

Summary
Xanthomonas campestris pv. campestris (Xcc), the causal agent of black rot in crucifers, produces a membrane‐bound yellow pigment called xanthomonadin to protect against photobiological and peroxidative damage, and uses a quorum‐sensing mechanism mediated by the diffusible signal factor (DSF) family signals to regulate virulence factors production. The Xcc gene XCC4003, annotated as Xcc fabG3, is located in the pig cluster, which may be responsible for xanthomonadin synthesis. We report that fabG3 expression restored the growth of the Escherichia coli fabG temperature‐sensitive mutant CL104 under non‐permissive conditions. In vitro assays demonstrated that FabG3 catalyses the reduction of 3‐oxoacyl‐acyl carrier protein (ACP) intermediates in fatty acid synthetic reactions, although FabG3 had a lower activity than FabG1. Moreover, the fabG3 deletion did not affect growth or fatty acid composition. These results indicate that Xcc fabG3 encodes a 3‐oxoacyl‐ACP reductase, but is not essential for growth or fatty acid synthesis. However, the Xcc fabG3 knock‐out mutant abolished xanthomonadin production, which could be only restored by wild‐type fabG3, but not by other 3‐oxoacyl‐ACP reductase‐encoding genes, indicating that Xcc FabG3 is specifically involved in xanthomonadin biosynthesis. Additionally, our study also shows that the Xcc fabG3‐disrupted mutant affects Xcc virulence in host plants.
Keywords: 3‐ketoacyl‐acyl carrier protein reductase, xanthomonadin, Xanthomonas campestris pv. campestris
Introduction
The Xanthomonas genus is a ubiquitous group of plant‐associated bacterial pathogens that infect at least 124 monocotyledonous and 268 dicotyledonous plant species, many of which are economically important crops or plants (Bonas et al., 2000; Chan and Goodwin, 1999). A characteristic feature of the genus Xanthomonas is the production of yellow membrane‐bound pigments called xanthomonadins, which are mixtures of water‐insoluble, unusually brominated, aryl‐polyene esters (Starr and Stephens, 1964). Xanthomonadins are useful chemotaxonomic and diagnostic indicators of Xanthomonas (Starr et al., 1977). Moreover, xanthomonadins have roles in protecting against photobiological and peroxidative damage induced by host defence mechanisms and are required for epiphytic survival and host infection to maintain ecological fitness and virulence (Poplawsky et al., 2000; Rajagopal et al., 1997).
Xanthomonas campestris pv. campestris (Xcc) is the causal agent of black rot in crucifers (He and Zhang, 2008) and is also the producer of xanthan gum, a neutral and water‐soluble polysaccharide that is widely used in a variety of products including medicine, foods and cosmetics. Thus, Xcc has great commercial and biotechnological application values (Kumar et al., 2017). However, during the industrial production of xanthan gum, large amounts of ethanol are required to remove xanthomonadin impurities, so it is attractive to construct xanthomonadin‐deficient strains by the deletion of genes in pigment synthesis (Dai et al., 2018). In Xcc, the gene cluster for xanthomonadin biosynthesis (pig) contains seven transcriptional units, designated pigA to G, and consists of 22 open reading frames (ORF), encoding acyltransferase, ketoreductase, ketosynthase and dehydratase, which constitute part of a novel type II polyketide synthase pathway (He et al., 2011; Poplawsky and Chun, 1997). Xcc utilizes 3‐hydroxybenzoic acid (3‐HBA), a diffusible factor with regulatory function, as the precursor for xanthomonadin synthesis, and 3‐HBA is synthesized by a unique bifunctional chorismatase XanB2 (He et al., 2011; Wang et al., 2015; Zhou et al., 2013a, b). Recently, Cao et al. (2018) reported only 11 genes are responsible for xanthomonadin biosynthesis, and xanC encodes an acyl carrier protein (ACP) while xanA2 encodes the 3‐HBA:ACP ligase to initiate xanthomonadin biosynthesis. However, further studies are required to define the detailed mechanisms of xanthomonadin synthesis.
Xcc uses quorum‐sensing (QS) mechanisms mediated by molecules of the diffusible signal factor (DSF) family signals to regulate the expression of factors that contribute to virulence (He and Zhang, 2008; Zhou et al., 2015). At least four DSF family signals have been identified in Xcc: DSF (cis‐11‐methyl‐2‐dodecenoic acid [11‐Me‐C12:Δ2]), BDSF (cis‐2‐dodecenoic acid [C12:Δ2]), CDSF (cis‐11‐methyldodeca‐2,5‐dienoic acid [11‐Me‐C12: ∆ 2, 5]) and IDSF (cis‐10‐methyl‐2‐dodecenoic acid [10‐Me‐C12:Δ2]) (Deng et al., 2015, 2016; Zhou et al., 2015). RpfF, with both acyl‐ACP thioesterase and dehydratase activities, utilizes 3‐hydroxyacyl‐ACPs as substrates and is the key enzyme for DSF family signal biosynthesis. DSF family signals are involved in QS mechanisms that regulate pathogenicity by modulating the production of different virulence factors, such as extracellular enzymes, extracellular polysaccharides (EPS) and interspecies signalling, for example to influence the expression of a type III secretion system in Pseudomonas aeruginosa (Bi et al., 2012; Zhou et al., 2017).
Bacteria utilize a disassociated fatty acid synthase (FAS II) for de novo synthesis of fatty acids, which supply varieties of intermediates to other products, including lipids, vitamins (lipoic acid, biotin) and the 3‐hydroxyacyl‐ACPs for DSF family signals synthesis. The Xcc genome contains all of the genes required for FAS II, in which the key enzyme 3‐oxoacyl‐ACP reductase (OAR, FabG) is directly responsible for the synthesis of 3‐hydroxyacyl‐ACPs in the FAS II elongation cycle (Qian et al., 2005; Yu et al., 2016; Zhou et al., 2015). Four genes, fabG1 (XCC1018), fabG2 (XCC0416), fabG3 (XCC4003) and fabG4 (XCC0384), encode putative OARs in the Xcc genome (Qian et al., 2005). FabG1 plays the major role in catalysing the reduction of 3‐oxoacyl‐ACPs in fatty acid synthesis. FabG2 is a novel long‐chain specific OAR that catalyses the reduction of long‐chain (≥C8) 3‐oxoacyl‐ACPs but shows weak activity against shorter‐chain (C4, C6) substrates (Hu et al., 2018). The overexpression of fabG1 or fabG2 in Xcc led to a significant increase in the production of DSF family signals, while fabG2 deletion resulted in reduced production of DSF family signals, which indicated that Xcc modulates the activity of OAR to control DSF family signal production (Hu et al., 2018; Zhou et al., 2015). In contrast, FabG3 and FabG4 appear to be involved in xanthomonadin and biotin synthesis, respectively, but their functions will necessitate experimental evidence.
In the present report, we characterized the functions of FabG3 in the biosynthesis of xanthomonadins and DSF family signals using genetic complementation, biochemical analyses and gene deletion techniques. We found that FabG3 possesses OAR activity in vivo and in vitro, but fabG3 is not essential for growth or fatty acid synthesis. The fabG3 deletion strain did not produce xanthomonadin, reduced the amounts of DSF family signals sharply and attenuated virulence to host plants.
Results
Xcc fabG3 is not essential for growth, but is required for xanthomonadin biosynthesis
To identify the physiological functions of fabG3 in cell growth and fatty acid synthesis, we attempted to disrupt fabG3 with an in‐frame deletion in the wild‐type strain Xc1. First, the pK18mobsacB‐borne suicide plasmid pYYH‐4 used to delete fabG3 was constructed, and then it was introduced into the genome of Xcc wild‐type strain by conjugal transfer from Escherichia coli S17‐1. After negative selection mediated by Bacillus subtilis sacB, the fabG3 deletion mutant strain YH1(ΔfabG3) was obtained and confirmed by colony PCR using the primers listed in Table S1 and by sequencing the allelic gene in the YH1 strain (Fig. S1). The expression of the neighbouring genes XCC4002 and XCC4004 was confirmed using RT‐qPCR. The results showed expression of both genes was not affected in the mutant (Fig. S2A). Next, the growth of YH1 strain was tested in NYG medium. YH1 strain grew as well as the wild‐type strain (Fig. S2B) under these conditions, suggesting that fabG3 is not an essential gene for Xcc.
Xcc produces a mixture of organic solvent‐extractable pigments, xanthomonadins, which are incorporated into the cell membrane, rendering the cells yellow in colour (Poplawsky and Chun, 1997). Xcc fabG3 is located in the pig gene cluster, which is responsible for xanthomonadin synthesis. To determine whether fabG3 functions in xanthomonadin synthesis, we compared the colony colour of mutant strain YH1 with wild‐type strain on NYG plates. The colour of the wild‐type was yellow, while the mutant strain YH1 appeared colourless on agar plates, indicating that Xcc YH1 strain did not produce xanthomonadins. However, the complemented strain Xcc YH2, which carried the wild‐type fabG3 in plasmid pYYH‐5, turned yellow (Fig. 1A). The absorption spectra of the methanolic extracts were also recorded. Both the wild‐type strain and Xcc YH2 showed peaks at 445 nm, while Xcc YH1 did not show a peak, confirming that fabG3 mutant has little or no xanthomonadins (Fig. S3). The residual absorption seen at 445 nm for Xcc YH1 could be the result of the presence of intermediates in xanthomonadin synthesis (Rajagopal et al., 1997).
Figure 1.
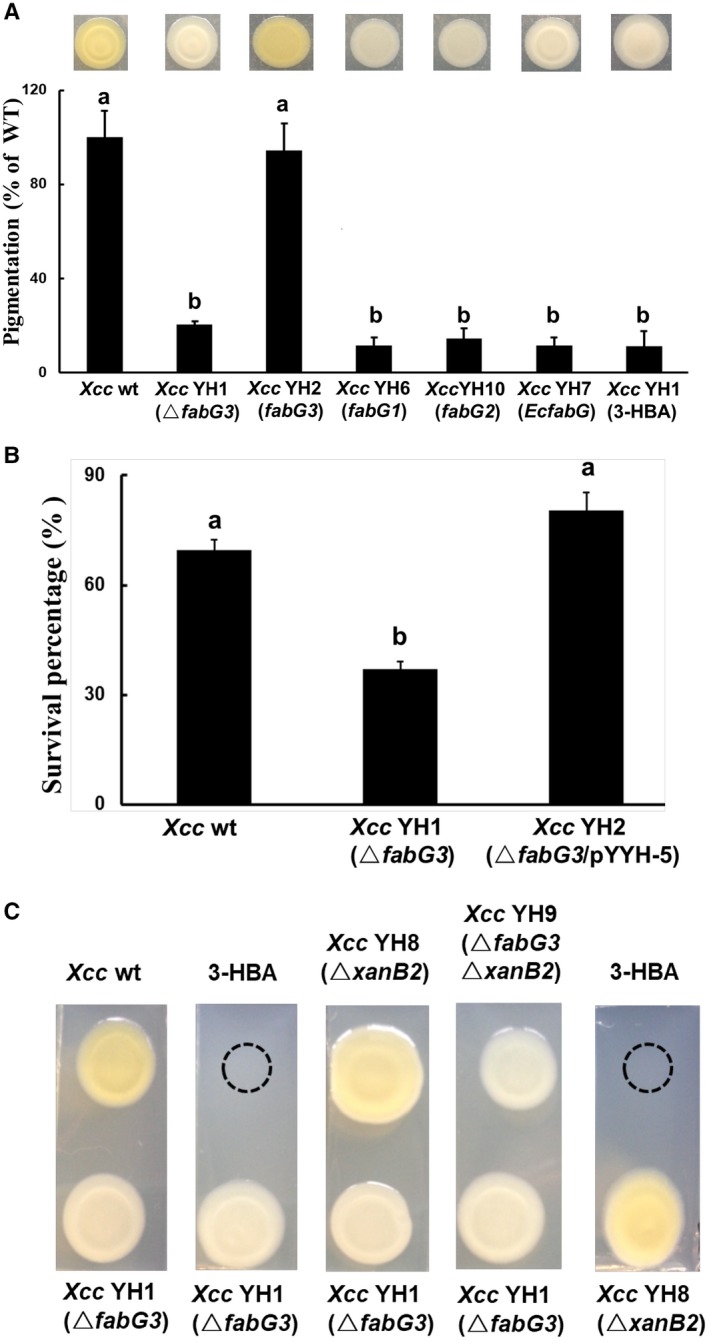
The reduced xanthomonadin production and H2O2 resistance of the Xcc YH1 (ΔfabG3) strain and restoration by Xcc fabG3. (A) Quantitative analysis of xanthomonadin production in Xanthomonas campestris pv. campestris (Xcc) strains. wt, wild‐type; fabG3, complemented with plasmid pSRK‐Km harbouring the gene Xcc fabG3; Xcc fabG1, complemented with plasmid pSRK‐Km harbouring the gene Xcc fabG1; Xcc fabG2, complemented with plasmid pSRK‐Km harbouring the gene Xcc fabG2; EcfabG, complemented with plasmid pSRK‐Km harbouring the gene Escherichia coli fabG; 3‐HBA, 3‐hydroxybenzoic acid. (B) Xcc cells with an optical density of 1.0 when measured at 600 nm were collected for H2O2 treatment. After 30 min of H2O2 treatment, the CFU value of each strain was determined on NYG plates. Values shown are the means ± standard deviations from three independent experiments. Different letters indicate significant difference between treatments based on the least significant difference at P = 0.05. (C) Diffusion plate assay showing the restoration of xanthomonadin production in ΔxanB2 following exposure to Xcc YH1(ΔfabG3) or 3‐HBA, and showing the failure to restore xanthomonadin production in Xcc YH1(ΔfabG3) following exposure to wide‐type, ΔxanB2 or 3‐HBA.
Previously, xanthomonadins were shown to protect Xanthomonas from peroxidation damage and, if xanthomonadins are reduced, Xanthomonas is more sensitive to oxidative stress (Zhou et al., 2013a). To assay oxidative stress resistance, hydrogen peroxide (H2O2) was added to cultures of Xcc YH1 strain at a final concentration of 880 μm and the survival percentage after H2O2 treatment was determined. The wild‐type strain maintained a c. 70% survival rate, while the fabG3 deletion strain Xcc YH1 decreased the bacterial resistance to oxidative stress, as indicated by the 37% survival rate after H2O2 treatment. The expression of fabG3 in Xcc YH1 restored H2O2 resistance to wild‐type levels (Fig. 1B). Thus, fabG3 is required for xanthomonadin synthesis, and the fabG3 deletion resulted in Xcc being sensitive to H2O2.
We also supplemented 3‐hydroxylbenzoic acid (3‐HBA), the precursor for xanthomonadin biosynthesis, and determined if it could restore Xcc YH1, as indicated by the colony turning yellow. 3‐HBA failed to turn the Xcc YH1 colonies yellow, which suggested that the failure of Xcc YH1 was not due to a lack of precursor (Fig. 1A). Because Xcc XanB2 catalyses 3‐HBA formation and plays a vital role in xanthomonadin biosynthesis (He et al., 2011), we further constructed the xanB2 deletion strain Xcc YH8 and the xanB2fabG3 double‐deletion strain Xcc YH9 using the homologous recombination method. As previously reported, Xcc YH8 (ΔxanB2) failed to produce xanthomonadin and colonies were colourless on agar plates, but addition of 3‐HBA in the vicinity of the colony turned Xcc YH8 colonies yellow (He et al., 2011). Xcc YH8 (ΔxanB2) also restored xanthomonadin production when exposed to Xcc YH1. However, Xcc YH9 (ΔxanB2ΔfabG3) could not be restored to the wild‐type phenotype by exposure to the Xcc YH1 strain (Fig. 1C). The diffusion plate assay also showed that Xcc YH1 did not turn yellow when grown in the vicinity of wild‐type or Xcc YH8 (ΔxanB2) (Fig. 1C). Thus, Xcc YH1 produced 3‐HBA, and Xcc FabG3 was involved downstream of 3‐HBA in xanthomonadin biosynthesis.
Xcc fabG3 complemented the growth of the E. coli fabG temperature‐sensitive mutant under non‐permissive temperature
Although Xcc fabG3 is located in the pig gene cluster and involved in xanthomonadin synthesis, protein sequence alignments showed that Xcc FabG3 is 42% identical with E. coli FabG (Lai and Cronan, 2004), and 40% identical with Xcc FabG1 (Fig. 2). Xcc FabG3 contains the short‐chain dehydrogenase/reductase (SDR) family’s catalytically active triad (Ser, Tyr and Lys) and the N‐terminal cofactor‐binding sequence (Gly motif [GlyXXGlyXXGly]) defined by the X‐ray crystal structure of E. coli FabG (Mao et al., 2016). Two E. coli FabG residues, Arg‐129 and Arg‐172, which play critical roles in facilitating the binding of the ACP moiety of the substrate, were also conserved in Xcc FabG3 (Feng et al., 2015) (Fig. 2). Thus, we hypothesized that Xcc fabG3 encodes a functional OAR. To test this hypothesis, Xcc fabG3 was inserted into the arabinose‐inducible vector pBAD24M to produce the expression construct pYYH‐2, which was further transferred into the E. coli fabG temperature‐sensitive strain CL104 (Lai and Cronan, 2004). The resulting transformant was tested for growth at a non‐permissive temperature (42 °C). The strain CL104 carrying pYYH‐2 grew well at 42 °C on rich broth (RB) plates in the presence of arabinose (Fig. 3A). However, in RB liquid medium with an arabinose inducer, the CL104 strain carrying pYYH‐2 showed a reduced growth rate compared with that of CL104 carrying Xcc fabG1 or E. coli fabG (Fig. 3B). These results preliminarily indicated that Xcc fabG3 could complement the E. coli fabG (ts) strain CL104, but Xcc FabG3 appeared to be less active than Xcc FabG1 or E. coli FabG.
Figure 2.

Alignment of Xanthomonas campestris pv. campestris (Xcc) FabG3 with Escherichia coli FabG (EcFabG) and Xcc FabG1. The two arginine residues thought to bind the acyl carrier protein (ACP) moiety are indicated by arrows and the catalytic triad residues are indicated by asterisks. The alignment was performed with ClustalW based on identical residues.
Figure 3.
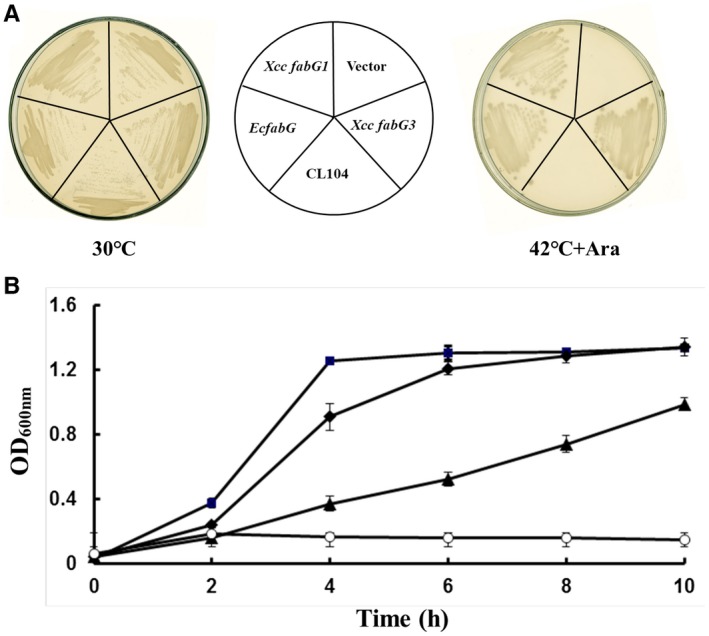
Growth of Escherichia coli fabG (ts) mutant CL104 transformants containing plasmids carrying Xanthomonas campestris pv. campestris (Xcc) fabG3, Xcc fabG1 and E. coli fabG. (A) E. coli CL104 derivatives, carrying the pBAD24M‐derived plasmids pHWG (pBAD‐EcfabG), pYYH‐1 (pBAD‐Xcc fabG1) and pYYH‐2 (pBAD‐Xcc fabG3), were grown at 30 °C or 42 °C on LB plates in the presence of 0.01% arabinose. (B) Transformants of E. coli fabG (ts) mutant CL104 with pBAD24M‐derived plasmids grown in LB liquid containing 0.01% arabinose. Squares, CL104/pHWG (pBAD‐EcfabG); diamonds, CL104/pYYH‐1 (pBAD‐XccfabG1); triangles, CL104/pYYH‐2 (pBAD‐Xcc fabG3); empty circles, CL104/pBAD24M. Data are means ± standard deviations of three independent assays.
Xcc FabG3 possesses OAR activity in vitro
To determine whether Xcc FabG3 possesses OAR activity in vitro, the N‐terminal 6 × His‐tagged Xcc FabG3 recombinant protein was expressed in E. coli BL21 (DE3) and purified by nickel chelate chromatography (data not shown). Other bacterial fatty acid biosynthetic proteins were also purified as described in the 'Experimental procedures' section.
First, the initiation step of the fatty acid synthesis reaction was reconstituted. Escherichia coli FabD, FabH, FabZ, FabI and OAR (E. coli FabG or Xcc FabG3) were added sequentially. After incubation for 1 h, the reaction mixture was analysed by conformationally sensitive gel electrophoresis. In the absence of OAR, only holo‐ACP was seen (Fig. 4A, Lane 4). The addition of Xcc FabG3 to the reaction mixture, like the reaction mixture containing E. coli FabG, resulted in the production of butyryl‐ACP (Mao et al., 2016) (Fig. 4A, lanes 2 and 3). Next, the enzymatic activity of Xcc FabG3 in the reduction of long‐chain 3‐ketoacyl‐ACP substrates was also determined. These reactions were reconstituted by adding malonyl‐ACP, octanoyl‐ACP, Ralstonia solanacearum RSp0194, OAR (E. coli FabG or Xcc FabG3), E. coli FabZ and FabI, and cofactors NADPH and NADH. In these reactions R. solanacearum RSp0194 condensed octanoyl‐ACP with malonyl‐ACP to produce 3‐oxodecanoyl‐ACP. Then, if Xcc FabG3 possessed OAR activity, 3‐oxodecanoyl‐ACP was reduced to 3‐hydroxyldecanoyl‐ACP. After dehydration catalysed by E. coli FabZ and reduction by FabI, 3‐hydroxyldecanoyl‐ACP was converted to decanoyl‐ACP. Both reaction mixtures, containing either E. coli FabG or Xcc FabG3, formed decanoyl‐ACP (Fig. 4B, lanes 2 and 3). These data further confirmed that, like E. coli FabG, Xcc FabG3 could reduce 3‐oxoacyl‐ACP to 3‐hydroxylacyl‐ACP in vitro and that Xcc fabG3 encoded a functional OAR.
Figure 4.
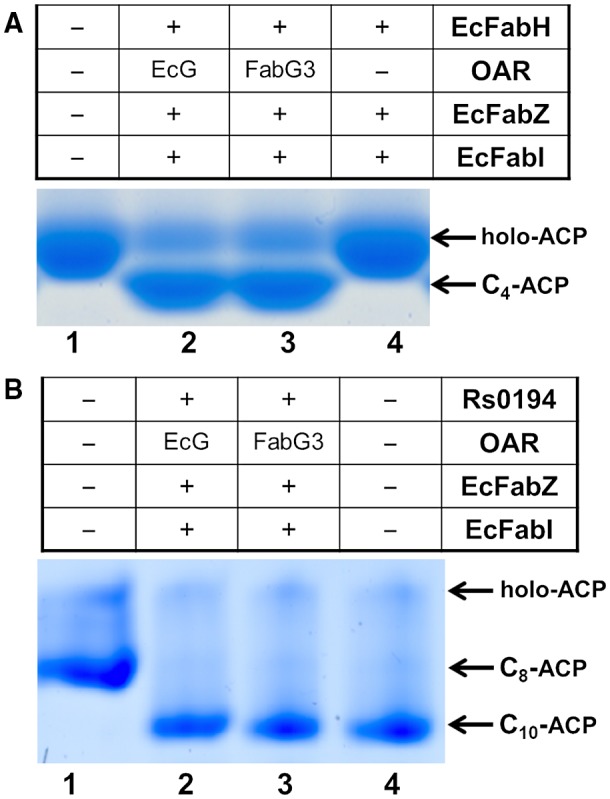
Assay of the in vitro fatty acid synthesis abilities of Xanthomonas campestris pv. campestris (Xcc) FabG3. (A) Function of Xcc FabG3 in the initial cycle of fatty acid synthesis. Lane 1, holo‐ACP; lane 2, reaction product containing E. coli FabG; lane 3, reaction product containing Xcc FabG3; lane 4, reaction product without 3‐ketoacyl‐ACP reductase. (B) Functions of Xcc XabG in the elongation cycle of fatty acid synthesis. Lane 1, product of octanoyl‐ACP synthesized by Vibrio harveyi AasS; lane 2, reaction product catalysed by E. coli FabG; lane 3, reaction product catalysed by Xcc FabG3; lane 4, product of decanoyl‐ACP synthesized by V. harveyi AasS. EcG, E. coli FabG; OAR, 3‐oxoacyl‐ACP reductase; EcFabH, E. coli 3‐ketoacyl‐ACP synthase III; EcFabZ, E. coli 3‐hydroxyacyl‐ACP dehydrase; EcFabI, E. coli enoyl‐ACP reductase.
The OAR activities of Xcc FabG3 were also assayed using various chains of 3‐oxoacyl‐ACPs as substrates, by monitoring the decrease in NADPH absorbance at 340 nm. Xcc FabG3 exhibited reductive activities with wide range of substrate specificities, from 3‐oxobutyryl ACP (C4) to 3‐oxododecanoyl ACP (C12). However, the reductive activity of Xcc FabG3 was much less than that of Xcc FabG1, from c. 10% for 3‐oxododecanoyl ACP (C12) to c. 50% for 3‐oxodecanoyl ACP (C10) (Table 1). These data explain why the E. coli CL104 strain carrying pYYH‐1 (with Xcc fabG3) grew much more slowly than the CL104 strain carrying pYYH‐1 (with Xcc fabG1) (Fig. 3B). We also compared the abilities of Xcc FabG1 and Xcc FabG3 to reduce the model substrate acetoacetyl‐CoA. Xcc FabG3 failed to reduce acetoacetyl‐CoA, while Xcc FabG1 reduced acetoacetyl‐CoA with a maximal rate of 610.0 ± 58.0 μmol/min.
Table 1.
Xanthomonas campestris pv. campestris (Xcc) FabG1 and Xcc FabG3 activities with various 3‐oxoacyl‐APCs*
| Substrate | Xcc FabG1 (μmoles/mg/min) | Xcc FabG3 (μmoles/mg/min) |
|---|---|---|
| 3‐oxobutyryl ACP | 14.69 ± 0.75 c | 4.65 ± 0.44 c |
| 3‐oxohexanoyl ACP | 24.15 ± 2.06 a | 8.10 ± 0.89 a |
| 3‐oxoctanoyl ACP | 22.31 ± 1.91 a | 7.76 ± 0.97 b |
| 3‐oxodecanoyl ACP | 17.94 ± 1.68 b | 8.53 ± 1.06 a |
| 3‐oxododecanoyl ACP | 17.21 ± 2.10 b | 1.74 ± 0.11 d |
The values are the means ± standard deviations of three independent experiments. The statistical analyses were performed in Microsoft Excel with P values between each pairwise comparison calculated by two‐tailed Student’s t‐tests. Significant differences are indicated by different letters (P < 0.05).
Xcc FabG3 is specific for xanthomonadin biosynthesis
A deletion of fabG3 caused Xcc to abolish the production of xanthomonadins. Xcc FabG3 possessed OAR activity, even though this activity was much lower than that of Xcc FabG1. Thus, we hypothesized that other OAR‐encoding genes might restore the fabG3 deletion mutant strain to produce xanthomonadins. To confirm this hypothesis, Xcc fabG1, Xcc fabG2 and E.coli fabG‐encoding plasmids were independently introduced into YH1 (ΔfabG3) strain, and the production of xanthomonadins was determined. Unfortunately, the OAR‐encoding genes did not restore xanthomonadin production in YH1 (ΔfabG3), suggesting that Xcc FabG3 plays a unique role in xanthomonadin biosynthesis and that other OAR enzymes might not functionally replace Xcc FabG3 in xanthomonadin biosynthesis (Fig. 1A). For further confirmation, the fatty acid composition of strain Xcc YH1 grown in NYG medium was determined by gas chromatography‐mass spectrometry (GC‐MS) (Table S2). The fatty acid composition of Xcc YH1 was similar to that of wild‐type strain, which suggested that Xcc FabG3 might not be involved in fatty acid synthesis and provided evidence to support Xcc FabG3 being specific for xanthomonadin biosynthesis.
Xcc fabG3 deletion mutant has attenuated virulence
To investigate the role of fabG3 in the virulence regulation of Xcc, the pathogenesis of fabG3 deletion mutant Xcc YH1 was determined on plants. A leaf clipping virulence assay using Chinese radish was conducted (Dow et al., 2003). The average lesion length caused by the wild‐type strain on a leaf of Chinese radish was 17.6 mm 2 weeks after inoculation (Fig. 5). The Xcc fabG3 deletion strain resulted in a significantly reduced average lesion length (9.5 mm), while the average lesion length of the fabG3‐complemented strain (14.7 mm) was not significantly different from that of the wild‐type (Fig. 5). We also determined the growth of Xcc strains in fully mature Chinese cabbage (Wongbok) extracts, and the fabG3 deletion mutant grew as well as the wild‐type (data not shown). These data showed that Xcc fabG3 contributed to virulence.
Figure 5.

The effects of the Xanthomonas campestris pv. campestris (Xcc) fabG3 deletion on the virulence of Xcc. (A) Pathogenicity test on Chinese radish with the Xcc wild‐type (wt) strain, Xcc fabG3‐deletion mutant strain Xcc YH1 and complementary strain Xcc YH2. (B) Test of the virulence of the Xcc strains by measuring lesion length after introducing bacteria into the vascular system of Chinese radish by leaf clipping. Values are expressed as the means and standard deviations of triplicate measurements, each comprising ten leaves. Different letters indicate significant differences between treatments (P = 0.05).
We also evaluated several pathogenicity‐related virulence factors produced by Xcc strains. The activities of extracellular enzymes, including cellulase, amylase and protease, were first tested. The production of the three extracellular enzymes was not statistically different between the fabG3 deletion mutant and the wild‐type (Fig. S4A). Next, extracellular polysaccharide (EPS) production by Xcc strains was determined. The amounts of EPS produced in wild‐type, Xcc YH1 (ΔfabG3) and complemented strain Xcc YH2 were not statistically different.
Xcc FabG3 is involved in DSF biosynthesis
Thus far we have shown that FabG3 is an OAR and the deletion of fabG3 causes Xcc to have a reduced virulence on host plants. To determine whether Xcc fabG3 affected the synthesis of DSF family signals, we analysed their production in the fabG3 deletion mutant strain. We tested the production of DSF family signals in Xcc YH1 using a bioassay in which Xcc FE58 (Wang et al., 2004) was the reporter strain. The blue halo that formed around Xcc YH1 was smaller than that around the wild‐type strain, indicating that YH1 decreased the production of DSF family signals (Fig. 6A). Furthermore, we extracted and purified the DSF family signals from the supernatants, and analysed them quantitatively using liquid chromatography‐mass spectrometry (LC‐MS). The results proved that BDSF and DSF production in the fabG3 mutant Xcc YH1 decreased to 46.9% and 35.3%, respectively, and the complemented strain Xcc YH2 recovered the DSF family signals production to the wild‐type level (Fig. 6B).
Figure 6.
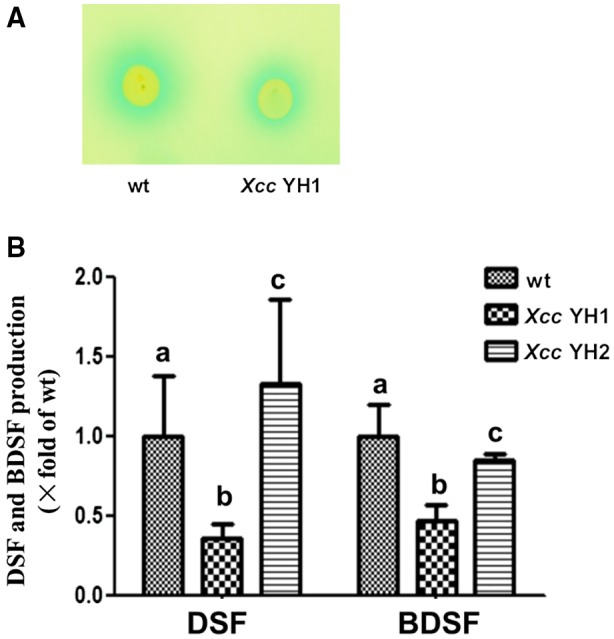
The effects of Xanthomonas campestris pv. campestris (Xcc) fabG3 deletion on the production of DSF family signals. (A) DSF family signal bioassay of Xcc strains. The formation of a blue halo owing to hydrolysis of 5‐bromo‐4‐chloro‐3‐indolyl‐β‐d‐glucuronic acid around the site of inoculation indicates the presence of DSF‐like activity. (B) DSF family signals produced by Xcc fabG3 deletion and complemented strains. Supernatants of 20 mL of Xcc strains grown in NA medium for 24 h were collected, and DSF family signals were detected with the liquid chromatography‐mass spectrometry (LC‐MS) method. In strain Xcc YH1 the Xcc fabG3 gene was deleted in the wild‐type strain; strain Xcc YH2 has Xcc fabG3 being complemented by the plasmid pSRK‐Km in Xcc YH2. The relative amounts of signal molecules were calculated on the basis of their peak areas. The data shown are the means of the results of three repeats and error bars indicate standard deviations. Different letters indicate significant differences between treatments based on the least significant difference at P = 0.05.
Because rpfC‐deletion strain XC1 (ΔrpfC) produced greater concentrations of DSF family signals (He et al., 2006), we further deleted fabG3 in XC1 (ΔrpfC), producing rpfC and fabG3 double deletion mutant Xcc YH3. The complemented strain Xcc YH4 harbouring plasmid pYYH‐5 (carrying fabG3) was also obtained. The DSF family signals in these Xcc strains were extracted and separated using high‐performance liquid chromatography (HPLC) (Fig. S5A). After incubation in NA medium for 24 h, DSF, BDSF and IDSF signal productions in Xcc YH3 (ΔfabG3) decreased sharply to 36.5%, 19.0% and 12.5%, respectively, compared with those produced by the parent strain ΔrpfC. The complemented strain even produced greater amounts of DSF family signals compared with those observed in ΔrpfC (Fig. S5B). Thus, Xcc FabG3 contributed to DSF family signals production. Moreover, to investigate whether expression of Xcc fabG3 in the parent strain ΔrpfC increased the production of DSF family signals, we introduced fabG3‐encoding plasmid pYYH‐5 into ΔrpfC strain and the production of DSF family signals was determined. The results confirmed that fabG3 overexpression led to the significantly increased production of DSF family signals in ΔrpfC (Fig. S5B). Because FabG3 showed OAR activity, this finding was consistent with previous studies in which the overexpression of two other OAR‐encoding genes (fabG1 and fabG2) in Xcc led to a significant increase in DSF family signals production (Hu et al., 2018; Zhou et al., 2015). Thus, it may be that the OAR activity in Xcc cells, rather than the presence of fabG3, is important for DSF family signals synthesis. We further examined the OAR activity in cell‐free extracts of Xcc strains using 3‐oxoacyl‐ACP as a substrate and confirmed the above conclusion. The activity of Xcc YH3 (ΔfabG3) (0.07 ± 0.02 μmol/mg per min) was only half the total OAR activity of ΔrpfC strain (0.15 ± 0.04 μmol/mg per min). The OAR activity in the complemented strain Xcc YH4 and the fabG3 overexpression strain Xcc YH5 increased to 0.21 ± 0.07 μmol/mg per min and 0.30 ± 0.06 μmol/mg per min, respectively (Fig. S5C). These results confirmed that Xcc modulated the total OAR activity in cells to control the production of DSF family signals.
Discussion
OAR catalyses the first reduction of 3‐oxoacyl‐ACP to 3‐hydroxylacyl‐ACP in the type II fatty acid synthase systems of bacteria (Magnuson et al., 1993). Although fabG is the only gene that encodes OAR activity in E. coli, multiple fabG paralogues have been reported in other species, and some are involved in fatty acid biosynthesis under special conditions (Feng et al., 2015; Mao et al., 2016). The Xcc genome encodes several FabG paralogues (Qian et al., 2005): FabG1 is the housekeeping OAR, while FabG2 is a novel OAR specific to reduce long‐chain 3‐oxoacyl‐ACP substrates (Hu et al., 2018). In this report, we confirmed that Xcc fabG3 encodes an OAR enzyme. First, fabG3 improved the growth of E. coli fabG(ts) strain CL104. Second, Xcc FabG3 catalysed the reduction of 3‐oxoacyl‐ACPs with varied chain lengths in the elongation cycle of FAS II in vitro. However, Xcc FabG3 seems distinct from Xcc FabG1. Xcc fabG1 is essential for growth and fatty acid synthesis, and cannot be deleted from the chromosome. However, Xcc fabG3 is not essential, and the fabG3 deletion strain was viable, producing similar species and amounts of fatty acids as the wild‐type strain. Additionally, the OAR activity of Xcc FabG3 was much lower than that of Xcc FabG, only ranging from 10% to 50% depending on the substrates. Moreover, Xcc FabG3 shows no reductive activity on the model substrate acetoacetyl‐CoA, but Xcc FabG1 possesses a high reductive ability.
The membrane‐bound yellow pigment xanthomonadins play essential roles in protecting bacteria from photobiological and peroxidative damage (Poplawsky and Chun, 1998; Rajagopal et al., 1997), but xanthomonadin biosynthesis mechanism needs further study. In our study, Xcc fabG3 deletion abolished xanthomonadin production, which indicated that FabG3 plays a vital role in xanthomonadin synthesis. Cao et al. (2018) identified 11 genes (named xanA2 to xanM, respectively) within the pig cluster as being essential for xanthomonadin synthesis, in which the ketoreductase‐coding gene xanH corresponds to fabG3 in this study. 3‐HBA, the precursor for xanthomonadin biosynthesis (Zhou et al., 2013b), could not restore the xanthomonadin production in the Xcc fabG3 mutant, implying that FabG3 catalyses the reduction of the 3‐keto group of an unknown intermediate downstream in the xanthomonadin synthetic pathway. At present, three OAR‐coding genes, fabG1, fabG2 and fabG3, have been identified in the Xcc genome; only fabG3 expression in the mutant could restore xanthomonadin production, but Xcc fabG1, fabG2 and E. coli fabG could not independently complement the xanthomonadin‐deficient phenotype. The results indicate that FabG3 plays a unique role in the xanthomonadin synthetic pathway, and its function cannot be replaced by other OARs. Our data revealed that the reductase activity of FabG1 is much greater than that of FabG3. Moreover, fabG1 is comparatively highly expressed and is a housekeeping gene that cannot be deleted directly. The white colony appearance of the fabG3 deletion mutant also demonstrated that FabG1 in the cell cannot functionally replace FabG3 in xanthomonadin synthesis. Based on the gene cluster, xanthomonadin is synthesized by a novel type II polyketide synthase pathway (He et al., 2011), and we propose a model reductive reaction catalysed by Xcc FabG3 in Fig. 7. However, the substrate(s) and catalytic mechanism of Xcc FabG3 need further study.
Figure 7.
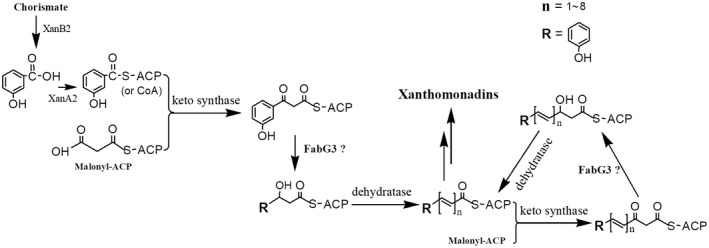
The proposed biosynthetic pathway for xanthomonadins in Xanthomonas campestris pv. campestris. The precursor 3‐hydroxylbenzoic acid is ligated to ACP (or CoA) by XanA2. Xcc3998 and Xcc4004 in the pig cluster are proposed to encode the keto synthase and dehydratase, respectively, involved in xanthomonadin biosynthesis.
Xcc RpfF, the key enzyme for the biosynthesis of the DSF family signals, utilizes 3‐hydroxyacyl‐ACPs as substrates (Zhou et al., 2015), while 3‐hydroxyacyl‐ACPs are the products of OAR in the bacterial fatty acid synthetic pathway (Rafferty et al., 1998). Thus, the production of DSF family signals is closely associated with OAR activity. In this report, we found Xcc FabG3 had OAR activity in vivo and in vitro, and it is reasonable that Xcc fabG3 null mutant decreased DSF family signals production, while a wild‐type fabG3‐encoding plasmid restored the high production level of DSF family signals. These results are consistent with our previous research. Hu et al. (2018) reported that Xcc fabG2 deletion mutant produced <50% of DSF family signals of wild‐type, and overexpression of fabG1 or fabG2 resulted DSF family signals levels 50% greater than wild‐type. Therefore, we concluded that Xcc might modulate the total OAR activity to control the production of DSF family signals.
In the study, Xcc fabG3 deletion resulted in a substantial reduction in virulence, which was restored to wild‐type level by complementation with plasmid expressing fabG3. During the system infection process, Xcc produces a variety of pathogenic factors, including EPS and extracellular enzymes (cellulose, amylase and protease), under the regulation of a DSF‐mediated QS system. However, the pathogenic factor productions were not statistically different between the fabG3 deletion mutant and wild‐type strain, indicating that the pathogenic factors’ reduction was not the cause for virulence attenuation. Because xanthomonadins protect Xcc from photooxidative damage, abolishment of xanthomonadins in the fabG3 deletion mutant is the most probable reason for virulence reduction. Moreover, the survival rate of fabG3 deletion mutant was much lower than that of wild‐type when exposed to oxidative stress (H2O2), which is a common environmental cue that pathogens encounter during systemic invasion. Similar findings were reported by Poplawsky and Chun (1998). When xanB2, an essential gene for xanthomonadins synthesis, was disrupted, the mutant virulence reduced sharply (He et al., 2011).
Experimental Procedures
Materials
Malonyl‐CoA, acetyl‐CoA, acetoacetyl‐CoA, fatty acids, NADH, NADPH and antibiotics were purchased from Sigma (USA). Takara Biotechnology Co. (Japan) provided molecular biology reagents. Novagen (USA) provided pET vectors. Invitrogen (USA) provided Ni‐agarose columns. GE Healthcare (USA) provided the HiTrap Q strong anion‐exchange column and Bio‐Rad (USA) provided the Quick Start Bradford dye reagent. All other reagents were of the highest available quality. Oligonucleotide primers were synthesized by TaKaRa Biotechnology Co.
Bacterial strains, plasmids and growth conditions
The strains and plasmids are listed in Table 2. Escherichia coli strains were grown in Luria‐Bertani (LB) medium at 37 °C. The phenotypes of E. coli fab strains were assessed on RB medium (Ulrich et al., 1983). The Xcc strains were grown at 30 °C in NYG medium (tryptone 5 g/L, yeast extract 3 g/L, glycerol 20 g/L) (Yu et al., 2016). When required, antibiotics and inducers were added as follows: sodium ampicillin 100 μg/mL, kanamycin sulphate 30 μg/mL, gentamycin 30 μg/mL for E. coli or 10 μg/mL for Xcc, rifampicin 50 μg/mL, arabinose 100 μg/mL, isopropyl‐β‐d‐thiogalactoside (IPTG) 240 μg/mL. Bacterial growth was determined by growing on solid media or by measuring the optical density at 600 nm.
Table 2.
Bacterial strains and plasmids used in this study
| Bacterial strain | Relevant characteristics* | Source |
|---|---|---|
| Escherichia coli | ||
| DH‐5α | F‐ deoR endA1 gyrA96 hsdR17(rK ‐mK +) recA1 relA1 supE 44 thi‐1 Δ(lacZYA‐argF)U169(φ80lacZΔM15) | Laboratory stock |
| BL21(DE3) | F‐ dcm ompT hsdS(rB ‐mB ‐) gal (λDE3) | Laboratory stock |
| S17‐1 | Tpr Smr recA thi pro hsdR (RP4‐2 Tc::Mu Km::Tn7), λpir | Laboratory stock |
| CL104 | fabG(ts) panD Cmr, Tetr, Kmr | Laboratory stock |
| MG1655 | Wild‐type | Laboratory stock |
| Xanthomonas campestris pv. campestris | ||
| XC1 | Rifr, wild‐type | Qian et al. (2005) |
| FE58 | Rifr, Tcr, rpfF::Tn5 lac, Biosensor for DSF signals | Wang et al. (2004) |
| YH1 | Rifr, ΔfabG3 | This study |
| YH2 | Rifr, Kmr, Xcc YH1/pYYH‐5 | This study |
| XC1(ΔrpfC) | Rifr, XC1ΔrpfC | Zhou et al. (2015) |
| YH3 | Rifr, XC1ΔrpfCΔfabG3 | This study |
| YH4 | Rifr, Kmr, Xcc YH3/pYYH‐5 | This study |
| YH5 | Rifr, Kmr, XC1ΔrpfC/pYYH‐5 | This study |
| YH6 | Rifr, Kmr, Xcc YH1/pYYH‐6 | This study |
| YH7 | Rifr, Kmr, Xcc YH1/pYYH‐7 | This study |
| YH8 | Rifr, Xcc 8004ΔXanB2 | This study |
| YH9 | Rifr, Xcc 8004ΔXanB2ΔfabG3 | This study |
| YH10 | Rifr, Kmr, Xcc YH1/pYYH‐10 | This study |
| Plasmids | ||
| pET‐28b | Kmr, T7 promoter‐based expression vector | Novagen |
| pMD19‐T | Ampr, TA cloning vector | Takara |
| pSRK‐Km | Kmr, broad‐host‐range expression vector containing lac promoter and lacI q, lacZα+ | Khan et al. (2008) |
| pK18mobscaB | Kmr, sacB‐based gene replacement vector | Schafer et al. (1994) |
| pBAD24M | Ampr; pBAD24 NcoI site replaced by NdeI site | Zhu et al. (2013) |
| pHWG | Ampr, EcfabG gene cloned into plasmid pBAD24M | Lab stock |
| pYYH‐1 | Ampr, Xcc fabG3 gene cloned into plasmid pMD19‐T | This study |
| pYYH‐2 | Ampr, Xcc fabG3 gene cloned into plasmid pBAD24M | This study |
| pYYH‐3 | Kmr, Xcc fabG3 cloned into plasmid pET‐28b | This study |
| pYYH‐4 |
Kmr, about 1000 bp Xcc fabG3 deletion DNA fragment inserted into pK18mobscaB between EcoRI/HindIII sites |
This study |
| pYYH‐5 | Kmr, fabG3 cloned into plasmid pSRK‐Km | This study |
| pYYH‐6 | Kmr, XccfabG1 cloned into plasmid pSRK‐Km | This study |
| pYYH‐7 | Kmr, E.coli fabG cloned into plasmid pSRK‐Km | This study |
| pYYH‐8 | Ampr, Xcc fabG1 gene cloned into plasmid pBAD24M | This study |
| pYYH‐9 |
Kmr, about 1000 bp Xcc xanB2 deletion DNA fragment inserted into pK18mobscaB between EcoRI/HindIII sites |
This study |
| pYYH‐10 | Kmr, XccfabG2 cloned into plasmid pSRK‐Km | This study |
Rifr, Tc r, Kmr and Ampr indicate resistance to rifampicin, tetracycline, kanamycin and ampicillin, respectively.
Plasmids construction
Xcc fabG3 was amplified from genomic DNA of wild‐type strain Xcc8004 with primers fabG3 P1 (NdeI) and fabG3 P1 (HindIII) listed in Table S1. The PCR was performed with Pfu DNA polymerase and the product was inserted into the pMD19‐T vector to obtain plasmid pYYH‐1. The Xcc fabG3 gene was confirmed by sequencing performed by Shanghai Sangon, Inc. (Shanghai, China). The Xcc fabG3 gene was digested with NdeI and HindIII, isolated and ligated into pBAD24M (Cheng et al., 2012) to create plasmid pYYH‐2. The fabG3 gene was also inserted into pET‐28b to produce pYYH‐3, and into pSRK‐Km (Khan et al., 2008) to produce pYYH‐5. The Xcc fabG1 gene was amplified and cloned into pSRK‐Km to create pYYH‐6 and into pBAD24M to create plasmid pYYH‐8.
Disruption of Xcc fabG3 or Xcc xanB2 in the chromosome
To disrupt the Xcc fabG3 gene, upstream (with an EcoRI site at the end) and downstream (with a HindIII site at the end) fragments of Xcc fabG3 (called fabG3 Up and fabG3 Dn, respectively) were amplified from Xcc genomic DNA with Pfu DNA polymerase, using the primer pairs fabG3 Up1/Up2 for fabG3 Up and fabG3 Down3/Down4 for fabG3 Dn (Table S1). The PCR products were purified and an overlap extension was carried out using fabG3 Up1 and fabG3 Down4 as primers. The fused fragment was then digested with EcoRI and HindIII and inserted into plasmid pK18mobsacB (Schafer et al., 1994) cut with the same enzymes to produce the suicide plasmid pYYH‐4.
Following the mating of derivatives of E. coli S17‐1 carrying pYYH‐4 with Xcc on NYG plates for 36 h at 30 °C, the cells were suspended in NYG medium and appropriate dilutions were spread onto NYG plates containing rifampicin plus kanamycin (resistant form of the plasmid pYYH‐4) to select for the single‐crossover integrants. Several single‐crossover integrant colonies were inoculated in NYG medium without kanamycin at 30 °C for 36 h and then appropriate dilutions were spread onto NYG plate containing a final concentration of 10% sucrose to select the deletion mutants. Colonies sensitive to kanamycin were screened by PCR with primers fabG3 Check Up and fabG3 Check Down, and the Xcc fabG3 deletion strain YH1 (ΔfabG3) was obtained.
The Xcc xanB2 deletion mutant YH8 was also obtained using a similar strategy. Moreover, the suicide plasmid pYYH‐9 with the upstream and downstream fragments of Xcc xanB2 was also transferred into the Xcc fabG3 deletion strain YH1 and the Xcc fabG3 xanB2 double‐deletion strain YH9 was constructed.
Protein expression and purification
The pET‐28b derived plasmid pYYH‐3 was introduced into E. coli BL21 (DE3) and Xcc FabG3 with a vector‐encoded N‐terminal hexahistidine (His)‐tag was highly expressed (data not shown). Xcc FabG3 was purified by nickel chelate chromatography as described previously (Yu et al., 2016). The E. coli FabD, FabH, FabG, FabZ, FabI and holo‐ACP, V. harveryi acyl‐ACP synthetase and R. solanacearum RSp0194 (Mao et al., 2015, 2016) were also purified as described previously (Zhu et al., 2013).
Assay of Xcc FabG3 activities in vitro
For the initiation reaction, the assay mixture contained 0.1 m sodium phosphate (pH 7.0), 0.1 μg each of EcFabD, EcFabH, EcFabG (or Xcc FabG3) and EcFabZ, 50 μm NADH, 50 μm NADPH, 1 mm β‐mercaptoethanol, 100 μm malonyl‐CoA, 50 μm holo‐ACP and 100 μm acetyl‐CoA in a final volume of 40 μL. During the long‐chain 3‐ketoacyl‐ACP reduction, EcFabH was replaced by Rsp0194 and acetyl‐CoA was replaced by octanoyl‐ACP, which was synthesized from octanoic acid, ATP and holo‐ACP using V. harveryi acyl‐ACP synthetase. The assay mixtures were incubated at 37 °C for 1 h and resolved by conformation‐sensitive gel electrophoresis on 20% polyacrylamide gels containing an optimized concentration of urea for separation. The gels were stained with Coomassie brilliant blue R250.
Spectrophotometric assay of OAR activity
UV‐visible spectrophotometry was used to measure OAR activity through the decrease in absorbance at 340 nm, using an NADPH extinction coefficient of 6220 m −1 (Qiu et al., 2005). The reaction mixture contained 0.5 mm acetoacetyl‐CoA, 0.2 mm NADPH, 0.1 m sodium phosphate buffer (pH 7.4) and 10 μg of a purified His‐tagged Xcc FabG1 or Xcc FabG3 protein. When testing the enzymatic abilities of Xcc FabG1 or Xcc FabG3, or cell‐free extract, to reduce various 3‐oxoacyl‐ACP chains, the reaction mixture contained acyl‐CoAs and malonyl‐CoA as substrates. Additionally, 2 μg of purified E. coli FabD, holo‐ACP and R. solanacearum RSp0194 were also added to the reaction mixture, in a final volume of 100 μL. The reaction was initiated by adding 1 μg of Xcc FabG1 or Xcc FabG3 to the mixture, and the absorbance value was read at 340 nm.
Analysis of phospholipid compositions
The cultures were grown aerobically at 30 °C in NYG media for 2 days. Cells were harvested and washed three times with water. Fatty acid methyl esters were synthesized and extracted as described previously (Stead, 1989). Briefly, cellular lipids were saponified by the addition of 1 mL sodium hydroxide/methanol solution at 100 °C for 40 min with shaking (800 rpm). Then, fatty acids were methylated by the addition of 2 mL hydrochloric acid/methanol solution at 80 °C for 30 min and immediately cooled to below 20 °C. Fatty acid methyl esters were obtained by three extractions each with 1 mL of petroleum ether. The solvent was removed under a stream of nitrogen, and the residue was analysed by GC‐MS.
Extraction and purification of DSF family signals from Xcc culture supernatant
The method for extraction and purification was described previously by He et al. (2010). To quantify DSF and BDSF production in the culture of the wild‐type strain XC1 with LC‐MS, 20 mL of the supernatant was collected. Its crude ethyl acetate extract was passed through a 0.45‐µm Minisart filter unit and then condensed to 0.1 mL for LC‐MS (Zhou et al., 2015, 2015b). Two microliters of the condensed samples was applied to an AB SCIEX 6500 QTRAP system (USA) and eluted with methanol/water (80:20, v/v) at a flow rate of 0.3 mL/min in a diode array detector. BDSF and DSF levels in the culture supernatant were quantified using peak intensity (PI) in the extracted ion chromatogram.
To quantify DSF and BDSF production in the culture of strain ΔrpfC with HPLC, 50 mL of the supernatant was collected (He et al., 2010). The crude ethyl acetate extract was passed through a 0.45‐µm Minisart filter unit and then condensed to 0.1 mL. Then 3 μL of the extract was applied to HPLC on a C18 reverse‐phase column (4.6 × 150 mm, Agilent) and eluted with water in methanol (23:77, v/v, 0.1% formic acid) at a flow rate of 1 mL/min in an Agilent Technologies 1260 Infinity system with a DAD G1315D VL detector (USA).
Quantification of xanthomonadin pigment production
Pigments were extracted from Xcc using previously described procedures (He et al., 2011). In brief, Xcc cells were collected from 10 mL suspension by centrifugation and mixed with 1 mL methanol. The mixtures were then incubated for 10 min in darkness in a rotating shaker and subsequently centrifuged at 12 000 g for 5 min. The xanthomonadin pigments present in the supernatant were quantified using their absorbance at 445 nm.
Measurement of extracellular enzymatic activity and EPS production
Relative activities of extracellular enzymes were assayed as described previously (Chao et al., 2008). Briefly, 2 μL of each Xcc strain culture (OD600 ≈ 1.0) was spotted onto NYG agar plates containing 1% (w/v) skim milk (for protease) (Sangon, Shanghai, China), 0.5% (w/v) carboxymethylcellulose (for cellulase) (Sangon) or 0.1% (w/v) starch (for amylase) (Sangon) and incubated at 30 °C for 24–48 h. Plates were stained where necessary. Zones of clearance around the spots due to the degradation of the substrate were photographed. Three plates were inoculated in each experiment, and each experiment was repeated three times. The relative activities of the enzymes were indicated by the diameter of the clear zones.
The EPS production was measured as described previously. Each Xcc strain culture (2 mL, OD600 ≈ 1.0) was used to inoculate 100 mL of NYG containing 4% glucose with shaking at 180 rpm for 4 days at 30 °C. The EPS was precipitated from the culture supernatant by addition of 4 volumes of 100% ethanol. The pelleted EPS was washed with 70% ethanol, air dried and weighed. Three flasks were inoculated in each experiment and each experiment was repeated three times.
Statistical analysis
An analysis of variance for the experimental datasets was performed using JMP software v. 5.0 (SAS Institute Inc., Cary, NC, USA). Significant effects of treatment were determined by the F value (P = 0.05). When a significant F test was obtained, a separation of means was accomplished by Fisher’s protected least significant difference at P = 0.05.
Supporting information
Fig. S1 (A) Strategy for isolation of Xcc YH1 mutant strain. (B) Genetic organization of the fabG3 region in Xcc wild‐type (a) or Xcc YH1 (b). (C) PCR analysis of genomic DNA from strains in (B). CH, chromosome; Up, upstream fragment of Xcc fabG3; Dn, downstream fragment of Xcc fabG3.
Fig. S2 (A) RT‐qPCR analysis of the fabG3 neighbouring genes XCC4002 and XCC4004 . (B) Xcc strains grown in NYG liquid medium at 30 °C. After growth, the OD600 was monitored using the Bioscreen‐C Automated Growth Curves Analysis System (OY Growth Curves FP‐1100‐C, Helsinki, Finland).
Fig. S3 Light absorption spectra (400–500 nm) of the pigments extracted from various Xcc strains.
Fig. S4 (A) The relative activity of extracellular enzymes produced by Xcc strains in NYG. The black columns indicate the Xcc wild‐type strain, the grey columns indicate the mutant strain Xcc YH1(ΔfabG3) and the white columns indicate the complementary strain Xcc YH2. (B) The amount of extracellular polysaccharide (EPS) produced by the Xcc strains. Data are the mean ± standard deviation of triplicate measurements. The different letters in each data column indicate significant differences at P = 0.05.
Fig. S5 The effects of Xcc fabG3 deletion and expression on the production of DSF family signals and an analysis of OAR activities in the cell‐free extracts of Xcc ΔrpfC strains. (A) HPLC analysis of the DSF family signals extracted from different strains. (B) DSF family signals produced by Xcc fabG3 deletion and expression strains. Supernatants of 50 mL of Xcc strains grown in NA medium for 36 h were collected and DSF family signals were detected by HPLC. Strain Xcc YH3 referred to Xcc fabG3 gene was deleted in Xcc XC1(∆rpfC); strain Xcc YH4 referred to Xcc fabG3 complemented in plasmid pSRK‐Km in Xcc YH3; strain Xcc YH5 referred to Xcc fabG3 expressed from plasmid pSRK‐Km in Xcc XC1(∆rpfC). The relative amounts of signal molecules were calculated on the basis of their peak areas. (C) 3‐oxoacyl‐ACP reductase activity analyses in the cell‐free extracts of Xcc strains. The activities were monitored by the decrease in the absorbance at 340 nm using an NADPH extinction coefficient of 6220 m –1.The reaction mixture contained octanoyl‐CoA, malonyl‐CoA, E. coli FabD, E. coli ACP and R. solanacearum RSp0194. The data shown are the means of the results of three repeats and error bars indicate standard deviations. Different letters indicate significant differences between treatments based on the least significant difference at P = 0.05.
Table S1 Sequences of the PCR primers used.
Table S2 Fatty acid composition of total lipid extracts from Xcc strain grown on NYG medium.
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (grants 31601601 and 31671987), the National Key Project for Basic Research (grant 2015CB150600), the Natural Science Foundation of Guangdong Province (grants 2014A030313455 and 2015A030312005) and the Science Foundation of Guangdong Food & Drug Vocational College (grant 2017ZR006). The authors declare no conflict of interest.
References
- Bi, H.K. , Christensen, Q.H. , Feng, Y.J. , Wang, H.H. and Cronan, J.E. (2012) The Burkholderia cenocepacia BDSF quorum sensing fatty acid is synthesized by a bifunctional crotonase homologue having both dehydratase and thioesterase activities. Mol. Microbiol. 83, 840–855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonas, U. , Van den Ackerveken, G. , Buttner, D. , Hahn, K. , Marois, E. , Nennstiel, D. , Noel, L. , Rossier, O. and Szurek, B. (2000) How the bacterial plant pathogen Xanthomonas campestris pv. vesicatoria conquers the host. Mol. Plant Pathol. 1, 73–76. [DOI] [PubMed] [Google Scholar]
- Cao, X.Q. , Wang, J.Y. , Zhou, L. , Chen, B. , Jin, Y. and He, Y.W. (2018) Biosynthesis of the yellow xanthomonadin pigments involves an ATP‐dependent 3‐hydroxybenzoic acid: acyl carrier protein ligase and an unusual type II polyketide synthase pathway. Mol. Microbiol. 110, 16–32. [DOI] [PubMed] [Google Scholar]
- Chan, J.W. and Goodwin, P.H. (1999) The molecular genetics of virulence of Xanthomonas campestris . Biotechnol. Adv. 17, 489–508. [DOI] [PubMed] [Google Scholar]
- Chao, N.X. , Wei, K. , Chen, Q. , Meng, Q.L. , Tang, D.J. , He, Y.Q. , Lu, G.T. , Jiang, B.L. , Liang, X.X. , Feng, J.X. , Chen, B.S. and Tang, J.L. (2008) The rsmA‐like gene rsmA Xcc of Xanthomonas campestris pv. campestris is involved in the control of various cellular processes, including pathogenesis. Mol Plant–Microbe Interact. 21, 411–423. [DOI] [PubMed] [Google Scholar]
- Cheng, J.L. , Ma, J.C. , Lin, J.S. , Fan, Z.C. , Cronan, J.E. and Wang, H.H. (2012) Only one of the five Ralstonia solanacearum long‐chain 3‐ketoacyl‐acyl carrier protein synthase homologues functions in fatty acid synthesis. Appl. Environ. Microbiol. 78, 1563–1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai, X.H. , Gao, G. , Wu, M.M. , Wei, W.Y. , Qu, J.M. , Li, G.Q. and Ting, M. (2018) Construction and application of a Xanthomonas campestris CGMCC15155 strain that produces white xanthan gum. Microbiol. Open, e00631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng, Y.Y. , Liu, X.L. , Wu, J.E. , Lee, J. , Chen, S.H. , Cheng, Y.Y. , Zhang, C.Y. and Zhang, L.H. (2015) The host plant metabolite glucose is the precursor of diffusible signal factor (DSF) family signals in Xanthomonas campestris . Appl. Environ. Microbiol. 81, 2861–2868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng, Y.Y. , Wu, J.E. , Yin, W.F. , Li, P. , Zhou, J.N. , Chen, S.H. , He, F. , Cai, J. and Zhang, L.H. (2016) Diffusible signal factor family signals provide a fitness advantage to Xanthomonas campestris pv. campestris in interspecies competition. Environ. Microbiol. 18, 1534–1545. [DOI] [PubMed] [Google Scholar]
- Dow, J.M. , Crossman, L. , Findlay, K. , He, Y.Q. , Feng, J.X. and Tang, J.L. (2003) Biofilm dispersal in Xanthomonas campestris is controlled by cell‐cell signaling and is required for full virulence to plants. Proc. Natl. Acad. Sci. USA. 100, 10995–11000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng, S.X. , Ma, J.C. , Yang, J. , Hu, Z. , Zhu, L. , Bi, H.K. , Sun, Y.R. and Wang, H.H. (2015) Ralstonia solanacearum fatty acid composition is determined by interaction of two 3‐ketoacyl‐acyl carrier protein reductases encoded on separate replicons. BMC Microbiol. 15, 223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He, Y.W. and Zhang, L.H. (2008) Quorum sensing and virulence regulation in Xanthomonas campestris . FEMS Microbiol. Rev. 32, 842–857. [DOI] [PubMed] [Google Scholar]
- He, Y.W. , Wang, C. , Zhou, L. , Song, H. , Dow, J.M. and Zhang, L.H. (2006) Dual signaling functions of the hybrid sensor kinase RpfC of Xanthomonas campestris involve either phosphorelay or receiver domain‐protein interaction. J Biol. Chem. 281, 33414–33421. [DOI] [PubMed] [Google Scholar]
- He, Y.W. , Wu, J.E. , Cha, J.S. and Zhang, L.H. (2010) Rice bacterial blight pathogen Xanthomonas oryzae pv. oryzae produces multiple DSF‐family signals in regulation of virulence factor production. BMC Microbiol. 10, 187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He, Y.W. , Wu, J.E. , Zhou, L. , Yang, F. , He, Y.Q. , Jiang, B.L. , Bai, L.Q. , Xu, Y.Q. , Deng, Z.X. , Tang, J.L. and Zhang, L.H. (2011) Xanthomonas campestris diffusible factor is 3‐hydroxybenzoic acid and is associated with xanthomonadin biosynthesis, cell viability, antioxidant activity, and systemic invasion. Mol. Plant–Microbe. Interact. 24, 948–957. [DOI] [PubMed] [Google Scholar]
- Hu, Z. , Dong, H.J. , Ma, J.C. , Yu, Y.H. , Li, K.H. , Guo, Q.Q. , Zhang, C. , Zhang, W.B. , Cao, X.Y. , Cronan, J.E. and Wang, H.H. (2018) Novel Xanthomonas campestris long‐chain‐specific 3‐oxoacyl‐acyl carrier protein reductase involved in diffusible signal factor synthesis. mBio, 9, e00596–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan, S.R. , Gaines, J. , Roop, R.M. 2nd and Farrand, S.K. (2008) Broad‐host‐range expression vectors with tightly regulated promoters and their use to examine the influence of TraR and TraM expression on Ti plasmid quorum sensing. Appl. Environ. Microbiol. 74, 5053–5062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar, A. , Deepak, Sharma S. , Srivastava, A. and Kumar, R. (2017) Synthesis of xanthan gum graft copolymer and its application for controlled release of highly water soluble levofloxacin drug in aqueous medium. Carbohydr. Polym. 171, 211–219. [DOI] [PubMed] [Google Scholar]
- Lai, C.Y. and Cronan, J.E. (2004) Isolation and characterization of beta‐ketoacyl‐acyl carrier protein reductase (fabG) mutants of Escherichia coli and Salmonella enterica serovar Typhimurium. J. Bacteriol. 186, 1869–1878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magnuson, K. , Jackowski, S. , Rock, C.O. and Cronan, J.E. (1993) Regulation of fatty acid biosynthesis in Escherichia coli . Microbiol. Rev. 57, 522–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao, Y.H. , Ma, J.C. , Li, F. , Hu, Z. and Wang, H.H. (2015) Ralstonia solanacearum RSp0194 encodes a novel 3‐keto‐acyl carrier protein synthase III. PLoS One, 10, e0136261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao, Y.H. , Li, F. , Ma, J.C. , Hu, Z. and Wang, H.H. (2016) Sinorhizobium meliloti functionally replaces 3‐oxoacyl‐acyl carrier protein reductase (FabG) by overexpressing NodG during fatty acid synthesis. Mol. Plant–Microbe. Interact. 29, 458–467. [DOI] [PubMed] [Google Scholar]
- Poplawsky, A.R. and Chun, W. (1997) pigB determines a diffusible factor needed for extracellular polysaccharide slime and xanthomonadin production in Xanthomonas campestris pv. campestris . J. Bacteriol. 179, 439–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poplawsky, A.R. and Chun, W. (1998) Xanthomonas campestris pv. campestris requires a functional pigB for epiphytic survival and host infection. Mol. Plant–Microbe. Interact. 11, 466–475. [DOI] [PubMed] [Google Scholar]
- Poplawsky, A.R. , Urban, S.C. and Chun, W. (2000) Biological role of xanthomonadin pigments in Xanthomonas campestris pv. campestris . Appl. Environ. Microbiol. 66, 5123–5127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian, W. , Jia, Y. , Ren, S.X. , He, Y.Q. , Feng, J.X. , Lu, L.F. , Sun, Q. , Ying, G. , Tang, D.J. , Tang, H. , Wu, W. , Hao, P. , Wang, L. , Jiang, B.L. , Zeng, S. , Gu, W.Y. , Lu, G. , Rong, L. , Tian, Y. , Yao, Z. , Fu, G. , Chen, B. , Fang, R. , Qiang, B. , Chen, Z. , Zhao, G.P. , Tang, J.L. and He, C.Z. (2005) Comparative and functional genomic analyses of the pathogenicity of phytopathogen Xanthomonas campestris pv. campestris . Genome Res. 15, 757–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu, X. , Choudhry, A.E. , Janson, C.A. , Grooms, M. , Daines, R.A. , Lonsdale, J.T. and Khandekar, S.S. (2005) Crystal structure and substrate specificity of the beta‐ketoacyl‐acyl carrier protein synthase III (FabH) from Staphylococcus aureus . Protein Sci. 14, 2087–2094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rafferty, J.B. , Fisher, M. , Langridge, S.J. , Martindale, W. , Thomas, N.C. , Simon, J.W. , Bithell, S. , Slabas, A.R. and Rice, D.W. (1998) Crystallization of the NADP‐dependent beta‐keto acyl carrier protein reductase from Escherichia coli . Acta Crystallogr. D. Biol. Crystallogr. 54, 427–429. [DOI] [PubMed] [Google Scholar]
- Rajagopal, L. , Sundari, C.S. , Balasubramanian, D. and Sonti, R.V. (1997) The bacterial pigment xanthomonadin offers protection against photodamage. FEBS Lett. 415, 125–128. [DOI] [PubMed] [Google Scholar]
- Schafer, A. , Tauch, A. , Jager, W. , Kalinowski, J. , Thierbach, G. and Puhler, A. (1994) Small mobilizable multi‐purpose cloning vectors derived from the Escherichia coli plasmids pK18 and pK19: selection of defined deletions in the chromosome of Corynebacterium glutamicum . Gene, 145, 69–73. [DOI] [PubMed] [Google Scholar]
- Starr, M.P. and Stephens, W.L. (1964) Pigment and taxonomy of the genus Xanthomonas . J. Bacteriol. 87, 293–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starr, M.P. , Jenkins, C.L. , Bussey, L.B. and Andrewes, A.G. (1977) Chemotaxonomic significance of the xanthomonadins, novel brominated aryl‐polyene pigments produced by bacteria of the genus Xanthomonas . Arch. Microbiol. 113, 1–9. [DOI] [PubMed] [Google Scholar]
- Stead, D.E. (1989) Grouping of Xanthomonas campestris pathovars of cereals and grasses by fatty acid profiling. EPPO Bull. 19, 57–68. [Google Scholar]
- Ulrich, A.K. , de Mendoza, D. , Garwin, J.L. and Cronan, J.E. (1983) Genetic and biochemical analyses of Escherichia coli mutants altered in the temperature‐dependent regulation of membrane lipid composition. J. Bacteriol. 154, 221–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, L.H. , He, Y. , Gao, Y.F. , Wu, J.E. , Dong, Y.H. , He, C.Z. , Wang, S.X. , Weng, L.X. , Xu, J.L. , Tay, L. , Fang, R.X. and Zhang, L.H. (2004) A bacterial cell‐cell communication signal with cross‐kingdom structural analogues. Mol. Microbiol. 51, 903–912. [DOI] [PubMed] [Google Scholar]
- Wang, J.Y. , Zhou, L. , Chen, B. , Sun, S. , Zhang, W. , Li, M. , Tang, H.Z. , Jiang, B.L. , Tang, J.L. and He, Y.W. (2015) A functional 4‐hydroxybenzoate degradation pathway in the phytopathogen Xanthomonas campestris is required for full pathogenicity. Sci. Rep. 5, 18456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu, Y.H. , Hu, Z. , Dong, H.J. , Ma, J.C. and Wang, H.H. (2016) Xanthomonas campestris FabH is required for branched‐chain fatty acid and DSF‐family quorum sensing signal biosynthesis. Sci. Rep. 6, 32811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, L. , Huang, T.W. , Wang, J.Y. , Sun, S. , Chen, G. , Poplawsky, A. and He, Y.W. (2013a) The rice bacterial pathogen Xanthomonas oryzae pv. oryzae produces 3‐hydroxybenzoic acid and 4‐hydroxybenzoic acid via XanB2 for use in xanthomonadin, ubiquinone, and exopolysaccharide biosynthesis. Mol. Plant–Microbe. Interact. 26, 1239–1248. [DOI] [PubMed] [Google Scholar]
- Zhou, L. , Wang, J.Y. , Wang, J.H. , Poplawsky, A. , Lin, S.J. , Zhu, B.S. , Chang, C.Q. , Zhou, T.L. , Zhang, L.H. and He, Y.W. (2013b) The diffusible factor synthase XanB2 is a bifunctional chorismatase that links the shikimate pathway to ubiquinone and xanthomonadins biosynthetic pathways. Mol. Microbiol. 87, 80–93. [DOI] [PubMed] [Google Scholar]
- Zhou, L. , Yu, Y.H. , Chen, X.P. , Diab, A.A. , Ruan, L. , He, J. , Wang, H.H. and He, Y.W. (2015) The multiple DSF‐family QS signals are synthesized from carbohydrate and branched‐chain amino acids via the FAS elongation cycle. Sci. Rep. 5, 13294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, L. , Wang, X.Y. , Sun, S. , Yang, L.C. , Jiang, B.L. and He, Y.W. (2015b) Identification and characterization of naturally occurring DSF‐family quorum sensing signal turnover system in the phytopathogen Xanthomonas . Environ. Microbiol. 17, 4646–4658. [DOI] [PubMed] [Google Scholar]
- Zhou, L. , Zhang, L.H. , Camara, M. and He, Y.W. (2017) The DSF family of quorum sensing signals: diversity, biosynthesis, and turnover. Trends Microbiol. 25, 293–303. [DOI] [PubMed] [Google Scholar]
- Zhu, L. , Bi, H.K. , Ma, J.C. , Hu, Z. , Zhang, W.B. , Cronan, J.E. and Wang, H.H. (2013) The two functional enoyl‐acyl carrier protein reductases of Enterococcus faecalis do not mediate triclosan resistance. mBio, 4, e00613–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1 (A) Strategy for isolation of Xcc YH1 mutant strain. (B) Genetic organization of the fabG3 region in Xcc wild‐type (a) or Xcc YH1 (b). (C) PCR analysis of genomic DNA from strains in (B). CH, chromosome; Up, upstream fragment of Xcc fabG3; Dn, downstream fragment of Xcc fabG3.
Fig. S2 (A) RT‐qPCR analysis of the fabG3 neighbouring genes XCC4002 and XCC4004 . (B) Xcc strains grown in NYG liquid medium at 30 °C. After growth, the OD600 was monitored using the Bioscreen‐C Automated Growth Curves Analysis System (OY Growth Curves FP‐1100‐C, Helsinki, Finland).
Fig. S3 Light absorption spectra (400–500 nm) of the pigments extracted from various Xcc strains.
Fig. S4 (A) The relative activity of extracellular enzymes produced by Xcc strains in NYG. The black columns indicate the Xcc wild‐type strain, the grey columns indicate the mutant strain Xcc YH1(ΔfabG3) and the white columns indicate the complementary strain Xcc YH2. (B) The amount of extracellular polysaccharide (EPS) produced by the Xcc strains. Data are the mean ± standard deviation of triplicate measurements. The different letters in each data column indicate significant differences at P = 0.05.
Fig. S5 The effects of Xcc fabG3 deletion and expression on the production of DSF family signals and an analysis of OAR activities in the cell‐free extracts of Xcc ΔrpfC strains. (A) HPLC analysis of the DSF family signals extracted from different strains. (B) DSF family signals produced by Xcc fabG3 deletion and expression strains. Supernatants of 50 mL of Xcc strains grown in NA medium for 36 h were collected and DSF family signals were detected by HPLC. Strain Xcc YH3 referred to Xcc fabG3 gene was deleted in Xcc XC1(∆rpfC); strain Xcc YH4 referred to Xcc fabG3 complemented in plasmid pSRK‐Km in Xcc YH3; strain Xcc YH5 referred to Xcc fabG3 expressed from plasmid pSRK‐Km in Xcc XC1(∆rpfC). The relative amounts of signal molecules were calculated on the basis of their peak areas. (C) 3‐oxoacyl‐ACP reductase activity analyses in the cell‐free extracts of Xcc strains. The activities were monitored by the decrease in the absorbance at 340 nm using an NADPH extinction coefficient of 6220 m –1.The reaction mixture contained octanoyl‐CoA, malonyl‐CoA, E. coli FabD, E. coli ACP and R. solanacearum RSp0194. The data shown are the means of the results of three repeats and error bars indicate standard deviations. Different letters indicate significant differences between treatments based on the least significant difference at P = 0.05.
Table S1 Sequences of the PCR primers used.
Table S2 Fatty acid composition of total lipid extracts from Xcc strain grown on NYG medium.