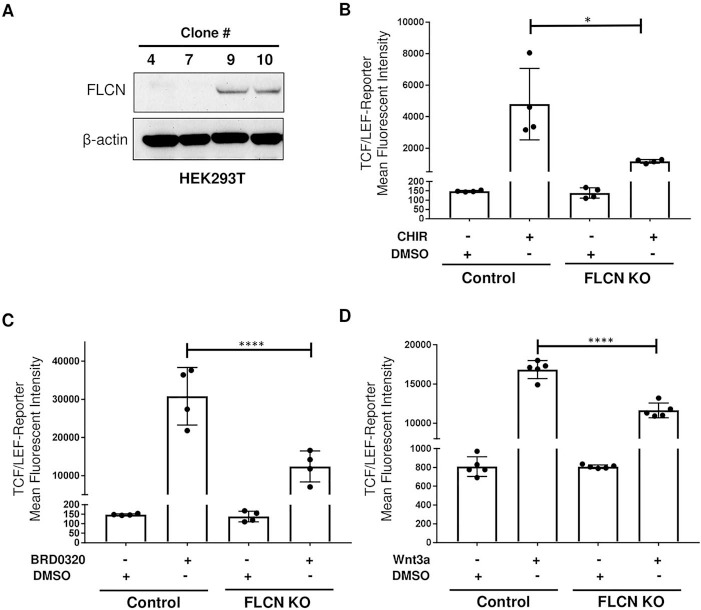
Canonical WNT signaling activity is decreased in FLCN-deficient HEK293T cells. (A) Western blot of HEK293T clones generated after transient transfection, flow sorting and single-cell selection. FLCN was knocked out in clones 4 and 7, but not in clones 9 and 10, which were used as controls. β-actin is a loading control. The data are representative of three independent experiments. (B–D) Mean fluorescence intensity of control or FLCN-CRISPR HEK293T cells infected with lentivirus containing the TCF/LEF-GFP reporter construct (clone 7) and treated for 48 h with the GSK-3α/β inhibitors CHIR99021 (CHIR, 1 μΜ) (B) or BRD0320 (5 μM) (C) or the canonical WNT ligand WNT3a (150 ng/ml) (D). GFP fluorescence was measured with flow cytometry. Data are representative of three independent experiments and presented as mean ± SD, *P < 0.05, ****P < 0.0001 one-way ANOVA.
Canonical WNT signaling and β-catenin levels are decreased in FLCN-deficient lung fibroblasts
We next sought to determine the impact of FLCN downregulation in human lung fibroblasts. We transiently transfected MRC-5 cells, which are derived from human fetal lung fibroblasts, with our CRISPR/Cas-9 construct, and generated single-cell clones with FLCN KO. We found that total β-catenin and active β-catenin (ABC) (as assessed using an antibody specific for β-catenin dephosphorylated on Ser37 or Thr41) were decreased at the protein level in these cells (Fig. 3A). RNA-sequencing of FLCN KO and control MRC-5 cells revealed differential expression of multiple transcripts in the WNT pathway (Fig. 3B and Supplementary Material, 2). WNT2 was downregulated ~ 98-fold (P adj = 5.48−43) and WNT7b was downregulated ~ 33-fold (P adj = 4.24−20), while WNT16 was upregulated ~ 6 fold (P adj = 1.44−22) and FZD5 was upregulated ~ 5-fold (P adj = 7.83−24). The WNT7a transcript was not detected in these control or KO cells. GPNMB was upregulated in the FLCN KO cells by ~ 28-fold (P adj = 4.12−198), as observed earlier in the Flcn KO MEFs. TCF/LEF-GFP reporter lines were generated from two FLCN KO clones (clone 4 and clone 6), from the parental MRC-5 cells, and from one clone (clone 10) of MRC-5 cells that had gone through the transfection, cell sorting and single-cell cloning process but had not deleted the FLCN transcript. Again, we saw a ~ 60% decrease in TCF/LEF-GFP reporter activity after CHIR stimulation in FLCN KO cells compared with the control cells (P = 0.001, one-way ANOVA) (Fig. 3C).
Figure 3.
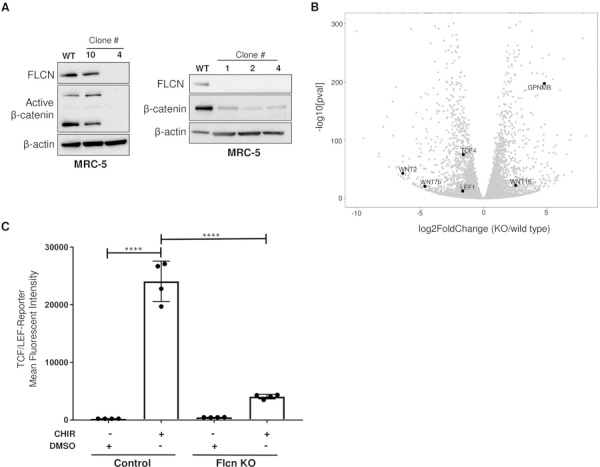
Canonical WNT signaling is decreased in FLCN-deficient MRC-5 cells. (A) Western blot of human fetal lung fibroblasts (MRC-5) transiently transfected with CRISPR/Cas9 plasmid and single cell sorted by FACS. β-actin is a loading control. The data are representative of three independent experiments. (B) Volcano plot generated from RNA-Seq differential expression analysis using MRC-5 wild-type and KO (clone 5) RNA. Scale is Log2 fold change; WNT2 = −6.37, WNT7b = −4.91. (C) Mean fluorescence intensity of control or FLCN-KO MRC-5 (clone 4) cells infected with lentivirus containing the TCF/LEF-GFP reporter construct and treated for 48 h with control or 3.3 μM GSK-3α/β inhibitor CHIR99021 (CHIR). GFP fluorescence was measured by flow cytometry. Data are representative of three independent experiments and presented as mean ± SD, ****P < 0.0001.
Multiple components of the recently characterized WNT enhanceosome are downregulated in FLCN CRISPR KO cells
The TCF/LEF family of transcription factors, which is comprised of four members (TCF-1, TCF-3, TCF-4 and LEF-1), recruits ABC to WNT target genes. Coactivation by ABC drives transcription of WNT signaling target genes (39). In the absence of a WNT signal, these proteins act as repressors of WNT target gene transcription through interactions with the Groucho/TLE protein complex. The TCF/LEF family, together with other proteins including the chromatin binding protein Pygo and the scaffold protein BCL9, form the Wnt enhanceosome. The Wnt enhanceosome is thought to repress WNT signaling at baseline and to undergo a conformational change triggered by the migration of ABC to the nucleus and subsequent Groucho/TLE ubiquitination by UBR5 and dissociation by VCP/p97 (40).
To better understand the mechanism by which WNT signaling is decreased in FLCN KO cells, we examined a panel of transcripts known to be involved in the WNT signaling pathway using the Nanostring nCounter Vantage 3D Wnt panel, which analyzes 189 WNT pathway genes. RNA was hybridized from control MRC-5 cells treated with DMSO or CHIR (3.3 μM for 48 h), and FLCN KO MRC-5 cells treated with DMSO or CHIR (3.3 μM for 48 h), both in triplicate. One hundred sixty-nine genes were valid for analysis after processing (nSolver 4.0 software). The changes seen in WNT signaling-associated mRNA transcript levels including decreases in TCF4, PDGFRα and IL6 served as further validation of the RNA-Seq results described in the previous section (Fig. 4A). As expected based on our reporter assay data, classic WNT pathway target transcripts including AXIN2 and BMP4 were decreased in the FLCN KO cells after CHIR treatment (Fig. 4A, B). We also noted decreases in many members of the WNT enhanceosome including LEF1, TCF7, TCF4 and BCL9. The enhanceosome corepressor TLE1 was increased in FLCN KO cells as compared to control MRC-5 cells in the absence of CHIR, but the transcript increased to the same degree after CHIR treatment in this experiment. Interestingly, TLE2 decreased by 81% and TLE3 decreased by 67% between control MRC-5 cells and FLCN KO MRC-5 cells in the RNA-Seq experiment (TLE2 and TLE3 transcripts were not included in the Nanostring panel). Many transcripts in the Nanostring panel were increased in FLCN KO cells after CHIR treatment when compared to wild-type cells including Cyclin D1 and Cyclin D2, which have been previously shown to be upregulated in FLCN KO cell lines (18,25,41,42), and the putative WNT2 receptor FZD5 (43) (Fig. 4C, D).
Figure 4.
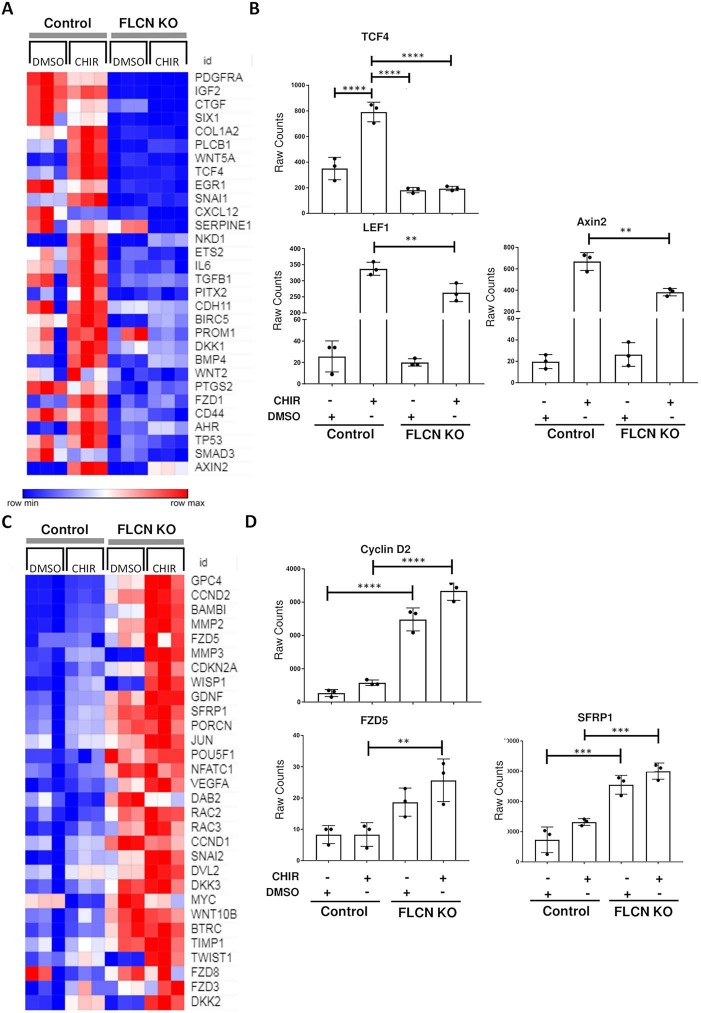
FLCN-dependent WNT pathway changes in MRC-5 cells. (A) Top thirty transcripts downregulated by GSK3-α/β inhibition (CHIR99021, 3 μM, 48 h) in FLCN KO versus control MRC-5 cells (n = 3). (B) Raw transcript counts for TCF4, LEF1 and AXIN2 from same experiment. (C) Top 30 upregulated transcripts by GSK3-α/β inhibition (CHIR99021, 3 μM, 48 h) of FLCN KO (clone 4) versus control MRC-5 cells (n = 3). (D) Raw transcript counts for Cyclin D2, WNT2 receptor FZD5 and SFRP1. **P < 0.01, ***P < 0.001, ****P < 0.0001, one-way ANOVA.
Finally, we examined total protein levels of LEF1 in these cells in response to GSK3-α/β inhibition. After CHIR treatment, we found a substantially lesser degree of LEF-1 upregulation in the FLCN KO cells, by both immunofluorescence (Fig. 5A) and western blot (Fig. 5B), in comparison to the control cells.
Figure 5.
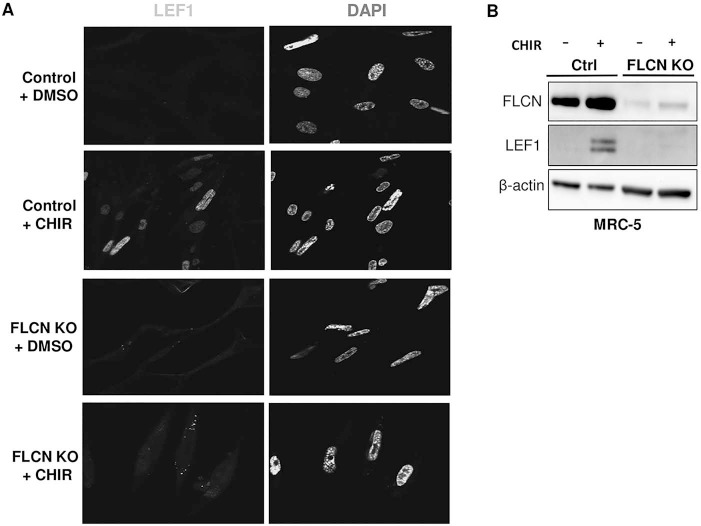
LEF1 is not induced by CHIR99021 in FLCN-deficient cells. (A) Immunofluorescent images of control and FLCN knockout MRC-5 (clone 5) cells treated with DMSO or CHIR99021 (3 μM, 48 h). Blue is DAPI and green is LEF1 staining, 60× magnification. The data are representative of three independent experiments. (B) Western blot of control and FLCN-deficient MRC-5 cells (clone 5) treated with control or CHIR (3 μM, 48 h). β-actin is a loading control.
Over-expression of ABC or silencing of TFE3 rescues the canonical WNT signaling decrease seen in FLCN-deficient cells
To determine if the decrease in canonical WNT signaling in FLCN-deficient cells is β-catenin dependent, a constitutively active beta-catenin (ABC) (S33Y) construct was transfected into wild-type and FLCN-deficient HEK293T TCF/LEF-GFP reporter cells. Upregulation of ABC was observed by western blot (Fig. 6A), but only a partial rescue of the decrease in WNT reporter activity was seen (Fig. 6B). This suggested that the decrease in WNT signaling is not completely dependent on levels of ABC, as we had hypothesized from our finding of loss of ABC protein after FLCN knock out in the MRC-5 cells.
Figure 6.
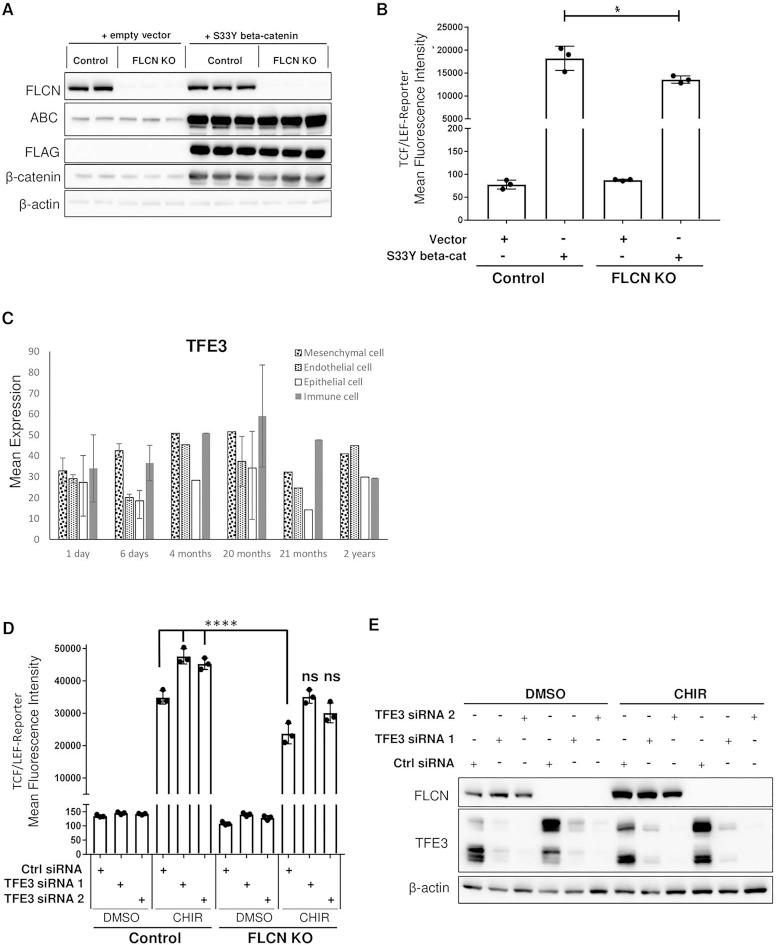
ABC overexpression or TFE3 silencing rescues the canonical WNT signaling decrease seen in FLCN-deficient cells. (A) Western blot of wild-type or FLCN-deficient HEK293T cells transfected with either empty vector or constitutively ABC (S33Y). β-actin is a loading control. The data are representative of 3 independent experiments. ABC is ABC. (B) Mean fluorescence intensity of control or FLCN-deficient HEK293T cells with either empty vector or constitutively ABC (S33Y) and stably expressing the TCF/LEF-GFP reporter construct (clone 7). Data are representative of three independent experiments and presented as mean ± SD, *P < 0.05, one-way ANOVA. (C) RNA-Seq data obtained from LungMAP database showing TFE3 expression pattern by cell type in children from the age of 1 day to 2 years. (D) Mean fluorescence intensity of control or FLCN-deficient HEK293T cells with control siRNA or two TFE3 siRNAs and stably expressing the TCF/LEF-GFP reporter construct (clone 7). Cells were either treated with DMSO or with the GSK-3α/β inhibitor CHIR99021 (CHIR, 3.3 μM) for 48 h. Data are representative of three independent experiments and presented as mean ± SD, ****P ≤ 0.0001, one-way ANOVA, ns indicates no significant changes as compared to wild-type cells treated with CHIR. (E) Western blot of wild-type or FLCN-deficient HEK293T cells transfected with either control siRNA or two TFE3 siRNAs and treated with DMSO or with the GSK-3α/β inhibitor CHIR99021 (CHIR, 3.3 μM) for 48 h. β-actin is a loading control. The data are representative of three independent experiments.
TFE3 is a member of the Micropthalmia transcription factor (MiT) family together with MiTF, TFEB and TFEC. A distinct subset of RCCs, termed translocation RCC result from translocations of the genomic locus for TFE3 or TFEB (44). Loss of FLCN has been shown to increase nuclear TFE3 in a RCC cell line (UOK257) and in MEFs (45). Interestingly, TFE3 has a similar expression pattern to FLCN in the LungMAP database (Figure 6C). MiTF has been shown to physically bind to LEF1 in melanoma cells through a basic helix-loop-helix/leucine zipper (bHLH/LZ) domain contained in the protein. This same domain is present in TFE3 and functional cooperation between TFE3 and LEF1 has been demonstrated (46). MiTF has also been shown to bind β-catenin at the basic helix-loop-helix (bHLH) domain shared by all family members and redirect transcription away from β-catenin/LEF1 dependent targets to MiTF-specific target promoters (47).
Given these data, we silenced TFE3 in our canonical WNT signaling reporter model. siTFE3 completely rescued the decrease in WNT reporter activity seen in FLCN knock out cells (Fig. 6D,E).
Discussion
The cellular mechanisms underlying cystic lung disease and pneumothorax in individuals with BHD syndrome are incompletely understood (9,48). Using RNA sequencing, we identified Wnt2 as the most downregulated gene in Flcn-deficient mouse fibroblasts. This was associated with decreased levels of Wnt reporter activity in Flcn-deficient fibroblasts after stimulation with SB216763, a GSK3-α/β inhibitor. WNT2 was also among the most downregulated transcripts in FLCN KO MRC-5 cells, which are derived from human fetal lung fibroblasts. WNT reporter activity was decreased in both FLCN-deficient mouse and human fibroblasts and in FLCN KO HEK293 cells. We also found that ABC and total β-catenin levels are decreased in FLCN-deficient MRC-5 cells. The mechanism of the decreased WNT signaling activity in FLCN-deficient cells may involve the inability to upregulate LEF1 upon GSK3-α/β inhibition. LEF1 is a final effector of the WNT enhanceosome, driving WNT pathway-dependent transcription. We also found that we could completely rescue the phenotype we uncovered by silencing the transcription factor TFE3. TFE3 translocates to the nucleus in FLCN-deficient cells and functionally cooperates with LEF1 to drive transcription (21,45,46). TFE3 is a MiTF family member and MiTF has been shown to bind β-catenin directly at the bHLH domain and divert it from WNT target promoters to MiTF target promoters (47). We hypothesize that in FLCN-deficient cells, TFE3 is acting in an inhibitory manner in the nucleus of cells through either direct or indirect interaction with the enhanceosome member LEF1 to depress canonical WNT signaling (Fig. 7). It is also possible to hypothesize that nuclear TFE3 may be binding β-catenin directly to redirect it from WNT-specific promoters to TFE3 specific gene promoters. This might be the reason that transfection with a constitutively ABC construct only partially rescues this phenotype. Further definition of the exact events involved will require additional studies.
Figure 7.
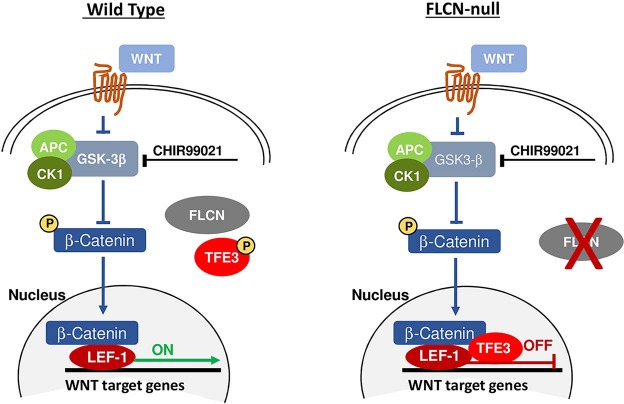
Proposed working model for role of TFE3 in inhibition of canonical Wnt signaling in FLCN-deficient cells.
Three previous studies have suggested a role for FLCN in the Wnt pathway. In a FLCN-null metastatic thyroid carcinoma line, decreased transcriptional levels of FZD1, SFRP4, LRP5 and increased CCND2 (cyclin D2) were observed (42), but no assays of Wnt activity were performed. In FLCN-deficient mouse kidney epithelial cells (25), increased levels of β-catenin protein and increased levels of Axin2 transcripts were seen (opposite from our findings), but no functional assessments of Wnt activity was performed. Finally, of interest from a developmental standpoint, a very recent study showed that FLCN-deficient human embryonic stem cells upregulate multiple WNT ligands, and this axis is involved in exit from pluripotency (22). To our knowledge, therefore, ours is the first study directly focused on FLCN-dependent canonical WNT signaling activity, and the first evidence that FLCN regulates WNT activity in mesenchymal-lineage cells.
We chose to focus on the mesenchyme cell lineage for two reasons: first, because previously published in vivo and in vitro work on the function of FLCN has focused primarily on the epithelium (9,18); and second for the known key roles played by mesenchyme during lung development and bulk RNA-Seq data from newborn and toddlers (LungMAP) showing high levels of FLCN expression in pulmonary mesenchyme (Fig. 1A). The decreased WNT activity in FLCN-deficient mesenchymal cells is of particular interest given recent data further delineating the critical impact of Wnt2 secreted by lung mesoderm in the induction of Nkx2.1 and formation of lung buds during early development. Wnt2 is first expressed at E9.5 in the mesoderm on the ventral side of the anterior foregut in mice (43). Wnt2 and β-catenin have been shown to be necessary and sufficient to specify lung endoderm progenitor cells from the developing anterior foregut and without Wnt2/2b expression, mouse embryos failed to express Nkx2.1, the earliest marker of lung progenitor cells (49). WNT5a, another WNT ligand that is produced by early mesenchyme, has recently been shown to effect alveolar epithelial repair (50). We also observed FLCN-dependent changes in WNT5a after CHIR stimulation (Figure 4A). Recent single-cell RNA-Seq and lineage trace experiments have identified multiple populations of lung mesenchymal cells that occupy the niche around alveolar type II (ATII) pneumocytes. A population marked by Axin2/PDGFRα double positivity represents the ‘mesenchymal alveolar niche cell (MANC)’ that secretes factors including BMP-4, IL-6 and FGF7 to support the ATII (51). In proximity, but distinct from these MANC cells, are Wnt2 positive mesenchyme cells, which may function as support to the MANC. Lgr5 and Lgr6 have also been reported to mark distinct mesenchymal populations in the adult lung with Lgr5+ lung mesenchyme found to induce the alveolar lineage and Lgr6+ mesenchyme cells found to support bronchiolar lineage differentiation (52). Flcn loss in ATII cells results in increased distal air space diameter (18); the effect of Flcn loss in lung mesenchymal cells has not yet been reported. Given the consistent downregulation of canonical WNT activity and WNT2 ligand that we see in our FLCN KO mesenchyme models, it is enticing to hypothesize that a defect in this developmental axis during lung development or after injury, may lead to pulmonary cyst formation in BHD patients.
This proposed developmental model of the pathogenesis of cystic disease in BHD augments the prior ‘stretch hypothesis’ in which enhanced cell–cell adhesion among epithelium was proposed to underlie cyst formation in BHD patients (9,23,48). As mentioned earlier, although the pulmonary cysts in BHD were initially thought to arise in the late 3rd or 4th decade of life, there have been reports of lower lobe cysts in pediatric BHD patients (32) and a case report of lung cysts identified by ultrasound in a fetus whose mother and grandmother were known to carry FLCN mutations (33). Furthermore, results from at least three small cohort studies confirm the clinical impression that pulmonary cysts in BHD do not increase in size or number over time, in contrast to cysts in other diseases such as lymphangioleiomyomatosis (LAM; OMIM #606690) and Pulmonary Langerhans Cell Histiocytosis (OMIM 604856) (53–55). While certainly not conclusive, these observations are consistent with a developmental origin of lung cysts in BHD.
In conclusion, we report here that FLCN-deficient mesenchymal-lineage cells have defective WNT signaling activity. Precise WNT signaling between mesenchyme and epithelium is critical to lung development beginning at the time of the specification of the first Nkx2.1-positive lung progenitors from the anterior foregut endoderm at around embryonic week 4 in humans (56,57). Dysregulation of this highly orchestrated sequence of signaling events is thought to contribute to developmental and chronic lung diseases in humans (50). We hypothesize here that the cause of lung cysts in BHD patients is at least partially due to defective WNT-dependent cell-cell communication during lung development.
Materials and Methods
Cell lines and culture conditions
MEFs were derived from a Flcnfl/fl: Rosa26 Cre-ERT2 mouse embryo and treated with 4-hydroxytamoxifen (2 μM, Sigma-Aldrich) for 3 days resulting in loss of Flcn at the protein and mRNA levels. The MEFs were then maintained in Tamoxifen-free media and single-cell cloned. HEK293T cells (ATCC CRL-3216) and MRC-5 cells (ATCC CCL-171) were obtained from American Type Culture Collection (Manassas, VA). Cells were cultured in DMEM supplemented with 10% FBS (MEFs and HEK293T) or EMEM (MRC-5) supplemented with 10% FBS.
Immunoblotting
For all western blot analyses, cells were lysed on ice in 1× RIPA (Cell Signaling Technology) containing phosphatase and protease inhibitors, homogenized by passing through the 25G needle 10 times and centrifuged for 15 min at 12000 rpm. Lysates were normalized by total protein concentration, resolved on 4–12% Bis-Tris gels (Thermo Fisher) and transferred to a nitrocellulose membrane. Blots were blocked with 5% milk and incubated with primary and secondary antibodies. Chemiluminescence was captured with Syngene G-Box gel documentation system.
FLCN knockout and stable WNT reporter cell line generation
FLCN knockout HEK293T and MRC-5 cell lines were generated using a guide (CAGGGGGCACCATGAATGCCCATCG) selected using the Zhang Lab prediction tool (http://crispr.mit.edu) and cloned into the pSpCas9(BB)-2A-GFP (PX458) backbone. The plasmid was transiently transfected and GFP-expressing single cells were then sorted and expanded. FLCN knockout was confirmed by Sanger sequencing and/or immunoblot. To generate WNT reporter lines, cells were stably infected with a lentiviral packaged TCF/LEF-GFP expression construct (pCIGNAL, Qiagen).
RNA-Seq and Nanostring experiments
RNA was prepared for mRNAseq (polyA enriched) library preparation with single-end 75 bp sequencing on a NextSeq system (Illumina) with 16 pooled barcoded samples, followed by VIPER pipeline analysis (Molecular Biology Core Facility, Dana-Farber Cancer Institute). Four technical replicates of each sample were analyzed. Nanostring analysis was performed using the Nanostring nCounter Vantage 3D Wnt panel, which analyzes 189 WNT pathway genes (Boston Children’s Hospital Molecular Genetics Core Facility). Data was analyzed using nSolver 4.0 software (NanoString Technologies Inc.) and displayed as heat maps using Morpheus (https://software.broadinstitute.org/morpheus).
Antibodies, RT-PCR probes and drugs
The following antibodies were used: FLCN (D14G9), Total β-catenin (D10A8), LEF1 (C12A5) (all from Cell signaling Technology), FLCN ab124885 (Abcam), β-Actin (Sigma-Aldrich) and ABC (Anti-ABC) Clone 8E7 05–665 (Millipore). TaqMan Probes for Flcn (assay ID Mm00840973_m1, 75 bp amplicon length), Wnt2 (assay ID Mm00470018_m1, 145 bp amplicon length), Wnt7a (assay ID Mm00437356_m1, 77 bp amplicon length) and β-actin control (cat# 4351315) were obtained from ThermoFisher. The GSK3-α/β inhibitors were obtained from Sigma (SB216763) and Selleckchem (CHIR99021). BRD0320 was the gift of Florence Wagner, The Broad Institute. Recombinant mouse Wnt3a was obtained from R&D Systems (1324-WN).
Flow cytometry analysis of WNT reporter activity
Following drug treatment as specified in the text, cells were resuspended in cold PBS and filtered through a 12 × 75 mm cell strainer. Fluorescence was assessed by flow cytometry (BD FACS Canto II, BD Biosciences) and analyzed with FlowJo_V10 analytical software (FlowJo; LLC). Mean fluorescence intensity of FITC (GFP) was measured.
Immunofluorescence and confocal microscopy
Cells were rinsed with PBS, fixed with 2% paraformaldehyde for 10 min and permeabilized with Triton (0.02%) at room temperature for 5 min, before a final wash and incubation with primary antibody in 2.5% BSA overnight at 4°C. The following day cells were washed again three times in PBS before incubation with secondary antibody (Alexa Fluor 488, Donkey anti-rabbit, Invitrogen) and DAPI. Images were captured with a FluoView FV-10i Olympus Laser Point Scanning Confocal Microscope using a 60x objective.
Experimental reproducibility
All experiments were repeated at least three times (n = 3) and included three or more replicates.
Statistical analyses
Normally distributed data were analyzed for statistical significance with Student’s unpaired t-test. Multiple comparisons were made with one-way ANOVAs with Bonferroni correction. Data is presented as mean +/− standard deviation. GraphPad Prism version 6 was used. Statistical significance was defined as P < 0.05.
Supplementary Material
{kind=link}
Acknowledgements
The results presented here are in part based upon data generated by the LungMAP Consortium (U01HL122642) and downloaded from (www.lungmap.net), on February 8, 2019. The authors wish to thank Xi He and Yan Xu for helpful discussions about Wnt activity assays and the LungMAP data sets, respectively. We would also like to thank Florence Wagner for the GSK3 inhibitors developed in her laboratory.
Conflict of Interest statement. None declared.
Funding
This work was supported by the National Institutes of Health (1RO1HL141352-01 to W.S. and T32HL007633-31 to J.C.K.).
Author Contributions
Conception and design: J.C.K., D.K. and E.P.H.; performed experiments: J.C.K., D.K., T.H.; analysis and interpretation: J.C.K., D.K., T.H., W.S. and E.P.H.; drafting the manuscript for important intellectual content: J.C.K., D.K., J.N. and EP.H.; responsibility for the integrity of the data and accuracy of the data analysis: all authors.
References
- 1. Birt A.R. and H.G., Dube WJ (1977) Hereditary multiple fibrofolliculomas with trichodiscomas and acrochordons. Arch. Dermatol., 113, 1674–1677. [PubMed] [Google Scholar]
- 2. Nickerson M.L., Warren M.B., Toro J.R., Matrosova V., Glenn G., Turner M.L., Duray P., Merino M., Choyke P., Pavlovich C.P. et al. (2002) Mutations in a novel gene lead to kidney tumors, lung wall defects, and benign tumors of the hair follicle in patients with the Birt-Hogg-Dube syndrome. Cancer Cell, 2, 157–164. [DOI] [PubMed] [Google Scholar]
- 3. Gupta N., Kopras E.J., Henske E.P., James L.E., El-Chemaly S., Veeraraghavan S., Drake M.G. and McCormack F.X. (2017) Spontaneous pneumothoraces in patients with Birt-Hogg-Dube syndrome. Ann. Am. Thorac. Soc., 14, 706–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Toro J.R., Pautler S.E., Stewart L., Glenn G.M., Weinreich M., Toure O., Wei M.H., Schmidt L.S., Davis L., Zbar B. et al. (2007) Lung cysts, spontaneous pneumothorax, and genetic associations in 89 families with Birt-Hogg-Dube syndrome. Am. J. Respir. Crit. Care Med., 175, 1044–1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Houweling A.C., Gijezen L.M., Jonker M.A., van M.B., Oldenburg R.A., van K.Y., Leter E.M., van T.A., van N.C., Jaspars E.H. et al. (2011) Renal cancer and pneumothorax risk in Birt-Hogg-Dube syndrome; an analysis of 115 FLCN mutation carriers from 35 BHD families. Br. J. Cancer, 105, 1912–1919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gupta N., Vassallo R., Wikenheiser-Brokamp K.A. and McCormack F.X. (2015) Diffuse cystic lung disease. Part I. Am J. Respir. Crit. Care Med., 191, 1354–1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Boone P.M., Scott R.M., Marciniack S.J., Henske E.P. and Raby B.A. (2019) Genetics of pneumothorax. Am. J. Respir. Crit. Care Med., 199, 1344–1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. van M., Khabibullin D., Hartman T.R., Nicolas E., Kruger W.D. and Henske E.P. (2007) The Birt-Hogg-Dube and tuberous sclerosis complex homologs have opposing roles in amino acid homeostasis in Schizosaccharomyces pombe. J. Biol. Chem., 282, 24583–24590. [DOI] [PubMed] [Google Scholar]
- 9. Khabibullin D., Medvetz D.A., Pinilla M., Hariharan V., Li C., Hergrueter A., Laucho Contreras M., Zhang E., Parkhitko A., Yu J.J. et al. (2014) Folliculin regulates cell-cell adhesion, AMPK, and mTORC1 in a cell-type-specific manner in lung-derived cells. Physiol. Rep., 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Tsun Z.Y., Bar-Peled L., Chantranupong L., Zoncu R.,Wang T., Kim C., Spooner E. and Sabatini D.M. (2013) The folliculin tumor suppressor is a GAP for the RagC/D GTPases that signal amino acid levels to mTORC1. Mol. Cell., 52, 495–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Petit C.S., Roczniak-Ferguson A. and Ferguson S.M. (2013) Recruitment of folliculin to lysosomes supports the amino acid-dependent activation of rag GTPases. J. Cell Biol., 202, 1107–1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Baba M., Hong S.B., Sharma N., Warren M.B., Nickerson M.L., Iwamatsu A., Esposito D., Gillette W.K., Hopkins R.F. 3rd, Hartley J.L. et al. (2006) Folliculin encoded by the BHD gene interacts with a binding protein, FNIP1, and AMPK, and is involved in AMPK and mTOR signaling. Proc. Natl. Acad. Sci. USA, 103, 15552–15557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hudon V., Sabourin S., Dydensborg A.B., Kottis V., Ghazi A., Paquet M., Crosby K., Pomerleau V., Uetani N. and Pause A. (2010) Renal tumour suppressor function of the Birt-Hogg-Dube syndrome gene product folliculin. J. Med. Genet., 47, 182–189. [DOI] [PubMed] [Google Scholar]
- 14. Hartman T.R., Nicolas E., Klein-Szanto A., Al-Saleem T., Cash T.P., Simon M.C. and Henske E.P. (2009) The role of the Birt-Hogg-Dube protein in mTOR activation and renal tumorigenesis. Oncogene, 28, 1594–1604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Cash T.P., Gruber J.J., Hartman T.R., Henske E.P. and Simon M.C. (2011) Loss of the Birt-Hogg-Dube tumor suppressor results in apoptotic resistance due to aberrant TGFbeta-mediated transcription. Oncogene, 30, 2534–2546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hasumi H., Baba M., Hasumi Y., Huang Y., Oh H., Hughes R.M., Klein M.E., Takikita S., Nagashima K., Schmidt L.S. et al. (2012) Regulation of mitochondrial oxidative metabolism by tumor suppressor FLCN. J. Natl. Cancer Inst., 104, 1750–1764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Dunlop E.A., Seifan S., Claessens T., Behrends C., Kamps M.A., Rozycka E., Kemp A.J., Nookala R.K., Blenis J., Coull B.J. et al. (2014) FLCN, a novel autophagy component, interacts with GABARAP and is regulated by ULK1 phosphorylation. Autophagy, 10, 1749–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Goncharova E.A., Goncharov D.A., James M.L., Atochina-Vasserman E.N., Stepanova V., Hong S.B., Li H., Gonzales L., Baba M., Linehan W.M. et al. (2014) Folliculin controls lung alveolar enlargement and epithelial cell survival through E-cadherin, LKB1, and AMPK. Cell Rep., 7, 412–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yan M., Gingras M.C., Dunlop E.A., Nouet Y., Dupuy F., Jalali Z., Possik E., Coull B.J., Kharitidi D., Dydensborg A.B. et al. (2014) The tumor suppressor folliculin regulates AMPK-dependent metabolic transformation. J. Clin. Invest., 124, 2640–2650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hong S.B., Oh H., Valera V.A., Stull J., Ngo D.T., Baba M., Merino M.J., Linehan W.M. and Schmidt L.S. (2010) Tumor suppressor FLCN inhibits tumorigenesis of a FLCN-null renal cancer cell line and regulates expression of key molecules in TGF-beta signaling. Mol. Cancer, 9, 160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Betschinger J., Nichols J., Dietmann S., Corrin P.D., Paddison P.J. and Smith A. (2013) Exit from pluripotency is gated by intracellular redistribution of the bHLH transcription factor Tfe3. Cell, 153, 335–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mathieu J., Detraux D., Kuppers D., Wang Y., Cavanaugh C., Sidhu S., Levy S., Robitaille A.M., Ferreccio A., Bottorff T. et al. (2019) Folliculin regulates mTORC1/2 and WNT pathways in early human pluripotency. Nat. Commun., 10, 632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Nahorski M.S., Seabra L., Straatman-Iwanowska A., Wingenfeld A., Reiman A., Lu X., Klomp J.A., Teh B.T., Hatzfeld M., Gissen P. et al. (2012) Folliculin interacts with p0071 (plakophilin-4) and deficiency is associated with disordered RhoA signalling, epithelial polarization and cytokinesis. Hum. Mol. Genet., 21, 5268–5279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Medvetz D.A., Khabibullin D., Hariharan V., Ongusaha P.P., Goncharova E.A., Schlechter T., Darling T.N., Hofmann I., Krymskaya V.P., Liao J.K. et al. (2012) Folliculin, the product of the Birt-Hogg-Dube tumor suppressor gene, interacts with the adherens junction protein p0071 to regulate cell-cell adhesion. PLoS One, 7, e47842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Luijten M.N., Basten S.G., Claessens T., Vernooij M., Scott C.L., Janssen R., Easton J.A., Kamps M.A., Vreeburg M., Broers J.L. et al. (2013) Birt-Hogg-Dube syndrome is a novel ciliopathy. Hum. Mol. Genet., 22, 4383–4397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Laviolette L.A., Mermoud J., Calvo I.A., Olson N., Boukhali M., Steinlein O.K., Roider E., Sattler E.C., Huang D., Teh B.T. et al. (2017) Negative regulation of EGFR signalling by the human folliculin tumour suppressor protein. Nat. Commun., 8, 15866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Nookala R.K., Langemeyer L., Pacitto A., Ochoa-Montano B., Donaldson J.C., Blaszczyk B.K., Chirgadze D.Y., Barr F.A., Bazan J.F. and Blundell T.L. (2012) Crystal structure of folliculin reveals a hidDENN function in genetically inherited renal cancer. Open Biol., 2, 120071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Meng J. and Ferguson S.M. (2018) GATOR1-dependent recruitment of FLCN-FNIP to lysosomes coordinates Rag GTPase heterodimer nucleotide status in response to amino acids. J. Cell. Biol., 217, 2765–2776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Peli-Gulli M.P., Sardu A., Panchaud N., Raucci S. and De C. (2015) Amino acids stimulate TORC1 through Lst4-Lst7, a GTPase-activating protein complex for the Rag family GTPase Gtr2. Cell Rep, 13, 1–7. [DOI] [PubMed] [Google Scholar]
- 30. Kumasaka T., Hayashi T., Mitani K., Kataoka H., Kikkawa M., Tobino K., Kobayashi E., Gunji Y., Kunogi M., Kurihara M. et al. (2014) Characterization of pulmonary cysts in Birt-Hogg-Dube syndrome: histopathological and morphometric analysis of 229 pulmonary cysts from 50 unrelated patients. Histopathology, 65, 100–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Furuya M., Tanaka R., Koga S., Yatabe Y., Gotoda H., Takagi S., Hsu Y.H., Fujii T., Okada A., Kuroda N. et al. (2012) Pulmonary cysts of Birt-Hogg-Dube syndrome: a clinicopathologic and immunohistochemical study of 9 families. Am. J. Surg. Pathol., 36, 589–600. [DOI] [PubMed] [Google Scholar]
- 32. Geilswijk M., Bendstrup E., Madsen M.G., Sommerlund M. and Skytte A.B. (2018) Childhood pneumothorax in Birt-Hogg-Dube syndrome: a cohort study and review of the literature. Mol. Genet. Genomic. Med., 6, 332–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sundaram S., Tasker A.D. and Morrell N.W. (2009) Familial spontaneous pneumothorax and lung cysts due to a Folliculin exon 10 mutation. Eur. Respir. J., 33, 1510–1512. [DOI] [PubMed] [Google Scholar]
- 34. Baba M., Toyama H., Sun L., Takubo K., Suh H.C., Hasumi H., Nakamura-Ishizu A., Hasumi Y., Klarmann K.D., Nakagata N. et al. (2016) Loss of folliculin disrupts hematopoietic stem cell quiescence and homeostasis resulting in bone marrow failure. Stem. Cells., 34, 1068–1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Pham D.L., Trinh T.H., Ban G.Y., Kim S.H. and Park H.S. (2017) Epithelial folliculin is involved in airway inflammation in workers exposed to toluene diisocyanate. Exp. Mol. Med., 49, e395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kawai A., Kobayashi T. and Hino O. (2013) Folliculin regulates cyclin D1 expression through cis-acting elements in the 3′ untranslated region of cyclin D1 mRNA. Int. J. Oncol., 42, 1597–1604. [DOI] [PubMed] [Google Scholar]
- 37. Furuya M., Hong S.B., Tanaka R., Kuroda N., Nagashima Y., Nagahama K., Suyama T., Yao M. and Nakatani Y. (2015) Distinctive expression patterns of glycoprotein non-metastatic B and folliculin in renal tumors in patients with Birt-Hogg-Dube syndrome. Cancer Sci., 106, 315–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wagner F.F., Benajiba L., Campbell A.J., Weiwer M., Sacher J.R., Gale J.P., Ross L., Puissant A., Alexe G., Conway A. et al. (2018) Exploiting an asp-Glu "switch" in glycogen synthase kinase 3 to design paralog-selective inhibitors for use in acute myeloid leukemia. Sci. Transl. Med., 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Flack J.E., Mieszczanek J., Novcic N. and Bienz M. (2017) Wnt-dependent inactivation of the Groucho/TLE co-repressor by the HECT E3 ubiquitin ligase Hyd/UBR5. Mol Cell, 67(181–193), e185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. van L.M., Mieszczanek J., Fiedler M., Rutherford T.J. and Bienz M. (2017) Constitutive scaffolding of multiple Wnt enhanceosome components by legless/BCL9. Elife, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Preston R.S., Philp A., Claessens T., Gijezen L., Dydensborg A.B., Dunlop E.A., Harper K.T., Brinkhuizen T., Menko F.H., Davies D.M. et al. (2011) Absence of the Birt-Hogg-Dube gene product is associated with increased hypoxia-inducible factor transcriptional activity and a loss of metabolic flexibility. Oncogene, 30, 1159–1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Reiman A., Lu X., Seabra L., Boora U., Nahorski M.S., Wei W. and Maher E.R. (2012) Gene expression and protein array studies of folliculin-regulated pathways. Anticancer Res., 32, 4663–4670. [PubMed] [Google Scholar]
- 43. Miller M.F., Cohen E.D., Baggs J.E., Lu M.M., Hogenesch J.B. and Morrisey E.E. (2012) Wnt ligands signal in a cooperative manner to promote foregut organogenesis. Proc. Natl. Acad. Sci. USA, 109, 15348–15353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Damayanti N.P., Budka J.A., Khella H.W.Z., Ferris M.W., Ku S.Y., Kauffman E., Wood A.C., Ahmed K., Chintala V.N., Adelaiye-Ogala R. et al. (2018) Therapeutic targeting of TFE3/IRS-1/PI3K/mTOR axis in translocation renal cell carcinoma. Clin. Cancer Res., 24, 5977–5989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hong S.B., Oh H., Valera V.A., Baba M., Schmidt L.S. and Linehan W.M. (2010) Inactivation of the FLCN tumor suppressor gene induces TFE3 transcriptional activity by increasing its nuclear localization. PLoS One, 5, e15793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Yasumotos K., Takeda K., Saito H., Watanabe K., Takahashi K. and Shibahara S. (2002) Microphthalmia-associated transcription factor interacts with LEF-1, a mediator of Wnt signaling. EMBO J., 21, 2703–2714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Schepsky A., Bruser K., Gunnarsson G.J., Goodall J., Hallsson J.H., Goding C.R., Steingrimsson E. and Hecht A. (2006) The microphthalmia-associated transcription factor Mitf interacts with beta-catenin to determine target gene expression. Mol. Cell. Biol., 26, 8914–8927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kennedy J.C., Khabibullin D. and Henske E.P. (2016) Mechanisms of pulmonary cyst pathogenesis in Birt-Hogg-Dube syndrome: the stretch hypothesis. Semin. Cell. Dev. Biol., 52, 47–52. [DOI] [PubMed] [Google Scholar]
- 49. Goss A.M., Tian Y., Tsukiyama T., Cohen E.D., Zhou D., Lu M.M., Yamaguchi T.P. and Morrisey E.E. (2009) Wnt2/2b and beta-catenin signaling are necessary and sufficient to specify lung progenitors in the foregut. Dev. Cell., 17, 290–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Baarsma H.A., Skronska-Wasek W., Mutze K., Ciolek F., Wagner D.E., John-Schuster G., Heinzelmann K., Gunther A., Bracke K.R., Dagouassat M. et al. (2017) Noncanonical WNT-5A signaling impairs endogenous lung repair in COPD. J. Exp. Med., 214, 143–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Zepp J.A., Zacharias W.J., Frank D.B., Cavanaugh C.A., Zhou S., Morley M.P. and Morrisey E.E. (2017) Distinct mesenchymal lineages and niches promote epithelial self-renewal and myofibrogenesis in the lung. Cell, 170, 1134–1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Lee J.H., Tammela T., Hofree M., Choi J., Marjanovic N.D., Han S., Canner D., Wu K., Paschini M., Bhang D.H. et al. (2017) Anatomically and functionally distinct lung mesenchymal populations marked by Lgr5 and Lgr6. Cell, 170, 1149–1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Tobino K., Gunji Y., Kurihara M., Kunogi M., Koike K., Tomiyama N., Johkoh T., Kodama Y., Iwakami S., Kikkawa M. et al. (2011) Characteristics of pulmonary cysts in Birt-Hogg-Dube syndrome: thin-section CT findings of the chest in 12 patients. Eur. J. Radiol., 77, 403–409. [DOI] [PubMed] [Google Scholar]
- 54. Johannesma P.C., Houweling A.C., van J.H., van R.J., Starink T.M., Menko F.H. and Postmus P.E. (2014) The pathogenesis of pneumothorax in Birt-Hogg-Dube syndrome: a hypothesis. Respirology, 19, 1248–1250. [DOI] [PubMed] [Google Scholar]
- 55. Ayo D.S., Aughenbaugh G.L., Yi E.S., Hand J.L. and Ryu J.H. (2007) Cystic lung disease in Birt-Hogg-Dube syndrome. Chest, 132, 679–684. [DOI] [PubMed] [Google Scholar]
- 56. Herriges M. and Morrisey E.E. (2014) Lung development: orchestrating the generation and regeneration of a complex organ. Development, 141, 502–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Snoeck H.W. (2015) Modeling human lung development and disease using pluripotent stem cells. Development, 142, 13–16. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.