

Abstract
Vasculitis poses a great diagnostic, investigative and therapeutic challenge to the treating physician. The classification of vasculitides itself still eludes universal acceptance. Comprehensive management comprises establishing the diagnosis of true vasculitis after ruling out vasculitis mimics, finding the etiology if feasible, assessing the caliber of the vessels involved, deciphering the pathological process of vessel damage, investigating for the existence and extent of systemic involvement and finally planning the therapy in the background of co-morbidities. Successful management also entails regular monitoring to foresee complications arising from the disease process itself as well as complications of immunosuppressive treatment. Although steroids remain first line drug, biologics are emerging as popular agents in the treatment of immune-mediated vasculitis. Triphasic treatment is the best plan of action comprising induction, maintenance of remission and treatment of relapses.
Keywords: Biomarkers, classification, management, vasculitis
Introduction
Vasculitides is a heterogeneous disease entity of variable causes in which immunologically mediated inflammatory reaction of the blood vessel wall leads to vessel wall damage and weakening (aneurysm, rupture) or obstruction of lumen, leading to infarction of tissue.[1]
Vasculitis affects nearly 38-40 persons per million population.[2] Cutaneous vasculitis is predominantly due to infections in 22%, drugs in 20%, connective tissue disorders in 12%, Henoch Schonlein purpura (HSP) in 10% and <5% each due to malignancy, primary systemic vasculitis or systemic inflammatory disease.[3,4] Yet, exact etiology may not be established in spite of exhaustive work up in many case scenarios and idiopathic nature predominates. The mean age of onset is 7 years in children, 47 years in adults, vasuclitis being, commoner in adults than children.[5] Successful management of the patient with vasculitis syndrome depends on good history taking, diligent physical examination and relevant investigations to confirm the diagnosis of vasculitis and assess systemic involvement.[6] The article attempts to cater this need providing a checklist for dermatologists to approach a case of vasculitis and treatment update.
Clinical Approach: Concept of ‘Vasculitis as a Syndrome’
Vasculitis is a syndrome with an array of clinical features as localized/systemic symptoms, visceral signs due to stratified dysfunction at specific cellular, tissue or organ involvement, specific and non-specific inflammatory symptoms to be put together for final diagnosis evolving over weeks to months [Figure 1]. In primary vasculitis (inflammation of vessel wall as an initial event in the absence of recognized precipitating disease or associated disease), auto immune mechanism is believed to play a key role.[7] Both humoral and cell-mediated immunity are implicated. Genetics, environmental factors, immune regulatory mechanisms render the patient susceptible to develop vasculitis.[4,5] Selective involvement of few vessels may be explained by the distribution of the antigen, local immune and inflammatory cascade.
Figure 1.

Algorithm for syndromic approach of symptoms in vasculitis
The pathological basis for the clinical manifestations of vasculitis is related to compromise in vessel function and breakdown of vessel wall due to inflammation.[8] This leads to localized tissue reaction in the interstitium to organ dysfunction.[8] The localized symptoms may appear independent at first glance, but systematic approach leads to unmask sequence of events in the course of disease and helps to understand the pathogenetic process.
Local symptoms are characterized by the simultaneous (or sequential) appearance of symptoms from different organs in a patient with vasculitis syndrome. Systemic symptoms caused by inflammation include: Fever (spiking fever of 38-39°C), weight loss, weakness, general malaise, arthralgia, myalgia.[8] Localized manifestations result from insufficient blood supply caused by the inflammation of blood vessels and local tissue damage. Biopsy is recommended in cases of localized myalgia. The aforementioned systemic symptoms in elderly warrant screening examinations for malignant tumors.[8,9]
Visceral signs
Skin is directly affected in both SVV and MVV and also in LVV (secondarily due to associated underlying disease state). In SVV palpable purpura, urticaria, vesiculobullous lesions and targetoid lesions dominate whereas in MVV, subcutaneous nodules, livedo reticularis, ulcers, infarcts, digital pitted scars and gangrene occur[4,5] [Figures 2–6].
Figure 2.
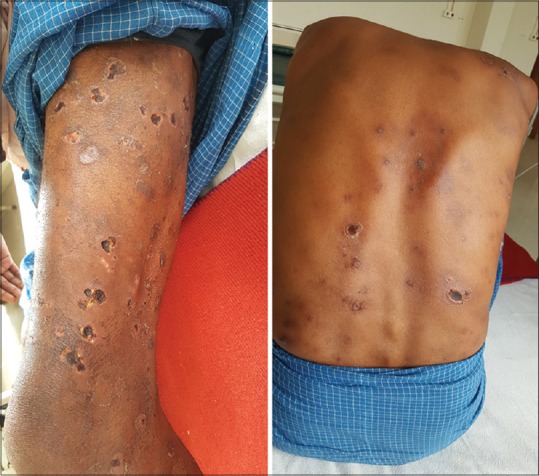
Erythema nodosum necroticans showing ulcerative lesions
Figure 6.
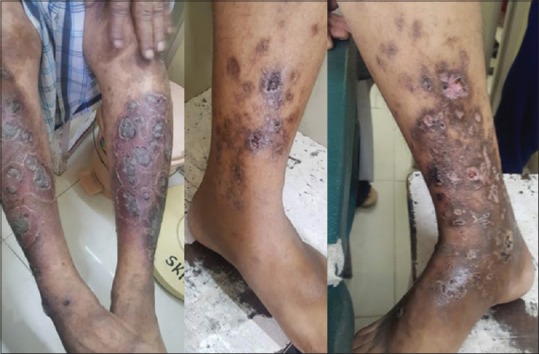
Cutaneous polyarteritis nodosa (medium vessel vasculitis) showing ulcerative lesions
Figure 3.
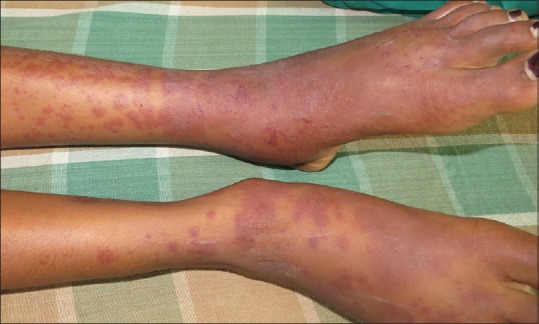
IgA vasculitis (HSP) shows palpable purpura over the lower extremities
Figure 4.
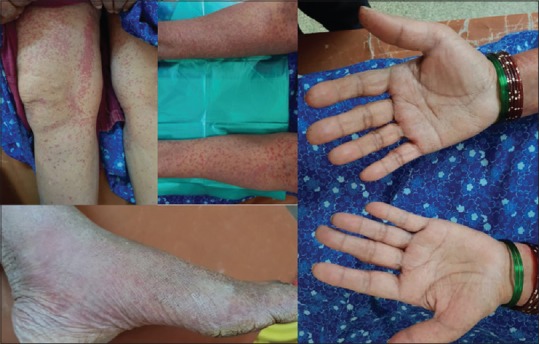
Leukoctocastic vasculitis
Figure 5.
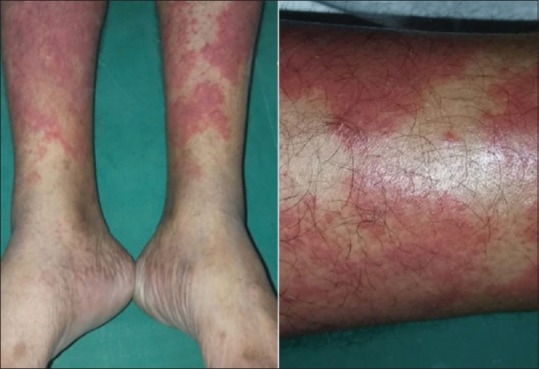
Urticarial vasculitis
Palpable purpura is a characteristic feature that develops in the lower limbs due to elevated hydrostatic pressure in dependent areas secondary to inflammatory damage. Mononeuritis multiplex is due to vasculitis of medium-sized arteries that feed the affected nerves. In the early stage, it may manifest as sensory disturbance, such as hyperesthesia or hypoesthesia, and, if progressive, leads to wrist drop and foot drop.[8] Renal manifestations in SVV include those of nephritis, such as hematuria, proteinuria and cylindruria. In large-vessel vasculitis, a decrease in aortic blood pressure severely affects blood flow to all downstream arteries. Bloody and foamy sputum occurs in pulmonary alveolar hemorrhage caused by arteriolitis or venulitis in the lungs. Pulse deficit, jaw claudication, loss of vision, and acute abdomen also occur in accordance to vessels affected.[8]
Thus, clinically vasculitic syndromes range from a benign, regionally restricted process to a life-threatening systemic disorder. Approaching individual case as a syndrome of symptoms, bracketed sequentially helps in confirming vasculitis, identifies prognostic factors, long-term outcome and best therapeutic options. The course can be acute and self limited (resolves in less than 6 months).
Differential diagnoses to be considered are vasculitic mimics which include infections such as infective endocarditis, syphilis, rickettsial infections, drug toxicity due to cocaine, malignancies like lymphoma, thrombotic and thromboembolic conditions, miscellaneous conditions such as sarcoidosis and amyloidosis.[10]
Good history taking is crucial in arriving at final diagnosis. History suggestive of infection, drug intake, existence of comorbid conditions, obstetric history of recurrent abortions, history of cigarette smoking, unprotected exposure to sexually transmitted infections all play an important role in the proper assessment of the patient.
Investigations
Table 1 enumerates a checklist of investigations to be evaluated in any suspected case of vasculitis.
Table 1.
Showing a checklist of investigations in vasculitis.
| Baseline investigations: |
| Complete hemogram with ESR |
| Acute phase reactants: CRP |
| Urinalysis for blood, protein, casts |
| Liver and Renal function tests |
| Antibody and complement levels: |
| C3, C4, ANA, dsDNA (SLE, Sjogren syndrome, HUV) |
| EIA of Proteinase3, MPO are moe useful in WG, CSS, MPA and drug induced vasculitis. |
| Cryoglobulins |
| Bacterial culture, HBV, HCV, HIV serology, VDRL |
| IgE, eosinophil count |
| Radiology: |
| Xray, HRCT chest, CECT abdomen with CT angiography (mesentric, celiac arteries for MVV), |
| 2D ECHO, magnetic resonance angiogram of thorax for Takayasu arteritis |
| Mantoux test (When long term immunosuppresants/biologics are to be considered) |
| Endoscopy :GI endoscopy in HSP in case of GI bleed |
| Skin biopsy and Organ biopsy |
| Pregnancy test is mandatory in female patients of child bearing age |
Hemogram may be abnormal in almost all patients with vasculitis. It is usually characterized by normochromic, normocytic anaemia, leukocytosis (eosinophilia >10% in CSS) and raised ESR and CRP.[1] Proteinuria, cylindruria and hematuria are also common findings. ANCA-associated vasculitis (WG, MPA and idiopathic pauciimmune crescentic glomerulonephritis) and HSP show active nephritic urinary sediment with red cell casts. Classic PAN is not associated with active sediment while, TA and GCA being large vessels vasculitis do not involve kidneys usually.[1] Also, these changes are seen often in MPA, whereas similar abnormalities are found in the late stages of PAN, GPA. PAN can cause hematuria due to renal infarct and proteinuria by hypertension.
Renal and hepatic function tests
They help in determining the disease extent, organ damage and therapeutic intervention. Elevated serum creatinine, BUN and decreased creatinine clearance occurs in PAN, GPA and MPA.
Serum proteins
Elevated γ globulin levels are common. The IgG type gammaglobulinaemia is often seen, but in WG and HSP, IgA type also occurs and IgE levels are increased in CSS. Hypocomplementemia occurs in immune complex vasculitis, for example, cryoglobulinaemia, PAN and HSP. Cryoglobulins are present in essential cryoglobulinemic vasculitis.[1]
Antineutrophil cytoplasmic autoantibodies (ANCA)
ANCA staining patterns seen in indirect immunoflourescence are given below:
Cytoplasmic ANCA (c-ANCA): Diffuse, coarse and granular cytoplasmic staining. Antibodies are directed specifically against proteinase - 3 (PR-3). This pattern is seen in active systemic WG (55% with limited WG and 88% with systemic WG).[11] Specificity of anti PR-3 (cANCA) is 99%, c-ANCA may be absent if patient has limited or inactive WG.[12] ANCA levels may correlate with disease activity[13]
Perinuclear ANCA (p-ANCA): Staining of the nuclear or perinuclear area is seen. These antibodies are directed against myeloperoxidase enzyme (MPO). Anti MPO antibodies are found in MPA, CSS and in drug-induced vasculitis.[1] p-ANCA may be found in minority of patients with anti-GBM disease (Goodpasture's syndrome),[14] primary sclerosing cholangitis, classic PAN, ulcerative colitis and in <5% patients with WG, SLE, RA, Sjogren's syndrome, Felty's syndrome, Still's disease and Kawasaki's disease[1]
Atypical staining (A or x-ANCA): Mixed pattern of fluorescence is seen, that is, cytoplasmic as well as perinuclear. Antibodies are directed against lactoferrin, cathepsin G, elastase, lysozyme and bacterial permeability increasing protein. Antibodies against lactoferrin and elastase show high association with drug-induced vasculitis.[15]
If ANCA is positive, specific antigenic reactivity of ANCA, that is, anti PR-3 or anti MPO should be assessed by enzyme-linked immunosorbent assay (ELISA). ANCA should always be assessed if patient has pulmonary hemorrhage, systemic vasculitis, rapidly progressive glomerulonephritis, multiple leg nodules, chronic destructive disease of upper airways, long standing sinusitis or otitis, subglottic tracheal stenosis, mononeuritis multiplex, other peripheral neuropathy or retro-orbital mass.[16]
Skin biopsy
Organ biopsy remains the gold standard for diagnosis of vasculitis.[1] Apart from skin biopsy, other sites such as renal, muscle, lung, and heart can also be biopsied.[1]
Cutaneous vasculitis may be diagnosed by taking full thickness biopsy from involved site as MVV can be easily missed if dermis and subcutaneous fat is absent. It usually presents with vessel wall inflammation along with perivascular involvement with or without leukocytoclasis. There are no histological features that are absolutely pathognomonic of any type of vasculitis.
For histopathology, lesions less than 48 hours (18-36 hours) old, ideally within 6 hours of appearance of lesions should be biopsed[17,18] and the lesions should be less than 8-12 hours old for direct immunofluorescence (DIF IgA, IgM, C3, C1), which is performed if patient has symptoms of systemic involvement or even otherwise[18] [Figures 7–10].
Figure 7.
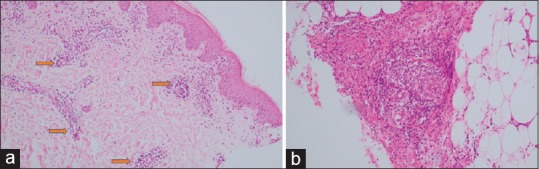
(a and b) Leucocytic vasculitis: inflammation is vasculocentric including neutrophils and nuclear dust, with fibrinoid necrosis of the vessel wall and endothelial swelling. H and E staining (a) low magnification 10× (b) high magnification 45×
Figure 10.
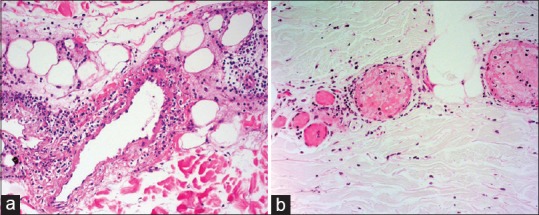
(a) Lymphocytic vasculitis: Fibrinoid necrosis of vessel wall and lymphocytic infiltration (b) Disseminated Intravascular Coagulation: Intravascular fibrin thrombi and leukocytoclasis with no fibrinoid necrosis. H and E staining; high magnification 45×
Figure 8.
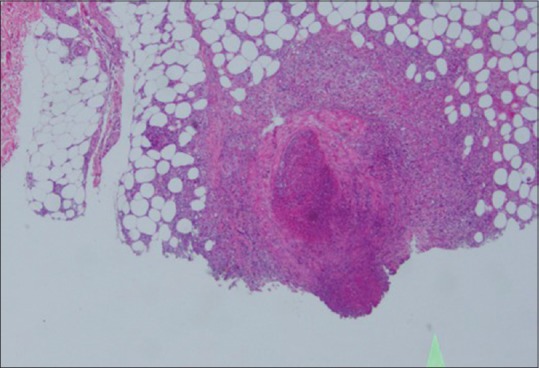
Poyarteritis nodosa: medium vessel vasculitis. H and E staining; High magnification 45×
Figure 9.
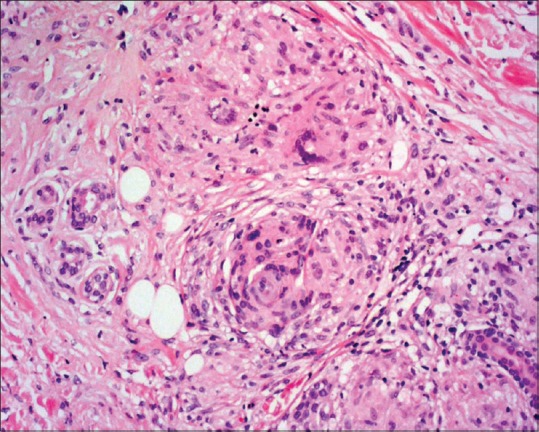
Granulomatous vasculitis showing granuloma formation in perivascular area. H and E staining; high magnification 45×
Diagnostic criteria
Endothelial swelling, fibrinoid necrosis, leukocytoclasia, extravasation of RBCs is needed for diagnosis of LCV.[4]
Verhoff van Gieson stain is necessary to differentiate arteritis from thrombophlebitis in suspected case of MVV. One should also look for panniculitis. If extensive panniculitis is associated, Fite, and PAS stains may be necessary to look for mycobacteria and fungi, respectively. Small vessel vasculitis and presence of vessel wall IgA is a distinct feature of HSP [Figure 11].
Figure 11.
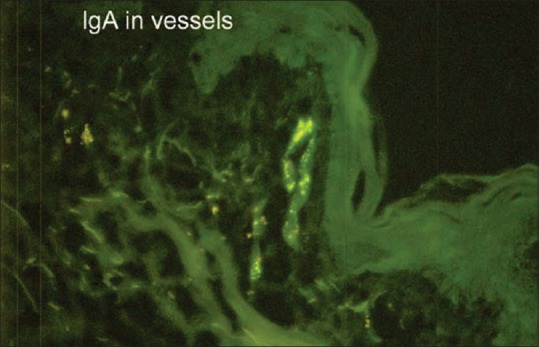
Direct immunofluorescence showing IgA deposition in the perivascular area in HSP (FITC, 100x)
Other organ biopsy
The choice for biopsy site depends on pattern of organ involvement. Biopsy specimen should only be obtained from involved accessible tissue. The yield of blind biopsy is usually low. Nodular skin lesions and involved muscles are preferred biopsy sites for PAN. If peroneal neuropathy is seen on electromyography (EMG), a sural nerve biopsy is indicated. Pulmonary tissue obtained by open lung biopsy is highly specific for WG. Temporal artery is easily accessible for biopsy in suspected cases of GCA. Biopsy is taken from subclavian artery in TA. Since the inflammation is segmental in nature, serial sections are recommended.
In PAN, necrotizing angitis is seen. Since arterioles, capillaries and venules are not involved, glomerulonephritis is not reported. In GPA, destructive granulomatous lesions involving upper respiratory tract are seen (nose, paranasal sinuses and palate) which contain giant cells, along with glomerulonephritis, if there is renal involvement. EGPA shows granulomatous vasculitis with predominant eosinophilic infiltration involving peripheral nerves, muscles and lungs.
Serological tests ELISA and RIA are quite sensitive as well as specific tests to detect serum anti GBM antibodies. In anti GBM disease, deposits of IgG and C3 are seen in capillary walls. Crescentric glomerulonephritis is a characteristic finding.
Birmingham vasculitis activity score (BVAS)[19] may be employed to quantify severity of the disease which has 66 features and 9 organ systems. Scoring for ongoing vasculitic damage can be carried out by employing Vasculitis Damage Index (VDI) and Disease Extent Index (DEI).
Radiology in vasculitis
Angiography is important, especially when large- and medium-sized vessels are involved. In PAN, multiple aneurysmal lesions along hepatic, coeliac, mesenteric, renal and other medium sized arteries; and in TA and GCA decreased lumen, or complete occlusion of large arteries are found. Gadolinium enhanced MR angiography in TA will detect aortic wall structural abnormalities, thus indicating ischemia. F-Fluorodeoxyglucose positron emission tomography (FDG-PET) scan will help in detection of aortic wall inflammation.[20,21,22,23,24]
X-ray, CT scanning and MRI of thorax may be helpful in differential diagnosis of vasculitis with pulmonary involvement such as WG, MPA, CSS and Goodpasteur's syndrome.[23] There are fixed cavitatory nodules in WG, and migrating non-cavitatory nodules in CSS. 2D echocardiography is done to rule out left ventricular dysfunction, myxoma.
Electrophysiological studies like ECG, EEG, electromyography (EMG) help in the diagnosis and aid in assessing the severity of the disease. Subclinical inflammation can also be detected with EEG and EMG.[24]
Treatment
Proper counseling and consent are important before initiation of therapy with immunosuppressive drugs. General measures are enumerated in Table 2.
Table 2.
General measures in vasculitis
| General measures |
|---|
| Bed rest, foot end elevation. |
| Avoidance of smoking. Naproxen or ibuprofen (10 mg/kg/day) is administered to relieve pain |
| Onset of new symptoms and problems encountered as a result of changes in medication or anytime during course of treatment should be immediately notified. |
| In multidisciplinary consultations like dental check-up, patient should provide details of treatment to consulting doctors especially when on immunosuppressants. |
| Live vaccines such as yellow fever, typhoid and measles avoided in active disease states. Flu and Pneumonia vaccines are recommended for vasculitis. For any other types of vaccination it is essential to question the need or reason for these, what they contain and discuss any concerns with the physician. |
| Pre-conception counselling should be done for women with vasculitis wanting children so that any risks to the mother, pregnancy or future baby can be discussed and appropriate plans made. |
Specific therapy
Large-vessel vasculitis treatment options are enumerated in Table 3.
Table 3.
Treatment options in large vessel vasculitis
| Type of vasculitis | Treatment options (precautions and adverse effects) |
|---|---|
| Large vessel vasculitis: GCA and TK | Steroids are the first line agent. |
| EULAR guidelines suggest 1 month of high-dose glucocorticoid therapy (prednisolone 1 mg/kg/day, maximum 60 mg/day). | |
| BSR guidelines: | |
| Uncomplicated GCA (without visual loss or jaw claudication): Prednisolone 40-60 mg (at least 0.75 mg/kg) daily until the resolution of symptoms and laboratory abnormalities. | |
| Complicated GCA (visual loss or a history of amaurosis fugax): 500 mg-1 g of intravenous methylprednisolone per day for 3 days; and at least 60 mg prednisolone daily for patients with established visual loss | |
| Slow tapering of steroids. Rapid relapse is inevitable when daily prednisolone dose reaches 5-10 mg. | |
| Steroid-sparing agents: | |
| Methotrexate: initial dose of 10-15 mg/week, p.o. maximum dose of 25 mg/week | |
| Cyclophosphamide | |
| Mycophenolate mofetil: | |
| Biologicals: | |
| Tocilizumab (IL-6 receptor antagonist): 4 mg/kg every 4 weeks followed by 8 mg/kg intravenously[25] | |
| Infliximab is 3-10 mg/kg every 4 weeks, Adalimumab 40 mg every 2 weeks and etanercept 50 mg/week. | |
| Abatacept: 10 mg/kg IV on days 1, 15, 29, week 8 together with prednisone |
GCA=Giant cell arteritis; TK=Takayasu arteritis; EULAR=European League Against Rheumatism; BSR=British Society for Rheumatology; IL 6=Interleukin 6
Behcet's disease
Oral colchicine along with topical steroids is the initial mode of therapy. When this fails one can prescribe azathioprine, IFN alpha, thalidomide or TNF alpha antagonists.[26] Recently, apremilast 30 mg twice daily was found quite effective though not recommended in EULAR guidelines.[27]
ANCA-associated vasculitis (AAV)
Induction phase
Cyclophosphamide (CYC) is a time-tested standard drug which suppresses B cell. Rituximab (RTX) has specific activity against B cell and is quite effective. Both agents induce prolonged remission in majority of the patients. Opportunistic infections are equally common both with CYC and RTX. RTX is supposed to be effective in inducing remission in patients with relapse.
Maintenance phase
Rituximab 0.5-1 gm every 6 months is a viable option to maintain remission and prevent relapses. Duration of maintenance therapy is highly individualized. Co-trimoxazole has been employed in GPA which prevents respiratory infection induced relapses and also as prophylaxis in pnemocystis pneumonia. Avacopan (CCX168) is an oral small molecule C5a receptor (C5aR) antagonist that blocks neutrophil activation. Avacopan 30 mg, twice daily dosage, can replace high-dose glucocorticoids effectively and safely.[28,29,30,31]
Cryoglobulinemic vasculitis (CV)
In Hep C induced CV, treatment with ribavirin 200-1400 mg/d, combined with sofosbuvir 400 mg/d not only brings down viral load, but also lessens the production of cryoglobulins.[32] Steroids and immunosuppressants have role only in severe disease like mononeuritis, CNS involvement. In Hep C negative patients, rituximab scores over steroids, however, both are used in conjunction, along with CYC, azathioprine as far as efficacy is concerned, especially in patients with severe GN, skin necrosis, multiple neuritis and in patients with life-threatening complications.[33,34,35]
Hypersensitivity vasculitis
Basic investigations include CBC, ESR, CRP, RFT, LFT and urinalysis. Skin biopsy for histopathology and DIF is mandatory to look for immune-mediated disease. Advanced investigations are anti-streptolysin O titers, Hep B and C panels, HIV serology, ANA and complement levels. Up to 10% of patients may have chronic disease lasting between 2 and 4 years. Mild cases may respond to rest, leg elevation and antihistamines. If not, one may need to start oral prednisolone 1-2 mg/kg tapered over a period of 8-12 weeks. Colchicine may be a good option: usual dose is 0.5-1.5 mg/day as needed or as tolerated.[36,37,38]
Dapsone is an often-used option for those with cutaneous disease. Dose range varies from 50 to 150 mg/day in adults. Precautions include a screening G6PD test, starting at a lower dose (25-50 mg/day for adults) and frequent monitoring (weekly until maximum tolerated dose, then monthly for 3 months and eventually quarterly for maintenance). Hydroxychloroquine may be of value in patients not responding or intolerant to dapsone or colchicine. Standard dose of 200-400 mg/day (adults) is used; baseline eye examination and yearly (retinal) examinations are recommended.
Prednisolone is still the most often used drug for those patients with more symptoms or more severe cutaneous disease. Dose is typically 0.5-1.0 mg/kg/day until the cutaneous lesions improve and then slowly tapered over several weeks to months.
Azathioprine may be used as a monotherapy or as a steroid-sparing agent. Dose is most often 50-100 mg/day. A baseline thiopurine methyltransferase (TPMT) enzyme activity level is desirable as well as periodic monitoring of blood cell counts and liver function tests.
Mycophenolate mofetil is a good choice for patients who have become steroid resistant or dependent. The adult dose is most often 500-2500 mg/day in two divided doses. Methotrexate has been reported to be effective as a steroid-sparing agent in some cases but is not used as often as azathioprine. Oral dose ranges from 5 to 25 mg/week. Adding folic acid 1 mg/day is recommended. Cyclosporin A has been effective in patients either intolerant of prednisolone or in those in whom prednisolone is contraindicated. It is used most often in the early stage of disease and tapered in weeks. Dose range in adults is 2.5-5 mg/kg/day given in two divided doses.
Intravenous immune globulin (IVIG) has been used uncommonly because of the cost and non-availability. It is recommended when the disease is severe and where infection or immune deficiency may be present. Precautions in the use of IVIG include checking for IGA deficiencies and the doses are titrated depending upon the renal status.
Plasmapheresis has been used in severe cases refractory to other treatments. Exchanges can vary from 2 to 7 in number depending on response. This is a useful adjuvant therapy in patients with renal involvement
Henoch Schonlein purpura (IgA vasculitis)
Investigations include CBC, ESR, CRP and urinalysis. Thrombocytosis is seen and hematuria, proteinuria indicate renal pathology. Skin biopsy with DIF will help in the diagnosis with the demonstration of IgA1 and C3 deposits in the post capillary venules.[39] Renal biopsy will reveal IgA deposits in glomeruli.[40]
Prednisolone is given in patients with severe cutaneous and joint disease, pulmonary hemorrhage, stroke, nephritic syndrome, and gastrointestinal hemorrhage in the dose of 1-2 mg/kg/day tapered over 8-12 weeks. Steroid is also combined with oral cyclophosphamide 100-200 mg/day for a period of 6-12 months.[39]
Dapsone
It is found effective as a steroid sparing agent as it does not mask intestinal hemorrhage. It is effective in ameliorating cutaneous and joint manifestations. It decreases the synthesis of IgA and Prostaglandin D2.[41] It has no effect on renal disease.
Azathioprine is a second line agent in HSP after DDS and colchicine. It has a steroid sparing advantage. Idiosyncratic reaction, bone marrow toxicity and hepatotoxicity are the commonly observed drawbacks.[42]
Rituximab
It is emerging as a frontline drug in HSP with renal involvement. It is given intravenously. Two doses, 1 g each are given, second dose being administered, 2 weeks after first dose. It gives prolonged remission.[43,44]
Mycophenolate mofetil
It is employed in steroid dependent, steroid resistant cases of HSP. It is given orally at the dosage of 30 mg/kg/day in two divided doses. It is effective in HSP patients with renal complications.
Plasmapheresis
It is reserved for the patients with progressive nephropathy, not responding to steroids and other immunosuppressives. It should be started within 2 weeks of onset of renal complications.[45]
Cutaneous and joint manifestations are managed by NSAIDs and dapsone and steroids. Renal involvement is treated by steroid + CYC, mycophenolate mofetil, rituximab, plasmapheresis, dialysis and finally renal transplant in that sequence.
Urticarial vasculitis (UV)
CBC, ESR, CRP and urinalysis are the initial investigations. C3, C4, C1q, anti C1q antibodies, CH50, ANA, ds DNA are also carried out, if facilities are available.[46,47] Skin biopsy with DIF will demonstrate IgG and C3 at basement membrane zone if associated SLE is present. RFT, chest x-ray, ECG and eye examination will reveal renal, pulmonary, cardiac and ocular involvement, respectively. UV can disclose purpuric dots or globules in a patchy orange-brown background dermatoscopically corresponding to extravasation and degradation of red blood cells due to leukocytoclastic vasculitis.
Treatment of Normocomplementemic UV (NUV) and Hypocomplementemic UV (HUV) includes using prednisone and colchicine which can be effective to gain control of the disease. The use of glucocorticoids combined with dapsone, colchicine or hydroxychloroquine to aid in their systemic disease have proven to be beneficial during the initial stages in patients who have mild to moderate disease.[47,48,49] Additionally, biological agents that interrupt the IL-1 pathway may also be of benefit. These include anakinra (monoclonal recombinant IL-1 receptor antagonist protein. Canakinumab (monoclonal antibody against IL-1β) has also been shown to be beneficial.[50] Rituximab (monoclonal antagonist directed against the CD20 molecule on B lymphocytes) will be an exciting option.
Mycophenolate mofetil has also been shown to be beneficial. Methotrexate used as steroid sparing is also effective. Azathioprine in combination with prednisone has shown to have significant improvement in patients with nephropathy and well as skin involvement in patients with HUV. Cyclosporine has also been effective in treating HUV especially in patients suffering from pulmonary and renal involvement and used to taper patients off glucocorticoids.
Kawasaki disease
The most effective regimen includes high dosage (2 g/kg) of intravenous immunoglobulin (IVIG) infusion with aspirin (80-100 mg/kg/day) to achieve anti-inflammatory and antiplatelet effect.[51] IVIG should be infused slowly over 8-12 hours and repeated if fever persists 36-48 hours after the first infusion.[51] High dosage aspirin is recommended during the first 48 hours. Later, aspirin dosage (5 mg/kg/day) is indicated until long-term follow up is completed.[51,52]
Baseline echocardiographic coronary evaluation helps to stratify the coronary risk and also determines the antithrombotic treatment schedule. Z scores <2 implies no coronary artery lesions and low dosage aspirin for 4-6 weeks is given, with repeated echocardiographic evaluations in 2 weeks and 6-8 weeks after the onset of illness.[53,54] Z scores >2.5 has higher coronary risks with dilated coronary artery diameters need a lifelong follow-up for surveillance of worsening aneurysms and antithrombotic treatment based on their coronary risk.[53,54]
Conclusion
Vasculitis syndrome needs comprehensive approach for its successful management. Detailed history taking, thorough cutaneous and systemic examination followed by basic and relevant advanced investigations will help in arriving at diagnosis. Treatment needs to be individualized based on severity of cutaneous and systemic pathology.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
Acknowledgements
We would like to acknowledge Dr Eswari L, Associate professor, Department of Dermatology, Bangalore Medical college and research institute and Dr Sahana MS, consultant dermatologist, Indira Gandhi institute of child health for providing the clinical and histopathological photographs.
References
- 1.Saigal R, Agrawal A, Dadhich D. Vasculitis syndrome: An approach. J Assoc Physicians India. 2004;54:645–8. [PubMed] [Google Scholar]
- 2.Vasculitis Treatment in India, Stem cell therapy for vasculitis. [Last retrieved on 2018 Apr 17]. Available from: https://www.giostar.com/Therapy/vasculitis .
- 3.Khetan P, Sethuraman G, Khaitan BK, Sharma VK, caR, Dinda AK. An etiological and clinicopathological study on cutaneous vasculitis. Indian J Med Res. 2012;135:107–13. doi: 10.4103/0971-5916.93432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Carlson JA, Ng BT, Chen KR. Cutaneous vasculitis update: Diagnostic criteria, classification, epidemiology, etiology, pathogenesis, evaluation and prognosis. Am J Dermatopathol. 2005;27:504–28. doi: 10.1097/01.dad.0000181109.54532.c5. [DOI] [PubMed] [Google Scholar]
- 5.Carlson JA, Cavaliere LF, Grant-Kels JM. Cutaneous vasculitis: Diagnosis and management. Clin Dermatol. 2006;24:414–29. doi: 10.1016/j.clindermatol.2006.07.007. [DOI] [PubMed] [Google Scholar]
- 6.Jennette JC, Falk RJ, Bacon PA, Basu N, Cid MC, Ferrario F, et al. 2012 Revised International chapel hill consensus conference nomenclature of vasculitides. Arthritis Rheum. 2013;65:1–11. doi: 10.1002/art.37715. [DOI] [PubMed] [Google Scholar]
- 7.Lintermans LL, Stegeman CA, Heeringa P, Abdulahad WH. T cells in vascular inflammatory diseases. Front Immunol. 2014;5:504. doi: 10.3389/fimmu.2014.00504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Okazaki T, Shinagawa S, Mikage H. Vasculitis syndrome: Diagnosis and therapy. J Gen Fam Med. 2017;18:72–8. doi: 10.1002/jgf2.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fain O, Hamidou M, Cacoub P, Godeau B, Wechsler B, Paries J, et al. Vasculitides associated with malignancies: Analysis of sixty patients. Arthritis Rheum. 2007;57:1473–80. doi: 10.1002/art.23085. [DOI] [PubMed] [Google Scholar]
- 10.Khasnis A, Molloy E. Mimics of primary systemic vasculitides. Int J Clin Rheumatol. 2009;4:597–609. [Google Scholar]
- 11.Gross WL, Schmitt WH, Csernok E. ANCA and associated diseases: Immunodiagnostic and pathogenetic aspects. Clin and Exp Immunol. 1993;91:1–12. doi: 10.1111/j.1365-2249.1993.tb03345.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hagen EC, Daha MR, Hermans J, Andrassy K, Csernok E, Gaskin G, et al. Diagnostic value of standardized assays for anticytoplasmic antibodies in idiopathic systemic vasculitis. Clin Nephrol. 1998;53:743–53. doi: 10.1046/j.1523-1755.1998.00807.x. [DOI] [PubMed] [Google Scholar]
- 13.Nolle B, Specks U, Ludemann J, Rohrback MS, Deremee RA, Gross WL. Anticytoplasmic autoantibodies: Their immunodiagnsotic value in Wegner's granulomatosis. Ann Intern Med. 1998;111:28–40. doi: 10.7326/0003-4819-111-1-28. [DOI] [PubMed] [Google Scholar]
- 14.Falk RJ, Jennete JC. Antineutrophil autoantibodies with specificity for myeloperoxidase in patients with systemic vasculitis and idiopathic necrotising and crescentic glomerulonephritis. N Eng J Med. 1988;318:1651–57. doi: 10.1056/NEJM198806233182504. [DOI] [PubMed] [Google Scholar]
- 15.Choi HK, Merkel PA, Walker AM, Niles JL. Drug induced antineutrophil cytoplasmic antibody positive vasculitis. Arthritis Rheum. 2000;43:405–13. doi: 10.1002/1529-0131(200002)43:2<405::AID-ANR22>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 16.Malaviya AN. Vasculitis - an update. J Indian Rheumat Assoc. 2001;9:3. [Google Scholar]
- 17.Chen KR, Carlson JA. Clinical approach to cutaneous vasculitis. Am J Clin Dermatol. 2008;9:71–92. doi: 10.2165/00128071-200809020-00001. [DOI] [PubMed] [Google Scholar]
- 18.Poornimambaa M, Asokan N, Augustine J. Utility of direct immunofluroscence in small vessel vasculitis of the skin. Indian Dermatol Online J. 2017;8:515–7. doi: 10.4103/idoj.IDOJ_298_16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kermani TA, Cuthbertson D, Carette S, Hoffman GS, Khalidi NA, Koening CL, et al. The Birmingham vasculitis activity score as a measure of disease activity in patients with giant cell arteritis. J Rheumatol. 2016;43:1078–84. doi: 10.3899/jrheum.151063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Muratore F, Pipitone N, Salvarani C, Schmidt WA. Imaging of vasculitis: State of the art. Best Pract Res Clin Rheumatol. 2016;30:688–706. doi: 10.1016/j.berh.2016.09.010. [DOI] [PubMed] [Google Scholar]
- 21.Schmidt WA, Kraft HE, Vorpahl K, Volker L, Gromnica EJ. Color duplex ultrasonography in the diagnosis of temporal arteritis. N Engl J Med. 1997;337:1336–42. doi: 10.1056/NEJM199711063371902. [DOI] [PubMed] [Google Scholar]
- 22.Roman MJ, Naqvi TZ, Gardin JM, Hermann GM, Jaff M, Mohler E. Clinical application of non-invasive vascular ultrasound in cardiovascular risk stratification: A report from the American society of echocardiography and the society for vascular medicine and biology. Vasc Med. 2006;11:201–11. doi: 10.1177/1358863x06070511. [DOI] [PubMed] [Google Scholar]
- 23.Aschwanden M, Daikeler T, Kesten F, Baldi T, Benz D, Tyndall A, et al. Temporal artery compression sign-a novel ultrasound finding for the diagnosis of giant cell arteritis. Ultraschall Med. 2013;34:47–50. doi: 10.1055/s-0032-1312821. [DOI] [PubMed] [Google Scholar]
- 24.Schmidt WA. Technology Insight: The role of color and power. Doppler ultrasonography in rheumatology. Nat Clin Pract Rheumatol. 2007;3:35–42. doi: 10.1038/ncprheum0377. [DOI] [PubMed] [Google Scholar]
- 25.Mollan SP, Horsburgh J, Dasgupta B. Profile of tocilizumab and its potential in the treatment of giant cell arteritis. Eye Brain. 2018;10:1–11. doi: 10.2147/EB.S127812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Eleftheriou D, Melo M, Marks SD, Tullus K, Sills J, Cleary G, et al. Biologic therapy in primary systemic vasculitis of the young. Rheum (Oxford) 2009;48:978–86. doi: 10.1093/rheumatology/kep148. [DOI] [PubMed] [Google Scholar]
- 27.Saleh Z, Arayssi T. Update on the therapy of behcets disease. Ther Adv Chronic Dis. 2014;5:112–34. doi: 10.1177/2040622314523062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ntatsaki E, Carruthers D, Chakravarty K, D’Cruz D, Harper L, Jayne D, et al. BSR and BHPR guideline for the management of adults with ANCA-associated vasculitis. Rheumatology. 2014;5:2306–9. doi: 10.1093/rheumatology/ket445. [DOI] [PubMed] [Google Scholar]
- 29.Yates M, Watts RA, Bajema IM, Cid MC, Crestani B, Hauser T, et al. EULAR/ERA-EDTA recommendations for the management of ANCA-associated vasculitis. Ann Rheum Dis. 2016;75:1583–94. doi: 10.1136/annrheumdis-2016-209133. [DOI] [PubMed] [Google Scholar]
- 30.Stone JH, Merkel PA, Spiera R, Seo P, Langford CA, Hoffman GS, et al. Rituximab versus cyclophosphamide for ANCA-associated vasculitis. N Engl J Med. 2010;363:221–32. doi: 10.1056/NEJMoa0909905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jones RB, Tervaert JW, Hauser T, Luqmani R, Morgan MD, Peh CA, et al. Rituximab versus cyclophosphamide in ANCA-associated renal vasculitis. N Engl J Med. 2010;363:211–20. doi: 10.1056/NEJMoa0909169. [DOI] [PubMed] [Google Scholar]
- 32.Sise ME, Bloom AK, Wisocky J, Lin MV, Gustafson JL, Lundquist AL, et al. Treatment of hepatitis C virus-associated mixed cryoglobulinemia with direct-acting antiviral agents. Hepatology. 2016;63:408–17. doi: 10.1002/hep.28297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mukhtyar C, Guillevin L, Cid MC, Dasgupta B, de Groot K, Gross W, et al. European vasculitis study group EULAR recommendations for the management of primary small and medium vessel vasculitis. Ann Rheum Dis. 2009;68:310–7. doi: 10.1136/ard.2008.088096. [DOI] [PubMed] [Google Scholar]
- 34.Ferri C, Ramos-Casals M, Zignego AL, Arcaini L, Roccatello D, Antonelli A, et al. International diagnostic guidelines for patients with HCV-related extrahepatic manifestations. A multidisciplinary expert statement. Autoimmun Rev. 2016;15:1145–60. doi: 10.1016/j.autrev.2016.09.006. [DOI] [PubMed] [Google Scholar]
- 35.Ostojic P, Jeremic IR. Managing refractory cryoglubulinemic vasculitis: Challenges and solutions. J Inflamm Res. 2017;10:49–54. doi: 10.2147/JIR.S114067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Baigrie D, Crane JS. StatPearls [Internet] Treasure Island (FL): StatPearls Publishing; 2018. Jan, [Last retrieved on 2018 Apr 25]. Leukocytoclastic Vasculitis (Hypersensitivity Vasculitis) [Updated 2018 Feb 17] Available from: https://www.ncbi.nlm.nih.gov/books/NBK482159/ [Google Scholar]
- 37.Gota CE, Calabrese LH. Diagnosis and treatment of cutaneous leukocytoclastic vasculitis. Int J Clin Rheumatol. 2013;8:49–60. [Google Scholar]
- 38.Sais G, Vidaller A, Juccgla A, Gallardo F, Jordi P. Colchicine in the treatment of cutaneous leukocytoclastic vasculitis: Results of a prospective, randomized controlled trial. Arch Dermatol. 1995;13:1399–402. [PubMed] [Google Scholar]
- 39.Pasha AB, Zhou GP. Recent advances in the treatment of Henoch-Schönlein purpura. Sci Lett. 2017;5:131–7. [Google Scholar]
- 40.Fauci AS. The vasculitis syndromes. In: Braunwald E, Isselbacher KJ, Petersdorf RG, Wilson JD, Martin JB, Fauci AS, editors. Harrison's Book of Internal Medicine. 11th ed. New York NY: McGraw-Hill; 1987. p. 1441. [Google Scholar]
- 41.Iqbal H, Evans A. Dapsone therapy for Henoch-Schonlein purpura: A case series. Arch Dis Child. 2005;90:985–6. doi: 10.1136/adc.2004.061598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shin JI, Park JM, Shin YH, Kim JH, Lee JS, Kim PK, et al. Can Azathioprine and steroids alter the progression of severe Henoch-Schönlein nephritis in children? Pediatr Nephrol. 2005;20:1087–92. doi: 10.1007/s00467-005-1869-x. [DOI] [PubMed] [Google Scholar]
- 43.Bellan M, Pirisi M, Sainaghi PP. Long-term remission of corticosteroid- and cyclophosphamide-resistant Henoch-Schönlein purpura with rituximab. Scand J Rheumatol. 2015;27:1–2. doi: 10.3109/03009742.2015.1058417. [DOI] [PubMed] [Google Scholar]
- 44.Kattah AG, Fervenza FC, Roccatello D. Rituximab-based novel strategies for the treatment of immune-mediated glomerular diseases. Autoimmun Rev. 2013;12:854–9. doi: 10.1016/j.autrev.2012.09.002. [DOI] [PubMed] [Google Scholar]
- 45.Shenoy M, Ognjanovic MV, Coulthard MG. Treating severe Henoch-Schönlein and IgA nephritis with plasmapheresis alone. Pediatr Nephrol. 2007;22:1167–71. doi: 10.1007/s00467-007-0498-y. [DOI] [PubMed] [Google Scholar]
- 46.Davis MDP, Daoud MS, Kirby B, Gibson LE, Rogers RS. Clinicopathologic correlation of hypocomplementemic and normocomplementemic urticarial vasculitis. J Am Acad Derm. 1998;38:899–905. [PubMed] [Google Scholar]
- 47.Bulva J, Simon R. Hypocomplementemic urticarial vasculitis (HUV) and Hypocomplementemic urticarial vasculitis syndrome (HUVS); background, pathogenesis, diagnosis, laboratory testing, management and treatment. J Autoimmun Res. 2017;4:1028. [Google Scholar]
- 48.Mehregan DR, Hall MJ, Gibson LE. Urticarial vasculitis: A histopathologic and clinical review of 72 cases. J Am Acad Dermatol. 1992;26:441–8. doi: 10.1016/0190-9622(92)70069-r. [DOI] [PubMed] [Google Scholar]
- 49.Venzor J, Lee WL, Huston DP. Urticarial vasculitis. Clin Rev Allergy Immunol. 2002;23:201. doi: 10.1385/CRIAI:23:2:201. [DOI] [PubMed] [Google Scholar]
- 50.Krause K, Mahamed A, Weller K, Metz M, Zuberbier T, Maurer M. Efficacy and safety of canakinumab in urticarial vasculitis: An open-label study. J Allergy Clin Immunol. 2013;132:751–4. doi: 10.1016/j.jaci.2013.04.008. [DOI] [PubMed] [Google Scholar]
- 51.Newburger JW, Takahashi M, Gerber MA, Gewitz MH, Tani LY, Burns JC, et al. Diagnosis, treatment, and long-term management of Kawasaki disease: A statement for health professionals from the committee of rheumatic fever, endocarditis and Kawasaki disease, council on cardiovascular disease in the young, American heart association. Circulations. 2004;110:2747–71. doi: 10.1161/01.CIR.0000145143.19711.78. [DOI] [PubMed] [Google Scholar]
- 52.Ayusawa M, Sonobe T, Uemura S, Ogawa S, Nakamura Y, Kiyosawa N, et al. Revision of diagnostic guidelines for Kawasaki disease (the 5th revised edition) Pediatr Int. 2005;47:232–4. doi: 10.1111/j.1442-200x.2005.02033.x. [DOI] [PubMed] [Google Scholar]
- 53.Newburger JW, Takahashi M, Burns JC. Kawasaki disease. J Am Coll Cardiol. 2016;67:1738–49. doi: 10.1016/j.jacc.2015.12.073. [DOI] [PubMed] [Google Scholar]
- 54.Manlhiot C, Millar K, Golding F, McCrindle BW. Improved classification of coronary artery abnormalities based only on coronary artery z-scores after Kawasaki disease. Pediatric cardiology. 2010;31:242–9. doi: 10.1007/s00246-009-9599-7. [DOI] [PubMed] [Google Scholar]