

Abstract
Amyotrophic lateral sclerosis (ALS) is a progressive neurodegenerative disease that affects motor nerve cells in the brain and the spinal cord. Etiological mechanisms underlying the disease remain poorly understood; recent studies suggest that deregulation of p25/Cyclin-dependent kinase 5 (Cdk5) activity leads to the hyperphosphorylation of Tau and neurofilament (NF) proteins in ALS transgenic mouse model (SOD1G37R). A Cdk5 involvement in motor neuron degeneration is supported by analysis of three SOD1G37R mouse lines exhibiting perikaryal inclusions of NF proteins and hyperphosphorylation of Tau. Here, we tested the hypothesis that inhibition of Cdk5/p25 hyperactivation in vivo is a neuroprotective factor during ALS pathogenesis by crossing the new transgenic mouse line that overexpresses Cdk5 inhibitory peptide (CIP) in motor neurons with the SOD1G37R, ALS mouse model (TriTg mouse line). The overexpression of CIP in the motor neurons significantly improves motor deficits, extends survival and delays pathology in brain and spinal cord of TriTg mice. In addition, overexpression of CIP in motor neurons significantly delays neuroinflammatory responses in TriTg mouse. Taken together, these data suggest that CIP may serve as a novel therapeutic agent for the treatment of neurodegenerative diseases.
Introduction
Amyotrophic lateral sclerosis (ALS) is an adult-onset neurological disorder characterized by the progressive loss of upper motor neurons in the cortex and lower motor neurons in the brainstem and the spinal cord, resulting in severe muscle wasting and fatal respiratory failure (1). Missense mutations in the gene coding for the Cu/Zn superoxide dismutase 1 (SOD1), located on chromosome 21, account for 20% cases of familial ALS (2,3). The SOD1 protein is a cytosolic metalloenzyme that catalyzes the conversion of superoxide anions to hydrogen peroxide. Transgenic mice expressing mutant SOD1 develop motor neuron disease, resembling ALS through an unidentified mechanism (4,5,6).
Abnormal accumulations of neurofilaments (NFs) have been observed in the perikaryon or axon of spinal motor neurons in ALS patients (7,8,9) and in mice overexpressing ALS-linked SOD1 mutants (4,6). Nguyen et al. (10) also reported the importance of NF abnormalities that contribute to the pathogenesis of human ALS. The overexpression of wild-type NF subunits in mice can provoke ALS-like NF accumulations in the perikaryon of motor neurons, axonal atrophy and sometimes motor dysfunction caused by altered ionic conductance, but it does not cause significant motor neuron death (10,11). The NF-H protein is a highly phosphorylated protein, and its KSP (Lys-Ser-Pro) repeat-enriched tail domain constitutes a target for several protein kinases, including stress-activated protein kinase-g (SAPK-g; 12), extracellular signal regulated kinases (ERK 1/2; 13), glycogen synthase kinase 3 (GSK-3; 14,15) and Cdk5.
It is intriguing that in several neurodegenerative disorders (Alzheimer’s disease, ALS, Parkinson’s disease and Huntington’s disease) a hyperactive Cdk5 is detected in the brain specimen obtained at the autopsy (16,10) and has been shown to be upstream of the pathways leading to plaques, tangles and inflammation in cultured neurons and mouse models (17,18). Elevated levels of p25 have been reported in brains of AD (19) and ALS (20,21). The abnormal subcellular localization of Cdk5 has been observed in several neurodegenerative diseases, including ALS (22), Alzheimer’s disease (23,24), Parkinson’s disease (25), as well as in canine motor neuron disease (26). Moreover, recent studies suggest that a deregulation of Cdk5 activity may directly contribute to pathogenesis. Patrick et al. (19) reported the accumulation of p25, a truncated form of p35, in the brains of patients with Alzheimer’s disease. Their results show that p25, which is not targeted to the plasma membrane like p35, sequesters Cdk5 away from normal compartments of p35/Cdk5 and deregulates its activity. Expression of the Cdk5/p25 complex in cultured cells leads to apoptosis (19,16). Moreover, overexpression of p25 in the CNS of mice caused hyperphosphorylation of tau and NFs, cytoskeletal disruption and behavioral deficits (27). Nguyen et al. (10) reported that deregulation of Cdk5 activity associated with the hyperphosphorylation of tau and NF proteins in mice expressing a mutant superoxide dismutase (SOD1G37R).
Clearly, Cdk5/p25 is a potential therapeutic target for neurodegeneration (18,28–31). Although roscovitine and related compounds have been proposed and evaluated, their effects are nonspecific as they bind the common ATP site shared by other Cdks and most other kinases. Our laboratory has taken a different approach based on a study of truncated fragments of the p35 regulator (32). Two peptides were identified, Cdk5 inhibitory peptide (CIP; 126 a.a.) and a smaller peptide p5 (24 a.a.), derived from the p25 domain of the parent sequence, exhibited vigorous inhibition of Cdk5/p35 and Cdk5/p25 activities in test tube experiments (32,33,34). Surprisingly, however, in cultured cortical neurons, the peptides inhibited only the Cdk5/p25 complex and spared the Cdk5/p35 kinase that retained most of its activity (28–33,35). E18 cortical neurons stressed by toxic A-beta display an AD phenotype, hyperactive Cdk5/p25, hyperphosphorylated tau and NFs, A-beta accumulation and apoptosis. These effects are reduced when cells are incubated in different concentrations of p5 or CIP (32–34). The specificity of peptide inhibition was dramatic; whereas Cdk5/p25 activity was inhibited; Cdk5/p35 activity was unaffected as were the activities of cyclin-dependent kinases (36).
To study the therapeutic potential of the CIP peptide in vivo, mutants with p25Tg AD-like phenotype were crossed with normally appearing CIP double transgenic (producing TetraTg- CIP mice controlled by a CAMK2a promoter) overexpressing p25 in a background of CIP inhibitor peptide overexpression in the brain (37). Doxycycline removal for 1 week initiated overexpression of p25 and Cdk5 hyperactivation in forebrains of p25Tg mice that were diminished in the TetraTg CIP × p25 Tg-expressing mice as was inflammation, tau phosphorylation and amyloid deposition; AD pathology was significantly reduced as were neuronal cell loss and neurocognitive defects. This is the first successful therapeutic targeting of Cdk5/p25 hyperactivity in vivo while sparing effects on Cdk5/p35 activity. To extend further the use of CIP in the ALS we are showing upregulation of p25 in brain and spinal cord of five ALS patients with matched controls (Supplementary Material, Fig. S4), together with new transgenic mouse line that overexpresses CIP (Chat Cre/CIP) in motor neurons with the SOD1G37R. We reported here that overexpression of CIP in the motor neurons significantly extends survival, improves motor deficits and delays pathology in the spinal cord and in the brain of SOD1G37R mice. Furthermore, overexpression of CIP in motor neurons significantly delays neuroinflammatory response and improves muscle pathology. Taken together, these data suggest that CIP may serve as a novel therapeutic agent for the treatment of neurodegenerative diseases.
Results
Generation and characterization of mice overexpressing CIP transgene
The original CIP (CIP floxed) transgenic breeder mice were a generous gift from Dr Sashi Kesavapany and have been maintained on a pure C57BL6 background. The CIPTg construct contains a 3′-FLAG-tagged CIP transgene incorporated in to the ROSA26 locus whose 5′ regulatory elements were separated from the coding region with a floxed stop sequence. An frt-flanked neomycin resistance (Neo) cassette was inserted next to the transgene before the 3′ homology arm (37). To express the CIP in the motor neurons, the CIP floxed mice were crossed with ChAT-IRES-Cre transgenic mice (Stock no 006410; Jackson Laboratory, Bar Harbor, Maine). To study the ability of CIP inhibit the Cdk5/p25 hyperactivation-mediated motor neurodegeneration in ALS, in vivo, we generated a TriTransgenic (TriTg, CIP ChAT, Cre + ALS, SOD1G37R) mouse that overexpresses CIP under the direction of the Chat promoter, CIP Chat Cre mice crossed ALS mouse, SOD1G37R 42Dpr/J (Stock no 008342; Jackson Laboratory). To examine the CIP transgene expression pattern in CIPTg (CIP ChAT-Cre) and TriTg (CIP ChAT, Cre + ALS, SOD1G37R) mice, Quantitative PCR (q-PCR), (Fig. 1A and B), western blot analyses (Fig. 1C and D) and immunohistochemistry (Fig. 1E) were performed on the brain and spinal cord samples. Results confirmed the expression of CIP in the brain and spinal cord of CIPTg and TriTg (Fig. 1A–E).
Figure 1.
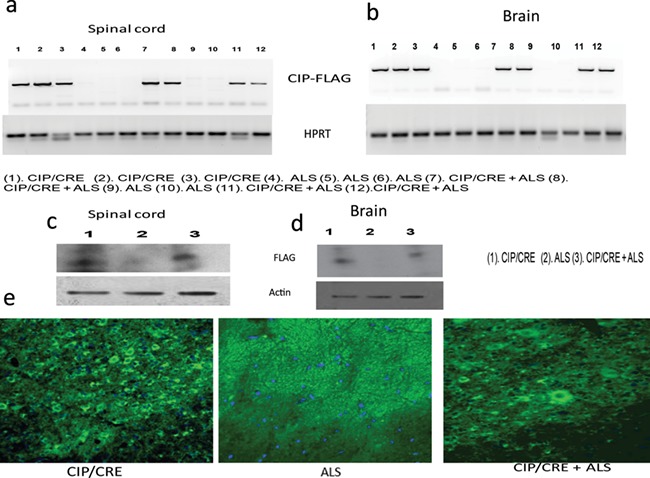
Characterization of CIP Tg (CIP/Chat CRE) and TriTg (CIP/Chat CRE +ALS) mice. The original CIP (CIP floxed) transgenic breeder mice were a generous gift from Dr Sashi Kesavapany and have been maintained on a pure C57BL6 background. The CIPTg construct contains a 3′-FLAG-tagged CIP transgene incorporated in to the ROSA26 locus whose 5′ regulatory elements were separated from the coding region with a floxed stop sequence. An frt-flanked neomycin resistance (Neo) cassette was inserted next to the transgene before the 3′ homology arm (43). The CIP floxed mice crossed with ChAT-IRES-Cre transgenic mice (Stock no 006410; Jackson Laboratory). The CIP Chat CRE mice then crossed with ALS mouseSOD1G37R) (Stock no 008342; Jackson Laboratory). (A and B) Representative RT-PCR images of spinal cord and brain from 21.4-week-old CIPTg, ALS and TriTg. (C and D) Immunoblot analyses of brain and spinal cord lysates from CIPTg, ALS and TriTg mice using anti-FLAG antibodies. Equal amounts of protein loading were confirmed by reprobing the membrane with anti-tubulin antibody. (E) Representative confocal images of spinal cord from 21.4-week-old CIPTg, ALS and TriTg mice. The sections were immunostained with anti-FLAG antibody (green), and nuclei were counterstained with DAPI (blue).
CIP overexpression in the motor neurons specifically inhibits Cdk5/p25 hyperactivation and prevents the motor neuronal death in TriTg mice
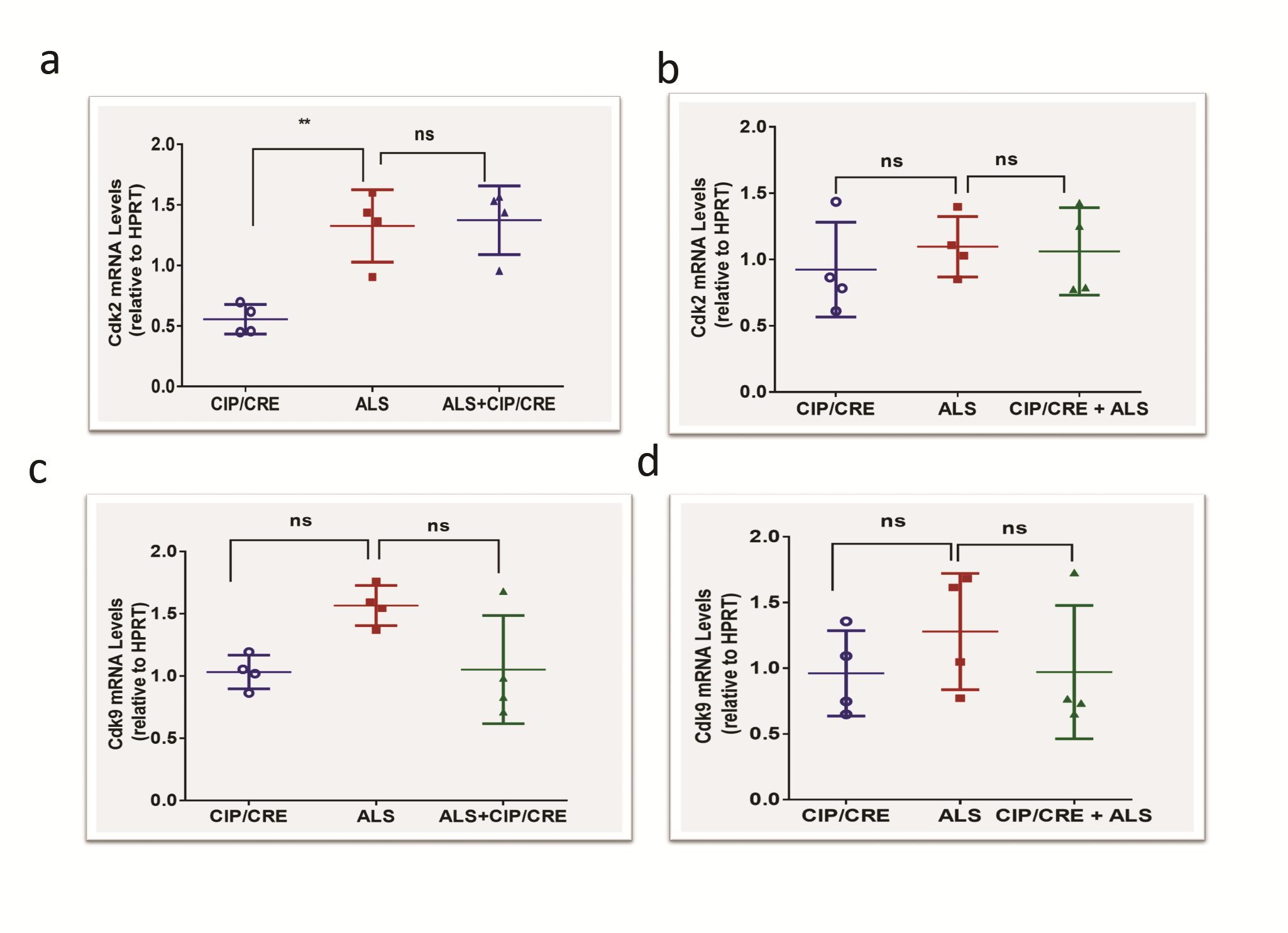
To study the ability of CIP to inhibit the Cdk5/p25 hyperactivation-mediated motor neurodegeneration in SOD1G37R mice, we generated a TriTg mouse that overexpresses CIP under the direction of the Chat promoter crossing the CIPTg mice with the SOD1G37R transgenic mice (Jackson Laboratory, Stock no 008342). To examine the effect of CIP overexpression on the Cdk5 hyperactivation, we performed q-PCR, western blot analyses and in vitro kinase assays on the samples from 12 week SOD1G37R, TriTg mice and age-matched CIPTg mouse brain and spinal cord. No significant changes in the Cdk5 mRNA and protein expression levels in the brain and spinal cord regions were observed among all the samples (Supplementary Material, Fig. S1A and C, spinal cord; S1D and E, brain). p35 mRNA levels were also not changed in the groups (Supplementary Material, Fig. S1B, spinal cord; S1F, brain) but significant amount of p25 generation was observed in the SOD1 and TriTg mouse spinal cord and brain (Supplementary Material, Fig. S1G, brain; S1H, spinal cord). In addition to that, there was a significant increase in Cdk5 activity in SOD1G37R mice spinal cord and brain compared with the CIPTg mice (Fig. 2A, B and E, spinal cord; 2C, D and F, brain). This observation agreeing with the previous study showed deregulated Cdk5 activity associated with ALS pathogenesis in SOD1G37R mice (10). In contrast, Cdk5/p25 hyperactivity was reduced in both spinal cord and brain of TriTg mice compared with the SOD1G37R mice (Fig. 2A–F). Collectively, these results suggest that Cdk5 hyperactivation in the SOD1G37R mice was effectively inhibited by motor neuronal CIP overexpression in TriTg mice. We also checked if motor neuronal CIP overexpression affected expression of other CDKs in brain and spinal cord. We analyzed Cdk2 and Cdk9 mRNA expression levels between the groups. No significant changes in the mRNA levels in the brain and spinal cord were observed, compared SOD1G37R with TriTransgenic mice (Supplementary Material, Fig. S5A and C, spinal cord; B and D, brain).
Figure 2.
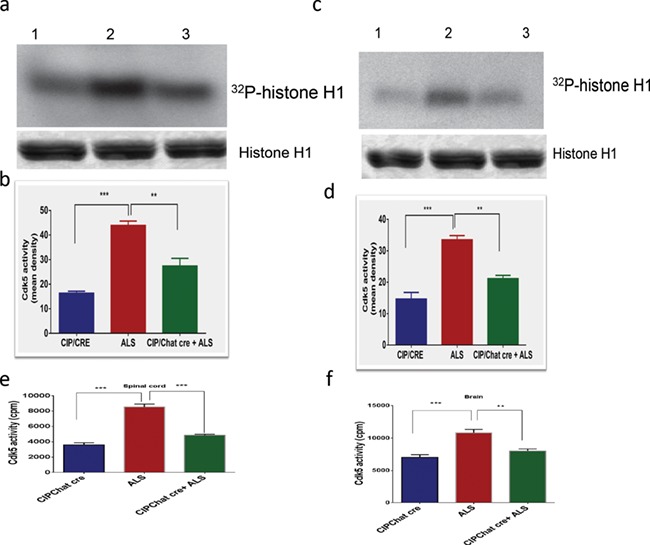
CIP overexpression in motor neurons specifically inhibits Cdk5/p25 hyperactivation in TriTg mice. (A and B) Representative in vitro kinase assays using active kinase (Cdk5) immunoprecipitated from the brain and (C and D) spinal cord of CIPTg, ALS and TriTg mice (**P < 0.001, ***P < 0.0001). (E) Immunoprecipitates were also subjected to in vitro kinase pad assays spinal cord (n = 8) and (F) brain (n = 8) of CIPTg, ALS and TriTg mice. Activity, as counts/minute, was quantified and summarized in the bar graphs (**P < 0.001; ***P < 0.0001). Data are presented as mean ± SEM.
We further investigated whether motor neuronal overexpression of CIP from early development can delay or prevent motor neuron degeneration by crossing CIPTg mice with SOD1G37R mice. We analyzed offspring from this cross of all three genotypic combinations for pathological parameters. Motor neuronal overexpression of CIP improves motor neurons survival in TriTg mice compared with SOD1G37R. Representative images of Nissl-stained motor neurons in matching lumbar spinal cord cross-sections at Day 150 (Fig. 3A and B) show significant decrease of motor neuron survival counts in lumbar spinal cord cross-sections of SOD1G37R mice compared to CIPTg mice. The motor neuronal overexpression of CIP prevents the motor neuronal death in TriTg mice (Fig. 3A and C; values are mean ± SEM (n = 3–4 per genotype; **P < 01, ***P < 0.001). We also confirmed that expression of the CIP in the motor neurons, we performed the double immunohistochemistry and Chat is the motor neuronal marker and confirm that CIP is expressed in the motor neurons (Fig. 3C). We then quantified motor neurons, which express Chat and CIP (flag tagged) survival in the lumbar spinal cord at Days 80 and 150 to determine whether the delayed neuropathology. Importantly, while no significant differences in motor neuron survival were observed between the three genotypes at Day 80 (data not shown), motor neurons viability is significantly improved in TriTg mice when compared to that of SOD1G37R mice at Day 150 (P < 0.0001), with motor neuron numbers in TriTg mice (Fig. 3C and D).
Figure 3.
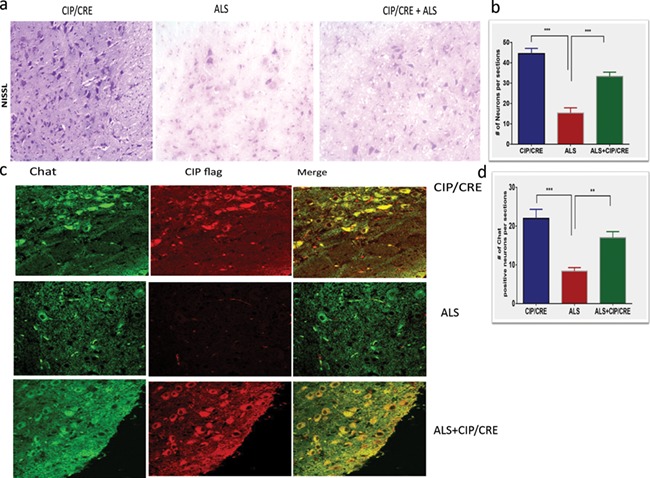
Motor neuronal overexpression of CIP improves motor neuron survival in TriTg mice. (A) Representative images of Nissl-stained motor neurons in matching lumbar spinal cord cross-sections at Day 150. (B and D) Motor neuron survival counts in lumbar spinal cord cross-sections at Day 150; values are mean ± SEM (n = 3–4 per genotype; **P < 0.001, ***P < 0.0001; one-way ANOVA, with Tukey’s post hoc tests). Scale bar = 100 mm. Immunohistochemical staining for Chat (motor neuronal marker) and FLAG for CIP, in the spinal cord demonstrating CIP expression in motor neurons at Day 150. Scale bar = 30 mm (C).
Motor neurons overexpression of CIP reduces hyperphosphorylation of Tau and NF proteins in TriTg mice
Previous studies already proved that the increased Cdk5 activity in SOD1G37R mice was associated with hyperphosphorylation of Cdk5 cytoskeletal substrates such as tau and NF proteins (10). We then examined if motor neurons CIP overexpression can decrease the phosphorylation of NF and tau in spinal cord and brain of TriTg mice. We used the AT-8 and PHF-1 antibodies that recognize three phosphoserines (199, 202 for AT-8 and 396 for PHF-1) in the tau protein that can be phosphorylated by Cdk5. The hyperphosphorylation of these sites has been associated with Alzheimer’s disease (23,24). Spinal cord and brain extracts from CIPTg, SOD1G37R and TriTg were fractionated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) followed by immunoblotting with AT-8 or PHF-1 antibodies. Significant AT-8 and PHF-1 immunoreactivities were detected in samples from SOD1G37R mice spinal cord and brain compared to CIPTg (Fig. 4A–I). However, in extracts from TriTg mice, significant decrease of hyperphosphorylation of tau in these brain and spinal cord extracts, compared to SOD1G37R mouse, was observed (Fig. 4G, spinal cord); line 3, h line 3, e and the total tau remains unchanged in all these groups. These combined results with the three anti-tau antibodies indicate that tau is hyperphosphorylated in spinal cord and brain of SOD1G37R mice significantly decreased in the TriTg mice.
Figure 4.
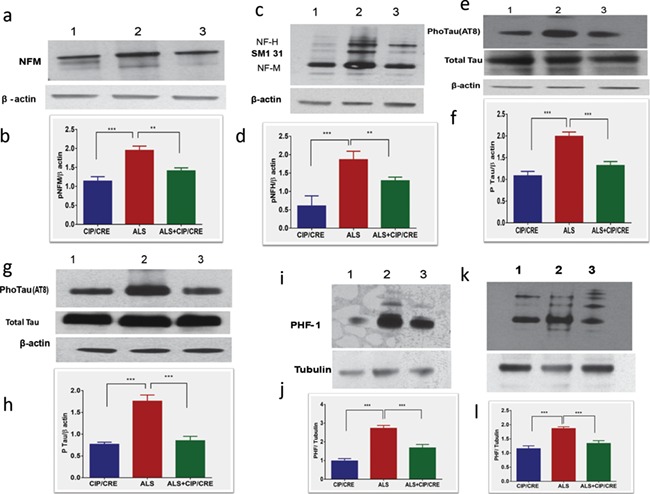
Inhibition of hyperphosphorylation of Tau and NF proteins in motor neurons of TetraTg mice. (A) Hyperphosphorylated NF and tau protein levels were analyzed by western blot analyses and quantification of anti-Neurofilament M (n = 6), (C) anti-160 kD Neurofilament H (n = 6) spinal cord and using AT8, (G) brain, (E and F) spinal cord and PHF1 antibodies, (I) brain, (K) spinal cord. Quantification of immunoblots in by densitometric scanning B, D, H, J and I (**P < 0.001, ***P < 0.0001).
The NF-H and NF-M proteins were also abnormally hyperphosphorylated in the SOD1G37R mice (10). In SOD1G37R mice, significant increase of hyperphosphorylation in NF-H and NF-M proteins (Fig. 4A, C, I and K, line 2) agreeing with the previous results and motor neuronal CIP overexpression significantly decreases the hyperphosphorylation in spinal cord and brain of TriTg mice (Fig. 4A, C, I and K, line 3 and B, D, J and L). This again confirmed, in SOD1G37R mice, robust immunostaining of perikarya in the SMI31 antibodies that recognized the hyperphosphorylated forms of NF-H and NF-M, respectively (Supplementary Material, Fig. S2B, panels). In contrast, in TriTg mice, insignificant immunostaining of neuronal perikarya was obtained in cell body of motor neurons, demonstrating the normal NF-H and NF-M proteins (Fig. 2C, panels). Double immunofluorescence microscopy confirmed the co-localization of Cdk5 and perikaryal NF protein accumulations in the SOD1G37R (Supplementary Material, Fig. S2A–C). Such co-localization of perikaryal NF proteins with Cdk5 activators was not observed in spinal cord sections from CIPTg and TriTg (Supplementary Material, Fig. S2C panels). Collectively our results showed that motor neuronal CIP overexpression effectively reduces the hyperphospho tau and NF neuropathological hallmarks in TriTg mice.
Motor neurons CIP overexpression reduces neuroinflammation in TriTg mice
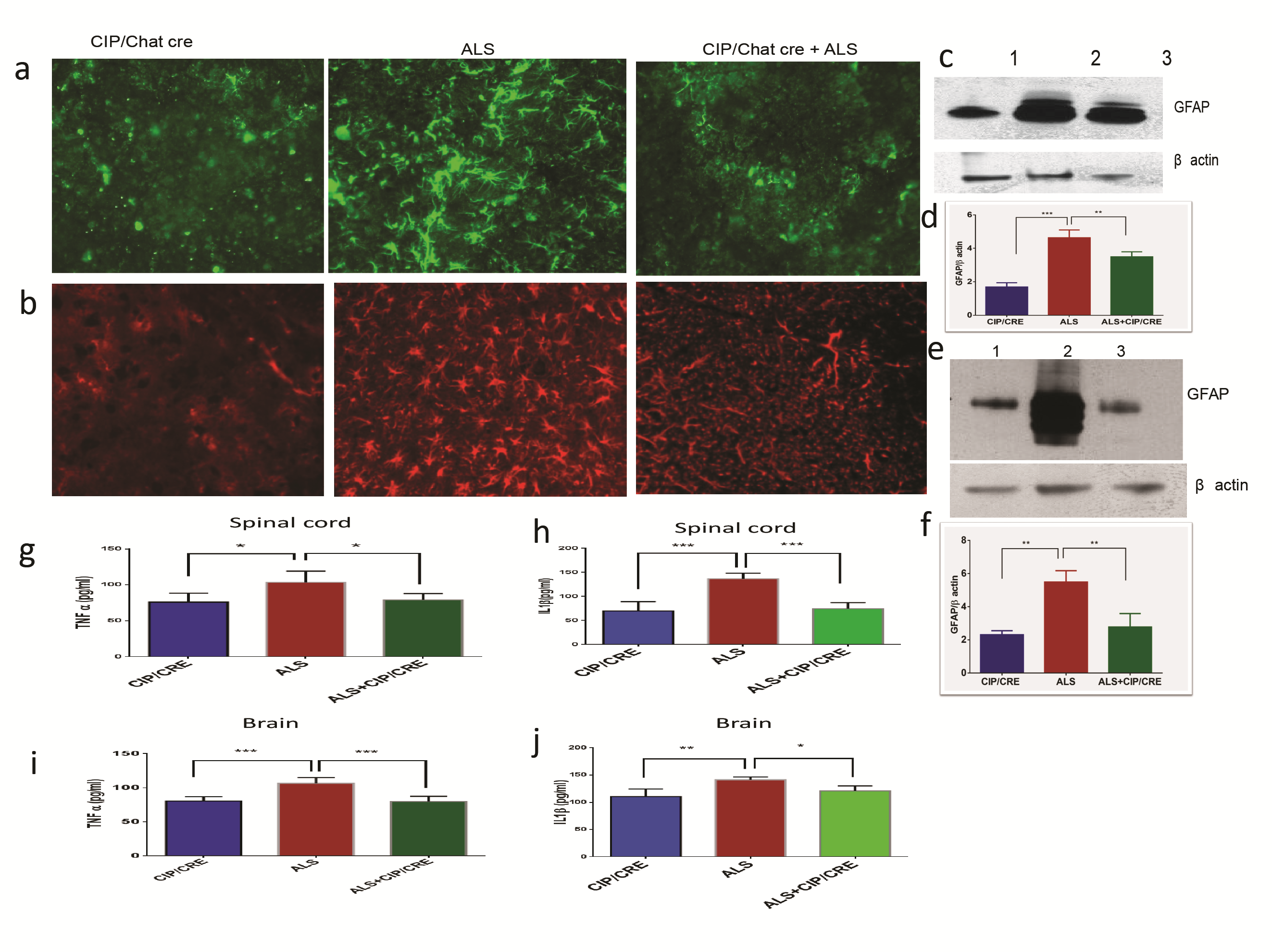
Previous studies have shown robust neuroinflammation in SOD1G37R mice. To examine the effect of motor neurons CIP expression on astrogliosis and microgliosis, we preformed immunohistochemistry and western blot analyses using GFAP (marker for astrocytes) and Cd11b (marker for microglia) antibodies on the samples from 12 week SOD1G37R, TriTg and age-matched CIPTg mice spinal cord and brains. Motor neurons CIP expression on its own did not induce any astrogliosis and microgliosis. Whereas SOD1G37R mice had elevated GFAP and Cd11b expression levels in spinal cord and brain, astrogliosis and microgliosis were significantly reduced in TriTg mice compared with SOD1G37R mice (Supplementary Material, Fig. S3A–E). To further validate this phenomenon, we chose five other markers of proinflammatory cytokines and macrophage activation markers, Mpeg1, IL1β, IL-13, TNF-α and INF-γ. We analyzed the expression levels of these genes in all the groups of spinal cord and brain of Day 150 to assess the extent of candidate gene deregulation during disease progression. Quantitative real-time PCR analysis confirmed that Mpeg1, IL1β, IL-13, TNF-α and INF-γ significantly upregulated in the spinal cord and brain of SOD1G37R mice compared to that of TriTg (Fig. 5A–P). To quantify some of the proinflammatory cytokines upregulation, we performed Enzyme-Linked Immunosorbent Assay (ELISA) for TNF-α and IL1β in all three groups. Significant increase of these cytokines in the brain and spinal cord of SOD1G37R mouse was observed compared to CIPTg; in contrast levels of these proinflammatory cytokines were significantly decreased in TriTg compared to SOD1G37R mouse (Supplementary Material, Fig. S3G–J). Together, these data prove that motor neuronal CIP overexpression is sufficient to decrease pathological neuroinflammation in the spinal cord and brain of TriTg mice.
Figure 5.
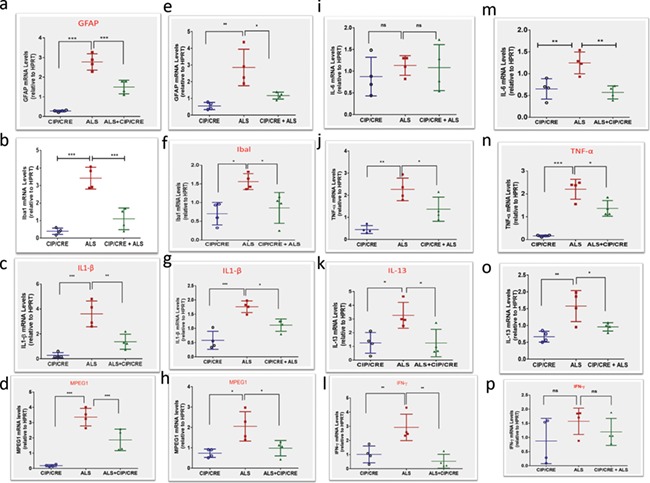
Motor neurons CIP overexpression decreases neuroinflammatory markers transcriptome levels in TriTg mice spinal cord and brain. (A, B, C, D, E, I, J, K and L) ALS (SODG37R)-induced expression of GFAP, Ibal, IL1β, Mpeg1, IL-6, TNF-α, IL-13,INF-γ, markers of neuroinflammation, in the spinal cord and (B, F, G, H, M, N, O and P) brain were reduced in TriTg mice at Day 150 as shown by quantitative PCR.
CIP overexpression in motor neurons extends survival of TriTg mice
The correlation between Cdk5 activity and severity of disease in SOD1G37R models has been previously reported. The deregulated Cdk5/p25 activity causes the decreased lifespan in the SOD1G37R mice (10). We investigated whether motor neurons overexpression of CIP from early development can extend the lifespan crossing CIPTg mice with SOD1G37R mice. We analyzed offspring from this cross of all two genotypic combinations for survival rates. We did not compare the SOD1G37R and CIPTg because CIPTg mouse lives normal lifespan similar to any wild-type mouse (C57black). Therefore, we have compared the TriTg mouse with SOD1G37R mouse. We obtained survival rates for SOD1G37R and TriTg mice. Strikingly, in TriTg mouse, median survival is significantly increased by 16% (P < 0.0001), from 163 days for SOD1G37R mice to 189 days for TriTg animals (Fig. 6A). These data demonstrate that motor neuron-specific CIP overexpression improves survival in TriTg mice.
Figure 6.
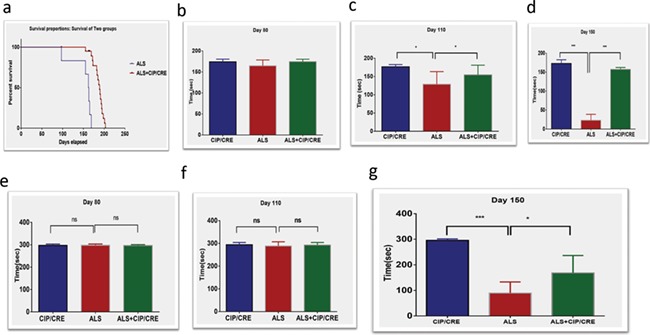
Motor neurons overexpression of CIP extends lifespan and improves motor function in SOD1G37R mice. (A) Kaplan–Meier log rank test for survival, showing CIP overexpression increases median survival from 163 days for SOD mouse (n = 16) to 189 days for TriTg mice (CIP/CRE+ALS, n = 35). (C and D) TriTg mice show significantly improved muscle strength on a grip strength test when compared with SOD1G37R at (C) Day 110 and (D) Day 150, but not at (B) Day 80. Values are mean ± SEM (n = 12–24 per genotype; ***P5 < 0.001, *P5 < 0.05 Mann-Whitney U-tests). (E–G) Motor neurons overexpression of CIP improves motor function SOD1G37R mice. TriTg mice have improved motor performance on the accelerating Rotarod at (G) Day 150, but not at (E) Day 80 and (F) Day 110 when compared with SOD1G37R. Values are mean ± SEM of motor performance (seconds, s) for mice still alive at each respective time point (n = 9–11) per animal per genotype at Days 60–120 and (n = 6–11) per sex per genotype at Day 105 due animals reaching end stage; *P < 0.05, **P < 0.001; Mann-Whitney U-tests).
Motor neurons CIP overexpression improves motor function in TriTg mouse
As muscle dysfunction and atrophy are downstream consequences of motor neurons loss in ALS patients and SOD1 mice, we investigated whether motor neurons overexpression of CIP improves muscle pathology in TriTg mice and delays disease progression. To measure muscle function, we analyzed performance of mice on a grip strength monitor first (Fig. 6B–D) and found that TriTg mice have significantly improved muscle strength compared with SOD1G37R animals (***P < 0.001, *P < 0.05) from Days 110 and 150 (Fig. 6B–D). Then we analyzed the motor function. TriTg mouse have significantly improved motor performance on the accelerating Rotarod at Day 150 (Fig. 6G), but not at Days 80 (Fig. 6E) and 110 (Fig. 6F) compared with SOD1G37R. In addition, we also did the open field study; before tremor onset, SOD1G37R mice would exhibit motor performance deficits when allowed to roam freely versus forced motor behavior tests to check motor neuronal CIP overexpression delays the progression. Freely behaving SOD1G37R transgenic mice displayed motor-related deficits during the pre-symptomatic phase; animals were placed in an open field apparatus for 20 min, beginning at postnatal Day 80 to postnatal Day 160 (P80–P160; Fig. 7A–X). In general, performance of SOD1G37R transgenic mice on all open field measures was below that of CipTg mice (Fig. 7). Compared to CIPTg, SOD1G37R mice showed significant deficits across time (P80–P160) on open field motor performance evaluations including total distance traveled, rotation of the animal body and total time freezing episodes. Taken together these results, motor neuronal CIP overexpression delays the diseases progression in TriTg mouse compared with SOD1G37R mouse (Fig. 7; *P < 0.05, **P < 0.01, ***P < 0.001).
Figure 7.
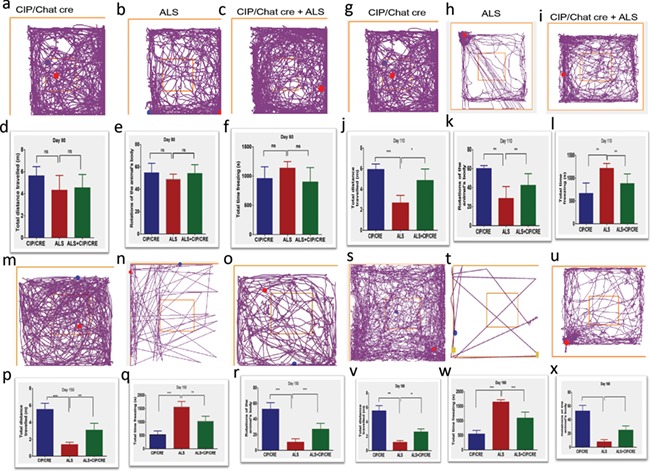
CIP overexpression delays progression of motor deficits in SOD mice. Before tremor onset, SOD1G37R mice would exhibit motor performance deficits when allowed to roam freely versus forced motor behavior tests and CIP overexpression delays the progression. To investigate the likelihood that freely behaving SOD1G37R transgenic mice displayed motor-related deficits during the pre-symptomatic phase, animals were placed in an open field apparatus for 20 min, beginning at postnatal Day 80 to postnatal Day 160 (P80–P160). In general, performance of SOD1G37R transgenic mice on all open field measures was below that of control mice (Fig. 7). Compared to control mice, SOD1G37R mice showed significant deficits across time (P80–P160) on open field motor performance evaluations including total distance traveled, rotation of the animal body and total time freezing episodes. CIP overexpression delays the diseases progression. Values are mean ± SEM of motor performance (total distance traveled, meter m, rotation of the animal body and total time freezing episodes in seconds s) for mice still alive at each respective time point (n = 9–11) per animal per genotype at Days 60–120 and n = 6–11 per animal per genotype at Days 150 and 160 due animals reaching end stage; *P < 0.05, **P < 0.01,***P < 0.001, Mann-Whitney U-tests).
Motor neurons overexpression of CIP delays muscle pathology and prevents the cell death in TriTg mice
Previous studies on human ALS patients demonstrated skeletal muscle changes with clear signs of denervation and reinnervation. Hematoxylin and eosin (H&E) staining from all the groups at Day 150 reveals morphological changes with increasingly disorganized muscle fibers including increased number of atrophic fibers in SOD1G37R mice compared with CIPTg and TriTg. The SOD1G37R mouse showed a high degree of atrophy, and areas of macrophage infiltration at Day 150. Furthermore, increased number of inflammatory cells as well as nuclear clumps is present between the muscle fibers at Day 150. These changes are rescued in the TriTg mouse. Gastrocnemius muscle atrophy of TriTg mice is significantly reduced at Day 150, compared to that of SOD1G37R mice (Fig. 8I–K). These data demonstrate that motor neuronal overexpression of CIP delays atrophy in gastrocnemius muscles.
Figure 8.
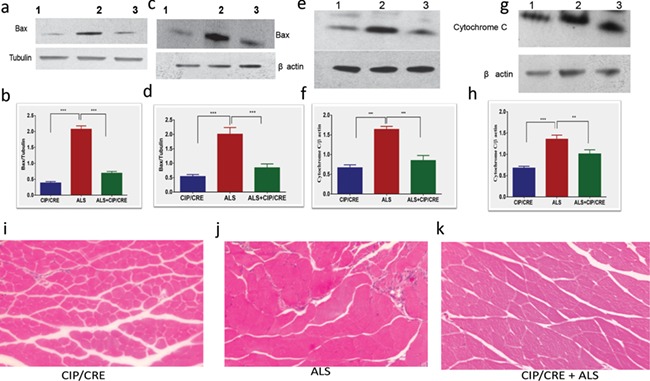
Motor neurons overexpression of CIP prevents the cell death and delays muscle pathology in TriTg mice. (A and B) CIP overexpression inhibits Bax up regulation in brain, (C and D) spinal cord and (E and F) cytochrome c release in brain and (G and H) spinal cord. TriTg mice showed significantly improved muscular atrophy. Representative images of H&E stained gastrocnemius muscle fibers at Day 150. Values are mean ± SEM (n = 4–6 per genotype; ***P < 0.001,**P < 0.01, Mann-Whitney U-tests).
We previously reported that CIP reduces neuronal apoptosis in vitro (33). We measured the anti-apoptosis effects of CIP to prevent the motor neuronal death in vivo. Cytochrome c release from the mitochondria into the cytoplasm is a potent physiological stimulus for caspase-9 and caspase-3 activation. CIP overexpression in the motor neurons significantly inhibited release of cytochrome c in brain and spinal cord of SOD1G37R mice compared with TriTg (Fig. 8A–H). We also measure the Bax expression in all the three groups. TriTg mouse showed the significantly lower expressions of Bax in the spinal cord and brain compared to the SOD1G37R mouse. These data suggest the motor neuronal CIP overexpression decrease apoptosis in TriTg mouse.
Discussion
In this study, we show that in vivo motor neurons expression of CIP effectively inhibits the hyperactivation of Cdk5/p25 and rescues the ALS-related pathology in SOD1G37R mouse. These data are an extension to the in vitro findings detailing CIP protection in primary neurons (34) as well as in vivo findings of reduced neurodegeneration in p25Tg AD-like phenotype in mouse model (37). In addition, this is the first time Cdk5/p25 aberrant hyperactivation has been selectively targeted in the motor neurons to rescue the ALS pathology in mouse model.
Our previous studies identified a central fragment of p35 (residues 154–279), called CIP, that can effectively inhibit Cdk5 hyperactivity in vitro (32,38). Additional studies identified CIP as a potent, selective inhibitory peptide toward Cdk5/p25 without affecting p35/Cdk5 activity in vivo (34). As an extension, CIPTg mouse was created where CIP expressed in the forebrain using Camk2a promoter (37) reduced neurodegeneration in p25Tg AD-like phenotype in mouse model. Based on previous in vitro and in vivo evidence, we believed that normal Cdk5 activity would not be affected in the CIPTg mice (34,37,39) and our results confirmed that CIPTg mice were phenotypically identical to WT C57BL/6 mice in terms of nervous system development, body weight and behavior (37). Similar results were obtained from our present study; specific motor neuronal CIP level was stable under the Chat promoter and mice were phenotypically identical to WT C57BL/6 mice in terms of nervous system development, body weight and behavior.
Previously, Nguyen et al. (10) extensively characterized the SOD1G37R mice, model of ALS where deregulation of Cdk5 activity is associated with disease caused by mutant SOD1, and their results revealed a mislocalization of Cdk5 in the cytoplasm of motor neurons, an elevated ratio of p25 to p35 and an increase of Cdk5 activity in SOD1G37R mice. The deregulated Cdk5 activity in SOD1G37R mice is accompanied by the hyperphosphorylation of tau and NF proteins. A contribution of Cdk5 to neurodegeneration is suggested from their finding of a correlation between Cdk5 activity and longevity of ALS mice. We could also reproduce their results in our SOD1G37R mice colonies and limited number of postmortem human ALS brain and spinal cord, elevated ratio of p25 to p35 (Supplementary Material, Fig. S4 and Fig. 2). In addition, hyper activation of Cdk5/p25 activity in the brain and spinal cord of both human ALS patients as well as mouse model of ALS were also seen (Supplementary Material, Fig. S4 and Fig. 2A–C). No significant changes in the Cdk5 mRNA and protein expression level in the brain and spinal cord regions were observed among all the samples (Supplementary Material, Fig. S1) but there was a significant increase in Cdk5/p25 activity in SOD1G37R mice compared with the CIPTg mice (Fig. 2A, B and E, spinal cord; 2C, D and F, brain). In addition, Cdk5/p25 hyperactivity was significantly reduced in TriTg mice compared with the SOD1G37R mice (Fig. 2A–F). These results suggest that Cdk5/p25 hyperactivation in the SOD1G37R mice was effectively inhibited by motor neurons CIP overexpression in TriTg mice. The present study also clearly shows that the deregulated Cdk5/p25 activity in SOD1G37R mice is accompanied by the hyperphosphorylation of tau and NF proteins in the SOD1G37R mice. These effects were ameliorate in the TriTransgenic mice where CIP is specifically overexpressed in the motor neurons (Fig. 4 and Supplementary Material, Fig. S2).
While recent studies suggest that a deregulation of Cdk5 activity can induce cytoskeletal abnormalities and neuronal death (27,40), our results are also in agreements with facts that the deregulated Cdk5 activity in SOD1G37R mice is accompanied by the hyperphosphorylation of tau and NF proteins (33). CIP overexpression in the motor neurons of ALS mouse significantly protects from degeneration (Fig. 3A–D). Our observations involving motor neurons-specific CIP expression are consistent with other reports indicating that disease initiates in neurons. In this regard, motor neurons-specific overexpression of Thy-1.2 promoter driven mutant hSOD1G93A produced motor neurons disease in mice (41) and deletion of mutant hSOD1G37R protein in motor neurons delayed disease onset (42). Neuron-specific overexpression of calpastatin (CAST), the highly selective endogenous inhibitor of calpains, accounts for their longer time of survival and neuroprotective in an ALS mouse model (43); moreover overexpression of OXR1 in neurons significantly delays dysregulation of genes that are involved in diverse molecular functions altered in ALS (44).
The correlation between Cdk5 activity and longevity of ALS mice has been reported earlier; our results showed that motor neurons overexpression of CIP from early development can extant the lifespan of SOD1G37R ALS mice. Inhibiting deregulated Cdk5/p25 activity in the TriTg mouse, median survival is significantly increased by 16%, from 163 days for SOD1G37R mice to 189 days for TriTg animals (Fig. 6A). These data again demonstrate that motor neurons-specific CIP overexpression improves survival in TriTg mice and is in agreement with previous study that mentioned the role of deregulated Cdk5 in longevity of ALS mice (43). Neuroinflammation has been inextricably linked to ALS disease progression, and activated microglia and astrocytes substantially contribute to motor neuron death (40,45,46). Neuroinflammation was extensively decreased in both microgliotic and astrogliotic paradigms in TriTg mice. The levels of proinflammatory cytokines are significantly decreased in TriTg mice compared to SOD1G37R mice (Supplementary Material, Fig. S3G–J). Together, these data prove that motor neurons CIP overexpression is sufficient to decrease pathological neuroinflammation in the spinal cord and brain of TriTg mice. Previous studies also noted that specific deletion of mutant SOD1 from either astrocytes or microglia significantly slows disease progression in SOD1G37R mice, while monoclonal antibody treatment against CD-40L, a T cells surface ligand that activates the immune response, or inhibiting NF-KB activation in microglia significantly extends survival in SOD1G37R mice (42,47,48). Moreover, transplantation of SOD1G37R glial-restricted progenitors into the spinal cord of wild-type rats induces astrogliosis and microgliosis, motor dysfunction and motor neurons death, demonstrating non-cell autonomous toxicity in ALS (49). As similar to previous study (36), to ensure that the reductions of Cdk5/p25 activity, phosphorylated tau and phosphorylated NF manifested in neuroprotection and behavior benefit, we used different behavioral assessments at different time period of the diseases. Together, this study is the extension and proof of concept for the overexpression of CIP in the ALS model rescued the motor neuronal death, decrease inflammation, amelioration in the behavior impairments and extend the lifespan.
Materials and Methods
Animal handling
All animal experiments were performed according to approved protocols by the Institutional Animal Care and Use Committee of the National Institutes of Health, USA.
In the present study, both male and female mice and their age-matched littermates were used. Genotyping was performed by PCR analysis of tail DNA. All the behavior experiments were conducted in a blinded fashion with respect to the genotype and experimental conditions of the mice. Mice were housed and bred in accordance with the U.S. National Institutes of Health Guide for Care and Use of Laboratory Animals. Mice were group housed with a 12 h light/dark cycle and had ad libitum access to food and water.
Generation of CIPTg mice (CIP/Chat CRE) and Tri Transgenic (CIP/Chat Cre+ ALS)
The original CIP (CIP floxed) transgenic breeder mice were a generous gift from Dr Sashi Kesavapany and have been maintained on a pure C57BL6 background. The CIPTg construct contains a 3′-FLAG-tagged CIP transgene incorporated in to the ROSA26 locus whose 5′ regulatory elements were separated from the coding region with a floxed stop sequence. An frt-flanked neomycin resistance (Neo) cassette was inserted next to the transgene before the 3′ homology arm (36). The germ-line founders (CIP floxed) were then crossed with ChAT-IRES-Cre transgenic mice (Stock no 006410; Jackson Laboratory) to get the bitransgenic mice (CIPTg mice), which express CIP in the motor neurons using the CRE/LoxP recombination system. The CIPTg mice were then crossed with ALS mouse, SOD1G37R) 42Dpr/J (Stock no 008342; Jackson Laboratory) to get the TriTg mice (CIP/Chat CRE +ALS) that overexpress the CIP in the SOD1 (SOD1G37R mouse). Littermates of the same sex were used for comparison whenever possible.
Antibodies
Antibodies used for both western blot analyses and immunohistochemistry were mouse monoclonal anti-FLAG (1:200; Sigma, St. Louis, Missouri), rabbit polyclonal anti-Cdk5 (C8, 1:500; Santa Cruz Biotechnology, St. Louis, Missouri), mouse anti-α-tubulin (1:10000; Sigma), rabbit polyclonal anti-p35 (C-19, 1:1000; Santa Cruz Biotechnology, Dallas, Texas), anti-phospho-Neurofilament H mouse (SMI-31, 1:1000; Millipore, Temecula, California), anti-phospho-Neurofilament M rabbit monoclonal (1:1000 Millipore), rabbit polyclonal anti-Cytochrome c (1:1000; Cell Signaling Technology, Danvers, Massachusetts), Bax Monoclonal (6A7, 1:100; Thermo Fisher Scientific, Waltham, CA, USA), mouse monoclonal anti-GFAP (1:1000; Sigma), mouse monoclonal anti-Cd11b (1:200; Millipore), mouse monoclonal anti-paired helical filaments (PHF)–tau antibodies (clones AT8 and AT180, 1:500; Pierce) and sheep polyclonal anti-Choline Acetyltransferase antibody (1:100; Abcam, Cambridge, UK). Secondary fluorescence-conjugated antibodies Alexa Fluor 488 and Alexa Fluor 594 (Invitrogen, Camarillo, California) were used at 1:200 dilutions for immunohistochemistry. Horseradish peroxidase-conjugated mouse or rabbit secondary antibodies (GE Healthcare, Marlborough, Massachusetts) were used at 1:1000 dilutions for western blot analyses.
RNA extraction and quantitative real-time PCR
Spinal cords and brains were dissected out from CIPTg (n = 4), ALS (n = 4) and TriTg (n = 4) mice. All tissues were immediately frozen and kept at −80°C. For real-time PCR analysis, four mice per groups but six mice per group for western blot were included.
Total RNA was extracted using TRIZOL reagent (Thermo Fisher) according to the manufacturer’s instructions. RNA from each sample was reverse transcribed using a High Capacity cDNA Reverse Transcription Kit (Thermo Fisher). q-PCR reactions were conducted using Assays on Demand and TaqMan Universal Master Mix (both Thermo Fisher Scientific). All samples were run in duplicate using the Real-time PCR System 7500 (Thermo Fisher). The p35, Cdk5, GFAP, Iba1, IL-1β, Mpeg1, IL-6, TNF-α, IL-13, INF-γ, Cdk2 and Cdk9 levels were normalized to the levels of HPRT using the comparative threshold method.
Western blot analyses
Spinal cord and brain lysates from CIPTg (n = 6), ALS (n = 6) and TriTg (n = 6) mice were prepared as described previously (30). Polyacrylamide gel running, nitrocellulose membrane transfer and detection were performed as reported previously (30).
Cdk5 assay
Spinal cord and brain lysates from CIPTg (n = 6), ALS (n = 6) and TriTg (n = 6) mice were prepared and kinase assays were performed as described previously (30). Cdk5 was immunoprecipitated with the polyclonal C8 antibody for 2 h at 4°C, and immunoglobulin was isolated using protein A–Sepharose beads for 2 h at 4°C. Immunoprecipitates were washed three times with lysis buffer and then once with 1× kinase buffer containing 20 mm Tris-Cl, pH 7.4, 1 mm Ethylenediaminetetraacetic acid (EDTA) 1 mm ethylene glycol tetraacetic acid, 10 mm MgCl2, 10 mm sodium fluoride and 1 mm sodium orthovanadate. The samples were added to the reaction mix containing kinase buffer, 50 μm ATP, 20 μg of histone H1 and 0.5 Ci of [32P] ATP and incubated at 30°C for 1 h. Reactions were halted by the addition of loading buffer, and samples were then electrophoresed on 12% SDS-PAGE gels. Histone bands were visualized by Coomassie blue staining. Gels were dried, and then autoradiographs were scanned on a PhosphorImager. Radioactive band density was analyzed using ImageJ software (National Institutes of Health, Bethesda, MD), and statistical analysis was performed. In pad assays, 25 μl aliquots of the incubation mixture were placed on a Whatman p81 paper square, air-dried and washed five times for 15 min each in 75 mm phosphoric acid and once in 95% ethanol. After air-drying, squares were transferred to vials containing Bio-Safe II scintillation fluid for counting.
ELISA
Spinal cord and brain lysates from CIPTg (n = 6), ALS (n = 6) and TriTg (n = 6) mice were prepared as described previously (30). For cytokine assays, the concentrations of TNF-α, and IL-1β, in the brain and spinal cord lysates were measured with an ELISA kit (Thermo Scientific), according to the manufacturer’s instructions. Absolute concentrations were derived by comparison with a standard curve.
Histochemical studies
Ten micrometer cryostat sections of brain, spinal cord and gastrocnemius muscle CIPTg (n = 6), ALS (n = 6) and TriTg (n = 6) mice were collected on slides and prepared for immunohistochemistry, which was performed according to our previously published protocol (30).
Disease course analysis and behavior tests
Disease onset was retrospectively defined as the age when mice reached maximum body weight as previously described (5,6). Disease end stage was defined by the age when mice suffered from functional paralysis of both hind limbs; this phenotype has recently been established experimentally as an earlier and more humane but predictable and reproducible endpoint for transgenic mouse models of ALS, limiting the duration of disease exposure. Disease progression was retrospectively defined as the number of days between disease onset and disease end stage for each mouse. To test motor function, mice CIPTg (n = 12), ALS (n = 12) and TriTg (n = 12) were placed on a grooved plastic beam of a Rotarod device, which revolves at a default 14 rpm, facing in an orientation opposite to the rotation. The time latency to fall from the rod or complete two rotations on the rod without an attempt to run was recorded; a single trial was carried out per day over 3 days in total at each experimental time point and the recorded values were averaged.
Hanging-wire test
Neuromuscular strength was tested by the hanging-wire test in CIPTg (n = 12), ALS (n = 12) and TriTg (n = 12) mice. Each mouse was placed on a wire lid of a conventional housing cage and the lid was turned upside down. The latency from the beginning of the test until the mouse stood with at least two limbs on the lid was timed. The animals had three attempts to stand for a maximum of 180 s per trial, and the longest latency was recorded.
Open field study
Open field study was performed as described previously (50). A multiple unit open field maze consisting of four activity chambers was used for this analysis. Each chamber measured 50 cm (length) × 50 cm (width) × 38 cm (height) and was made from white high density and non-porous plastic. Texture the floors of the maze for traction during ambulation while maze walls were smooth. Maze quadrants were completely empty for this test. Wipe the chamber with a 95% Ethanol prior to use and before subsequent tests to remove any scent clues left by the previous subject mouse. Allow the ethanol to evaporate completely prior to testing mice. In the open field experiment, mice were monitored for horizontal activity, vertical activity, total distance traveled (centimeters), total movement time (seconds), total time freezing (seconds) and rotation of the animal body over a 30 min test session. For this analysis, CIPTg (n = 12), ALS (n = 12) and TriTg (n = 12) mice were used, and the Video Tracking software from Any Maze recorded and evaluated the mouse movement.
Statistics
All values are expressed as the mean of at least three determinations ± SEM. Data were analyzed by one-way Analysis of variance (ANOVA) and P value < 0.05 was considered to indicate statistical significance. For open field analyses, were plotted by the mice over the entire test sessions averaged and subjected to one-way ANOVA, followed by Whitney U-tests.
Funding
Intramural Research Programs of the National Institutes of Health; National Institute of Neurological Disorders and National Institute of Dental and Craniofacial Research, USA; and Council of Scientific & Industrial Research (CSIR), India.
Author contributions
B.K. and H.C.P. conceived of the presented idea. B.K. and M.P. carried out the experiments. B.K. wrote the manuscript with support from H.C.P. S.S. and N.D. contributed to behavior and kinase assay. S.K. generated CIP transgenic mouse. P.G., A.B.K. and V.K. contributed to the final version of the manuscript. All authors provided critical feedback and helped shape the research, analysis and manuscript.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
References
- 1. Hardiman O., van den Berg L.H. and Kiernan M.C. (2011) Clinical diagnosis and management of amyotrophic lateral sclerosis. Nat. Rev. Neurol., 7, 639–649. [DOI] [PubMed] [Google Scholar]
- 2. Cudkowicz M.E., McKenna-Yasek D., Sapp P.E., Chin W., Geller B., Hayden D.L., Schoenfeld D.A., Hosler B.A., Horvitz H.R. and Brown R.H. (1997) Epidemiology of mutations in superoxide dismutase in amyotrophic lateral sclerosis. Ann. Neurol., 41, 210–221. [DOI] [PubMed] [Google Scholar]
- 3. Rosen D.R. (1993) Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature, 362, 59–62. [DOI] [PubMed] [Google Scholar]
- 4. Bruijn L.I., Beal M.F., Becher M.W., Schulz J.B., Wong P.C., Price D.L. and Cleveland D.W. (1997) Elevated free nitrotyrosine levels, but not protein-bound nitrotyrosine or hydroxyl radicals, throughout amyotrophic lateral sclerosis (ALS)-like disease implicate tyrosine nitration as an aberrant in vivo property of one familial ALS-linked superoxide dismutase 1 mutant. Proc. Natl. Acad. Sci. U. S. A., 94, 7606–7611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bruijn L.I., Becher M.W., Lee M.K., Anderson K.L., Jenkins N.A., Copeland N.G., Sisodia S.S., Rothstein J.D., Borchelt D.R., Price D.L. et al. (1997) ALS-linked SOD1 mutant G85R mediates damage to astrocytes and promotes rapidly progressive disease with SOD1-containing inclusions. Neuron, 18, 327–338. [DOI] [PubMed] [Google Scholar]
- 6. Wong P.C., Pardo C.A., Borchelt D.R., Lee M.K., Copeland N.G., Jenkins N.A., Sisodia S.S., Cleveland D.W. and Price D.L. (1995) An adverse property of a familial ALS-linked SOD1 mutation causes motor neuron disease characterized by vacuolar degeneration of mitochondria. Neuron, 14, 1105–1116. [DOI] [PubMed] [Google Scholar]
- 7. Carpenter S. (1968) Proximal axonal enlargement in motor neuron disease. Neurology, 18, 841–851. [DOI] [PubMed] [Google Scholar]
- 8. Fukuyama Y. and Hirano Y. (1984) Multiple sclerosis and related disorders. Rinsho Shinkeigaku, 24, 1221–1224. [PubMed] [Google Scholar]
- 9. Rouleau G.A., Clark A.W., Rooke K., Pramatarova A., Krizus A., Suchowersky O., Julien J.P. and Figlewicz D. (1996) SOD1 mutation is associated with accumulation of neurofilaments in amyotrophic lateral sclerosis. Ann. Neurol., 39, 128–131. [DOI] [PubMed] [Google Scholar]
- 10. Nguyen M.D., Larivière R.C. and Julien J.P. (2001) Deregulation of Cdk5 in a mouse model of ALS: toxicity alleviated by perikaryal neurofilament inclusions. Neuron, 30, 135–147. [DOI] [PubMed] [Google Scholar]
- 11. Meier J., Couillard-Després S., Jacomy H., Gravel C. and Julien J.P. (1999) Extra neurofilament NF-L subunits rescue motor neuron disease caused by overexpression of the human NF-H gene in mice. J. Neuropathol. Exp. Neurol., 58, 1099–1110. [PubMed] [Google Scholar]
- 12. Giasson B.I. and Mushynski W.E. (1996) Aberrant stress-induced phosphorylation of perikaryal neurofilaments. J. Biol. Chem., 271, 30404–30409. [DOI] [PubMed] [Google Scholar]
- 13. Veeranna, Amin N.D., Ahn N.G., Jaffe H., Winters C.A., Grant P. and Pant H.C. (1998) Mitogen-activated protein kinases (Erk1,2) phosphorylate Lys-Ser-Pro (KSP) repeats in neurofilament proteins NF-H and NF-M. J. Neurosci., 18, 4008–4021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bajaj N.P. and Miller C.C. (1997) Phosphorylation of neurofilament heavy-chain side-arm fragments by cyclin-dependent kinase-5 and glycogen synthase kinase-3alpha in transfected cells. J. Neurochem., 69, 737–743. [DOI] [PubMed] [Google Scholar]
- 15. Guidato S., Tsai L.H., Woodgett J. and Miller C.C. (1996) Differential cellular phosphorylation of neurofilament heavy side-arms by glycogen synthase kinase-3 and cyclin-dependent kinase-5. J. Neurochem., 66, 1698–1706. [DOI] [PubMed] [Google Scholar]
- 16. Lee M.S., Kwon Y.T., Li M., Peng J., Friedlander R.M. and Tsai L.H. (2000) Neurotoxicity induces cleavage of p35 to p25 by calpain. Nature, 405, 360–364. [DOI] [PubMed] [Google Scholar]
- 17. Cruz J.C. and Tsai L.H. (2004) Cdk5 deregulation in the pathogenesis of Alzheimer's disease. Trends Mol. Med., 10, 452–458. [DOI] [PubMed] [Google Scholar]
- 18. Tsai L.H., Lee M.S. and Cruz J. (2004) Cdk5, a therapeutic target for Alzheimer's disease? Biochim. Biophys. Acta, 1697, 137–142. [DOI] [PubMed] [Google Scholar]
- 19. Patrick G.N., Zukerberg L., Nikolic M., de la Monte S., Dikkes P. and Tsai L.H. (1999) Conversion of p35 to p25 deregulates Cdk5 activity and promotes neurodegeneration. Nature, 402, 615–622. [DOI] [PubMed] [Google Scholar]
- 20. Bajaj N.P. (2000) Cyclin-dependent kinase-5 (CDK5) and amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Other Motor Neuron Disord., 1, 319–327. [DOI] [PubMed] [Google Scholar]
- 21. Shah K. and Lahiri D.K. (2017) A tale of the good and bad: remodeling of the microtubule network in the brain by Cdk5. Mol. Neurobiol., 54, 2255–2268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bajaj N.P., Al-Sarraj S.T., Anderson V., Kibble M., Leigh N. and Miller C.C. (1998) Cyclin-dependent kinase-5 is associated with lipofuscin in motor neurones in amyotrophic lateral sclerosis. Neurosci. Lett., 245, 45–48. [DOI] [PubMed] [Google Scholar]
- 23. Yamaguchi H., Ishiguro K., Uchida T., Takashima A., Lemere C.A. and Imahori K. (1996) Preferential labeling of Alzheimer neurofibrillary tangles with antisera for tau protein kinase (TPK) I/glycogen synthase kinase-3 beta and cyclin-dependent kinase 5, a component of TPK II. Acta Neuropathol., 92, 232–241. [DOI] [PubMed] [Google Scholar]
- 24. Pei J.J., Grundke-Iqbal I., Iqbal K., Bogdanovic N., Winblad B. and Cowburn R.F. (1998) Accumulation of cyclin-dependent kinase 5 (cdk5) in neurons with early stages of Alzheimer's disease neurofibrillary degeneration. Brain Res., 797, 267–277. [DOI] [PubMed] [Google Scholar]
- 25. Brion J.P. and Couck A.M. (1995) Cortical and brainstem-type Lewy bodies are immunoreactive for the cyclin-dependent kinase 5. Am. J. Pathol., 147, 1465–1476. [PMC free article] [PubMed] [Google Scholar]
- 26. Green S.L., Vulliet P.R., Pinter M.J. and Cork L.C. (1998) Alterations in cyclin-dependent protein kinase 5 (CDK5) protein levels, activity and immunocytochemistry in canine motor neuron disease. J. Neuropathol. Exp. Neurol., 57, 1070–1077. [DOI] [PubMed] [Google Scholar]
- 27. Ahlijanian M.K., Barrezueta N.X., Williams R.D., Jakowski A., Kowsz K.P., McCarthy S., Coskran T., Carlo A., Seymour P.A., Burkhardt J.E. et al. (2000) Hyperphosphorylated tau and neurofilament and cytoskeletal disruptions in mice overexpressing human p25, an activator of cdk5. Proc. Natl. Acad. Sci. U. S. A., 97, 2910–2915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Binukumar B.K. and Pant H.C. (2016) TFP5/TP5 peptide provides neuroprotection in the MPTP model of Parkinson's disease. Neural Regen. Res., 11, 698–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Binukumar B.K., Pelech S.L., Sutter C., Shukla V., Amin N.D., Grant P., Bhaskar M., Skuntz S., Steiner J. and Pant H.C. (2016) Profiling of p5, a 24 amino acid inhibitory peptide derived from the CDK5 activator, p35 CDKR1 against 70 protein kinases. J. Alzheimers Dis., 54, 525–533. [DOI] [PubMed] [Google Scholar]
- 30. Binukumar B.K., Shukla V., Amin N.D., Grant P., Bhaskar M., Skuntz S., Steiner J. and Pant H.C. (2015) Peptide TFP5/TP5 derived from Cdk5 activator P35 provides neuroprotection in the MPTP model of Parkinson's disease. Mol. Biol. Cell, 26, 4478–4491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Glicksman M.A., Cuny G.D., Liu M., Dobson B., Auerbach K., Stein R.L. and Kosik K.S. (2007) New approaches to the discovery of cdk5 inhibitors. Curr. Alzheimer Res., 4, 547–549. [DOI] [PubMed] [Google Scholar]
- 32. Zheng Y.L., Li B.S., Amin N.D., Albers W. and Pant H.C. (2002) A peptide derived from cyclin-dependent kinase activator (p35) specifically inhibits Cdk5 activity and phosphorylation of tau protein in transfected cells. Eur. J. Biochem., 269, 4427–4434. [DOI] [PubMed] [Google Scholar]
- 33. Zheng Y.L., Amin N.D., Hu Y.F., Rudrabhatla P., Shukla V., Kanungo J., Kesavapany S., Grant P., Albers W. and Pant H.C. (2010) A 24-residue peptide (p5), derived from p35, the Cdk5 neuronal activator, specifically inhibits Cdk5-p25 hyperactivity and tau hyperphosphorylation. J. Biol. Chem., 285, 34202–34212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zheng Y.L., Kesavapany S., Gravell M., Hamilton R.S., Schubert M., Amin N., Albers W., Grant P. and Pant H.C. (2005) A Cdk5 inhibitory peptide reduces tau hyperphosphorylation and apoptosis in neurons. EMBO J., 24, 209–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Binukumar B.K., Zheng Y.L., Shukla V., Amin N.D., Grant P. and Pant H.C. (2014) TFP5, a peptide derived from p35, a Cdk5 neuronal activator, rescues cortical neurons from glucose toxicity. J. Alzheimers Dis., 39, 899–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Amin N.D., Zheng Y., Bk B., Shukla V., Skuntz S., Grant P., Steiner J., Bhaskar M. and Pant H.C. (2016) The interaction of Munc 18 (p67) with the p10 domain of p35 protects in vivo Cdk5/p35 activity from inhibition by TFP5, a peptide derived from p35. Mol. Biol. Cell, 27, 3221–3232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sundaram J.R., Poore C.P., Sulaimee N.H., Pareek T., Asad A.B., Rajkumar R., Cheong W.F., Wenk M.R., Dawe G.S., Chuang K.H. et al. (2013) Specific inhibition of p25/Cdk5 activity by the Cdk5 inhibitory peptide reduces neurodegeneration in vivo. J. Neurosci., 33, 334–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Amin N.D., Albers W. and Pant H.C. (2002) Cyclin-dependent kinase 5 (cdk5) activation requires interaction with three domains of p35. J. Neurosci. Res., 67, 354–362. [DOI] [PubMed] [Google Scholar]
- 39. Shukla V., Seo J., Binukumar B.K., Amin N.D., Reddy P., Grant P., Kuntz S., Kesavapany S., Steiner J., Mishra S.K. et al. (2017) TFP5, a peptide inhibitor of aberrant and hyperactive Cdk5/p25, attenuates pathological phenotypes and restores synaptic function in CK-p25Tg mice. J. Alzheimers Dis., 56, 335–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ferraiuolo L., Kirby J., Grierson A.J., Sendtner M. and Shaw P.J. (2011) Molecular pathways of motor neuron injury in amyotrophic lateral sclerosis. Nat. Rev. Neurol., 7, 616–630. [DOI] [PubMed] [Google Scholar]
- 41. Jaarsma D., Teuling E., Haasdijk E.D., De Zeeuw C.I. and Hoogenraad C.C. (2008) Neuron-specific expression of mutant superoxide dismutase is sufficient to induce amyotrophic lateral sclerosis in transgenic mice. J. Neurosci., 28, 2075–2088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Boillée S., Yamanaka K., Lobsiger C.S., Copeland N.G., Jenkins N.A., Kassiotis G., Kollias G. and Cleveland D.W. (2006) Onset and progression in inherited ALS determined by motor neurons and microglia. Science, 312, 1389–1392. [DOI] [PubMed] [Google Scholar]
- 43. Rao M.V., Campbell J., Palaniappan A., Kumar A. and Nixon R.A. (2016) Calpastatin inhibits motor neuron death and increases survival of hSOD1(G93A) mice. J. Neurochem., 137, 253–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Liu K.X., Edwards B., Lee S., Finelli M.J., Davies B., Davies K.E. and Oliver P.L. (2015) Neuron-specific antioxidant OXR1 extends survival of a mouse model of amyotrophic lateral sclerosis. Brain, 138, 1167–1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ilieva H., Polymenidou M. and Cleveland D.W. (2009) Non-cell autonomous toxicity in neurodegenerative disorders: ALS and beyond. J. Cell Biol., 187, 761–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Bowerman M., Vincent T., Scamps F., Perrin F.E., Camu W. and Raoul C. (2013) Neuroimmunity dynamics and the development of therapeutic strategies for amyotrophic lateral sclerosis. Front. Cell. Neurosci., 7,–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Lincecum J.M., Vieira F.G., Wang M.Z., Thompson K., De Zutter G.S., Kidd J., Moreno A., Sanchez R., Carrion I.J., Levine B.A. et al. (2010) From transcriptome analysis to therapeutic anti-CD40L treatment in the SOD1 model of amyotrophic lateral sclerosis. Nat. Genet., 42, 392–399. [DOI] [PubMed] [Google Scholar]
- 48. Frakes A.E., Ferraiuolo L., Haidet-Phillips A.M., Schmelzer L., Braun L., Miranda C.J., Ladner K.J., Bevan A.K., Foust K.D., Godbout J.P. et al. (2014) Microglia induce motor neuron death via the classical NF-κB pathway in amyotrophic lateral sclerosis. Neuron, 81, 1009–1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Papadeas S.T., Kraig S.E., O'Banion C., Lepore A.C. and Maragakis N.J. (2011) Astrocytes carrying the superoxide dismutase 1 (SOD1G93A) mutation induce wild-type motor neuron degeneration in vivo. Proc. Natl. Acad. Sci. U. S. A., 108, 17803–17808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Seibenhener M.L. and Wooten M.C. (2015) Use of the open field maze to measure locomotor and anxiety-like behavior in mice. J. Vis. Exp., 96, e52434. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.