

Summary
Disturbances in autophagy and stress granule dynamics have been implicated as potential mechanisms underlying inclusion body myopathy (IBM) and related disorders. Yet, the roles of core autophagy proteins in IBM and stress granule dynamics remain poorly characterized. Here, we demonstrate that disrupted expression of the core autophagy proteins ULK1/2 in mice causes a vacuolar myopathy with ubiquitin and TDP-43–positive inclusions, similar to that caused by mutations in VCP, the most common cause of familial IBM. Mechanistically, we show that ULK1/2 localize to stress granules and phosphorylate VCP, thereby increasing VCP’s activity and ability to disassemble stress granules. These data suggest that VCP dysregulation and defective stress granule disassembly contribute to IBM-like disease in Ulk7/2-deficient mice. In addition, stress granule disassembly is accelerated by an ULK1/2 agonist, suggesting ULK1/2 as targets for exploiting higher-order regulation of stress granules for therapeutic intervention of IBM and related disorders.
eTOC Blurb
Wang et. al, demonstrate that the autophagy-inducing kinases ULK1 and ULK2 regulate stress granule disassembly by phosphorylating and activating the AAA+ ATPase VCP. Defects in this pathway contribute to the development of inclusion body myopathy-like disease in mice.
Graphical Abstract
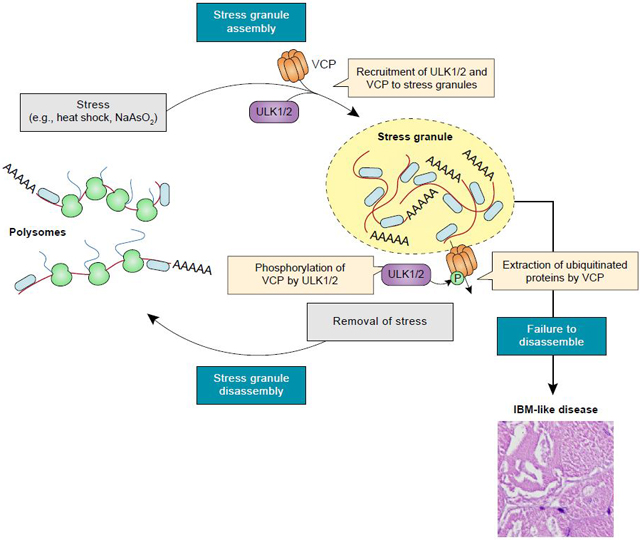
INTRODUCTION
Aberrant stress granule dynamics and defects in autophagy are thought to underlie the pathogenesis of a related group of degenerative diseases, including inclusion body myopathy (IBM), amyotrophic lateral sclerosis (ALS), frontotemporal dementia (FTD), and the overlap syndrome multisystem proteinopathy (MSP, also known as IBM with early-onset Paget disease and FTD) (Taylor, 2015). This family of diseases shares causative mutations in genes that fall largely into two functional categories: those that encode RNA-binding proteins (e.g., TDP-43, hnRNP A1, hnRNP A2, MATR3, FUS, TIA1) and those that encode ubiquitin-binding proteins involved in autophagy (e.g., VCP/p97, SQSTM1/p62, UBQLN2, OPTN) (Taylor, 2015). IBM, MSP, and many subtypes of ALS/FTD are characterized by the intracellular accumulation of ubiquitin-positive aggregates that are also positive for disease-associated RNA-binding proteins and autophagy-related proteins in affected tissues (Meyer and Weihl, 2014; Taylor, 2015).
Studies of disease-associated mutations in RNA-binding proteins have revealed that these mutations alter the dynamics of multiple membrane-less organelles, including stress granules (Mackenzie et al., 2017; Molliex et al., 2015; Taylor et al., 2016), increasing the propensity of certain stress granule proteins (e.g., TDP-43) to transition to pathologic, amyloid-like fibrils (Mackenzie et al., 2017). Nondynamic stress granules may be a central feature of disease, as supported by recent data indicating that persistent stress granules are intrinsically cytotoxic and evolve over time into the characteristic cytoplasmic inclusions seen in patients with IBM, ALS/FTD, or MSP (Zhang et al., 2018b).
Several lines of evidence have suggested that ubiquitin-dependent autophagy regulates the remodeling and/or clearance of stress granules and that disease-causing mutations in patients with IBM or a related disease may impinge on this process. First, an unbiased genetic screen performed in yeast highlighted the importance of Cdc48 (the yeast homologue of VCP) and selective autophagy in the degradation of stress granules (Buchan et al., 2013). VCP is a AAA-ATPase that regulates diverse cellular processes by interacting with ubiquitinated proteins via cofactors and facilitating their extraction from membranes or protein complexes for degradation or recycling (Meyer et al., 2012; Meyer and Weihl, 2014). Disease-causing mutations in VCP impair its autophagy-related functions and cause the abnormal persistence of stress granules (Buchan et al., 2013; Seguin et al., 2014; Turakhiya et al., 2018). Second, in addition to VCP, TBK1, SQSTM1, UBQLN2, and OPTN are all proteins that regulate ubiquitin-dependent autophagy and whose autophagy-related functions are disrupted by disease-causing mutations in patients with TDP-43–positive pathology (Deng et al., 2017; Majcher et al., 2015). Moreover, like VCP, SQSTM1 and UBQLN2 have been implicated in the regulation of stress granule dynamics. While it remains to be determined whether these proteins play a role in autophagic clearance of stress granules, it was recently reported that both SQSTM1 (Chitiprolu et al., 2018) and UBQLN2 (Dao et al., 2018) localize to stress granules and undergo ubiquitin-regulated phase separation (Dao et al., 2018; Sun et al., 2018a).This finding suggests a mechanism whereby these ubiquitin-binding autophagy adapters are recruited to stress granules and, upon binding polyubiquitin chains, extract ubiquitinated clients from stress granules, perhaps facilitating their subsequent delivery to nascent autophagosomes.
Together, these observations suggest a possible direct link between the two groups of proteins mutated in patients with IBM, ALS, FTD, or MSP, in which the persistence of stress granules in affected tissues may be caused by disturbances in stress granule dynamics (by mutations in RNA-binding proteins) or by a failure to clear stress granules or aggregation-prone stress granule components (by mutations in autophagy-related proteins). If this relation is true, one would expect that genetic manipulation of core components of the autophagy pathway might lead to the persistence of stress granules in cultured cells and the development of TDP-43–and ubiquitin-positive pathology in model organisms. Yet, the roles of core components of the autophagy machinery, such as ULK/Atg1, in the dynamics or degradation of stress granules in mammalian cells and their contributions to the pathogenesis of the IBM/ALS/FTD/MSP spectrum of diseases remain poorly characterized. ULK1 and ULK2 (hereafter “ULK1/2”) are druggable members of the ULK/Atg1 family of kinases that exert higher-order regulation over macroautophagy in mammals by phosphorylating substrates such as ATG13, VPS34, BECN1, ATG14, and SQSTM1 (Mercer et al., 2018; Wang and Kundu, 2017). In their canonical role in inducing autophagy, ULK1/2 function within complexes consisting of at least three additional partners (ATG13, RB1CC1, and ATG101), and recent studies have identified the protein encoded by C9ORF72 as a variable addition to this core complex (Webster et al., 2016; Yang et al., 2016).
To investigate the role of ULK1/2 in homeostatic mechanisms that may be disrupted in the IBM/ALS/FTD/MSP spectrum of diseases, we examined the physiologic consequences of Ulk1/2 deficiency in mice. We demonstrate that mice deficient in Ulk1/2 develop IBM-like disease with inclusions that are positive for ubiquitin, as well as the stress granule markers TIA1 and TDP-43. Pursuing the underlying mechanism of this phenotype using unbiased proteomic analyses, we show that the ULK1 interactome is enriched in stress granule proteins, including VCP. Furthermore, we show that unlike other autophagy-related proteins, ULK1/2 regulate the disassembly of stress granules by phosphorylating VCP. Finally, we demonstrate that enhancing ULK1/2 activity not only accelerates the normal dissolution of stress granules but also promotes the resolution of persistent stress granules caused by the expression of disease-associated mutant RNA-binding proteins.
RESULTS
Ulk1/2 Deficiency Leads to IBM-Like Disease in Mice
Consistent with their partially redundant functions, mice lacking either Ulk1 or Ulk2 survive to older than 1 year with nondebilitating deficits (Cheong et al., 2014; Kundu et al., 2008), whereas mice lacking both Ulk1 and Ulk2 die prenatally or shortly after birth (Cheong et al., 2014; Wang et al., 2017), similar to those with conventional germline knockout of nonredundant autophagy genes (e.g., Atg13, Rb1cc1, Becn1, Atg5, and Atg7) (Gan et al., 2006; Kaizuka and Mizushima, 2016; Komatsu et al., 2005; Kuma et al., 2004; Yue et al., 2003). These findings necessitated the use of targeted conditional knockout approaches to investigate the role of ULK1/2 in specific tissues. While generating and analyzing Ulk1/2 conditional double knockout (cDKO) animals (Joo et al., 2016), we observed that tissues from Ulk2flox/flox mice expressed significantly lower levels of Ulk2 mRNA (Figure S1A) with no overt change in splice junction usage compared to tissues from Ulk2+/+ mice (data not shown). This suggests that the introduction of the loxP sites flanking exons 1–3 of Ulk2 rendered it hypomorphic. Ulk1−/−Ulk2−/flox(hereafter “Ulk1/2 hypomorph”) mice retaining a single floxed Ulk2 allele showed retarded growth and shortened life spans compared to Ulk1+/−Ulk2−/flox (hereafter “control”) and Ulk1−/−Ulk2+/− (“control 2”) mice (Figures S1B and S1C). Necropsies performed on these animals provided a unique opportunity to identify tissues and organ systems particularly sensitive to the loss of Ulk1/2 expression. These studies revealed pathologic alterations in muscle, brain, spinal cord, liver, and kidney from 14-month-old Ulk1/2 hypomorphs that were not present in age-matched controls (Figure S1D). In addition to vacuoles in skeletal muscle fibers and dystrophic axons in deep cerebellar nuclei and spinal cord, we observed hepatocyte hypertrophy and pigmentation similar to that reported in liver-specific Ulk1/2 cDKO mice (Sun et al., 2018b), splenomegaly with extramedullary hematopoiesis (data not shown), and a defect in reticulocyte maturation (data not shown), similar to the phenotypes observed in Ulk1−/− mice (Kundu et al., 2008).
The muscle pathology observed in Ulk1/2 hypomorph mice was reminiscent of pathology found in patients with IBM, prompting us to pursue more detailed characterization of the muscular phenotype in these mice. Hallmarks of the muscle disease in patients with MSP or sporadic IBM include muscle weakness, myopathy with vacuolar degeneration, inclusions that are positive for ubiquitin, SQSTM1, TDP-43, and other markers (e.g., phosphorylated tau, amyloid, LC3B), and tubulofilamentous aggregates by electron microscopy (Bucelli et al., 2015; Watts et al., 2004; Weihl et al., 2015). Many of these features are also found in mice expressing mutant forms of VCP (Badadani et al., 2010; Custer et al., 2010; Weihl et al., 2007), the most common cause of MSP.
We began by examining serum creatine kinase levels and readouts of muscle function in Ulk1/2 hypomorphs. Similar to findings from patients with IBM (Hilton-Jones and Brady, 2016), Ulk1/2 hypomorph mice showed a mild increase in serum creatine kinase levels compared to controls at early time points (i.e., <16 weeks of age) (Figure S1E). Ulk1/2 hypomorphs also showed a significant deficit in grip strength, as assessed by the hanging wire test, at over 24 weeks of age (Figure S1F).
Pathologic examination of muscle tissues in these mice revealed that the phenotypes were associated with inclusions in fast-twitch fibers, as revealed by fiber typing performed on epaxial (data not shown) and quadriceps muscles (Figure S1G ) using antibodies against type-specific myosin heavy chains. Given our finding that inclusions were restricted to type II (fast) myofibers, we restricted our subsequent analyses to quadriceps muscles due to the prominence of type II fibers in those muscles.
Deeper characterization of quadriceps muscles in Ulk1/2 hypomorph mice confirmed further similarities between these mice, mice expressing mutant VCP, and patients with IBM. First, hematoxylin and eosin–stained (H&E) sections of quadriceps from 20-week-old Ulk1/2 hypomorph mice demonstrated the presence of vacuoles, which were positively labeled by modified Gomori trichrome staining (Figure 1A), similar to the “rimmed” vacuoles typically found in patients with IBM and mice expressing mutant VCP. These sections also showed evidence of myogenic myopathy, including some irregularity in fiber size and central nuclei (Figures 1A and S1H). Second, immunohistochemical analyses revealed that the vacuoles in skeletal muscles of Ulk1/2 hypomorph mice contained inclusions that were positive for ubiquitin, SQSTM1, and LC3B (Figure 1B). Furthermore, the RNA-binding proteins TDP-43 and TIA1, whose expression is typically restricted to nuclei in normal muscle, were aberrantly localized to sarcoplasmic patches (Figures 1B and 1C, box 1) and SQSTM1-positive inclusions (Figure 1C, box 2). Other RNA-binding proteins, such as hnRNP A2B1 and FUS, showed normal nuclear localization in the muscles of Ulk1/2 hypomorphs (Figure 1D). Reactivity to antibodies that detect phosphorylated tau (SMI-31) and β-amyloid (1–42) was also present in the inclusion bodies (Figure S1I). Third, ultrastructural studies showed disruption of the normal myofiber architecture by an infiltrate composed of membranous structures with electron-dense material, as well as an expansion of subsarcolemmal mitochondria, occasionally swollen and filled with degenerative cisternae (Figure 1E). Aggregates of tubulofilamentous material were also identified within muscle fibers of Ulk1/2 hypomorphs (Figure 1E), similar to the filamentous structures often observed in affected tissues of patients with IBM or ALS (Hilton-Jones and Brady, 2016). Together, these findings highlight the similarities between the muscle disease in Ulk1/2 hypomorphs, mice expressing mutant VCP, and patients with IBM.
Figure 1. Ulk1/2 Deficiency Causes IBM-Like Disease in Mice.
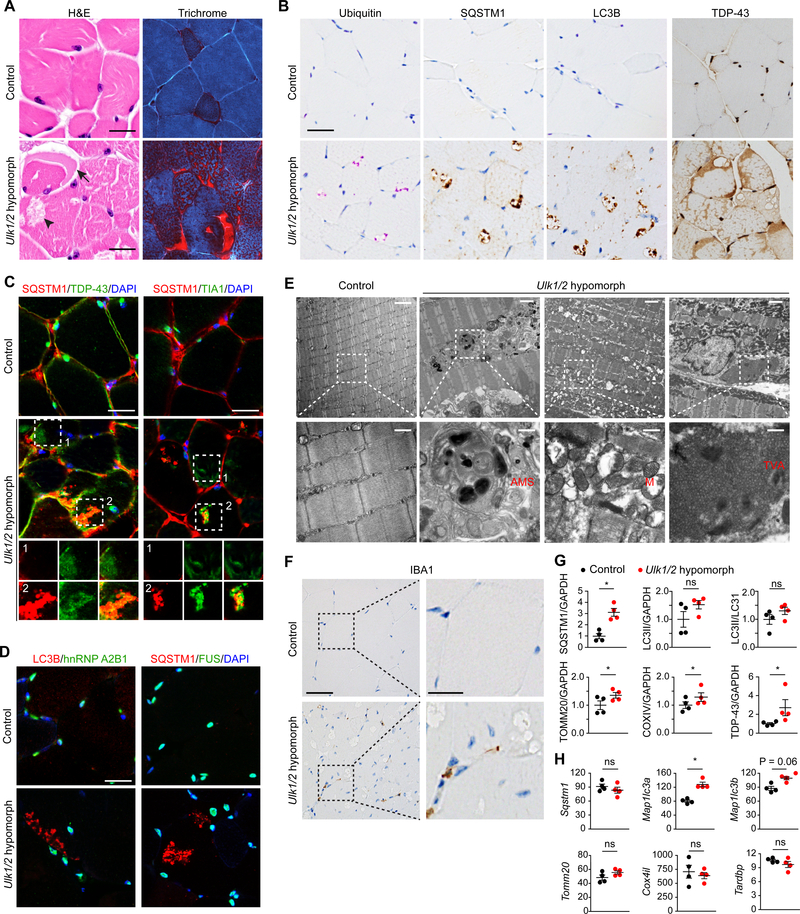
(A) Cross-sections of quadriceps from control (Ulk1+/−Ulk2−/flox) and Ulk1/2 hypomorph (Ulk1−/− Ulk2−/flox) mice at 24 weeks were stained with H&E and modified Gomori trichrome staining. Arrow indicates a crescentic inclusion and arrowhead indicates a vacuole. Scale bars, 25 μm.
(B) Cross-sections of quadriceps from control and Ulk1/2 hypomorph mice at 24 weeks were stained with antibodies against ubiquitin, SQSTM1, LC3B, and TDP-43. Scale bar, 25 μm.
(C-D) Frozen sections of quadriceps from 24-week-old control and Ulk1/2 hypomorph mice were costained with SQSTM1 and TDP-43 (C), SQSTM1 and TIA1 (C), LC3B and hnRNP A2B1 (D), or SQSTM1 and FUS (D). Scale bars, 25 μm.
(E) Ultrastructural studies on quadriceps of 24-week-old control and Ulk1/2 hypomorph mice. Top scale bars, 1 μm; bottom scale bars, 500 nm. AMS, abnormal membranous structures; M, abnormal mitochondria; TVA, tubulovesicular aggregates.
(F) Representative images of immunostaining against IBA1 on sections of quadriceps from control and Ulk1/2 hypomorph mice at 24 weeks of age. Left scale bar, 25 μm; right scale bar, 10 μm.
(G) Protein lysates prepared from quadriceps of control and Ulk1/2 hypomorph mice at 24 weeks of age were analyzed by immunoblotting against SQSTM1, LC3B, TOMM20, COXIV, and TDP-43. Relative band intensities were quantified by densitometry and are presented as mean ± SEM. ns, not significant; *P <0.05 by Student’s t-test.
(H) The mRNA levels (FPKM) of Sqstm1, Map1/c3a, Map1/c3b, Tomm20, Cox4il, and Tardbp were determined by RNA sequencing in quadriceps prepared from 24-week-old control and Ulk1/2 hypomorph mice. Data are presented as mean ± SEM. ns, not significant; *P <0.05 by Student’s t-test.
See also Figure S1.
Inflammatory infiltrates enriched in cytotoxic T cells are often present in muscle biopsies of patients with sporadic IBM but are not a feature of the myopathy in patients with MSP. We observed a mild increase (0.69/mm2 vs 3.62/mm2) in IBA1-positive activated macrophages in sections from 14-month-old Ulk1/2 hypomorphs (Figure 1F) but failed to detect an increase in CD3-positive T cells or PAX5-positive B cells (data not shown). The increase in IBA1 positivity was similar to that observed in mice expressing mutant VCP (Badadani et al., 2010).
The mitochondrial abnormalities observed by electron microscopy prompted us to perform additional studies, including cytochemical stains for COXIV, SDH (Figure S1J), and NADH (data not shown), which revealed higher levels of mitochondrial activity in the myofibers of Ulk1/2 hypomorphs, particularly in the type II fibers, compared to those in control animals. Consistent with a defect in ULK1/2-dependent autophagy and/or lysosomal function, immunoblot analyses showed increased steady-state levels of the mitochondrial proteins TOMM20 and COXIV, as well as SQSTM1 (Figures 1G and S1K), which were not due to increases in Tomm20, Cox4il, or Sqstm1 mRNA (Figure 1H). The similar increase in TDP-43 protein, but not Tardp mRNA, raised the possibility that TDP-43 turnover may also be regulated in an ULK1/2-dependent manner (Figures 1G, 1H, and S1K). Although the autophagy defect resulting from ULK1/2 deficiency is associated with reduced levels of lipidated LC3 (based on the ratio of LC3-II and LC3-I, or LC3-II normalized to a loading control such as GAPDH) in cultured cells (Joo et al., 2016; McAlpine et al., 2013), the levels of LC3-II were not significantly different in muscle extracts prepared from Ulk1/2 hypomorphs and controls (Figure 1G and S1K). This result may be due to a defect in lysosome-mediated turnover of LC3 and/or the observed increase in the levels of Map1lc3a and Map1lc3b mRNA in the Ulk1/2 hypomorphs compared to control mice (Figure 1H).
ULK1/2 Regulates Muscle Homeostasis in a Cell-Autonomous Manner via Autophagy-Dependent and Autophagy-Independent Mechanisms
To distinguish between cell-autonomous and non–cell-autonomous aspects of the muscle phenotype in Ulk1/2 hypomorphs and to exclude the possibility that expression of the floxed Ulk2 allele was contributing to the development of the IBM-like disease in this mouse model, we generated muscle-specific Ulk1/2 cDKO mice. Ulk1flox/floxUlk2−/− mice on a C57BL/6 background for more than 12 generations (Figure S2A) were crossed with C57BL/6 mice expressing Cre recombinase driven by the muscle creatine kinase promoter (Ckmm-Cre), which is expressed in skeletal muscle by embryonic day 13.5 (Bruning et al., 1998). To determine which aspects of the muscle phenotype could be attributed to defects in autophagy, we also generated mice with muscle-specific deletion of Atg7, a gene essential for autophagosome formation via the canonical autophagy pathway.
We began by correlating ULK1/2 and ATG7 expression with functional defects in autophagy. Immunoblot analyses of extracts prepared from Ulk1/2Ckmm-Cre cDKO (Ulk1flox/floxUlk2−/−; Ckmm-Cre) and Atg7Ckmm-CrecKO (Atg7flox/flox;Ckmm-Cre) quadriceps confirmed the loss of ULK1 and ATG7, respectively (Figure S2B). Ulk1/2Ckmm-CrecDKO and Atg7Ckmm-CrecKO mice were grossly indistinguishable in appearance from their aged-matched controls (Ulk1+/floxUlk2−/−; Ckmm-Cre, and Atg7+/flox;Ckmm-Cre) at birth and at the time of weaning, with no significant differences in body weights at 8 weeks of age (Figure 2A). To confirm that the decrease in Ulk1/2 expression was sufficient to cause a defect in autophagy, we examined autophagy levels in fed and starved 8-week-old control and Ulk1/2Ckmm-Cre cDKO mice, prior to the appearance of pathologic vacuoles and inclusions. As expected based on the defect in starvation-induced autophagy observed in murine embryonic fibroblasts (MEFs) prepared from Ulk1/2-deficient mice (Joo et al., 2016), the muscles of Ulk1/2Ckmm-Cre cDKO mice failed to show a starvation-induced increase in the number of LC3-positive puncta (Figures S2C and S2D) or in the level of LC3 conversion (i.e., LC3-II/LC3-I) (Figures S2E and S2F). The number of LC3-positive puncta (Figures S2C and S2D) and level of LC3 conversion (Figures S2E and S2F) were also both decreased in fed Ulk1/2Ckmm-Cre cDKO mice compared to controls and accompanied by a significant increase in the steady-state levels of the autophagy substrate SQSTM1 (Figures S2E and S2F). These results demonstrate that Ulk1/2Ckmm-Cre cDKO mice show the expected defects in autophagic function.
Figure 2. Disrupting Atg7 Expression Does Not Recapitulate the Pathology Caused by Ulk1/2 Deficiency in Muscle.
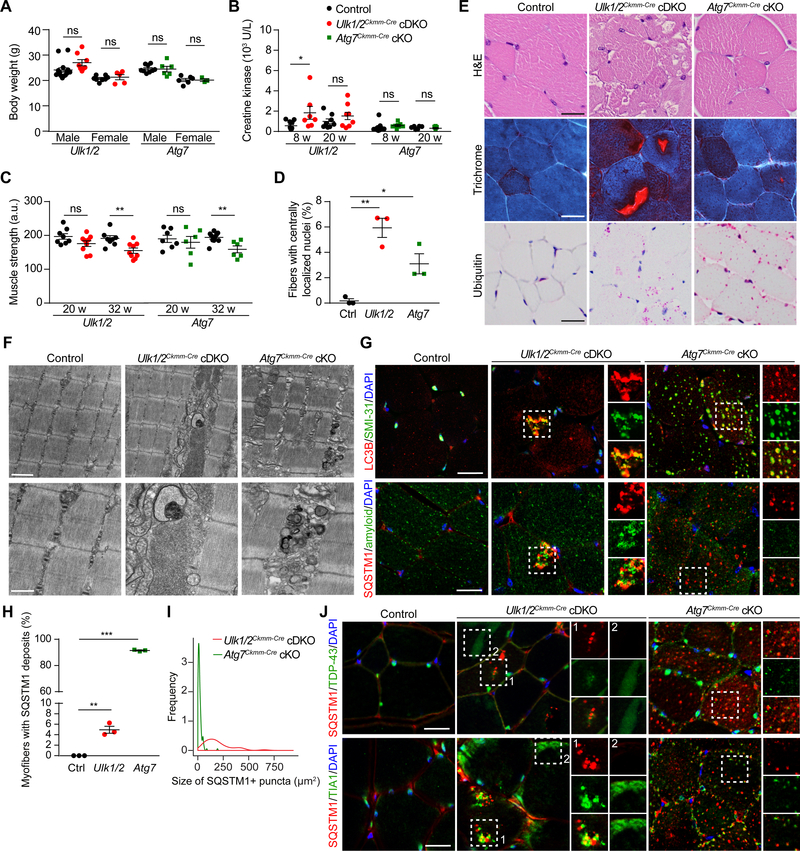
(A) Body weight of control, Ulk1/2Ckmm-Cre cDKO, and Atg7Ckmm-Cre cKO mice at 8 weeks of age.
(B) Serum creatine kinase levels of control, Ulk1/2Ckmm-Cre cDKO, and Atg7Ckmm-Cre cKO mice at 8 and 20 weeks of age. Creatine kinase levels were significantly elevated at 8 weeks in Ulk1/2Ckmm-Cre cDKO mice but not in Atg7Ckmm-Cre cKO mice.
(C) Muscle strength of control, Ulk1/2Ckmm-Cre cDKO, and Atg7Ckmm-Cre cKO mice at 20 and 32 weeks of age, showing progressive weakening of the Ulk1/2Ckmm-Cre cDKO and Atg7Ckmm-Cre cKO mice compared to control animals. a.u., arbitrary units.
(D) Quantification of myofibers with centralized nuclei in control, Ulk1/2Ckmm-Cre cDKO, and Atg7Ckmm-Cre cKO mice at 20 weeks of age. n = 3 for each genotype.
(E) Muscle cross sections of 20-week-old control, Ulk1/2Ckmm-Cre cDKO, and Atg7Ckmm-Cre cKO mice were stained with H&E, Gomori trichrome, and antibodies against ubiquitin. Scale bars, 25 μm.
(F) Ultrastructural studies of quadriceps from 20-week-old control, Ulk1/2Ckmm-Cre cDKO, and Atg7Ckmm-Cre cKO mice. Top scale bar, 1 μm; bottom scale bar, 500 nm.
(G) Frozen sections of quadriceps from 20-week-old control, UlK1/2Ckmm-Cre cDKO, and Atg7Ckmm-Cre cKO mice were costained for LC3B and SMI-31 (top) or SQSTM1 and amyloid (bottom). Scale bars, 25 μm.
(H) Percentages of myofibers with SQSTM1-positive deposits were quantified from control, Ulk1/2Ckmm-Cre cDKO, and Atg7Ckmm-Cre cKO mice at 20 weeks of age. n = 3 for each genotype.
(I) Size distribution of SQSTM1-positive deposits observed in Ulk1/2Ckmm-Cre cDKO and Atg7Ckmmm Cre cKO mice at 20 weeks of age. n = 3 for each genotype.
(J) Frozen sections of quadriceps from 20-week-old control, Ulk1/2Ckmm-Cre cDKO, and Atg7Ckmm-Cre cKO mice were costained for SQSTM1 and TDP-43 (top) or SQSTM1 and TIA1 (bottom). Scale bars, 25 μm.
For all comparisons, ns, not significant; *P <0.05; **P <0.01; ***P <0.001 by Student’s t-test.
All quantitative data are presented as mean ± SEM.
See also Figure S2.
The increase in SQSTM1 levels in the muscle of 8-week-old Ulk1/2Ckmm-Cre cDKO mice compared to littermate controls (Figures S2E and S2F) was also present in 20-week-old mice (Figures S2G and S2H), though the accumulation was not as pronounced as in Atg7Ckmm-Cre cKO mice (Figures S2G and S2H). Interestingly, we also observed changes in the accumulation of LC3 over the lifespan of Ulk1/2Ckmm-Cre mice. Whereas the level of LC3 conversion was reduced in 8-week-old Ulk1/2Ckmm-Cre cDKO mice compared to littermate controls (Figures S2E and S2F), this difference was not present in 20-week-old mice, in which we instead observed increased levels of LC3-II (Figures S2G and S2H). These findings indicate that over time, Ulk1/2 deficiency leads to the accumulation of LC3, perhaps due to a secondary defect in autophagosome maturation and/or lysosome function.
Next, we examined the Ulk1/2Ckmm-Cre cDKO and Atg7Ckmm-Cre cKO mice for the development of IBM-like disease similar to that observed in Ulk1/2 hypomorphs. Ulk1/2Ckmm-Cre cDKO mice but not Atg7Ckmm-Cre cKO mice had elevated levels of serum creatine kinase at 8 weeks of age (Figure 2B). Nevertheless, both sets of cKO mice showed progressive muscle weakness (Figure 2C) and histologic evidence of myogenic myopathy, with variation in fiber size and muscle fibers with central nuclei by 20 weeks (Figures 2D and 2E). The modified Gomori trichrome–positive vacuoles (i.e., rimmed vacuoles) characteristically found in patients with IBM were readily identified in the muscle sections from Ulk1/2Ckmm-Cre cDKO mice but not in those from Atg7Ckmm-Cre cKO mice (Figure 2E). Ultrastructural analyses highlighted the disruption of normal sarcomeric architecture in Ulk1/2Ckmm-Cre cDKO and Atg7Ckmm-Cre cKO mice by accumulations of abnormal membranous structures and debris in muscle (Figure 2F and data not shown), which are often detected in autophagy-defective tissues (Masiero et al., 2009; Raben et al., 2008). By contrast, tubulofilamentous structures like those in the Ulk1/2 hypomorphs were detected in the Ulk1/2Ckmm-Cre cDKO mice but not in the Atg7Ckmm-Cre cKO mice (Figure 2F).
To further define the relation between autophagy defects and the IBM-like phenotype observed in Ulk1/2-deficient mice, we performed additional histopathologic analysis comparing features of Ulk1/2-deficient and Atg7-deficient mice. Although the muscles from both Ulk1/2Ckmm-Cre cDKO and Atg7Ckmm-Cre cKO mice showed deposits positive for ubiquitin (Figure 2E) and SQSTM1 (Figure 2G, bottom panel), the SQSTM1-positive deposits in the Atg7Ckmm-Cre cKO mice were significantly smaller than those in the Ulk1/2Ckmm-Cre cDKO mice, albeit more numerous and widespread (Figures 2H and 2I). As described previously in autophagy-defective mice (Hara et al., 2006; Komatsu et al., 2006; Masiero et al., 2009), the SQSTM1- and ubiquitin-positive deposits in Ulk1/2Ckmm-Cre cDKO and Atg7Ckmm-Cre cKO mice were also positive for LC3B and LAMP1 (Figure S2I). Reactivity to phosphorylated tau (SMI-31) also colocalized with LC3B staining in both sets of cKO mice (Figure 2G, top panel).
Further immunofluorescence analyses revealed several additional features of the IBM phenotype in the Ulk1/2Ckmm-Cre cDKO mice that were not detected in the Atg7Ckmm-Cre cKO mice. First, we detected amyloid in the SQSTM1-positive inclusions of Ulk1/2Ckmm-Cre cDKO mice but not of Atg7Ckmm-Cre cKO mice (Figure 2G, bottom panel). Second, although a few small scattered TDP-43–positive and TIA1-positive foci were observed in the muscles of Atg7Ckmm-Cre cKO mice, the localization of TDP-43 and TIA1 to SQSTM1-positive inclusions (Figure 2J, box 1) and SQSTM1-negative sarcoplasmic patches (Figure 2J, box 2) was seen only in the Ulk1/2pkmm-Cre cDKO mice. Together, these studies suggest that although a defect in autophagy contributes to the ubiquitin-positive pathology and associated colocalization of autophagy-related markers (SQSTM1, LC3B, LAMP1), it is not sufficient to account for several of the histopathologic features of IBM that we observed in Ulk1/2-deficient (but not Atg7-deficient) animals, including the presence of vacuoles, amyloid, TDP-43–positive inclusions and sarcoplasmic patches, and tubulofilamentous aggregates.
ULK1/2 Activity Regulates Stress Granule Dynamics
We next used an unbiased proteomics approach to gain insight into the mechanism(s) underlying the development of the vacuolar myopathy in the Ulk1/2-deficient mice. We began by immunoprecipitating endogenous ULK1 using extracts from central nervous system tissue (cerebellum and cortex) from wild-type mice and MEFs prepared from wild-type and Atg13gt/gt mice (Kaizuka and Mizushima, 2016). Because ATG13 stabilizes the canonical autophagy-inducing ULK1–ATG13–RB1CC1 complex, the use of Atg13-deficient (i.e., Atg13gt/gt) MEFs permitted the identification of ULK1 interactions that could exist beyond the confines of the canonical complex. We used label-free quantitative mass spectrometry to generate a list of coisolated ULK1-interacting proteins, which we combined with ULK1-interacting proteins from public databases (STRING, BIOPLEX, InWeb, and BIOGRID) to create a comprehensive ULK1 interactome (Figure 3A and Tables S1-S3). In addition to the expected overrepresentation of autophagy proteins, stress granule proteins were among those showing significant enrichment in the ULK1 interactome (Figures 3A and S3A). Interactions between ULK1 and selected stress granule proteins (G3BP1, USP10, and CAPRIN1) were confirmed by immunoprecipitation and immunoblot analyses using extracts prepared from HeLa cells (Figure S3B). The interactome also highlighted the close relations of ULK1 to the RNA-binding and autophagy-related proteins mutated in patients with IBM and related degenerative disorders (Jain et al., 2016; Taylor et al., 2016), including interactions with VCP and SQSTM1 (Figure 3A).
Figure 3. ULK1/2 Interact with Stress Granule Proteins and Localize to Stress Granules.
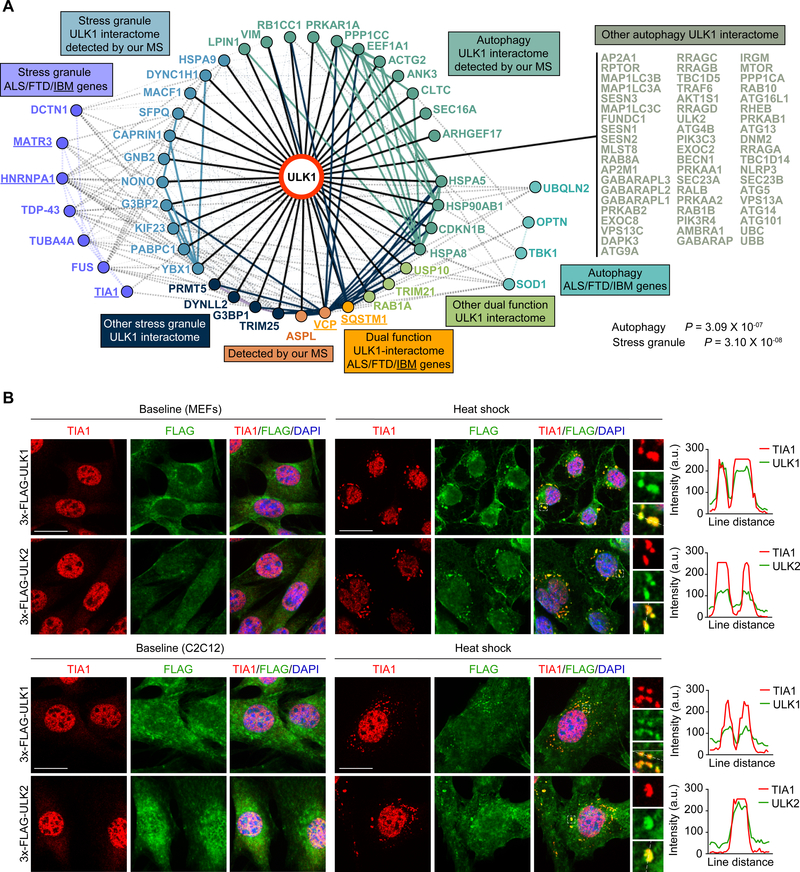
(A) Integrated ULK1 interactome from in-house–generated proteomic data and public databases (STRING, BIOPLEX, InWeb, and BIOGRID). Underlined proteins indicate genes that are mutated in patients with IBM. Pathway analysis for the ULK1 interactome was performed using ENRICHR and Fisher’s exact test.
(B) WT MEFs and C2C12 cells with 3×-FLAG tagged endogenous ULK1 and ULK2 were heat shocked at 43°C for 1 h and stained for FLAG and TIA1. Line scans indicate the degree of colocalization between ULK1 or ULK2 (green) and TIA1 (red) in lines drawn within the magnified images. Intensity profiles are presented as arbitrary units (a.u.). Scale bars, 20 μm.
The results of the ULK1 interactome analysis prompted us to examine the localization of ULK1 and ULK2 in response to heat shock, a stress that alters the dynamic properties of ribonucleoprotein assemblies (Anderson and Kedersha, 2002; Zhang et al., 2018a). For these experiments, we used CRISPR/Cas9 to tag the endogenous loci of Ulk1 and Ulk2 with 3×-FLAG in MEFs and C2C12 cells, which we confirmed via sequencing and immunoblotting (Figure S3C). As predicted, endogenous ULK1 and ULK2 were recruited into heat shock–induced TIA1-positive stress granules (Figure 3B). Transiently overexpressed GFP-tagged ULK1 and ULK2 also localized to stress granules (Figure S3D).
We next sought to examine the impact of inhibiting ULK1/2 activity on stress granule dynamics using both pharmacologic and genetic approaches. After confirming that the ULK1/2 inhibitor SBI-0206965 (Egan et al., 2015) reduced the phosphorylation of a well-established ULK1 substrate, ATG13 (S318) (Figure S4A), we used live-cell imaging to monitor the dynamic behavior of heat shock–induced stress granules in U2OS cells stably expressing an EGFP-tagged form of the stress granule marker G3BP1 in the presence or absence of SBI-0206965. Although treatment with the ULK1/2 inhibitor did not affect the kinetics of stress granule assembly, it significantly delayed their resolution during the recovery period: 2 hours after the heat shock, persistent stress granules were detected in approximately 50% of the SBI-0206965–treated cells but in fewer than 10% of the vehicle-treated cells (Figures 4A and 4B). The persistent stress granules in ULK1/2-inhibited cells stained positively for several stress granule proteins, including G3BP1, TIAR, PABP, TIA1, eIF3η, and TDP-43 (Figure 4C). By contrast, vehicle-treated cells showed the expected diffuse cytosolic staining of G3BP1, PABP, and eIF3η, and the expected nuclear-restricted pattern of the RNA-binding proteins TIAR, TIA1, and TDP-43 upon recovery (Figure S4B).
Figure 4. ULK1/2 Activity Regulates the Disassembly of Stress Granules.
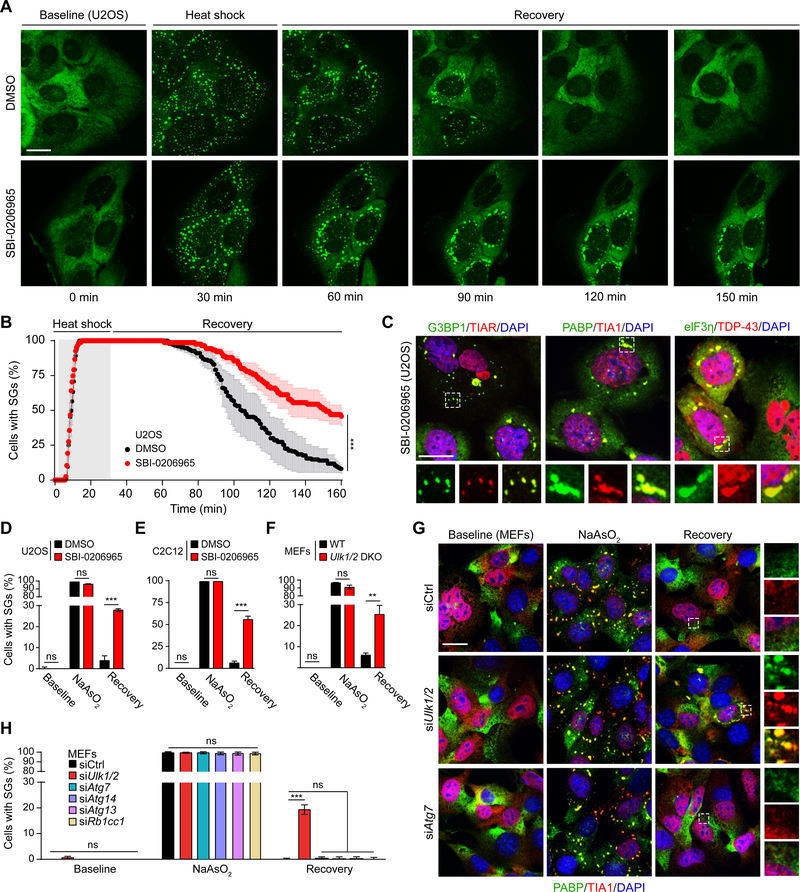
(A-B) Live-cell imaging of U2OS cells stably expressing G3BP1-GFP. Cells pretreated with DMSO or SBI-0206965 were heat shocked at 43°C for 30 min to induce stress granule formation, followed by recovery at 37°C for an additional 120 min. The percentage of cells with stress granules is shown in (B). Scale bar, 20 μm. ***P <0.001, chi-square test.
(C) U2OS cells pretreated with SBI-0206965 for 18 h were subjected to NaAsO2 for 1 h, allowed to recover for 2 h, then immunostained for stress granule components G3BP1, TIAR, PABP, TIA1, eIF3η, and TDP-43. Scale bar, 20 μm.
(D-F) U2OS (D) and C2C12 (E) cells pretreated with DMSO or SBI-0206965 and (F) WT or Ulk1/2 DKO MEFs were treated with NaAsO2 for 1 h, then allowed to recover in fresh media for 2 h. The percentage of cells with stress granules was quantified. ns, not significant; **P <0.01; ***P <0.001 by Student’s t-test.
(G) MEFs transfected with control siRNA or siRNA targeting Ulk1/2 or Atg7 were treated with NaAsO2 for 1 h followed by recovery in fresh media for 2 h, then immunostained for PABP and TIA1. Scale bar, 20 μm.
(H) MEFs transfected with control siRNA or siRNA against key autophagy genes (Atg7, Atg14, Atg13, Rb1cc1) were treated with NaAsO2 for 1 h and then allowed to recover for 2 h. The percentage of cells with stress granules at each stage is shown. ns, not significant; ***P <0.001 by one-way ANOVA with Dunnett’s multiple comparisons test.
All quantitative data are presented as mean ± SEM.
See also Figure S4.
These effects were not limited to stress granules induced by heat shock stress, as demonstrated by static imaging studies showing impaired resolution of stress granules by the ULK1/2 inhibitor in response to not only heat shock (Figure S4C) but also sodium arsenite treatment in U2OS cells (Figures 4D and S4D) and C2C12 myoblast cells (Figures 4E and S4E). Furthermore, genetic inhibition of ULK1/2 in MEFs using either knockout or knockdown approaches led to significant increases in the persistence of heat shock– and arsenite-induced stress granules (Figures 4F, 4H, and S4F-S4H).
To assess the contribution of autophagy to stress granule dynamics under our experimental conditions, Ulk1/2 and other autophagy genes (Atg7, Atg14, Atg13, and Rb1cc1) were silenced in parallel. Whereas RNAi-mediated silencing of Ulk1/2 caused a significant increase in the persistence of stress granules, the knockdown of the other autophagy genes did not (Figures 4G, 4H, S4I, and S4J). Moreover, treatment with the vacuolar-type H+-ATPase bafilomycin A, which inhibits lysosome-mediated clearance, did not impair the dissolution of stress granules (Figure S4K). Together, these data indicate that ULK1/2 regulate the disassembly of stress granules via a pathway that does not require canonical components of the autophagy machinery.
ULK1/2-Dependent Phosphorylation of VCP Is Required for Efficient Stress Granule Disassembly
We next pursued the surprising finding from our proteomics studies that VCP, the most common cause of inherited IBM (often as part of MSP) and a regulator of stress granule dynamics, interacts with ULK1 (Figure 3A and Table S1). We first confirmed the interaction between endogenous ULK1 and VCP in extracts prepared from wild-type muscle (Figure S5A), wild-type MEFs (Figure S5B), HeLa cells (Figure S5C), and U2OS cells (Figure S5D) by immunoprecipitating endogenous ULK1 and immunoblotting for the presence of VCP. The ULK1–VCP interaction was not strongly influenced by knockdown of Atg13 or Rb1cc1, which encode components of the canonical autophagy-inducing ULK/Atg1 kinase complex (Figure S5B). Although minimal amounts of endogenous VCP were detected in the anti-DDK immunoprecipitates prepared from unstressed HEK293T cells overexpressing DDK-tagged ULK1 or ULK2, the level of VCP pulled down with ULK1/2 increased dramatically after cells were subjected to heat shock (Figure 5A). The enhanced ULK1/2–VCP interaction was also confirmed by immunoprecipitation of endogenous ULK1, ULK2, and VCP (Figures 5B, S5E, and S5F). In line with the observed increase in ULK1/2–VCP complex formation, ULK1/2 colocalized with VCP in stress granules upon heat shock (Figure 5C).
Figure 5. ULK-Mediated Phosphorylation of VCP Promotes Stress Granule Disassembly.

(A) HEK293T cells were transfected with ULK1-DDK or ULK2-DDK and subjected to heat shock at 43°C for 1 h. Immediately following heat shock, cells were lysed and immunoprecipitated for DDK, followed by immunoblot against the indicated proteins.
(B) Representative immunoblots of endogenous VCP immunoprecipitated from WT MEFs subjected to heat shock at 43°C for 1 h.
(C) WT MEFs transfected with GFP-ULK1 or GFP-ULK2 were heat shocked at 43°C for 1 h, fixed, and stained for VCP and TIA1. Line scans indicate the degree of colocalization between ULK1 or ULK2 (green), VCP (red), and TIA1 (blue) in lines drawn within the magnified images. Intensity profiles are presented as arbitrary units (a.u.). Scale bar, 20 μm.
(D) Recombinant N-terminal ULK1 (1–649) and full-length VCP were used for an in vitro kinase assay at 37°C for 15 min. The reaction products were separated by SDS-PAGE and stained for Sypro Ruby (bottom panel); 32P incorporation was visualized by autoradiogram (top panel). Blot is representative of n = 3.
(E) Recombinant ULK1/2 and VCP were used for a cold in vitro kinase assay. VCP was analyzed by mass spectrometry to assess the stoichiometry of phosphorylated peptides containing S13, S282, and T761 over total peptide counts of VCP.
(F) FLAG-tagged VCP was coexpressed with ULK1 or ULK2 in HEK293T cells. Cells were treated with DMSO or NaAsO2 for 1 h. VCP was immunoprecipitated from the cell lysate and subjected to mass spectrometry analysis.
(G-H) MEFs stably expressing RNAi-resistant VCP WT, 3A, or 3D were subjected to heat shock at 43°C for 1 h, followed by recovery at 37°C for 2 h. Cells with stress granules were quantified at each stage (baseline, during heat shock, and during recovery) by staining for stress granule markers PABP and TIA1. Scale bar, 20 μm.
(I-J) MEFs stably expressing RNAi-resistant VCP WT or 3D were transfected with control or Ulk1 siRNA. Cells were treated with sodium arsenite for 1 h and fixed at 1 h and 2 h after recovery. Stress granules were visualized by eIF3η and TIAR staining and quantified at each time point. Scale bar, 20 μm.
(K) The in vitro ATPase activity levels of VCP WT and 3D were measured by determining the quantity of pi released over 30 min at room temperature with varying concentrations of ATP as substrates. Data are presented as mean ± SD.
For (G), and (I), data are shown as mean ± SEM. ns, not significant; ***P <0.001 by one-way ANOVA with Dunnett’s multiple comparisons test.
See also Figure S5.
Like several other ULK1/2-interacting proteins (Mercer et al., 2018; Wang and Kundu, 2017), purified recombinant VCP was phosphorylated by purified recombinant ULK1 or ULK2 in cell-free in vitro kinase assays (Figures 5D and S5G). Phosphosite mapping revealed three sites in VCP (i.e., S13, S282, and T761) that were phosphorylated in an ULK1/2-dependent manner in cell-free in vitro kinase reactions (Figure 5E) and HEK293T cells (Figures S5H and S5I). Moreover, quantitative mass spectrometry with synthetic peptide standards revealed a relative increase in the ULK1/2-dependent phosphorylation of VCP at these sites in response to sodium arsenite treatment (Figure 5F), suggesting that the stress-induced stabilization of the ULK1/2–VCP complex is associated with increased levels of VCP phosphorylation.
VCP regulates stress granule dynamics, which is consistent with our findings that inhibiting VCP function in U2OS cells by using RNAi-mediated knockdown of VCP or treatment with the VCP inhibitor Eer1 impaired stress granule disassembly (Figures S5J and S5K). Therefore, to explore the possibility that the ability of VCP to regulate stress granule disassembly is influenced by ULK1/2, we generated MEFs stably expressing FLAG-tagged wild-type, phospho-defective (S13A, S282A, T761A; hereafter “3A”) or phospho-mimetic (S13D, S282D, T761D; hereafter “3D”) human VCP, which were resistant to siRNAs directed against endogenous murine Vcp (Figure S5L). As expected, Vcp knockdown caused the persistence of heat shock–induced stress granules in a significantly higher proportion of MEFs compared to the nontargeting control siRNA (Figures 5G and 5H). The impaired disassembly of stress granules resulting from VCP depletion was rescued in cells expressing RNAi-resistant wild-type human VCP or the 3D mutant but not the 3A mutant (Figures 5G and 5H). Moreover, the 3D VCP mutant overcame the defect in stress granule disassembly caused by Ulk1 knockdown (Figures 5I and 5J). As ATP hydrolysis is required for VCP function (van den Boom and Meyer, 2018), we next examined the impact of ULK1/2-mediated phosphorylation of VCP on its ATPase activity. The phosphomimetic 3D mutant exhibited significantly enhanced ATPase activity compared to wild-type VCP (Figures 5K and S5M). Together, these results suggest that ULK1/2-mediated phosphorylation of VCP contributes to the disassembly of stress granules, at least in part by enhancing the ATPase activity of VCP.
Enhancing ULK1/2 Activity Promotes the Resolution of Stress Granules Caused by Expression of Disease-Associated Mutant Proteins
Because ULK1/2 activity is required for the disassembly of heat shock– and sodium arsenite–induced stress granules, we hypothesized that enhanced ULK1/2 activity might accelerate the resolution of stress granules induced by stress and/or expression of mutant proteins implicated in the pathogenesis of IBM and related disorders. To enhance ULK1/2 kinase activity, we turned to LYN1604, an ULK1/2 agonist that induces increased phosphorylation of ATG13 at S318 (Zhang et al., 2017), an effect we observed in our experimental setup (Figure S6). Indeed, treating U2OS cells stably expressing tdTomato-tagged G3BP1 with LYN1604 significantly decreased the time required for dissolution of heat shock–induced stress granules (Figure 6A, B). Cells expressing the phosphodefective VCP mutant did not show significant improvements in stress granule dissolution with the addition of the ULK1/2 activator (Figure 6C).
Figure 6. ULK1/2 Agonist Promotes the Disassembly of Aberrant Stress Granules in Cells Expressing Disease-Associated Mutant Proteins.

(A-B) Live-cell imaging of U2OS cells stably expressing G3BP1-tdTomato treated with DMSO or LYN-1604. Cells were subjected to heat shock at 43°C for 1 h followed by recovery at 37°C. Representative images are shown in (A), and the percentage of cells (mean ± SEM) containing stress granules is shown in (B). Scale bar, 20 μm. P <0.001, chi-square test.
(C) HeLa cells transfected with VCP WT or VCP 3A were treated with DMSO or LYN-1604 for 24 h. The cells were heat shocked at 43°C for 1 h, allowed to recover at 37°C for 2 h, and the percentage of cells with stress granules was calculated.
(D-E) HeLa cells transfected with HA-FUS R521C mutant were treated with DMSO or LYN-1604 for 24 h. The cells were stained with HA and eIF3η (E), and the percentage of cells containing stress granules is shown in (D). Scale bar, 20 μm.
(F) HeLa cells transfected with TIA1-GFP WT or A381T were treated with DMSO or LYN-1604 for 24 h. The cells were heat shocked at 43°C for 1 h, allowed to recover at 37°C for 2 h, and the percentage of cells with stress granules was calculated.
(G) HeLa cells transfected with VCP WT or VCP A232E were treated with DMSO or LYN-1604 for 24 h. The cells were heat shocked at 43°C for 1 h, allowed to recover at 37°C for 2 h, and the percentage of cells with stress granules was calculated.
For (C), (D), (F) and (G), data are presented as mean ± SEM. ns, not significant; *P <0.05; **P <0.01; ***P <0.001 by one-way ANOVA with Dunnett’s multiple comparisons test.
Mutations in FUS (e.g., R521C) and TIA1 (e.g., A381T) that negatively affect stress granule dynamics have been identified in patients with IBM and/or ALS/FTD (Dormann and Haass, 2013; Mackenzie et al., 2017). Treating cells with LYN-1604 significantly reduced the proportion of cells harboring spontaneous stress granules caused by expression of mutant FUS R521C (Figures 6D and E) or persistent heat shock–induced stress granules caused by expression of mutant (A381T) but not wild-type TIA1 (Figures 6F). Surprisingly, the persistent stress granules formed in cells expressing VCP A232E exhibited reduced sensitivity to the ULK1/2 agonist compared to those formed in cells expressing disease-associated RNA-binding proteins (Figure 6G). This effect may be due, in part, to reduced levels of ULK1/2-mediated phosphorylation at T761 in VCP A232E compared to VCP WT (Figure S6B). Together, these results raise the possibility that ULK1 and/or ULK2 could be targeted to treat diseases such as IBM/ALS/FTD.
DISCUSSION
Here, we demonstrate that disrupting ULK1/2 expression in mice results in an IBM-like phenotype with ubiquitin- and TDP-43–positive pathology, which is not solely attributable to a defect in autophagy. The phenotype is similar to that in mice expressing mutant forms of VCP, whose ability to extract proteins from macromolecular complexes and membranes is required for multiple cellular processes, including the disassembly of stress granules (Buchan et al., 2013; Turakhiya et al., 2018). Prompted by the enrichment of stress granule proteins and VCP within the ULK1 interactome, we found that in response to stresses that impair translation (e.g., heat shock and arsenite treatment), ULK1/2 localize to stress granules and phosphorylate VCP, thereby increasing its ATPase activity and ability to disassemble stress granules. Whereas inhibiting ULK1/2 activity impairs the disassembly of stress granules, an ULK1/2 agonist accelerates the process. The reduced sensitivity of the A232E mutant to activation by ULK1/2 may help explain the similarities in the phenotypes of the Ulk1/2 cDKO mice and the VCP A232E transgenic mice.
There are several possible explanations why neither ULK1 nor ULK2 has been reported in stress granule proteomes (Jain et al., 2016; Markmiller et al., 2018; Youn et al., 2018). Based on our observations, there may be some cell type–specific differences in the dynamics of ULK1/2 localization to stress granules. For example, although GFP-tagged ULK1 and ULK2 localize to stress granules in HeLa cells and MEFs, we were not able to detect this phenomenon in U2OS cells (data not shown). We suspect that the recruitment of ULK1/2 to stress granules in U2OS cells may occur with a rapid on/off rate in these cells. The reduced localization of ULK1/2 to stress granules in certain cell types may render it below the limits of sensitivity of detection by mass spectrometry.
ULK1/2 phosphorylate VCP at three sites: S13, S282, and T761. Extensive study of VCP structure by both x-ray crystallography and cryo-EM (van den Boom and Meyer, 2018) provides insight into possible consequences of site-specific phosphorylation. S282 resides at the end of an alpha-helix atop the central pore of VCP (Banerjee et al., 2016). Recent structural studies of a fungal VCP homologue demonstrate that the conserved NPL4–UFD1 substrate adaptor/cofactor anchors to the top of the D1 ring, above the central pore of VCP (Bodnar and Rapoport, 2017). The high degree of structural homology indicates that this interface likely encompasses the helix containing S282 in human VCP. Phosphorylation of S282, therefore, might increase binding of NPL4–UFD1 or other cofactors required for the interaction of ubiquitinated substrates with VCP (Hulsmann et al., 2018). T761 is located on the surface of the VCP hexamer, at the C-terminal end of the D2 AAA domain (Banerjee et al., 2016). D1 and D2 are arranged as two stacked rings, and like all AAA domains, they contain Walker A and B motifs that bind and hydrolyze ATP. A stable ionic bond formed between phosphorylated T761 in D2 of one VCP protomer and R741 in an adjacent VCP protomer, which is thought to contribute to the increased ATPase activity of VCP that has been phosphorylated at T761 compared to unphosphorylated VCP (Mori-Konya et al., 2009). Although S282 and T761 were among the residues phosphorylated in VCP purified from baculovirus-infected SF9 cells (Mori-Konya et al., 2009), the responsible kinase and functional significance of these sites were not known. To the best of our knowledge, this is the first study demonstrating that phosphorylation of VCP occurs in an ULK1/2-dependent manner and that these posttranslational modifications are required for efficient VCP-mediated stress granule disassembly. S13 is within an unstructured stretch of amino acids within the N-terminal domain of VCP, and the possible consequences of phosphorylation remain unclear.
One of the challenges to targeting VCP function for therapeutic purposes is the wide range of vital functions fulfilled by VCP substrates. We demonstrated that ULK1/2 interact with and phosphorylate VCP in response to specific cellular stresses, thereby providing a mechanism for spatially and temporally regulating VCP functions that are relevant to disease pathogenesis. Thus, ULK1/2 agonists and inhibitors may provide a unique opportunity for targeting specific VCP functions (e.g., stress granule disassembly). Our finding that the ULK1/2–VCP interaction is regulated in a stress-dependent manner may also help explain why under basal physiologic conditions the brains of Ulk1/2-deficient animals (i.e., Ulk1/2 hypomorphs and Ulk1/2Nestin-Cre cDKO mice) (Joo et al., 2016; Wang et al., 2017) failed to show ubiquitin- and TDP-43–positive pathology within their shortened life spans: the metabolically privileged status of the CNS may reduce the engagement of stress-related pathways regulated by ULK1/2, thus delaying the onset of such pathology.
Given the well-established role of both ULK1/2 and VCP in autophagy and the evidence from genetic studies highlighting the role of autophagy in the clearance of stress granules in yeast (Buchan et al., 2013) and germline P granules in worms (Zhang et al., 2018a), we were surprised that the persistence of stress granules caused by acute disruption of ULK1/2 expression or activity was not mirrored by the acute inhibition of autophagy. Indeed, neither RNAi-mediated knockdown of other canonical autophagy genes (i.e., Atg7, Atg13, Atg14, Rb1cc1) nor pharmacologic inhibition of lysosomal function significantly affected stress granule dynamics in cultured cells, suggesting that the majority of TDP-43– and TIA1-positive granules formed under our experimental conditions were not degraded by the autophagolysosomal system. Factors that decrease the dynamic properties of stress granules, such as the expression of mutant RNA-binding proteins, or an increase in the duration or intensity of instigating stress, increase the persistence of stress granules (Alberti et al., 2017; Mackenzie et al., 2017; Molliex et al., 2015). Thus, the persistence of stress granules in cultured Atg7−/− MEFs (Buchan et al., 2013) may relate to alterations in the dynamic properties and/or composition of the stress granules caused by cumulative effects of the prolonged autophagy defect in such cells. The persistent granules may be similar in nature and/or etiology to the small TDP-43– and TIA1-positive (but mostly SQSTM1-negative) foci that we observed in Atg7-deficient muscles.
Our studies suggest that autophagy plays a minor role in the clearance of stress granules and that a defect in autophagy alone is not sufficient to cause IBM-like pathology. Indeed, despite the profound autophagy defect caused by Atg7 deletion, the TDP-43–positive pathology and vacuolar degeneration characteristically found in patients with the IBM/ALS/FTD/MSP spectrum of diseases and in Ulk1/2-deficient mice were not present in Atg7Ckmm-Cre cKO mice. Thus, the autophagy-related proteins mutated in patients with IBM or related disorders probably have additional functions in the regulation of RNP granule dynamics. Notably, disease-associated mutations do not typically affect core components of the autophagy machinery but rather affect proteins like VCP, which are involved in ubiquitin signaling (Meyer et al., 2012; Meyer and Weihl, 2014). At least two of these autophagy-related proteins, UBQLN2 and SQSTM1, undergo phase transitions, enabling them to shuttle ubiquitinated proteins (possibly including aggregation-prone proteins) in and out of stress granules (Dao et al., 2018; Sun et al., 2018a). Nevertheless, given the ubiquitin-and SQSTM1-positive pathology typically observed in patients, including those with mutations in RNA-binding proteins, it remains likely that defects in the autophagolysosomal system, whether primary or secondary, contribute to the pathogenesis of IBM and related diseases. Thus, we speculate that the combined effect of ULK1/2 on VCP activity and stress granule dynamics, as well as on other canonical autophagy-related ULK1/2 targets, contributes to the development of IBM-like disease in Ulk1/2-deficient mice.
STAR Methods
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Mondira Kundu (Mondira.Kundu@stjude.org).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animals
The strategy for targeting the Ulk1 locus has been previously described (Kundu et al., 2008). Mice harboring the targeted Ulk1 allele (Ulk1flox-neo) were crossed with Ella-Cre transgenic mice (The Jackson Laboratory; stock 003724), and progeny with Ulk1flox alleles were crossed with WT mice to eliminate the expression of the Ella-Cre transgene. Generation of Ulk2flox/flox and Ulk2−/− mice has been described previously (Cheong et al., 2014; Joo et al., 2016). The Ckmm-Cre line was obtained from The Jackson Laboratory (stock 006475). Ulk1flox/flox;Ulk2−/− females were bred with Ulk1+/flox;Ulk2−/−;Ckmm-Cre males to generate Ulk1/2Ckmm-Cre cDKO (Ulk1flox/flox;Ulk2−/−;Ckmm-Cre) mice. Atg7flox/flox [a gift from Masaaki Komatsu, Tokyo Metropolitan Institute of Medical Science (Komatsu et al., 2005)] females were crossed with Ckmm-Cre males to generate Atg7Ckmm-Cre cKO mice. Ulk1/2Ckmm-Cre cDKO mice were bred in a C57BL/6 background. All other mice were maintained in a mixed background between C57BL/6 and 129. All animal experiments were approved by and performed in accordance with guidelines provided by the Institutional Animal Care and Use Committee at St. Jude Children’s Research Hospital.
Cell Culture
HEK293T (CRL-3216), U2OS (HTB-96), HeLa (CCL-2), and C2C12 (CRL-1772) cells were obtained from ATCC. Generation of WT and Ulk1/2 DKO MEFs and U2OS cells stably expressing G3BP1-GFP has been described previously (Figley et al., 2014; Joo et al., 2016). All cells were grown in DMEM (Invitrogen; 11995–065) supplemented with 10% FBS, penicillin/streptomycin (Invitrogen), and Glutamax (Invitrogen) at 37°C (5% CO2). To produce MEFs stably expressing wild-type or mutant forms of VCP, HEK293T cells were transfected with pMSCV-IRES-EGFP retroviral vectors containing different VCP cDNAs with packaging vector using FuGENE 6 (Promega). MEFs were incubated with cell-free retroviral supernatant fractions containing 8 μg/mL polybrene (Sigma-Aldrich; H9268–106), and transduced cells were FACS-sorted by the presence of GFP. CRISPR/Cas9 technology was used to tag endogenous G3BP1, Ulk1, and Ulk2 alleles. Briefly, cells were transiently transfected with gRNA expression plasmid (Addgene; 43860), Cas9 expression plasmid (Addgene; 43945), and donor plasmid via nucleofection (Lonza; 4D-Nucleofector X-unit) according to the manufacturer’s recommended protocol. Individual cells were sorted by FACS and clonally expanded. Clones were screened for the targeted integration event. Junctions were confirmed by Sanger sequencing.
METHODS DETAILS
Immunostaining and Histology
Mice were transcardially perfused with 4% PFA. Quadriceps were dissected and postfixed in 4% PFA for 1 h at 4°C. The tissues were then cryoprote cted using 30% sucrose in PBS overnight at 4°C and embedded in optimum cutting temperature (OCT) for cryosectioning. Frozen sections were washed with 0.2% Triton X-100 in Tris-buffered saline (TBST) and incubated in blocking solution (5% normal goat serum or normal donkey serum in TBST) for 1 h at room temperature. Sections were incubated with primary antibodies diluted in the blocking solution overnight at 4°C, washed with TBST, and incubated with Alexa Flu or–conjugated secondary antibodies (Invitrogen) diluted at 1:1000 in the blocking solution for 2 h at room temperature. Sections were mounted in ProLong Gold Antifade Reagent with DAPI (Invitrogen). The following primary antibodies were used: mouse anti-SQSTM1/p62 (Abnova; H00008878-M01), rabbit anti-ubiquitin (DAKO; Z0458), rabbit anti-LC3B (MBL; PM036), rat anti-LAMP1 (Developmental Studies Hybridoma Bank; 1D4B), mouse anti–SMI-31 (Covance; SMI-31R), rabbit anti–β-amyloid 1–42 (Millipore; AB5078P), rabbit anti-FUS (Bethyl Laboratories; IHC-00074), rabbit anti–TDP-43 (Proteintech; 10782–2-AP), goat anti-TIA1 (Santa Cruz Biotechnology; sc-1751), and mouse anti–hnRNP A2B1 (Santa Cruz Biotechnology; sc-53531).
For histologic analyses, tissues were embedded in paraffin, sectioned at 4 μm, and mounted on positively charged glass slides (Thermo Fisher Scientific; Superfrost Plus). Primary antibodies against SQSTM1/p62 (Abcam; ab194720), ubiquitin (Enzo Biochem; BML-PW8810–0500), LC3B (Millipore; L7543), TDP-43 (Proteintech; 10782–2-AP), IBA1 (Biocare Medical; CP290A), MHCf (Leica; NCL-MHCf), and MHCs (Leica; NCL-MHCs) were used for immunohistochemical analyses. Antigen retrieval for all targets (except SQSTM1) required pretreatments using Cell Conditioning Solution 1 (CC1) (Ventana Medical Systems) for 32 min on the automated Discovery Ultra platform (Ventana-Roche). Similarly, antigen retrieval for SQSTM1 required pretreatment using Cell Conditioning Solution (CC2) (Ventana Medical Systems). After antigen retrieval, tissues were incubated with primary antibodies with OmniMap anti-rabbit HRP kit (Ventana Medical Systems) used as secondary antibody. Primary antibody binding was detected using the DISCOVERY ChromoMap DAB Kit (Ventana Medical Systems), except for ubiquitin and MHCs, where the chromogen used was DISCOVERY Purple (Ventana Medical Systems) followed by hematoxylin counter-staining. The 12-μm cross sections were prepared from fresh-frozen muscle and stained with Gomori trichrome stain, cytochrome oxidase (COX), and succinate dehydrogenase (SDH), as previously described (Tanji and Bonilla, 2008). All histology images were examined by a pathologist blinded to the experimental groups. To quantify myofibers with centralized nuclei and SQSTM1-positive aggregates, muscle sections were stained with caveolin-3, SQSTM1, and DAPI and imaged at low magnification (20×). For each section, 4×4 fields were captured. For each animal, the mean value was obtained from two separate sections. n = 3 for each genotype. To quantify the area of SQSTM1, images were masked with SlideBook 6.0 based on SQSTM1 staining. The average area of SQSTM1-positive deposits was then calculated from at least 50 SQSTM1-positive objects for each genotype.
Muscle Strength Measurement
The force of forepaws and hindpaws was measured as previously described (Badders et al., 2018). Briefly, using a grip-strength meter (Bioseb), grip strength was measured as grams of force in six repeated measurements for forepaws and hindpaws of each animal. The mean muscle strength for each animal was calculated from four independent measurements, after excluding the maximal and minimal reads.
Blood Chemistry
Samples of blood were obtained by periorbital bleeding in anaesthetized mice. The levels of different chemicals, such as creatine kinase, in the serum were determined and analyzed by the Veterinary Pathology Laboratory Core at St. Jude Children’s Research Hospital.
Immunoblot Analyses
Tissues or cells were lysed in Triton-based cell lysis buffer. Proteins in cleared lysates were electrophoretically separated on 4%−12% Bis-Tris gels (Life Technologies; NP0335BOX). Proteins were then transferred to PVDF membranes. After incubation with a 5% skim milk block, blots were probed with antibodies against the following targets: ULK1 (Sigma Aldrich; A7481), SQSTM1/p62 (Sigma Aldrich; P0067), LC3B (MBL; PM036), TDP-43 (Proteintech; 10782–2-AP), COXIV (Abcam; ab16056), TOM20 (Santa Cruz Biotechnology; sc-11415), ATG7 (Cell Signaling; 8558S), ATG13 (Sigma Aldrich; SAB4200100), pATG13 (Rockland; 600–401-C49), RB1CC1 (Cell Signaling; 12436), ATG14 (MBL; PD026), VCP (Invitrogen; MA3–004), FLAG (Sigma Aldrich; A8592), G3BP1 (Proteintech; 13057–2-AP), Caprin-1 (Proteintech; 15112–1-AP), USP10 (Cell Signaling; 8501), and GAPDH (Sigma Aldrich; G9545). Membranes were then incubated with HRP-conjugated secondary antibodies, and bands were detected using chemiluminescence detection kits (Amersham; RPN2232).
Immunoprecipitation
Total protein lysates were extracted from animal tissues or cells using a Triton-based cell lysis buffer (40 mM HEPES, 120 mM NaCl, 1 mM EDTA, 1.5 mM Na3VO2, 50 mM NaF, 10 mM β-glycerophosphate, 20 mM MoO4, 0.5% Triton X-100, protease inhibitor, phosphatase inhibitor). The lysates were incubated with anti-ULK1 antibody (Santa Cruz Biotechnology; sc10900), or anti-VCP antibody (Santa Cruz Biotechnology; sc-57492) overnight at 4°C and precipitated with protein G agarose beads (Thermo Fisher Scientific; 20399). DDK IP was performed using anti-FLAG M2 affinity gel according to the manufacturer’s instructions (Sigma Aldrich; A2220). Eluate was electrophoretically separated on 4%−12% Bis-Tris gels (Life Technologies; NP0335BOX). Proteins were then transferred to PVDF membranes followed by standard immunoblot analyses as described above.
Quantitative Real-Time PCR
Total RNA was extracted with RNeasy Fibrous Tissue Mini Kit (Qiagen; 74704) according to the manufacturer’s instructions. The reverse-transcription reaction was carried out using the SuperScript III first-strand synthesis kit (Life Technologies; 18080051) according to the manufacturer’s instructions. TaqMan gene expression assays containing FAM-labeled primer/probe sets specific for Ulk1 (Mm-00437238_m1), Ulk2 (Mm-00497023_m1), and 18S were obtained from Applied Biosystems (4331182, 4331182, and 4333760F, respectively). The real-time PCR reactions were performed in a total reaction volume of 25 μL by using FastStart TaqMan Probe Master reagent (Roche; 04673409001), and results were analyzed using the ABI 7900 Real-Time PCR detection system (Applied Biosystems; 4351405). Relative expression was normalized to 18S RNA and calibrated to the respective controls.
Electron Microscopy
Mice were deeply anesthetized with CO2 and perfused transcardially with 10 mL phosphate buffer followed by 10 mL 2.5% glutaraldehyde/2% PFA in 0.1 M CaCO4. The quadriceps were removed, fixed in the same fixative, and postfixed in 2% osmium tetroxide in 0.1 M sodium cacodylate buffer with 0.3% potassium ferrocyanide for 2 h. The samples were then dehydrated through a series of graded ethanol-to-propylene oxide solutions, infiltrated and embedded in epoxy resin, and polymerized at 70°C overnight. Sem ithin (0.5-μm) sections were stained with toluidine blue for light microscope examination. Ultrathin (80-nm) sections were cut and imaged using an FEI Tecnai F 20 TEM FEG electron microscope with an AT XR41 camera.
Interactome Analysis and Phosphosite Mapping by Mass Spectrometry
A composite ULK1 protein–protein interaction network was generated based on STRING (v10) (Szklarczyk et al., 2015), BioPlex (Huttlin et al., 2015) (downloaded in January 2016), and InWeb_IM (Li et al., 2017). Edges with evidence of physical interactions (e.g., coIP or yeast two-hybrid) were used in the network construction. Protein–protein interactions were further appended by BioGRID (Chatr-Aryamontri et al., 2017), which includes both physical interactions and colocalization interactions. The ULK1 interactome was further annotated with autophagy-related proteins (HADb Human Autophagy Database; http://autophagy.lu/; accessed 2018/03/01) (Homma et al., 2011; Taylor et al., 2016) and stress granule-related proteins (Jain et al., 2016; Taylor et al., 2016). Pathway analysis for the ULK1 interactome was performed using ENRICHR (Kuleshov et al., 2016) and Fisher’s exact test.
The mass spectrometric (MS) analysis was performed using an optimized platform as previously described (Xu et al., 2009). ULK1-interacting proteins were visualized by performing GelCode Blue staining (Thermo Fisher Scientific; 24590) according to the manufacturer’s instructions. Proteins in gel bands were reduced by adding DTT and then alkylated by adding iodoacetamide. The gel bands were washed, dried in a speed vacuum, and rehydrated with a trypsin-containing buffer for overnight proteolysis. The digested peptides were extracted, dried, reconstituted, and loaded onto a capillary reverse-phase C18 column by an HPLC system (Waters ACQUITY UPLC). Peptides were eluted in a gradient, ionized by electrospray ionization, and detected by an in-line mass spectrometer (Thermo Fisher Scientific LTQ Orbitrap Elite). MS spectra were collected, and the top 20 abundant ions were sequentially isolated for MS/MS analysis. This process was cycled over the entire liquid chromatography gradient. The acquired MS/MS spectra were used to search protein databases to obtain possible peptide matches by our JUMP algorithm (Wang et al., 2014). All matched MS/MS spectra were filtered by mass accuracy and matching scores to reduce the protein FDR to less than 1%. Label-free quantitative comparison based on spectral counting (Pagala et al., 2015) was used to differentiate ULK1-interacting proteins from background proteins. A similar procedure was used for mapping phosphosites in VCP by geLC-MS/MS. The quantitative phosphosite analyses were performed with synthetic peptides (unphosphorylated and phosphorylated forms) spiked in the samples.
Protein Expression and Purification
All plasmids were generated using standard PCR-based cloning strategies. Human VCP/p97 protein with an N-terminal 6×-His tag followed by a TEV (Tobacco Etch Virus) protease cleavage site was expressed in E. coli BL21 (DE3) cells. Bacteria were grown in Luria Broth to an optical density of 0.6–0.8 at 37°C, followed by overnight induction with 1 mM isopropyl-β-D-thio-galactopyranoside (IPTG) at 18°C. Cells were p elleted and resuspended in lysis buffer (50 mM Tris pH 7.5, 500 mM NaCl, 2 mM β-mercaptoethanol, 30 mM imidazole) and lysed under pressure (25,000 psi) using an Avestin EmulsiFlex C3 homogenizer. Protein was purified using Ni-NTA affinity chromatography and eluted in lysis buffer containing 500 mM imidazole before being incubated with 6×-His tagged TEV protease and dialyzed overnight against cleavage buffer (50 mM Tris pH 7.5, 5 mM β-mercaptoethanol). The cleaved tags and TEV protease were separated from the proteins of interest by a second round of Ni-NTA affinity chromatography followed by size-exclusion chromatography on a Superdex S200 column (GE Healthcare) in size-exclusion buffer (25 mM sodium phosphate pH 7.5, 50 mM NaCl, 1 mM EDTA, 2 mM TCEP). Purified fractions were pooled, concentrated, and flash-frozen in liquid nitrogen.
In Vitro Kinase Assay
Recombinant human ULK1 (1–649) and human ULK2 (1–631) were purchased from Sigma Aldrich (SRP5096 and SRP5097). The reaction was conducted in kinase buffer (10 mM Tris pH 7.4, 15 mM NaCl, 10 mM MgCl2, and 0.5 mM DTT) in a final volume of 30 μL, with 5 μCi 32P-ATP or nonradioactive ATP and 0.3 μg of either ULK1 or ULK2, and varying amounts of purified recombinant VCP. Reactions were incubated at 37°C for 15 min, and then stopped with addition of 2× Laemmli sample buffer (Sigma; S3401). Half of the samples were subjected to gel electrophoresis in a 4%−12% Bis-Tris gel and stained with GelCode Blue Stain Reagent (Thermo Fisher Scientific; 24590), and the other half were separated by electrophoresis and visualized by autoradiography.
Immunofluorescence Microscopy
Cells growing on glass-bottom dishes (ibidi; 81156) were fixed in 4% PFA for 5 min at room temperature, permeabilized with 0.1% TBST, blocked in 3% BSA, and then incubated with one of the following primary antibodies at 4°C overnigh t: mouse anti-FLAG (also recognizes the “DDK” tag) (Sigma Aldrich; F3165), mouse anti-G3BP1 (BD Biosciences; 611126), chicken anti-GFP (Abcam; ab13970), goat anti-TIA1 (Santa Cruz Biotechnology; sc-1751), rabbit anti-TIAR (Cell Signaling; 8509S), goat anti-eIF3η (Santa Cruz Biotechnology; sc-16377), rabbit anti–TDP-43 (Proteintech; 12892–1-AP), rabbit anti-PABP (Abcam; ab21060), rabbit anti-HA (Cell Signaling; 3724). Cells were then incubated with secondary antibodies conjugated to Alexa-488, −555 or −647 (Invitrogen), and mounted with ProLong Gold Antifade Reagent with DAPI (Invitrogen; P36931). To detect endogenous ULK1 and ULK2, the signal was amplified using Alexa Fluor 488 Tyramide SuperBoost Kit (Thermo Fisher Scientific; B40941) per the manufacturer’s instructions. Quantification of cells with stress granules was performed as previously described (Buchan et al., 2013).
Plasmid Constructs
The pCMV6-DDK-human ULK1 (RC215643) and pCMV6-DDK-human ULK2 (RC206010) were purchased from Origene. The pMXs-IP-EGFP-ULK1 (38193) and pMXs-IP-EGFP-ULK2 (38201) were obtained from Addgene. FLAG-VCP WT (Tresse et al., 2010), FLAG-VCP A232E (Tresse et al., 2010), GFP-TIA1 WT (Mackenzie et al., 2017), GFP-TIA1 A381T (Mackenzie et al., 2017), and HA-FUS R521C (Guo et al., 2018) have been described previously. Constructs carrying point mutations were generated using a QuikChange II Site-Directed Mutagenesis Kit (Agilent Technologies; 200524) per the manufacturer’s instructions. To generate retroviral constructs, cDNA of Ulk1, Ulk2, and Vcp was amplified using a standard PCR-based approach. The amplified cDNA was cloned into pMSCV-IRES-EGFP retroviral vector (a gift from Joseph T. Opferman, St. Jude Children’s Research Hospital) using NEBuilder HiFi DNA Assembly Kit (New England Biolabs; E2621) per the manufacturer’s instructions.
Transfection, Gene Silencing, and Drug Treatment
To transiently overexpress cDNA constructs, cells were transfected with FuGENE 6 (Promega; E2691) or FuGENE HD (Promega; E2311) according to the manufacturer’s instructions. Knockdown experiments were performed using Lipofectamine RNAi Max (Life Technologies; 13778075) per the manufacturer’s instructions and with the following siRNA constructs obtained from Dharmacon: pooled nontargeting siRNA (D-001810–10-05), SMARTpool: ON-TARGETplus mouse Ulk1 siRNA (L-040155–00-0005), ON-TARGETplus mouse Ulk2 siRNA (L-040619–00-0005), ON-TARGETplus mouse Atg7 siRNA (L-049953–00-0005), ON-TARGETplus mouse Atg14 siRNA (L-172696–00-0005), ON-TARGETplus mouse Atg13 siRNA (L-053540–01-0005), ON-TARGETplus mouse Rb1cc1 siRNA (L-041191–01-0005), ON-TARGETplus mouse Vcp siRNA (L-057592–00-0005), ON-TARGETplus human VCP siRNA (L-008727–00-0005). Chemicals dissolved in DMSO were as follows: SBI-0206965 (20 μM, Sigma Aldrich), sodium arsenite (1 mM, Sigma Aldrich), bafilomycin A1 (0.5 μM; Sigma Aldrich), Eer1 (20 μM, Sigma Aldrich), CCCP (20 μM, Sigma Aldrich), and LYN-1604 (2 μM, Glixx Laboratories).
Live-Cell Imaging
Live-cell imaging using U2OS cells stably expressing G3BP1-GFP has been described previously (Mackenzie et al., 2017). Briefly, cells growing on cover slips were treated with DMSO or SBI-0206965 (20 μM) for 18 h prior to imaging. Using SlideBook software with definite focus engaged, multipoint images were taken every 30 s with the 488-nm laser at 30% power. Two minutes into imaging, both the Microaqueduct Slide and objective temperatures were raised to 43°C. At 30 min after heat shock, both we re lowered back to 37°C to alleviate the stress, and cells were imaged until granules disappeared or after 3–4 h had passed. Fresh media was perfused (250–500 μL/h) through the chamber throughout imaging. Live-cell imaging using U2OS cells stably expressing G3BP1-tdTomato was performed with Opterra microscopy (BRUKER). Briefly, cells growing on glass-bottom dishes were treated with DMSO or LYN-1604 (2 μM) for 24 h prior to imaging. Multipoint images were taken every 30 s with the 555-nm laser at 30% power. At 30 s into imaging, the objective temperatures were raised to 43°C for 60 min. After heat shock, the temperature was lowered back to 37°C to alleviate the stress, and cells were imaged until granules disappeared or after 2–3 h had passed.
ATPase Assay
Standard ATPase assays were carried out in assay buffer containing 25 mM Tris-HCl (pH 7.5), 50 mM NaCl, 5 mM DTT, 0.3 μM VCP, and a varying concentration of ATP for 30 min at room temperature. The inorganic phosphate released by ATP hydrolysis was measured using Biomol Green in 96-well plates (Enzo; BML-AK111–0250), and the absorbance at 635 nm was measured using a PerkinElmer 2300 EnSpire Multilabel Plate Reader. The average background absorbance obtained from reactions performed without VCP was subtracted from each reading. Reactions performed without VCP were included as negative controls and were subtracted from the experimental data gained by adding VCP. The phosphate released was calculated based on the standard curve established by phosphate standards. The assays were repeated at least three times, all of which showed similar trends.
QUANTIFICATION AND STATISTICAL ANALYSIS
All analyses were performed blinded to genotype. All quantitative data are shown as mean ± SEM from n ≥3 biological replicates unless otherwise specified. Statistical significance was determined by two-tailed Student’s t-tests or ANOVA as appropriate, and *P <0.05 was considered statistically significant. Statistical parameters are also reported in the Figures and Figure Legends.
Supplementary Material
The ULK1 protein-protein interaction network is shown. A group is defined as identified proteins that share one or more peptides (e.g., 01). The group is represented by the protein with maximal spectra count (e.g., 01.1). If other proteins in the same group are assigned with at least one uniquely identified peptide, these proteins are also counted as unique proteins (e.g., 01.2). Abundance: SC × 50 (KD) / protein size (KD). The P-value was derived by G-test. The summed SC of the control samples was compared with that of the test samples. FDR was corrected based on Benjamini–Hochberg procedure. 0.1 was added as pseudocount. A fold-enrichment cutoff of 2.0, a P-value cutoff of 5% P-value, and an FDR cutoff of 10% were applied. Fold-enrichment cutoff was calculated based on the control gene set revealed by K-means clustering on the global log2 fold change. An existing protein-protein interaction database of ULK1 was derived from public databases (STRING, BIOPLEX, InWeb, BIOGRID). Abbreviations: FDR, false discovery rate; SC, summed number of spectral counts (i.e., MS/MS scans) assigned to one identified protein; TP, total number of peptides identified for a protein.
Pathway analysis was performed based on the St. Jude–derived ULK1 interactome that satisfied cutoffs of adj.pval <0.05 and Combined Score >1. EnrichR (http://amp.pharm.mssm.edu/Enrichr/) was used to perform the analysis. For the autophagy and stress granule pathway, a custom database was generated and Fisher’s exact test was performed.
A composite ULK1 protein–protein interaction network was generated based on STRING (v10), BioPlex, InWeb_IM, and BioGRID. Edges are supported by either physical interactions or colocalization complex evidence. See Table S2 for details of SJ_CoIP.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse anti-SQSTM1/p62 | Abnova | Cat# H00008878-M01; RRID:AB_437085 |
| Rabbit anti-SQSTM1/p62 | Sigma-Aldrich | Cat# P0067; RRID:AB_1841064 |
| Rabbit anti-SQSTM1/p62 | Abcam | ab194720 |
| Rabbit anti-ubiquitin | DAKO | Cat# Z0458; RRID:AB_2315524 |
| Rabbit anti-ubiquitin | Enzo Life Sciences | Cat# BML-PW8810–0500; RRID:AB_2051891 |
| Rabbit anti-LC3B | MBL International | Cat# PM036; RRID:AB_2274121 |
| Rabbit anti-LC3B | Millipore | Cat# L7543; RRID:AB_796155 |
| Rat anti-LAMP1 | DSHB | Cat# 1 d4b; RRID:AB_2134500 |
| Mouse anti-SMI-31 | Covance | Cat# SMI-31 R-100; RRID:AB_10122491 |
| Rabbit anti–β-amyloid 1–42 | Millipore | Cat# AB5078P; RRID:AB_91677 |
| Rabbit anti-FUS | Bethyl | Cat# IHC-00074; RRID:AB_2247085 |
| Rabbit anti–TDP-43 | Proteintech | Cat# 10782–2-AP; RRID:AB_615042 |
| Rabbit anti–TDP-43 | Proteintech | Cat# 12892–1-AP; RRID:AB_2200505 |
| Goat anti-TIA1 | Santa Cruz Biotechnology | Cat# sc-1751; RRID:AB_2201433 |
| Mouse anti-hnRNP A2B1 | Santa Cruz Biotechnology | Cat#sc-53531; RRID:AB_2248245 |
| Rabbit anti-IBA1 | BIOCARE MEDICAL | Cat# CP290 A; RRID:AB_10578940 |
| Mouse anti-MHCf | Leica Microsystems | Cat# NCL-MHCf; RRID:AB_563899 |
| Mouse anti-MHCs | Leica Microsystems | Cat# NCL-MHCs; RRID:AB_563898 |
| Rabbit anti-ULK1 | Sigma-Aldrich | Cat# A7481; RRID:AB_1840703 |
| Goat anti-ULK1 | Santa Cruz Biotechnology | Cat# sc-10900; RRID:AB_2212515 |
| Rabbit anti-COXIV | Abcam | Cat# ab16056; RRID:AB_443304 |
| Rabbit anti-TOM20 | Santa Cruz Biotechnology | Cat# sc-11415; RRID:AB_2207533 |
| Rabbit anti-ATG7 | Cell Signaling Technology | Cat# 8558; RRID:AB_10831194 |
| Rabbit anti-ATG13 | Sigma-Aldrich | Cat# SAB4200100; RRID:AB_10602787 |
| Rabbit anti-pATG13 | Rockland | Cat# 600–401-C49; RRID:AB_11179920 |
| Rabbit anti-RB1CC1 | Cell Signaling Technology | Cat# 12436S; RRID: N/A |
| Rabbit anti-ATG14 | MBL International | Cat# PD026; RRID:AB_1953054 |
| Mouse anti-VCP | Thermo Fisher Scientific | Cat# MA3–004; RRID:AB_2214638 |
| Mouse anti-VCP | Santa Cruz Biotechnology | Cat# sc-57492; RRID:AB_793927 |
| Mouse anti-FLAG | Sigma-Aldrich | Cat# A8592; RRID:AB_439702 |
| Mouse anti-FLAG | Sigma-Aldrich | Cat# F3165; RRID:AB_259529 |
| Rabbit anti-G3BP1 | Proteintech | Cat# 13057–2-AP; RRID:AB_2232034 |
| Mouse anti-G3BP1 | BD Biosciences | Cat# 611126; RRID:AB_398437 |
| Rabbit anti-Caprin-1 | Proteintech | Cat# 15112–1-AP; RRID:AB_2070016 |
| Rabbit anti-USP10 | Cell Signaling Technology | Cat# 8501; RRID:AB_10949976 |
| Rabbit anti-GAPDH | Sigma-Aldrich | Cat# G9545; RRID:AB_796208 |
| Chicken anti-GFP | Abcam | Cat# ab13970; RRID:AB_300798 |
| Rabbit anti-TIAR | Cell Signaling Technology | Cat# 8509; RRID:AB_10839263 |
| Goat anti-eIF3η | Santa Cruz Biotechnology | Cat# sc-16377; RRID:AB_671941 |
| Rabbit anti-PABP | Abcam | Cat# ab21060; RRID:AB_777008 |
| Rabbit anti-HA | Cell Signaling Technology | Cat# 3724; RRID:AB_1549585 |
| Sheep anti-rabbit IgG–HRP conjugate | GE Healthcare | Cat# RPN4301; RRID:AB_2650489 |
| Sheep anti-mouse IgG–HRP conjugate | GE Healthcare | Cat# RPN4201; RRID: N/A |
| Bacterial and Virus Strains | ||
| E. coli DH5α | Thermo Fisher Scientific | Cat# C404003 |
| E. coli Rosetta (DE3) | Millipore | Cat# 70954 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| DMSO | Sigma Aldrich | Cat# D8418 |
| SBI-0206965 | Sigma Aldrich | Cat# SML1540 |
| Sodium arsenite | Sigma Aldrich | Cat# 1062771000 |
| Bafilomycin A1 | Sigma Aldrich | Cat# B1793 |
| Eer1 | Sigma Aldrich | Cat# E1286 |
| LYN-1604 | Glixx Laboratories | Cat# GLXC-09854 |
| Human ULK1 (1–649) | Sigma Aldrich | Cat# SRP5096 |
| Human ULK2 (1–631) | Sigma Aldrich | Cat# SRP5097 |
| Recombinant human VCP | This study | N/A |
| FuGENE 6 | Promega | Cat# E2691 |
| FuGENE HD | Promega | Cat# E2311 |
| Lipofectamine RNAi Max | Life Technologies | Cat# 13778075 |
| Polybrene | Sigma-Aldrich | Cat# H9268–106 |
| ProLong Gold Antifade Reagent with DAPI | Invitrogen | Cat# P36931 |
| Protein G agarose beads | Thermo Fisher Scientific | Cat# 20399 |
| 2× Laemmli sample buffer | Sigma | Cat# S3401 |
| Critical Commercial Assays | ||
| DISCOVERY ChromoMap DAB Kit | Roche | Cat# 760–159 |
| 4%−12% Bis-Tris gels | Life Technologies | Cat# NP0335B0X |
| ECL Western Blotting Detection | GE Healthcare | Cat# RPN2232 |
| RNeasy Fibrous Tissue Mini Kit | QIAGEN | Cat# 74704 |
| FastStart TaqMan Probe Master reagent | Roche | Cat# 04673409001 |
| SuperScript III first-strand synthesis kit | Life Technologies | Cat# 18080051 |
| NEBuilder HiFi DNA Assembly Kit | New England Biolabs | Cat# E2621 |
| Alexa Fluor 488 Tyramide SuperBoost Kit | Thermo Fisher Scientific | Cat# B40941 |
| Biomol Green | Enzo Life Sciences | Cat# BML-AK111–0250 |
| Deposited data | ||
| Original images and western blot | This paper | http://dx.doi.org/10.17632/zxpzhv2nkk.1 |
| Experimental Models: Cell Lines | ||
| HEK293T | ATCC | CRL-3216; RRID:CVCL_0063 |
| U2OS | ATCC | HTB-96 |
| HeLa | ATCC | CCL-2 |
| C2C12 | ATCC | CRL-1772 |
| G3BP1-GFP U2OS cells | (Mackenzie et al., 2017) | N/A |
| td-Tomato-G3BP1 U2OS cells | This study | N/A |
| 3×-FLAG-tagged ULK1 MEFs | This study | N/A |
| 3×-FLAG-tagged ULK2 MEFs | This study | N/A |
| 3×-FLAG-tagged ULK1 C2C12 | This study | N/A |
| 3×-FLAG-tagged ULK2 C2C12 | This study | N/A |
| Experimental Models: Organisms/Strains | ||
| Mouse: Ulk1flox-neo | (Kundu et al., 2008) | N/A |
| Mouse: Ulk2flox | (Joo et al., 2016) | N/A |
| Mouse: Ulk2−/− | (Joo et al., 2016) | N/A |
| Mouse: Ella-Cre | Jackson Laboratory | JAX: 003724 |
| Mouse: Ckmm-Cre | Jackson Laboratory | JAX: 006475 |
| Mouse: Atg7flox/flox | (Komatsu et al., 2005) | N/A |
| Oligonucleotides | ||
| Nontargeting siRNA | Dharmacon | D-001810–10-05 |
| ON-TARGETplus mouse Ulk1 siRNA | Dharmacon | L-040155–00-0005 |
| ON-TARGETplus mouse Ulk2 siRNA | Dharmacon | L-040619–00-0005 |
| ON-TARGETplus mouse Atg7 siRNA | Dharmacon | L-049953–00-0005 |
| ON-TARGETplus mouse Atg14 siRNA | Dharmacon | L-172696–00-0005 |
| ON-TARGETplus mouse Atg13 siRNA | Dharmacon | L-053540–01-0005 |
| ON-TARGETplus mouse Rb1cc1 siRNA | Dharmacon | L-041191–01-0005 |
| ON-TARGETplus mouse Vcp siRNA | Dharmacon | L-057592–00-0005 |
| ON-TARGETplus human VCP siRNA | Dharmacon | L-008727–00-0005 |
| Ulk1 TaqMan probe | Applied Biosystems | 4331182 |
| Ulk2 TaqMan probe | Applied Biosystems | 4331182 |
| 18 TaqMan probe | Applied Biosystems | 4333760F |
| Vcp-S13A Forward: 5′–GGTGATGACCTAGCCACAGCCATTCTCAAAC–3′ | This study | N/A |
| Vcp-S13A Reverse: 5′–GTTTGAGAATGGCTGTGGCTAGGTCATCACC–3′ | This study | N/A |
| Vcp-S282A Forward: 5′–AATTGGCTGGTGAGGCTGAGAGCAACCTTC–3′ | This study | N/A |
| Vcp-S282A Reverse: 5′–GAAGGTTGCTCTCAGCCTCACCAGCCAATT–3′ | This study | N/A |
| Vcp-T761A Forward: 5′–AGATGTTTGCCCAGGCCCTTCAGCAGAGTCG–3′ | This study | N/A |
| Vcp-T761A Reverse: 5′–CGACTCTGCTGAAGGGCCTGGGCAAACATCT–3′ | This study | N/A |
| Vcp-S13D Forward: 5′–GGTGATGACCTAGATACAGCCATTCTCAAAC–3′ | This study | N/A |
| Vcp-S13D Reverse: 5′–GTTTGAGAATGGCTGTATCTAGGTCATCACC–3′ | This study | N/A |
| Vcp-S282D Forward: 5′–AATTGGCTGGTGAGGATGAGAGCAACCTTC–3′ | This study | N/A |
| Vcp-S282D Reverse: 5′–GAAGGTTGCTCTCATCCTCACCAGCCAATT–3′ | This study | N/A |
| Vcp-T761D Forward: 5′–AGATGTTTGCCCAGGATCTTCAGCAGAGTCG–3′ | This study | N/A |
| Vcp-T761D Reverse: 5′–CGACTCTGCTGAAGATCCTGGGCAAACATCT–3′ | This study | N/A |
| 5gen.G3BP1 Forward 5′–GTACAGTACCATCTTTTCACACTGG–3′ | This study | N/A |
| 5junc.tdTomato Reverse 5′–TTGAAGCGCATGAACTCTTTGATGA–3′ | This study | N/A |
| 3junc.tdTomato Forward 5′–CACCTCCCACAACGAGGAC–3′ | This study | N/A |
| 3gen.G3BP1 Reverse 5′–TAATTCCCCACATACTCTGAACC–3′ | This study | N/A |
| mUlk1 sgRNA Target 5′–CAGGTTGGCAGCAGGTAGTCAGG–3′ | This study | N/A |
| mUlk1.g15.sense.ssODN 5′–TCCACAGGCAAGCTGTGCATTGAGAGGAGACTCTCGGCCCTGCTGAGTGGTGTCTATGCCGACTACAAAGACCATGACGGTGATTATAAAGATCATGACATCGATTACAAGGATGACGATGACAAGTGACTACCTGCTGCCAACCTGCAGGGTAGGGTCTGAGACCTGGCAGACTGTCCTCAACACTGA–3′ | This study | N/A |
| mUlk1.NGS Forward 5′–CACTCTTTCCCTACACGACGCTCTTCCGATCTAGGGGTTGCAGCACACTCTCACGGA–3′ | This study | N/A |
| mUlk1.NGS Reverse 5′–GTGACTGGAGTTCAGACGTGTGCTCTTCCGATCTAGGGACCCCTGGCACTCTGATGTCT–3′ | This study | SN/A |
| mUlk2 sgRNA Target 5′–ACTGCAACTGTGTGAGTAGCAGG–3′ | This study | N/A |
| mUlk2.g15.sense.ssODN 5′–CTAGATAAATGTAGTATTGAAAGAAGATTGTCAGCACTCTGCTGTAGCACTGCAACTGTGGACTACAAAGACCATGACGGTGATTATAAAGATCATGACATCGATTACAAGGATGACGATGACAAGTGAGTAGCAGGCTTGTCTGTGGACTGGCATGGAACAGGAGGTGATACATTTGGGATTACA–3′ | This study | N/A |
| mUlk2.NGS Forward 5′–CACTCTTTCCCTACACGACGCTCTTCCGATCTTGGGGCCAAGATAGAGCAGGAACCA–3′ | This study | N/A |
| mUlk2.NGS Reverse 5′–GTGACTGGAGTTCAGACGTGTGCTCTTCCGATCTTGAAGAGAAAGGAAAGGGGGAGTGACT–3′ | This study | N/A |
| Recombinant DNA | ||
| pCMV6-DDK-human ULK1 | Origene | RC215643 |
| pCMV6-DDK-human ULK2 | Origene | RC206010 |
| pMXs-IP-EGFP-ULK1 | Addgene | 38193 |
| pMXs-IP-EGFP-ULK2 | Addgene | 38201 |
| FLAG-VCP WT | (Tresse et al., 2010) | N/A |
| FLAG-VCP A232E | (Tresse et al., 2010) | N/A |
| pEGFP-TIA1 | (Mackenzie et al., 2017) | N/A |
| pEGFP-TIA1 A381T | (Mackenzie et al., 2017) | N/A |
| HA-FUS R521C | (Guo et al., 2018) | N/A |
| MLM3636 | Addgene | 43860 |
| p3s-Cas9HC | Addgene | 43945 |
| Software and Algorithms | ||
| ImageJ | NIH | https://imagej.nih.gov/ij/ |
| SlideBook 6.0 | 3i | https://www.intelligent-imaging.com/slidebook |
Highlights.
Ulk1/2 deficiency causes IBM-like disease with TDP-43+ and ubiquitin+ pathology
ULK1/2 promotes resolution of stress granules in an autophagy-independent manner
ULK1/2 localizes to stress granules and interacts with the AAA+ ATPase VCP
Phosphorylation of VCP by ULK1/2 is key to efficient disassembly of stress granules
ACKNOWLEDGMENTS
We are grateful to Noboru Mizushima (University of Tokyo) and Masaaki Komatsu (Tokyo Metropolitan Institute of Medical Science) for reagents. We are grateful to Natalia Nedelsky and Angela McArthur (St. Jude Children’s Research Hospital) for editing the manuscript. Most of the imaging data were acquired in the St. Jude Cell and Tissue Imaging Core. Electron microscopy samples were prepared and analyzed by Linda Horner and Sharon Frase in the St. Jude Cell and Tissue Imaging Core, and histologic samples were prepared by Pamela Johnson in the St. Jude Veterinary Pathology Core and Mariallan Shadle at the University of Tennessee Health Science Center. The St. Jude core facility resources are supported by Cancer Center Support Grant P30 CA021765 from the National Institutes of Health (NIH) and by ALSAC.
This work was partially supported by ALSAC (M.K., J.P., and J.P.T.), the NIH (R01 MH115058, HL114697 to M.K.; R01 GM114260 to J.P.; R35 NS097974 to J.P.T.), the Howard Hughes Medical Institute (J.P.T.), and the Robert Packard Center for ALS Research (M.K. and J.P.T.).
Footnotes
DECLARATION OF INTERESTS
J.P.T. is a consultant for Third Rock Ventures and Nido Biosciences.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DATA AND SOFTWARE AVAILABILITY
Source data for gel images and graphs can be found in Mendeley Data: http://dx.doi.org/10.17632/zxpzhv2nkk.1.
REFERENCES
- Alberti S, Mateju D, Mediani L, and Carra S. (2017). Granulostasis: Protein Quality Control of RNP Granules. Front Mol Neurosci 10, 84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson P, and Kedersha N. (2002). Stressful initiations. J Cell Sci 115, 3227–3234. [DOI] [PubMed] [Google Scholar]
- Badadani M, Nalbandian A, Watts GD, Vesa J, Kitazawa M, Su H, Tanaja J, Dec E, Wallace DC, Mukherjee J, et al. (2010). VCP associated inclusion body myopathy and paget disease of bone knock-in mouse model exhibits tissue pathology typical of human disease. PLoS One 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badders NM, Korff A, Miranda HC, Vuppala PK, Smith RB, Winborn BJ, Quemin ER, Sopher BL, Dearman J, Messing J, et al. (2018). Selective modulation of the androgen receptor AF2 domain rescues degeneration in spinal bulbar muscular atrophy. Nat Med 24, 427–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee S, Bartesaghi A, Merk A, Rao P, Bulfer SL, Yan Y, Green N, Mroczkowski B, Neitz RJ, Wipf P, et al. (2016). 2.3 A resolution cryo-EM structure of human p97 and mechanism of allosteric inhibition. Science 351, 871–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodnar NO, and Rapoport TA (2017). Molecular Mechanism of Substrate Processing by the Cdc48 ATPase Complex. Cell 169, 722–735e729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruning JC, Michael MD, Winnay JN, Hayashi T, Horsch D, Accili D, Goodyear LJ, and Kahn CR (1998). A muscle-specific insulin receptor knockout exhibits features of the metabolic syndrome of NIDDM without altering glucose tolerance. Mol Cell 2, 559–569. [DOI] [PubMed] [Google Scholar]
- Bucelli RC, Arhzaouy K, Pestronk A, Pittman SK, Rojas L, Sue CM, Evila A, Hackman P, Udd B, Harms MB, et al. (2015). SQSTM1 splice site mutation in distal myopathy with rimmed vacuoles. Neurology 85, 665–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchan JR, Kolaitis RM, Taylor JP, and Parker R. (2013). Eukaryotic stress granules are cleared by autophagy and Cdc48/VCP function. Cell 153, 1461–1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatr-Aryamontri A, Oughtred R, Boucher L, Rust J, Chang C, Kolas NK, O’Donnell L, Oster S, Theesfeld C, Sellam A, et al. (2017). The BioGRID interaction database: 2017 update. Nucleic Acids Res 45, D369–D379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheong H, Wu J, Gonzales LK, Guttentag SH, Thompson CB, and Lindsten T. (2014). Analysis of a lung defect in autophagy-deficient mouse strains. Autophagy 10, 45–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chitiprolu M, Jagow C, Tremblay V, Bondy-Chorney E, Paris G, Savard A, Palidwor G, Barry FA, Zinman L, Keith J, et al. (2018). A complex of C9ORF72 and p62 uses arginine methylation to eliminate stress granules by autophagy. Nat Commun 9, 2794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Custer SK, Neumann M, Lu H, Wright AC, and Taylor JP (2010). Transgenic mice expressing mutant forms VCP/p97 recapitulate the full spectrum of IBMPFD including degeneration in muscle, brain and bone. Hum Mol Genet 19, 1741–1755. [DOI] [PubMed] [Google Scholar]
- Dao TP, Kolaitis RM, Kim HJ, O’Donovan K, Martyniak B, Colicino E, Hehnly H, Taylor JP, and Castaneda CA (2018). Ubiquitin Modulates Liquid-Liquid Phase Separation of UBQLN2 via Disruption of Multivalent Interactions. Mol Cell 69, 965–978e966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng Z, Purtell K, Lachance V, Wold MS, Chen S, and Yue Z. (2017). Autophagy Receptors and Neurodegenerative Diseases. Trends Cell Biol 27, 491–504. [DOI] [PubMed] [Google Scholar]
- Dormann D, and Haass C. (2013). Fused in sarcoma (FUS): an oncogene goes awry in neurodegeneration. Mol Cell Neurosci 56, 475–486. [DOI] [PubMed] [Google Scholar]
- Egan DF, Chun MG, Vamos M, Zou H, Rong J, Miller CJ, Lou HJ, Raveendra-Panickar D, Yang CC, Sheffler DJ, et al. (2015). Small Molecule Inhibition of the Autophagy Kinase ULK1 and Identification of UlK1 Substrates. Mol Cell 59, 285–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Figley MD, Bieri G, Kolaitis RM, Taylor JP, and Gitler AD (2014). Profilin 1 associates with stress granules and ALS-linked mutations alter stress granule dynamics. J Neurosci 34, 8083–8097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gan B, Peng X, Nagy T, Alcaraz A, Gu H, and Guan JL (2006). Role of FIP200 in cardiac and liver development and its regulation of TNFalpha and TSC-mTOR signaling pathways. J Cell Biol 175, 121–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo L, Kim HJ, Wang H, Monaghan J, Freyermuth F, Sung JC, O’Donovan K, Fare CM, Diaz Z, Singh N, et al. (2018). Nuclear-Import Receptors Reverse Aberrant Phase Transitions of RNA-Binding Proteins with Prion-like Domains. Cell 173, 677–692e620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, Yokoyama M, Mishima K, Saito I, Okano H, et al. (2006). Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 441, 885–889. [DOI] [PubMed] [Google Scholar]
- Hilton-Jones D, and Brady S. (2016). Diagnostic criteria for inclusion body myositis. J Intern Med 280, 52–62. [DOI] [PubMed] [Google Scholar]
- Homma K, Suzuki K, and Sugawara H. (2011). The Autophagy Database: an all-inclusive information resource on autophagy that provides nourishment for research. Nucleic Acids Res 39, D986–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hulsmann J, Kravic B, Weith M, Gstaiger M, Aebersold R, Collins BC, and Meyer H. (2018). AP-SWATH Reveals Direct Involvement of VCP/p97 in Integrated Stress Response Signaling Through Facilitating CReP/PPP1R15B Degradation. Mol Cell Proteomics 17, 1295–1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huttlin EL, Ting L, Bruckner RJ, Gebreab F, Gygi MP, Szpyt J, Tam S, Zarraga G, Colby G, Baltier K, et al. (2015). The BioPlex Network: A Systematic Exploration of the Human Interactome. Cell 162, 425–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain S, Wheeler JR, Walters RW, Agrawal A, Barsic A, and Parker R. (2016). ATPase-Modulated Stress Granules Contain a Diverse Proteome and Substructure. Cell 164, 487–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joo JH, Wang B, Frankel E, Ge L, Xu L, Iyengar R, Li-Harms X, Wright C, Shaw TI, Lindsten T, et al. (2016). The Noncanonical Role of uLk/AtG1 in ER-to-Golgi Trafficking Is Essential for Cellular Homeostasis. Mol Cell 62, 491–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaizuka T, and Mizushima N. (2016). Atg13 Is Essential for Autophagy and Cardiac Development in Mice. Mol Cell Biol 36, 585–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komatsu M, Waguri S, Chiba T, Murata S, Iwata J, Tanida I, Ueno T, Koike M, Uchiyama Y, Kominami E, et al. (2006). Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 441, 880–884. [DOI] [PubMed] [Google Scholar]
- Komatsu M, Waguri S, Ueno T, Iwata J, Murata S, Tanida I, Ezaki J, Mizushima N, Ohsumi Y, Uchiyama Y, et al. (2005). Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J Cell Biol 169, 425–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuleshov MV, Jones MR, Rouillard AD, Fernandez NF, Duan Q, Wang Z, Koplev S, Jenkins SL, Jagodnik KM, Lachmann A, et al. (2016). Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res 44, W90–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuma A, Hatano M, Matsui M, Yamamoto A, Nakaya H, Yoshimori T, Ohsumi Y, Tokuhisa T, and Mizushima N. (2004). The role of autophagy during the early neonatal starvation period. Nature 432, 1032–1036. [DOI] [PubMed] [Google Scholar]
- Kundu M, Lindsten T, Yang CY, Wu J, Zhao F, Zhang J, Selak MA, Ney PA, and Thompson CB (2008). Ulk1 plays a critical role in the autophagic clearance of mitochondria and ribosomes during reticulocyte maturation. Blood 112, 1493–1502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li T, Wernersson R, Hansen RB, Horn H, Mercer J, Slodkowicz G, Workman CT, Rigina O, Rapacki K, Staerfeldt HH, et al. (2017). A scored human protein-protein interaction network to catalyze genomic interpretation. Nat Methods 14, 61–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackenzie IR, Nicholson AM, Sarkar M, Messing J, Purice MD, Pottier C, Annu K, Baker M, Perkerson RB, Kurti A, et al. (2017). TIA1 Mutations in Amyotrophic Lateral Sclerosis and Frontotemporal Dementia Promote Phase Separation and Alter Stress Granule Dynamics. Neuron 95, 808–816e809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majcher V, Goode A, James V, and Layfield R. (2015). Autophagy receptor defects and ALS-FTLD. Mol Cell Neurosci 66, 43–52. [DOI] [PubMed] [Google Scholar]
- Markmiller S, Soltanieh S, Server KL, Mak R, Jin W, Fang MY, Luo EC, Krach F, Yang D, Sen A, et al. (2018). Context-Dependent and Disease-Specific Diversity in Protein Interactions within Stress Granules. Cell 172, 590–604e513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masiero E, Agatea L, Mammucari C, Blaauw B, Loro E, Komatsu M, Metzger D, Reggiani C, Schiaffino S, and Sandri M. (2009). Autophagy is required to maintain muscle mass. Cell Metab 10, 507–515. [DOI] [PubMed] [Google Scholar]
- McAlpine F, Williamson LE, Tooze SA, and Chan EY (2013). Regulation of nutrient-sensitive autophagy by uncoordinated 51-like kinases 1 and 2. Autophagy 9, 361–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercer TJ, Gubas A, and Tooze SA (2018). A molecular perspective of mammalian autophagosome biogenesis. J Biol Chem 293, 5386–5395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer H, Bug M, and Bremer S. (2012). Emerging functions of the VCP/p97 AAA-ATPase in the ubiquitin system. Nat Cell Biol 14, 117–123. [DOI] [PubMed] [Google Scholar]
- Meyer H, and Weihl CC (2014). The VCP/p97 system at a glance: connecting cellular function to disease pathogenesis. J Cell Sci 127, 3877–3883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molliex A, Temirov J, Lee J, Coughlin M, Kanagaraj AP, Kim HJ, Mittag T, and Taylor JP (2015). Phase separation by low complexity domains promotes stress granule assembly and drives pathological fibrillization. Cell 163, 123–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori-Konya C, Kato N, Maeda R, Yasuda K, Higashimae N, Noguchi M, Koike M, Kimura Y, Ohizumi H, Hori S, et al. (2009). p97/valosin-containing protein (VCP) is highly modulated by phosphorylation and acetylation. Genes Cells 14, 483–497. [DOI] [PubMed] [Google Scholar]
- Pagala VR, High AA, Wang X, Tan H, Kodali K, Mishra A, Kavdia K, Xu Y, Wu Z, and Peng J. (2015). Quantitative protein analysis by mass spectrometry. Methods Mol Biol 1278, 281–305. [DOI] [PubMed] [Google Scholar]
- Raben N, Hill V, Shea L, Takikita S, Baum R, Mizushima N, Ralston E, and Plotz P. (2008). Suppression of autophagy in skeletal muscle uncovers the accumulation of ubiquitinated proteins and their potential role in muscle damage in Pompe disease. Hum Mol Genet 17, 3897–3908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seguin SJ, Morelli FF, Vinet J, Amore D, De Biasi S, Poletti A, Rubinsztein DC, and Carra S. (2014). Inhibition of autophagy, lysosome and VCP function impairs stress granule assembly. Cell Death Differ 21, 1838–1851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun D, Wu R, Zheng J, Li P, and Yu L. (2018a). Polyubiquitin chain-induced p62 phase separation drives autophagic cargo segregation. Cell Res 28, 405–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, Li TY, Song L, Zhang C, Li J, Lin ZZ, Lin SC, and Lin SY (2018b). Liver-specific deficiency of unc-51 like kinase 1 and 2 protects mice from acetaminophen-induced liver injury. Hepatology 67, 2397–2413. [DOI] [PubMed] [Google Scholar]
- Szklarczyk D, Franceschini A, Wyder S, Forslund K, Heller D, Huerta-Cepas J, Simonovic M, Roth A, Santos A, Tsafou KP, et al. (2015). STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res 43, D447–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanji K, and Bonilla E. (2008). Light microscopic methods to visualize mitochondria on tissue sections. Methods 46, 274–280. [DOI] [PubMed] [Google Scholar]
- Taylor JP (2015). Multisystem proteinopathy: intersecting genetics in muscle, bone, and brain degeneration. Neurology 85, 658–660. [DOI] [PubMed] [Google Scholar]
- Taylor JP, Brown RH Jr., and Cleveland DW (2016). Decoding ALS: from genes to mechanism. Nature 539, 197–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tresse E, Salomons FA, Vesa J, Bott LC, Kimonis V, Yao TP, Dantuma NP, and Taylor JP (2010). VCP/p97 is essential for maturation of ubiquitin-containing autophagosomes and this function is impaired by mutations that cause IBMPFD. Autophagy 6, 217–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turakhiya A, Meyer SR, Marincola G, Bohm S, Vanselow JT, Schlosser A, Hofmann K, and Buchberger A. (2018). ZFAND1 Recruits p97 and the 26S Proteasome to Promote the Clearance of Arsenite-Induced Stress Granules. Mol Cell 70, 906–919e907. [DOI] [PubMed] [Google Scholar]
- van den Boom J, and Meyer H. (2018). VCP/p97-Mediated Unfolding as a Principle in Protein Homeostasis and Signaling. Mol Cell 69, 182–194. [DOI] [PubMed] [Google Scholar]
- Wang B, Iyengar R, Li-Harms X, Joo JH, Wright C, Lavado A, Horner L, Yang M, Guan JL, Frase S, et al. (2017). The autophagy-inducing kinases, ULK1 and ULK2, regulate axon guidance in the developing mouse forebrain via a noncanonical pathway. Autophagy, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B, and Kundu M. (2017). Canonical and noncanonical functions of ULK/Atg1. Curr Opin Cell Biol 45, 47–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Li Y, Wu Z, Wang H, Tan H, and Peng J. (2014). JUMP: a tag-based database search tool for peptide identification with high sensitivity and accuracy. Mol Cell Proteomics 13, 3663–3673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watts GD, Wymer J, Kovach MJ, Mehta SG, Mumm S, Darvish D, Pestronk A, Whyte MP, and Kimonis VE (2004). Inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia is caused by mutant valosin-containing protein. Nat Genet 36, 377–381. [DOI] [PubMed] [Google Scholar]
- Webster CP, Smith EF, Bauer CS, Moller A, Hautbergue GM, Ferraiuolo L, Myszczynska MA, Higginbottom A, Walsh MJ, Whitworth AJ, et al. (2016). The C9orf72 protein interacts with Rab1a and the ULK1 complex to regulate initiation of autophagy. EMBO J 35, 1656–1676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weihl CC, Baloh RH, Lee Y, Chou TF, Pittman SK, Lopate G, Allred P, Jockel-Balsarotti J, Pestronk A, and Harms MB (2015). Targeted sequencing and identification of genetic variants in sporadic inclusion body myositis. Neuromuscul Disord 25, 289–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weihl CC, Miller SE, Hanson PI, and Pestronk A. (2007). Transgenic expression of inclusion body myopathy associated mutant p97/VCP causes weakness and ubiquitinated protein inclusions in mice. Hum Mol Genet 16, 919–928. [DOI] [PubMed] [Google Scholar]
- Yang M, Liang C, Swaminathan K, Herrlinger S, Lai F, Shiekhattar R, and Chen JF (2016). A C9ORF72/SMCR8-containing complex regulates ULK1 and plays a dual role in autophagy. Sci Adv 2, e1601167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youn JY, Dunham WH, Hong SJ, Knight JDR, Bashkurov M, Chen GI, Bagci H, Rathod B, MacLeod G, Eng SWM, et al. (2018). High-Density Proximity Mapping Reveals the Subcellular Organization of mRNA-Associated Granules and Bodies. Mol Cell 69, 517–532e511. [DOI] [PubMed] [Google Scholar]
- Yue Z, Jin S, Yang C, Levine AJ, and Heintz N. (2003). Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc Natl Acad Sci U S A 100, 15077–15082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang G, Wang Z, Du Z, and Zhang H. (2018a). mTOR Regulates Phase Separation of PGL Granules to Modulate Their Autophagic Degradation. Cell 174, 1492–1506e1422. [DOI] [PubMed] [Google Scholar]
- Zhang L, Fu L, Zhang S, Zhang J, Zhao Y, Zheng Y, He G, Yang S, Ouyang L, and Liu B. (2017). Discovery of a small molecule targeting ULK1-modulated cell death of triple negative breast cancer in vitro and in vivo. Chem Sci 8, 2687–2701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang P, Fan B, Yang P, Temirov J, Messing J, Kim HJ, and Taylor JP (2018b). OptoGranules reveal the evolution of stress granules to ALS-FTD pathology. bioRxiv, 348870. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The ULK1 protein-protein interaction network is shown. A group is defined as identified proteins that share one or more peptides (e.g., 01). The group is represented by the protein with maximal spectra count (e.g., 01.1). If other proteins in the same group are assigned with at least one uniquely identified peptide, these proteins are also counted as unique proteins (e.g., 01.2). Abundance: SC × 50 (KD) / protein size (KD). The P-value was derived by G-test. The summed SC of the control samples was compared with that of the test samples. FDR was corrected based on Benjamini–Hochberg procedure. 0.1 was added as pseudocount. A fold-enrichment cutoff of 2.0, a P-value cutoff of 5% P-value, and an FDR cutoff of 10% were applied. Fold-enrichment cutoff was calculated based on the control gene set revealed by K-means clustering on the global log2 fold change. An existing protein-protein interaction database of ULK1 was derived from public databases (STRING, BIOPLEX, InWeb, BIOGRID). Abbreviations: FDR, false discovery rate; SC, summed number of spectral counts (i.e., MS/MS scans) assigned to one identified protein; TP, total number of peptides identified for a protein.
Pathway analysis was performed based on the St. Jude–derived ULK1 interactome that satisfied cutoffs of adj.pval <0.05 and Combined Score >1. EnrichR (http://amp.pharm.mssm.edu/Enrichr/) was used to perform the analysis. For the autophagy and stress granule pathway, a custom database was generated and Fisher’s exact test was performed.
A composite ULK1 protein–protein interaction network was generated based on STRING (v10), BioPlex, InWeb_IM, and BioGRID. Edges are supported by either physical interactions or colocalization complex evidence. See Table S2 for details of SJ_CoIP.