

Abstract
Accumulating evidence suggested that long noncoding RNAs (lncRNAs) possess a potential role in prostate cancer (PCa) diagnosis and prognosis. Rapid biochemical recurrence (BCR) is considered as a sign for clinical recurrence metastasis and PCa-specific mortality. Hence, the aim of the present study was to identify a lncRNA signature that can predict BCR of PCa accurately. Bioinformatics analysis, Kaplan–Meier analyses, Cox regression analyses, and Gene Set Enrichment Analysis (GSEA) were performed in a publicly available database with 499 PCa tissues and 52 matched normal tissues. A signature was identified. All these lncRNAs were differentially expressed between tumor and normal tissues and differentially expressed between high Gleason score and low Gleason score tissues. Furthermore, we developed a seven lncRNAs signature that can predict PCa BCR. Patients classified into low-risk group showed better BCR survival significantly than the patients in the high-risk group (hazard ratio = 0.32, 95% CI: 0.20–0.52, concordance index = 0.63). The area under the curve was 0.68 for BCR. The signature also had good discrimination for BCR in men with Gleason 7 PCa. In conclusion, our results suggest that the seven lncRNAs signature is a new biomarker of BCR and high risk in PCa. In addition, the individual lncRNA warrants further study to uncover the associated mechanisms of PCa progression and the signature could be used to design direct clinical trials for adjuvant therapy.
Keywords: biochemical recurrence, biomarker, high risk, long noncoding RNA, prostate cancer
INTRODUCTION
Worldwide, prostate cancer (PCa) is one of the most common malignancies of males.1 Although surgery and/or radiation can cure the majority of patients with localized disease, many patients develop biochemical recurrence (BCR) and eventually progress to castration-resistant PCa.2,3 However, some patients have indolent tumors that do not progress or impact on quality of life. Thus, it is critical to distinguish between these patients before selecting treatments. Among the many clinical and pathological modalities, Gleason score is a dominant prognostic factor.
Previous studies have suggested that tumors with Gleason score ≤6 are nearly never life-threatening, while those with Gleason score ≥8 have a high risk of progression.4 Although histological evaluation can easily discriminate between tumors with Gleason score ≤6 and Gleason score ≥8, making risk assessments in patients with Gleason score 7 (3 + 4 or 4 + 3) is a significant challenge. Tumors with Gleason score 4 + 3 are more aggressive than those with Gleason score 3 + 4. Nonetheless, sampling error, subjectivity in assessing Gleason score, and interobserver variability are notable confounding factors.5 Hence, a molecular signature that can independently stratify PCa tumors into high- or low-risk groups is needed.
Previous studies have reported various genetic signatures that could provide independent prognostic information for patients with Gleason score 7 or distinguish tumors with Gleason score ≤6 from those with Gleason score ≥8.6 However, few studies have investigated the potential role of long noncoding RNAs (lncRNAs) as a diagnostic or prognostic signature in cancer. LncRNAs, which contain ≥200 nucleotides, have been extensively studied in recent years.7 A myriad of lncRNAs have been shown to play important roles in tumor initiation and progression, via processes such as cell proliferation, migration, invasion, and metastasis.8 LncRNAs could affect tumor development by acting as either oncogenes or tumor suppressors.9
Currently, bioinformatics analysis is widely applied in molecular biology experiments and clinical practice. Consequently, the aim of this study was to identify differentially expressed lncRNAs between PCa and normal tissues or between high and low Gleason score tissues using lncRNAs sequencing data from the Cancer Genome Atlas Project (TCGA) database. Then, a seven lncRNAs signature that could predict BCR was constructed. In addition, analyses of the pathways and functions of the seven lncRNAs can yield new insights into the underlying molecular mechanisms of PCa.
MATERIALS AND METHODS
Data preparation and processing
Raw lncRNAs sequencing data (fragments per kb of exon per million fragments mapped, FPKM) from PCa samples were obtained from TCGA data portal (https://cancergenome.nih.gov/, accessed on 13 Jan 2018). Then, lncRNA expression levels were converted into transcripts per kilobase million (TPM) values.
To identify differentially expressed lncRNAs between PCa and normal tissues, lncRNA sequencing data from both tumor and normal samples were included. To construct the lncRNAs signature, only tumor samples with clinical information and adequate BCR information were included. Differential lncRNA expression between PCa and normal tissues or between Gleason score ≤6 and Gleason score ≥8 was analyzed using limma package. Fold changes (FCs) were calculated and only lncRNAs with log2|FC| >2.0 and P < 0.05 were defined as significantly expressed lncRNAs. The overlapping significantly expressed lncRNAs were considered candidate lncRNAs for further analyses.
Developing the lncRNAs expression signature
The prognostic value for BCR of candidate lncRNAs was evaluated using Kaplan–Meier curve and the log-rank method. A total of 457 patients with sufficient BCR information were included in the analysis. The lncRNAs, significantly associated with BCR, were then subjected to Cox logistic regression analysis. Risk scores for each patient based on lncRNAs expression were calculated by their regression coefficients. Subsequently, a prognostic lncRNA signature was constructed. According to our seven-lncRNA signature, PCa patients were classified into high- or low-risk groups using the median risk score. Kaplan–Meier analysis of BCR was used to validate the prognostic value in the two groups.
Identifying a lncRNA signature associated with signaling pathways
The target miRNAs or genes of lncRNAs were predicted using online analysis tools, such as StarBase (http://starbase.sysu.edu.cn/), RBPDB (http://rbpdb.ccbr.utoronto.ca/), circlncRNAnet (http://app.cgu.edu.tw/circlnc/), and LncRNAMAP (http://lncrnamap.mbc.nctu.edu.tw/php/). Then, the overlapping target miRNAs or genes were identified using Venn diagrams. Gene Set Enrichment Analysis (GSEA) was performed to identify various gene sets that share common biological functions, chromosomal locations, or regulation. Thus, the biological pathways altered in our lncRNAs signature risk score were identified via Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analyses. The FDR <0.05 and P < 0.001 were set as the cutoff criteria.
Statistical analyses
Relationships between lncRNA expression and clinical features were assessed by the Wilcoxon rank-sum test for continuous variables and Chi-square test for categorical variables. To assess associations between lncRNA expression and BCR, Kaplan–Meier curves and the log-rank tests were used. Cox proportional hazards analysis was used to assess the relative impacts of different groups (high vs low level) on BCR. The concordance index (C-index) was used to evaluate the discriminatory powers of the signature. To assess predictive ability, the likelihood ratio test was used to compare the C-index of different signatures. All statistical analyses were performed using R (version 3.4.2, www.r-project.org). All statistical tests were 2-sided, and P < 0.05 was considered statistically significant.
RESULTS
Identifying lncRNAs with significantly altered expression in PCa
To identify target lncRNAs, 499 PCa tissues and 52 matched normal tissues were enrolled in this study. According to the definition (|log2 FC| >2.0 and P < 0.05), a total of 163 differentially expressed lncRNAs were identified between PCa and normal tissues, including 131 upregulated and 32 downregulated lncRNAs (Figure 1a). Another total of 22 lncRNAs were found to have significantly different expression levels between Gleason score ≤6 and Gleason score ≥8 tissues, including 5 upregulated and 17 downregulated lncRNAs (Figure 1b). Volcano plots were performed to more intuitively reflect these expressions data. Fifteen differentially expressed lncRNAs overlapped between the two analyses and were considered candidate lncRNAs. Based on differentially expressed lncRNAs patterns, hierarchical cluster analysis revealed that PCa tissues and tissues with high Gleason score could be distinguished from normal tissues and tissues with low Gleason score, respectively (Supplementary Figure 1 (1.2MB, tif) ).
Figure 1.
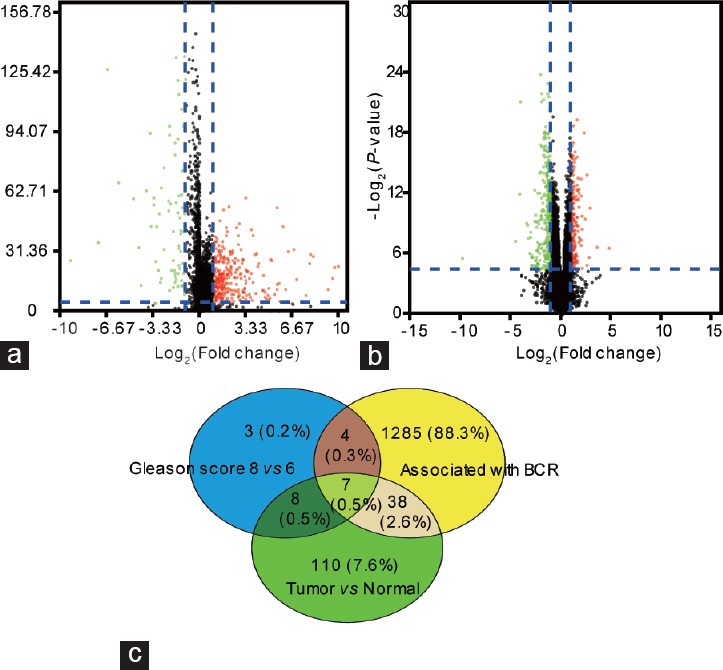
(a) Volcano plot of differentially expressed lncRNAs between tumor tissues and normal tissues. The red dot represents upregulated lncRNAs and green dot represents down-regulated lncRNAs. (b) Volcano plot of differentially expressed lncRNAs between tissues with Gleason score ≤6 and tissues with Gleason score ≥8. The red dot represents up-regulated lncRNAs and green dot represents downregulated lncRNAs. (c) Seven candidate lncRNAs were identified by Venn diagram. BCR: biochemical recurrence; lncRNAs: long noncoding RNAs.
Seven lncRNAs were associated with BCR in PCa
Associations between lncRNA expression and BCR were assessed by Kaplan–Meier analysis and the log-rank test. The average age of included patients was 60.8 years. We also evaluated detailed clinical characteristics, including diagnosis at age, metastasis, lymph node status, T stage, and Gleason score. Among this group, 181 patients had high Gleason scores (≥8) PCa and 43 patients had low Gleason scores (≤6) PCa. The remaining patients had Gleason score 7 PCa. Finally, 85 patients had BCR and 2 had metastases.
Our results indicated that a total of 1334 lncRNAs were significantly correlated with PCa BCR. Based on this analysis, seven of 15 candidate lncRNAs were associated with BCR and were used to construct the lncRNA signature (Figure 1c). Small nucleolar RNA host gene 1 (SNHG1) and colorectal neoplasia differentially expressed (CRNDE) were upregulated in the PCa tissues compared with normal tissues and were also upregulated in high Gleason score tissues compared with low Gleason score tissues. Conversely, the remaining five lncRNAs (RNA-Glu [CTC]-296K1.4, ubiquitin-binding protein domain protein 10 antisense RNA 1 [UBXN10-AS1], prostate androgen-regulated transcript 1 [PART1], CTC-296K1.3, and phosphoglucomutase 5 antisense RNA 1 [PGM5-AS1]) were all downregulated in PCa tissues and high Gleason score tissues. In addition, SNHG1 and CRNDE were positively associated with BCR, while the other five lncRNAS were negatively correlated with BCR.
The associations between seven lncRNAs and clinical features were evaluated in PCa patients (Table 1). The results showed that all seven lncRNAs were significantly associated with T stage (T1+T2 and T3+T4), lymph node status (N0 and N1), and Gleason score (<7, =7, and >7). In addition, SNHG1 and PART1 expression were significantly different between the groups with different ages at diagnosis (<60 years and ≥60 years).
Table 1.
Associations between seven lncRNAs and clinical features
| Variables | n | SNHG1 (mean±s.d.) | P | CRNDE (mean±s.d.) | P | CTC-296K1.4 (mean±s.d.) | P | UBXN10-AS1 (mean±s.d.) | P | PART 1 (mean±s.d.) | P | CTC-296K1.3 (mean±s.d.) | P | PGM5-AS1 (mean±s.d.) | P |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Age at diagnosis (year) | 0.02 | 0.52 | 0.24 | 0.24 | <0.01 | 0.12 | 0.46 | ||||||||
| <60 | 190 | 9.40±0.37 | 7.23±0.29 | 4.14±0.27 | 2.34±0.30 | 10.50±0.57 | 6.39±0.39 | 11.10±0.93 | |||||||
| ≥60 | 267 | 10.60±0.32 | 7.00±0.22 | 3.74±0.22 | 1.68±0.12 | 7.70±0.33 | 5.61±0.32 | 10.15±0.85 | |||||||
| T stage | <0.01 | <0.01 | 0.04 | <0.01 | <0.01 | 0.01 | <0.01 | ||||||||
| T1 + T2 | 175 | 8.89±0.30 | 6.09±0.22 | 4.34±0.29 | 2.72±0.33 | 10.26±0.47 | 6.77±0.41 | 14.22±1.22 | |||||||
| T3 + T4 | 277 | 10.92±0.34 | 7.76±0.25 | 3.61±0.21 | 1.47±0.11 | 7.99±0.41 | 5.38±0.31 | 8.11±0.65 | |||||||
| Lymph node status | <0.01 | <0.01 | 0.02 | 0.01 | 0.01 | 0.01 | <0.01 | ||||||||
| N0 | 317 | 9.78±0.28 | 6.74±0.19 | 3.96±0.20 | 1.83±0.13 | 8.98±0.39 | 5.88±0.29 | 10.56±0.75 | |||||||
| N1 | 73 | 11.98±0.73 | 9.25±0.60 | 2.92±0.33 | 1.11±0.19 | 6.65±0.70 | 4.27±0.48 | 5.05±0.84 | |||||||
| Metastasis | 0.36 | 0.99 | 0.20 | 0.50 | 0.45 | 0.16 | 0.30 | ||||||||
| M0 | 420 | 10.07±0.25 | 7.17±0.19 | 3.89±0.17 | 1.99±0.15 | 8.79±0.33 | 5.91±0.25 | 10.71±0.67 | |||||||
| M1 | 2 | 13.41±6.05 | 7.21±2.74 | 0.67±0.46 | 0.49±0.41 | 5.19±3.07 | 0.84±0.62 | 0.73±0.33 | |||||||
| Gleason score | <0.01 | <0.01 | <0.01 | <0.01 | <0.01 | <0.01 | <0.01 | ||||||||
| <7 | 43 | 8.90±0.55 | 5.59±0.34 | 5.78±0.72 | 4.15±1.07 | 10.40±0.91 | 9.26±1.06 | 20.54±3.09 | |||||||
| =7 | 233 | 8.74±0.28 | 6.45±0.21 | 4.11±0.24 | 2.15±0.17 | 9.97±0.47 | 6.26±0.34 | 11.69±0.84 | |||||||
| >7 | 181 | 12.14±0.44 | 8.28±0.33 | 3.20±0.24 | 1.18±0.11 | 7.07±0.42 | 4.72±0.34 | 6.68±0.78 |
s.d.: standard deviation; SNHG1: small nucleolar RNA host gene 1; CRNDE: colorectal neoplasia differentially expressed; PART 1: prostate androgen-regulated transcript 1; PGM5-AS1: phosphoglucomutase 5 antisense RNA 1; CTC: RNA-Glu; UBXN10-AS1: ubiquitin-binding protein domain protein 10 antisense RNA 1
Prognostic value of the seven lncRNAs signature risk score in PCa
The prognostic signature was constructed by integrating the expression of seven lncRNAs and their corresponding regression coefficients. The risk score formula was then expressed as the expression levels of each lncRNAs weighted by their regression coefficients as follows:
Risk score = (0.02117 × SNHG1 expression level) + (0.06256 × CRND expression level) + (0.17258 × CTC-296K1.4 expression level) + (−0.03433 × UBXN10-AS1 expression level) + (−0.02706 × PART1 expression level) + (−0.13507 × CTC-296K1.3 expression level) + (−0.00522 × PGM5-AS1 expression level).
Then, the 457 patients were classified into high-risk (n = 229) and low-risk group (n = 228) according to the median risk score. Time-dependent receiver operating characteristic (ROC) curves of BCR indicated that the area under the curves (AUC) at 1, 2, and 3 years were all 0.68 (Supplementary Figure 2 (1.3MB, tif) ). BCR-free survival analysis was performed using the Kaplan–Meier method. Patients with below-median scores had significantly longer BCR-free survival than patients with high scores. The BCR-free survival rates of patients in low-risk group were 95.4% for 1 year, 90.3% for 3 years, and 79.6% for 5 years, compared with 86.9%, 69.6%, and 53.6% in high-risk patients, respectively. The patients in low-risk group showed better BCR survival (Hazard Ratio [HR] = 0.32, 95% CI: 0.20–0.52, C-index = 0.63, Figure 2a). Furthermore, incorporating other clinical parameters into our seven-lncRNA signature provided better discriminatory power. When taking patient age and Gleason score into consideration, the AUCs at 1, 2, and 3 years were 0.71, 0.70, and 0.72, respectively (Supplementary Figure 2 (1.3MB, tif) ). The HR for patients in low-risk group was 0.26 (95% CI: 0.16–0.44, C-index = 0.63, Figure 2b).
Figure 2.
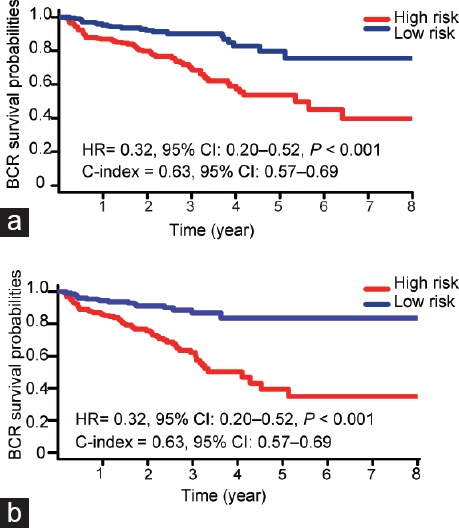
Kaplan–Meier survival curves of biochemical recurrence in prostate cancer patients. (a) The patients were stratified into high-risk group and low-risk group according to seven-lncRNAs signature. (b) The patients were stratified into high-risk group and low-risk group according to seven-lncRNAs signature incorporating clinical parameters (age and Gleason score). HR: hazard ratio; CI: credible interval; C-index: concordance index; BCR: biochemical recurrence; lncRNAs: long noncoding RNAs.
The same analysis was performed to evaluate the discriminatory powers of our seven lncRNAs signature in PCa patients with Gleason 7. The results suggested that our seven lncRNAs signature exhibited greater discrimination of BCR survival. The BCR survival of patients in low-risk group was significantly better than that of patients in high-risk group (HR = 0.39, 95% CI: 0.17–0.87, C-index = 0.60, Supplementary Figure 3 (638.7KB, tif) ).
Identifying pathways associated with seven lncRNAs signature
Target genes of the seven lncRNAs (SNHG1, CRNDE, CTC-296K1.4, UBXN10-AS1, PART1, CTC-296K1.3, and PGM5-AS1) were predicted using the online analysis tools lncRNABase, RBPDB, and circlncRNAnet, while target miRNAs were predicted with lncRNABase, circlncRNAnet, and LncRNAMAP. We then combined all of the identified target genes and miRNAs from each database (Figure 3 and Supplementary Table 1). GSEA in TCGA database was performed to identify biological signaling pathway associated with the seven lncRNAs. Significant gene sets (P < 0.001 and FDR <0.05) are displayed as enrichment plots (Supplementary Figure 4 (1.1MB, tif) and Supplementary Table 2). The lncRNAs associated gene sets regulated chromatin binding, protein serine/threonine kinase activity, RNA helicase activity, core promoter binding, and oxidoreductase activity.
Figure 3.
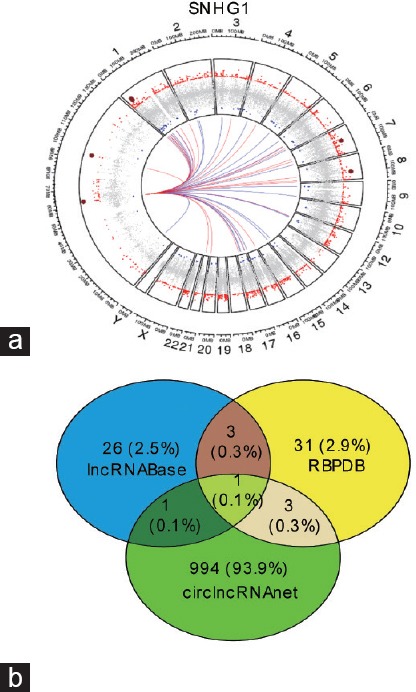
(a) Circos plot showed the most correlated and coexpressed genes of SNHG1 with respect to genomic coordinates. (b) Venn diagram showed the predicted target genes of SNHG1 by online tools. SNHG1: small nucleolar RNA host gene 1.
Table S1.
The seven-lncRNAs-associated target genes and miRNAs using online tools
| Target genes and miRNAs of seven-lncRNAs | |
|---|---|
| Gene | miRNAs |
| FUS | hsa-mir-3937 |
| IGF2BP1 | hsa-mir-4258 |
| QKI | hsa-mir-4739 |
| SFRS1 | hsa-mir-4712 |
| TIA1 | hsa-mir-3937 |
| ACO1 | hsa-mir-4739 |
| HNRNPA1 | hsa-mir-4712 |
| RBMX | hsa-mir-3937 |
| MBNL1 | hsa-mir-650 |
| PTBP1 | hsa-mir-3158 |
| PABPC1 | |
| NONO | |
Table S2.
The seven-lncRNAs-associated biological signaling pathway using GSEA
| ES | NES | P | FDR | |
|---|---|---|---|---|
| Chromatin binding | 0.515 | 2.011 | 0.000 | 0.020 |
| mitogen activated protein kinase binding | 0.683 | 2.003 | 0.000 | 0.021 |
| RNA helicase activity | 0.670 | 2.012 | 0.000 | 0.022 |
| demethylase activity | 0.726 | 1.988 | 0.000 | 0.023 |
| protein serine threonine kinase activity | 0.503 | 2.012 | 0.000 | 0.024 |
| purine ntp dependent helicase activity | 0.642 | 1.964 | 0.000 | 0.025 |
| receptor signaling complex scaffold activity | 0.676 | 2.015 | 0.000 | 0.025 |
| ubiquitin like protein specific protease activity | 0.579 | 1.969 | 0.000 | 0.025 |
| p53 binding | 0.585 | 2.023 | 0.000 | 0.027 |
| lysine n methyltransferase activity | 0.619 | 2.051 | 0.000 | 0.027 |
| kinesin binding | 0.682 | 2.058 | 0.000 | 0.031 |
| core promoter binding | 0.546 | 2.067 | 0.000 | 0.034 |
| protein complex scaffold | 0.510 | 1.921 | 0.000 | 0.037 |
| core promoter sequence specific dna binding | 0.515 | 1.917 | 0.000 | 0.038 |
| core promoter proximal region dna binding | 0.452 | 1.903 | 0.000 | 0.041 |
| RNA polymerase ii transcription cofactor activity | 0.478 | 1.893 | 0.000 | 0.044 |
| dynein binding | 0.643 | 1.882 | 0.000 | 0.045 |
| histone demethylase activity | 0.821 | 1.887 | 0.000 | 0.046 |
| glutathione peroxidase activity | -0.747 | -1.983 | 0.000 | 0.015 |
| oxidoreductase activity acting on nad P h quinone or similar compound as acceptor | -0.788 | -2.003 | 0.000 | 0.018 |
| electron carrier activity | -0.554 | -1.930 | 0.000 | 0.021 |
| oxidoreductase activity acting on a heme group of donors | -0.812 | -2.043 | 0.000 | 0.030 |
| disulfide oxidoreductase activity | -0.601 | -1.880 | 0.000 | 0.034 |
| hydrogen ion transmembrane transport | -0.652 | -2.166 | 0.000 | 0.016 |
| hydrogen transport | -0.568 | -2.098 | 0.000 | 0.030 |
| peptidyl lysine methylation | 0.664 | 2.201 | 0.000 | 0.045 |
DISCUSSION
Tumor risk stratification tools have significant clinical value for personalized medicine. However, using the current pathological parameters, risk stratification in PCa patients with Gleason 7 PCa remains a challenge.4 Similarly, establishing prediction models for PCa BCR also remains one of the most important problems that needs to be addressed. Most of the previous studies that developed molecular signatures have focused on gene expression. Here, we investigated the potential role of lncRNAs as novel signatures in this field.
LncRNAs play important roles in various cancers, including transcriptional regulation and mRNA posttranscriptional processing. Several studies have shown that lncRNAs differentially expressed in PCa.10,11 Evidence is mounting that lncRNAs may be useful diagnostic and prognostic biomarkers for cancer.11 Hence, lncRNAs were chosen as potential markers in our novel signature for predicting biochemical recurrence and high-risk in PCa.
In our study, seven lncRNAs (SNHG1, CRNDE, CTC-296K1.4, UBXN10-AS1, PART1, CTC-296K1.3, and PGM5-AS1) were identified to be significantly associated with PCa BCR and were differentially expressed not only between tumor and normal tissues, but also between tissues with Gleason score ≥8 and those with Gleason score ≤6. Moreover, our seven lncRNAs signature showed better discrimination of BCR survival curves and HRs both in all PCa patients and in patients with Gleason score 7 from TCGA database. In addition, our lncRNA signature predicted BCR well and provided novel tools for better clinical management of PCa patients with intermediate risk.
Previous studies suggested that active surveillance might be the most appropriate treatment for low-risk PCa patients, while high-risk patients should be treated aggressively.11 However, management decisions for patients with intermediate risk remain controversial.4 Approximately 20% of these patients will have BCR within 18 months after primary local therapy (radical prostatectomy or radiotherapy).12 Early BCR is considered a sign of clinical recurrence, metastasis, and PCa-specific mortality. Therefore, our signature that could classify patients into high or low risk of BCR may help select the most appropriate treatments and play an important role in the clinical management of PCa patients.13 Overestimating the risk of patients with low recurrence rates could lead to overtreatment. Identification of the real high-risk patients is key to precision medicine, preventing progression of PCa and reducing the burden of overtreatment.
Several studies have indicated that lncRNAs can be powerful predictors for survival in a variety of cancers.11,14 A previous gene molecular signature (HDDA10) that was established by TA Bismar showed significant performance for predicting PCa BCR with AUC = 0.65 in 2018.15 In comparison, our seven-lncRNA signature with AUC = 0.68 had relatively better discriminatory power. These results need to be prospectively validated, but provide evidence for the role of lncRNAs in the diagnosis and prognosis of PCa. If the negative predictive value (nNPV) is sufficiently high, the lncRNAs could be used to better identify patients that would benefit from adjuvant therapies in future randomized controlled trials (RCTs). Patients with a high risk of BCR should be enrolled into adjuvant or other systemic therapies, while low-risk patients should be entered into active surveillance. Subsequently, efficacy and safety evaluations of novel adjuvant therapies could be more accurately assessed.
Among our seven lncRNAs signature, SNHG1 had previously been identified, both alone and in combination with other lncRNAs as a biomarker for hepatocellular carcinoma, glioma, and lung cancer before.14,16 In addition, Li et al.17 found that SNHG1 could promote cell proliferation and cell cycle progression in PCa by binding miR-199a-3p.17 Similarly, previous studies demonstrated that PART1 was associated with poor prognosis in lung cancer.18 With regard to PCa, data published by Sun et al. suggested that PART1 promoted cell proliferation and blocked apoptosis via inhibiting TLR pathways.10 The roles of CRNDE and PGM5-AS1 as potential prognostic biomarkers were also validated in past findings.19,20,21 However, neither had been investigated in PCa. To date, there are few studies associated with the remaining three lncRNAs. From above results, SNHG1 and PART1 were identified as being involved in PCa carcinogenesis and development. CRNDE and PGM5-AS1 served as biomarkers and prognosis factors in other cancers. Hence, these studies suggested that our signature was reasonable and reliable. Future studies may focus on the other lncRNAs and investigate their function in PCa. Furthermore, these lncRNAs may have potential value in molecular targeted treatments.
Accumulating evidence has proved that lncRNAs participate in various biological processes by regulating mRNA expression at epigenetic, transcriptional, and posttranscriptional levels. However, most lncRNAs have not been functionally annotated in PCa. The present study indicated the associated biological signaling pathway of the seven lncRNAs through GSEA. Furthermore, previous studies have suggested that transcription factors associated with epithelial-to-mesenchymal transition (EMT) and androgen receptor (AR) splice variant-7 (AR-V7) may serve as biomarkers and play an important role in cancer progression and resistance to treatment. Our lncRNA signature was associated with the expression of zinc finger E-box binding homeobox 1 (ZEB1), vimentin (VIM, key EMT transcription factors), and AR (data not shown). Validation of the associations between the lncRNAs and actual signaling pathway is warranted. The potential molecular function of these lncRNAs, like protein serine threonine kinase activity, RNA helicase activity, core promoter binding, and oxidoreductase activity, may direct further studies that investigate mechanisms of PCa progression.
CONCLUSION
In summary, our study identified several lncRNAs that are significantly differentially expressed in PCa versus benign tissue and in high Gleason versus low Gleason score PCa. Furthermore, we developed a seven lncRNAs signature that can predict PCa BCR. The signature also has good discrimination for BCR in men with Gleason 7 PCa. Further studies are needed to validate our results and further RCTs can test the role of this signature for predicting the efficacy and safety of adjuvant therapy. In addition, functional studies are required to better understand the molecular mechanisms in PCa.
AUTHOR CONTRIBUTIONS
FNW and DWY conceived of the study and participated in its design and coordination. NS and YZ did the formal analysis. NS drafted the manuscript. YZ and DWY revised the manuscript. DWY reviewed the results and provided expert insight into the writing of the report. All authors read and approved the final manuscript.
COMPETING INTERESTS
The authors declared no competing interests.
Heat map (a) showed differentially expressed lncRNAs in prostate cancer between Gleason score ≤6 and Gleason score ≥8. Heat map (b) showed differentially expressed lncRNAs between prostate cancer tissues and normal tissues. lncRNAs: long noncoding RNAs.
Time-dependent ROC curves for seven lncRNAs signature at 1 (a), 2 (b) and 3 (c) years. ROC curves for seven lncRNAs signature incorporating age and Gleason score at 1 (d), 2 (e), and 3 (f) years. ROC: receiver operating characteristic.
Kaplan–Meier survival curves of biochemical recurrence in prostate cancer patients with Gleason score = 7. The patients were stratified into high-risk group and low-risk group according to seven-lncRNAs signature. HR: hazard ratios; CI: credible interval; C-index: concordance index; lncRNAs: long noncoding RNAs.
The Enrichment Map of seven-lncRNAs signature associated biological signaling pathway using GSEA (P < 0.001 and FDR < 0.05). lncRNAs: long noncoding RNAs; GSEA: Gene Set Enrichment Analysis.
ACKNOWLEDGMENTS
This study was supported by the National Natural Science Foundation of China (No. 81502192 to FNW, No. 81370073 to YZ and No. 81872099, 81472377 and 81672544 to DWY) and the Shanghai Rising Star Program (No. 16QA1401100).
Supplementary Information is linked to the online version of the paper on Asian Journal of Andrology website.
REFERENCES
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA Cancer J Clin. 2017;67:7–30. doi: 10.3322/caac.21387. [DOI] [PubMed] [Google Scholar]
- 2.Shao N, Wang Y, Jiang WY, Qiao D, Zhang SG, et al. Immunotherapy and endothelin receptor antagonists for treatment of castration-resistant prostate cancer. Int J Cancer. 2013;133:1743–50. doi: 10.1002/ijc.28162. [DOI] [PubMed] [Google Scholar]
- 3.Hirst CJ, Cabrera C, Kirby M. Epidemiology of castration resistant prostate cancer: a longitudinal analysis using a UK primary care database. Cancer Epidemiol. 2012;36:e349–53. doi: 10.1016/j.canep.2012.07.012. [DOI] [PubMed] [Google Scholar]
- 4.Yin Y, Zhang Q, Zhang H, He Y, Huang J. Molecular signature to risk-stratify prostate cancer of intermediate risk. Clin Cancer Res. 2017;23:6–8. doi: 10.1158/1078-0432.CCR-16-2400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dall'Era MA, Albertsen PC, Bangma C, Carroll PR, Carter HB, et al. Active surveillance for prostate cancer: a systematic review of the literature. Eur Urol. 2012;62:976–83. doi: 10.1016/j.eururo.2012.05.072. [DOI] [PubMed] [Google Scholar]
- 6.Penney KL, Sinnott JA, Fall K, Pawitan Y, Hoshida Y, et al. mRNA expression signature of Gleason grade predicts lethal prostate cancer. J Clin Oncol. 2011;29:2391–6. doi: 10.1200/JCO.2010.32.6421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang W, Zhao Z, Yang F, Wang H, Wu F, et al. An immune-related lncRNA signature for patients with anaplastic gliomas. J Neuro Oncol. 2018;136:263–71. doi: 10.1007/s11060-017-2667-6. [DOI] [PubMed] [Google Scholar]
- 8.Wilusz JE, Sunwoo H, Spector DL. Long noncoding RNAs: functional surprises from the RNA world. Gene Dev. 2009;23:1494–504. doi: 10.1101/gad.1800909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liang C, Zhang B, Ge H, Xu Y, Li G, et al. Long non-coding RNA CRNDE as a potential prognostic biomarker in solid tumors: a meta-analysis. Clin Chim Acta. 2018;481:99–107. doi: 10.1016/j.cca.2018.02.039. [DOI] [PubMed] [Google Scholar]
- 10.Sun M, Geng D, Li S, Chen Z, Zhao W. LncRNA PART1 modulates toll-like receptor pathways to influence cell proliferation and apoptosis in prostate cancer cells. Biol Chem. 2018;399:387–95. doi: 10.1515/hsz-2017-0255. [DOI] [PubMed] [Google Scholar]
- 11.Malik B, Feng FY. Long noncoding RNAs in prostate cancer: overview and clinical implications. Asian J Androl. 2016;18:568–74. doi: 10.4103/1008-682X.177123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lalonde E, Ishkanian AS, Sykes J, Fraser M, Ross-Adams H, et al. Tumour genomic and microenvironmental heterogeneity for integrated prediction of 5-year biochemical recurrence of prostate cancer: a retrospective cohort study. Lancet Oncol. 2014;15:1521–32. doi: 10.1016/S1470-2045(14)71021-6. [DOI] [PubMed] [Google Scholar]
- 13.Hansen J, Bianchi M, Sun M, Rink M, Castiglione F, et al. Percentage of high-grade tumour volume does not meaningfully improve prediction of early biochemical recurrence after radical prostatectomy compared with Gleason score. BJU Int. 2014;113:399–407. doi: 10.1111/bju.12424. [DOI] [PubMed] [Google Scholar]
- 14.Zhang M, Wang W, Li T, Yu X, Zhu Y, et al. Long noncoding RNA SNHG1 predicts a poor prognosis and promotes hepatocellular carcinoma tumorigenesis. Biomed Pharmacother. 2016;80:73–9. doi: 10.1016/j.biopha.2016.02.036. [DOI] [PubMed] [Google Scholar]
- 15.Abou-Ouf H, Alshalalfa M, Takhar M, Erho N, Donnelly B, et al. Validation of a 10-gene molecular signature for predicting biochemical recurrence and clinical metastasis in localized prostate cancer. J Cancer Res Clin. 2018;144:883–91. doi: 10.1007/s00432-018-2615-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang Q, Li Q, Zhou P, Deng D, Xue L, et al. Upregulation of the long non-coding RNA SNHG1 predicts poor prognosis, promotes cell proliferation and invasion, and reduces apoptosis in glioma. Biomed Pharmacother. 2017;91:906–11. doi: 10.1016/j.biopha.2017.05.014. [DOI] [PubMed] [Google Scholar]
- 17.Li J, Zhang Z, Xiong L, Guo C, Jiang T, et al. SNHG1 lncRNA negatively regulates miR-199a-3p to enhance CDK7 expression and promote cell proliferation in prostate cancer. Biochem Biophys Res Commun. 2017;487:146–52. doi: 10.1016/j.bbrc.2017.03.169. [DOI] [PubMed] [Google Scholar]
- 18.Li M, Zhang W, Zhang S, Wang C, Lin Y. PART1 expression is associated with poor prognosis and tumor recurrence in stage I-III non-small cell lung cancer. J Cancer. 2017;8:1795–800. doi: 10.7150/jca.18848. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 19.Ellis BC, Molloy PL, Graham LD. CRNDE: a long non-coding RNA involved in cancer, neurobiology, and development. Front Genet. 2012;3:270. doi: 10.3389/fgene.2012.00270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu T, Zhang X, Yang YM, Du LT, Wang CX. Increased expression of the long noncoding RNA CRNDE-h indicates a poor prognosis in colorectal cancer, and is positively correlated with IRX5 mRNA expression. Onco Targets Ther. 2016;9:1437–48. doi: 10.2147/OTT.S98268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhu H, Yu J, Zhu H, Guo Y, Feng S. Identification of key lncRNAs in colorectal cancer progression based on associated protein-protein interaction analysis. World J Surg Oncol. 2017;15:153. doi: 10.1186/s12957-017-1211-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Heat map (a) showed differentially expressed lncRNAs in prostate cancer between Gleason score ≤6 and Gleason score ≥8. Heat map (b) showed differentially expressed lncRNAs between prostate cancer tissues and normal tissues. lncRNAs: long noncoding RNAs.
Time-dependent ROC curves for seven lncRNAs signature at 1 (a), 2 (b) and 3 (c) years. ROC curves for seven lncRNAs signature incorporating age and Gleason score at 1 (d), 2 (e), and 3 (f) years. ROC: receiver operating characteristic.
Kaplan–Meier survival curves of biochemical recurrence in prostate cancer patients with Gleason score = 7. The patients were stratified into high-risk group and low-risk group according to seven-lncRNAs signature. HR: hazard ratios; CI: credible interval; C-index: concordance index; lncRNAs: long noncoding RNAs.
The Enrichment Map of seven-lncRNAs signature associated biological signaling pathway using GSEA (P < 0.001 and FDR < 0.05). lncRNAs: long noncoding RNAs; GSEA: Gene Set Enrichment Analysis.