

Abstract
The WD40-repeat protein WDR5 scaffolds various epigenetic writers and is a critical component of the mammalian SET/MLL histone methyltransferase complex. Dysregulation of the MLL1 catalytic function is associated with mixed-lineage leukemia, and antagonism of the WDR5-MLL1 interaction by small molecules has been proposed as a therapeutic strategy for MLL-rearranged cancers. Small molecule binders of the “WIN” site of WDR5 that cause displacement from chromatin have been additionally implicated to be of broader use in cancer treatment. In this study, a fragment screen with Surface Plasmon Resonance (SPR) was used to identify a highly ligand-efficient imidazole-containing compound that is bound in the WIN site. The subsequent medicinal chemistry campaign—guided by a suite of high-resolution cocrystal structures with WDR5—progressed the initial hit to a low micromolar binder. One outcome from this study is a moiety that substitutes well for the side chain of arginine; a tripeptide containing one such substitution was resolved in a high resolution structure (1.5 Å) with a binding mode analogous to the native tripeptide. SPR furthermore indicates a similar residence time (kd = ∼0.06 s−1) for these two analogs. This novel scaffold therefore represents a possible means to overcome the potential permeability issues of WDR5 ligands that possess highly basic groups like guanidine. The series reported here furthers the understanding of the WDR5 WIN site and functions as a starting point for the development of more potent WDR5 inhibitors that may serve as cancer therapeutics.
I. INTRODUCTION
A “cellular multitasker” with many binding partners, the scaffolding protein WDR5 is best studied in the context of histone methylation as part of the SET1/MLL (mixed-lineage leukemia) complexes.1 Each complex has a unique SET/MLL subunit and binds a consistent set of WRAD proteins (WDR5, RbBP5, Ash2L, and DPY30), which enables the methylation of lysine 4 of Histone 3 (H3K4me).2–4 This histone modification is highly enriched at active promoters near transcription start sites (TSSs) and positively correlated with transcription.5 WDR5 is key in this process, particularly for MLL1; the absence of WDR5 abolishes the structural integrity and activity of the MLL1 complex.2,4 Dysregulation of MLL1 is associated with acute myeloid and lymphoblastic leukemias, with MLL-rearranged (MLL-r) cancers retaining a wildtype copy of MLL1 whose activity has separately been ascertained as essential6,7 or nonessential8 for leukemogenesis.
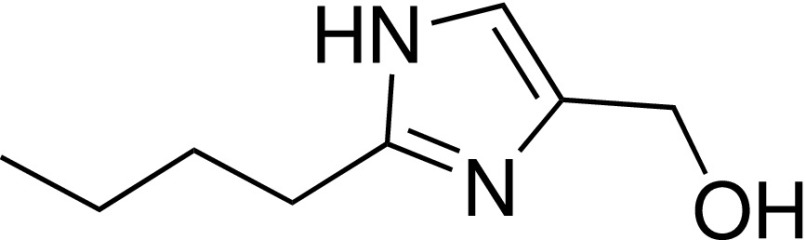
WDR5 comprises a 7-bladed β-propeller fold, each blade composed of a four-stranded antiparallel β-sheet (the WD40 motif).9–11 Cavities on each face, namely, the “WDR5-binding motif” (WBM) and “WDR5-interacting” (WIN) sites, are the interfaces key to its scaffolding role.12,13 In the context of the MLL1 complex, these sites bind RbBP5 and the MLL1 subunit, respectively.13,14 Although it is hypothesized that there are proteins that interact with the “sides” of WDR5,1 structurally interrogated binding partners to date relate only to the WBM and WIN sites. One or both of these sites are appropriate for the scaffolding of various complexes, including the NSL (nonspecific lethal),15 NuRD (nucleosome remodeling and deacetylase),16 and ATAC (Ada2a-containing) complexes.17 WDR5 is also known to bind MYC, an overexpressed oncogene transcription factor, via the WBM site; mutations of this interaction have been demonstrated to prevent MYC from associating with target genes.18 WBM-site inhibitors therefore have potential as anticancer agents—being able to directly disrupt the interaction of c-MYC to WDR5 and thus impede the MYC function in cancer cells.18,19 However, WDR5 inhibitors to date have consisted only of WIN-site binders. The WIN site is dominated by a conserved arginine pocket, where the guanidine headgroup sits in a “phenylalanine clamp”11 composed of Phe133 and Phe263. The “WIN-motif” has some flexibility regarding arginine-adjacent residues, but the arginine itself is critical; studies of peptide binding to WDR5 show a dramatic loss of affinity if the arginine is mutated.20,21 The first WDR5 inhibitors developed were accordingly peptides and peptidomimetics that built from this arginine-critical WIN-motif, guided by the co-crystal structures of WDR5 with MLL1 and H3 peptides. One such peptidomimetic, MM-401, was shown to be selective against MLL-r cancer cells and cell lines in vitro.22 Small molecules that bind the WIN site have focused on a series of N-methyl piperazines identified from a fluorescence polarization assay screen23 that led to OICR-9429.24–27 This compound inhibits the histone methyltransferase (HMT) activity of MLL1 in vitro and reduces the viability of primary human AML cells bearing C/EBPα mutations24 and inhibits “gain-of-function” p53 cell growth.26 Recently, an NMR-based fragment screen identified several high-affinity WIN-site binders.28,29 These compounds show cellular activity through displacement of WDR5 from chromatin, an effect that gives WDR5 inhibitors therapeutic relevance beyond MLL-r cancers.29 These studies have validated WDR5—in particular, the WIN site—as a promising anticancer target and advocate further research of novel inhibitors.30 The present study used a fragment screening approach with SPR (Surface Plasmon Resonance). Of the hits from this screen, one compound [(2-butyl-1H-imidazol-5-yl)methanol] was chosen as the most promising to advance. Cocrystal structures of the initial hit and subsequent derivatives enabled structure-guided optimization to 37 (KD = 4.5 μM), which represents a useful starting point for the development of clinically relevant WDR5 inhibitors. The structure-activity relationship (SAR) of this series in the context of the structural data is discussed. The imidazole-containing headgroup of this series was investigated as a replacement for arginine; one such imidazole-peptide hybrid was cocrystallized with WDR5 and found to maintain the binding mode of analogous peptides. This novel moiety thus potentially has additional utility as a general tool to overcome the poor permeability associated with highly basic groups like arginine.
II. RESULTS AND DISCUSSION
Fragment screening of WDR5 was performed using SPR, with compounds tested at a single concentration and potential hits re-tested in a dosage format. Screening focused on the Maybridge Ro3 and CSIRO fragment libraries,31 a total of 2256 compounds, with 12 of these identified as hits (0.53% hit rate, Fig. 1). Compound 1 presented a high ligand efficiency (KD = 480 μM, LE = 0.41 kcal·mol−1·heavy atom−1) and—following the confirmation of binding through cocrystallization with WDR5 (Table I)—was selected as the most promising hit to advance. Compound 1 was determined to bind in the WIN site, the imidazole ring stacking between F133 and F263 (the “phenylalanine clamp”11), thus occupying a similar space to the key arginine of the WIN motif (Fig. 2). The nitrogen atoms of the imidazole mimic the arginine headgroup, donating hydrogen bonds to the backbone of S91 and C261, while the hydroxyl moiety projects deeper into the pocket, hydrogen bonding directly to S175 and via a water molecule to V177. The hydroxyl displaces a water observed in the structures of WDR5 bound to MLL1 and H3K4 peptides.10,14,32 The butyl tail is oriented out of the pocket, thereby picking up additional hydrophobic interactions to S49, F133, and I305. The protein in the WDR5/1 complex displayed strong electron density from residue K32. A truncated WDR5 construct (32–334) was therefore expressed and used in crystallization. The majority of the structures described in this report are from this truncated construct, which routinely diffracted to high resolution (Table I and supplementary Table S1).
FIG. 1.

Chemical structures of selected hit compounds from the initial SPR screen.
TABLE I.
Summary of crystallographic data for WDR5 cocrystal structures.
| Compound | Resolution (Å) | Rwork/Rfree | PDB accession code |
|---|---|---|---|
| 1 | 2.04 | 17.3/20.6 | 6PG3 |
| 2 | 1.60 | 14.7/17.6 | 6PG4 |
| 16 | 1.67 | 18.7/20.7 | 6PG8 |
| 17 | 2.45 | 19.6/24.3 | 6PG7 |
| 24 | 1.76 | 18.2/21.3 | 6PGE |
| 29 | 1.68 | 23.3/26.9 | 6PG6 |
| 30 | 1.75 | 17.4/21.7 | 6PG9 |
| 31 | 1.73 | 17.4/21.7 | 6PGB |
| 32 | 2.45 | 19.7/25.0 | 6PGA |
| 35 | 1.99 | 15.6/20.2 | 6PG5 |
| 37 | 1.81 | 20.3/24.8 | 6PGC |
| 49 | 1.54 | 14.8/16.9 | 6PGF |
| 55 | 1.50 | 15.1/16.8 | 6PGD |
FIG. 2.

(a) Overlay of WDR5 cocrystal structures bound to the MLL peptide (3EG6,14 Ac-ARA-NH2 displayed), 1, and 2. Binding mode of (b) 1, (c) the MLL1 peptide, and (d) 2.
To probe the binding requirements of 1, prior to exploring elaborated derivatives, various fragment analogs were tested (Table II). Trimming the butyl tail of 1 to a methyl did not impair binding (2, KD = 260 μM), in contrast to the des-methyl (3, KD > 3 mM), indicating the (2-methyl-imidazol-4-yl)methanol core as a highly ligand efficient starting point (LE = 0.61). Various imidazoles and other heterocycles, as well as isosteres of the hydroxyl moiety, displayed a general loss of affinity. This suggested rigid requirements for the hydrogen bonding network, at least for this series' binding mode; changing the nitrogen pattern to remove the hydrogen bond at either C261 or S91 (4 and 5, respectively) greatly reduced affinity. Removing the methanol moiety entirely leaving 2-methyl imidazole also reduced binding (6, KD = 2.1 mM); however, this aligns with the reduced size of this compound and the high ligand efficiency of 2 is largely maintained (LE = 0.60). A cocrystal structure of 2 aligned well with 1 (ligand RMSD 0.2 Å) and placed the two solvent exposed positions—the 2-methyl and the hydroxyl groups—as the frontrunners for elaboration in the WDR5 channel.
TABLE II.
SAR of the fragment core.
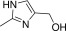
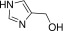
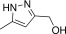
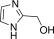
Mean ± SE.
Estimated from single concentration (100 μM) screen.
SE not reported (n = 1).
A. Derivatization of the hydroxymethyl substituent of 2
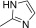
From the crystal structures of 1 and 2, it appeared possible to extend from the hydroxyl moiety, deeper into the WDR5 channel, with an eye to potentially form additional interactions with the hydrophobic residues F263, F219, or A176. Five analogs of 2 with hydrophobic substituents were tested to probe this vector (Table III and Scheme 1). Methyl, n-propyl, i-propyl, and 2,2,2,-trifluoroethyl groups all abrogated binding (7–10, KD > 3 mM); however, a benzyl moiety (11, KD = 950 μM) was better tolerated. No cocrystal structure was obtained for 11, and so a change in the binding orientation of the ligand cannot be ruled out. Further analogs would thus be required to convincingly demonstrate that extension from this vector could be tolerated with the appropriate functional group.
TABLE III.
SAR of ether-derivatized 2.
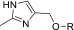 | ||
|---|---|---|
| Compound | R | KD (μM)a |
| 2 | H | 260 ± 20 |
| 7 | CH3 | > 3000 |
| 8 | n-propyl | > 3000 |
| 9 | i-propyl | > 3000 |
| 10 | CH2CF3 | > 3000 |
| 11 | Bn | 950b |
mean ± SE.
SE not reported (n = 1).
SCHEME 1.

Synthesis of ether derivatives 7–11.
B. Aromatic substituents in the 2-position of 4(5)–(hydroxymethyl)imidazole
Synthetic efforts subsequently focused on extending from the 2-position of the imidazole ring to fill the WIN site. Considering that the alkyl chain of 1 contributed little to binding, initial efforts were directed at assessing whether an aromatic ring was tolerated at varying distances (0–3 atoms) from the imidazole fragment (Table IV). Condensation of dihydroxyacetone with variously substituted amidines afforded access to the final compounds (Scheme 2).
TABLE IV.
SAR of derivatives of 2.
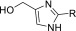 | ||
|---|---|---|
| Compound | R | KD (μM)a |
| 13 | 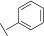 |
> 3000 |
| 14 | 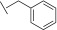 |
530b |
| 15 | 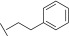 |
300 ± 40 |
| 16 | 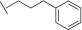 |
140 ± 30 |
| 17 | 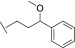 |
22 ± 6 |
| 23 | 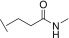 |
890b |
| 24 | 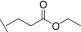 |
240 ± 10 |
| 28 | 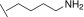 |
350 ± 40 |
| 29 | 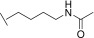 |
200 ± 10 |
| 30 | 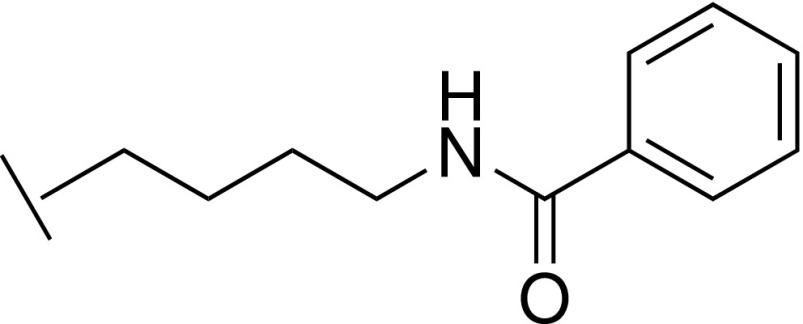 |
12 ± 1 |
| 31 | 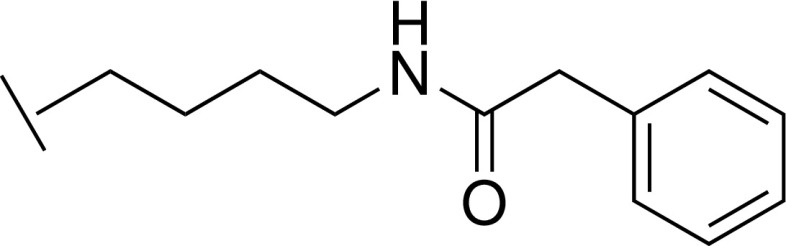 |
25 ± 3 |
| 32 | 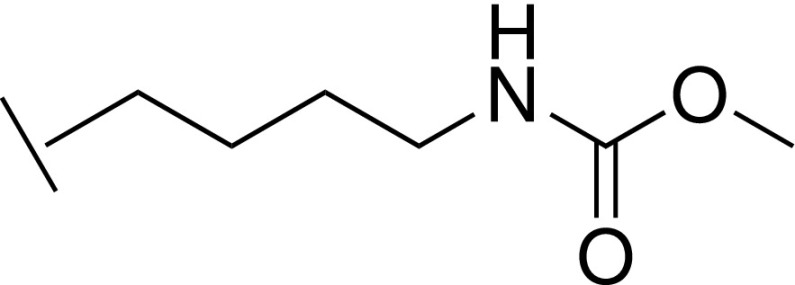 |
28.9 ± 0.5 |
| 35 | 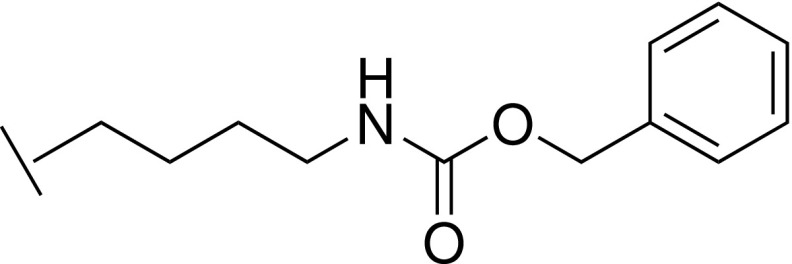 |
11 ± 1 |
| 37 | 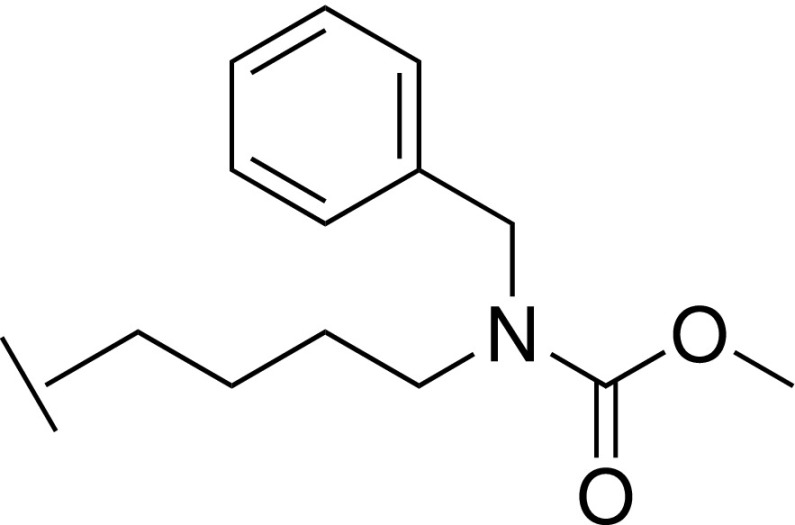 |
4.5 ± 0.3 |
| 40 | 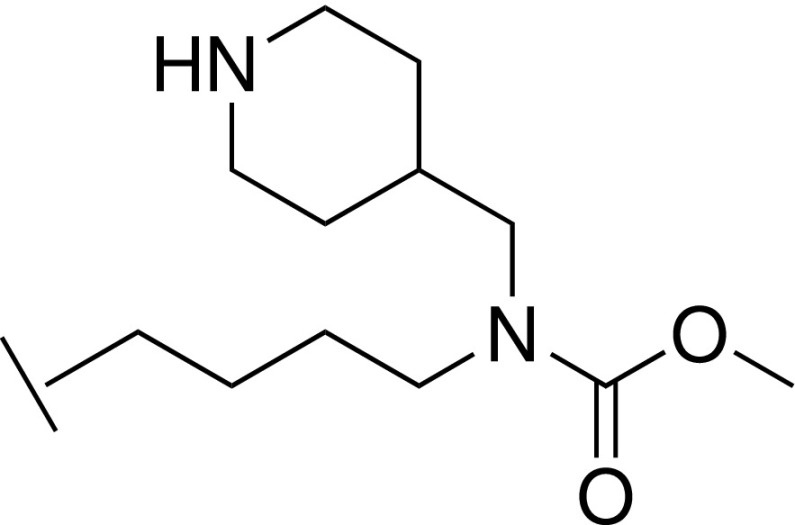 |
19 ± 3 |
| 45 | 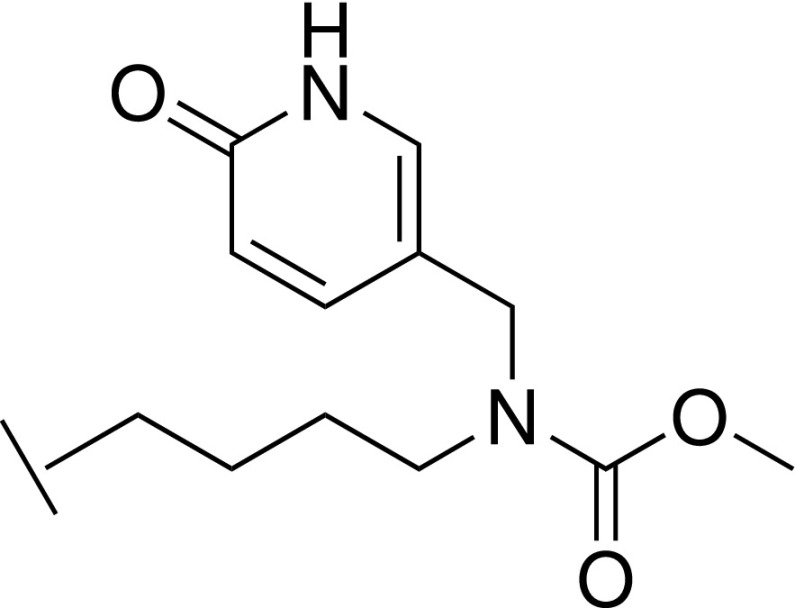 |
98.6 ± 0.6 |
| 47 | 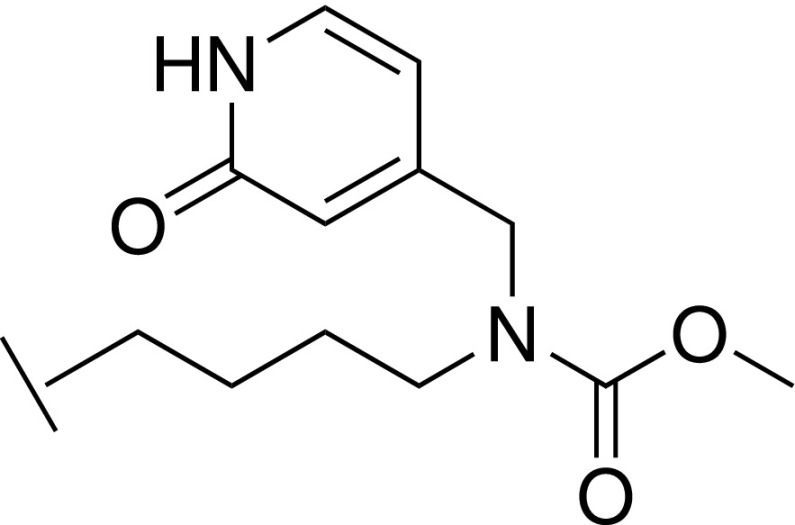 |
24.2 ± 0.2 |
mean ± SE.
SE not reported (n = 1).
SCHEME 2.

Synthesis of aryl derivatives 14–17.
While the zero atom linker showed little binding (13, KD = 5.2 mM), a linker length of one, two, or three methylene units was tolerated (14, KD = 530 μM, 15, KD = 300 μM, and 16, KD = 140 μM, respectively). Interestingly, the weakly binding fragment 13 is similar to the imidazole fragment hit reported by Wang et al.28 The subtle difference in orientation between the two series' imidazole rings appears key to avoid clashes with nearby residues S49 and I305. A cocrystal structure bound with 16 was solved, showing the introduced phenyl ring projecting into subpocket S4, toward residues A47, S49, A65, and L321 [Fig. 3(d)]. A methoxy substituent off the linker of 16 gave a decent improvement to binding, 17 (KD = 22 μM), ostensibly from an introduced water-mediated hydrogen bond to C261. Despite the improved binding of 17, the initial addition of the phenyl ring was noted to constitute a sizeable drop in ligand efficiency from the initial hit (16, LE = 0.33; 17, = 0.35).
FIG. 3.

(a) Overlay of the nine complexes presented and (b) rotated by 90°. The seven blades of the WD40 domain are numbered from 1 to 7. (c) Zoomed overlay detailing nearby residues. Protein atoms displayed for A–C are from the WDR5/35 complex. Cocrystal structures of WDR5 bound to (d) 16, (e) 17, (f) 24, (g) 29, (h) 30, (i) 31, (j) 32, (k) 35, and (l) 37. Dashed black lines represent hydrogen bonds. WDR5 is represented with a surface depiction, and residues within 4 Å of the ligand-substituent (i.e., excluding atoms analogous to fragment 2) are highlighted.
In an effort to improve the ligand efficiency of the 2-substituted imidazoles, two compounds that lacked the phenyl ring of 17 were synthesized (23 and 24, Scheme 3). These derivatives sought to retain the water-mediated hydrogen bond to C261 with a suitable hydrogen bond acceptor at a distance from the imidazole ring equivalent to the methoxy in 17 while also providing a synthetic handle for potential elaboration. While the ester-containing 24 marginally improved binding (KD = 240 μM) over the initial fragment hit, the amidic 23 lost binding (KD = 890 μM).
SCHEME 3.

Synthesis of compounds 23 and 24.
C. Use of a primary amine as a synthetic handle
In addition to the above derivatives, installation of a convenient synthetic handle onto initial hit 1 was pursued by condensing the azido-imidate 26 with dihydroxyacetone followed by a reduction to the primary amine 28 (Scheme 4). While binding of the butylamine was tolerated (28, KD = 350 μM), it also enabled the synthesis of a number of amides and carbamates with substituents aimed at more completely occupying the WIN binding site.
SCHEME 4.

Synthesis of 4(5)-(hydroxymethyl)imidazole derivatives. Reaction conditions for the scheme: (a) NaN3, DMSO, 50 °C; (b) 1. Pd/C, H2, MeOH and 2. Cbz-Cl, Et3N, DCM; (c) HCl(g), EtOH, 2 h; (d) 1. AcCl/EtOH and 2. NH3/MeOH; (e) dihydroxyacetone, NH4OH/i-PrOH, microwave, 90 °C, 1 h; (f) dihydroxyacetone, NH3.H2O; (g) Pd/C, H2, MeOH; (h) Ac2O, DCM, MeOH, rt; (i) ClCOR2, DCM, MeOH, Et3N; (j) R2COH, NaCNBH3, MeOH, AcOH; (k) ClCOR2, NaOH, H2O/DCM; (l) ClCO2CH3, DCM, TEA; (m) TBDPS-Cl, Et3N, DCM; (n) TBAF, THF; (o) TFA, DCM; and (p) HCl/dioxane.
Acetylating 28 to give 29 marginally improved binding (KD = 200 μM), cocrystallization revealing a binding mode that introduces a direct hydrogen bond to Cys261, and a vector oriented toward the hydrophobic subpocket S7 [Fig. 3(g)]. Addition of hydrophobic phenyl or benzyl moieties markedly improved affinity (30 and 31, KD = 12 and 25 μM, respectively). The phenyl group of 30 extends to form additional interactions to F133, F149, and Y191, while the benzyl group of 31 orients away from these residues instead to interact with the sidechains of K259 and Y260 (Fig. 3). A carbamate analog of 29 (32, KD = 28.9 μM, LE = 0.39) provided a compound that maintained the high ligand efficiency of the original fragment hit. A similar binding mode to 29 is observed, with 32 maintaining what appears to be a key hydrogen bond to Cys261 (Fig. 3). Similar to 29 vs 31, addition of an aromatic moiety to 32 further improved binding (35, KD = 11 μM), picking up interactions to F149 and Y191 (Fig. 3). Weaker electron density for the phenyl ring, relative to the rest of the molecule, suggests that this moiety maintains some mobility. Addition of the benzyl group instead to the carbamate nitrogen of 32 (37, KD = 4.5 μM) similarly improved binding, albeit with similarly weak electron density for the introduced ring.
D. N-substituted carbamate derivatives
With the crystal structure of 32 indicating a key H-bond between the carbamate and C261, further synthetic efforts were aimed at retaining this interaction while exploring N-substituents that could project toward A65. The cocrystal structure of 37 showed the benzyl substituent in close proximity to the side chain of E322, from the para position of the phenyl ring [Fig. 3(l)]. Replacing the phenyl of 37 with a 4-piperidine (40) afforded a hydrogen bond donor at this position to potentially interact with E322. It was noted that such a moiety could also potentially reorient toward residue D107. Such an interaction to D107 is present in the WDR5 inhibitor OICR-9429 through a pyridone ring.25 Pyridone analogs of 37 were additionally thus synthesized to mimic this interaction (45 and 47). Unfortunately, no improvement to affinity relative to compound 37 was observed for these three compounds (40, 45, and 47). In particular, compound 45 displayed a marked loss of affinity (KD = 98.6 μM, 20-fold weaker binding than 37), suggesting perhaps that the ring had limited scope to reorient and that electrostatic repulsion was occurring between E322 and the introduced oxygen atom of the pyridone. A study by Wang et al.28 showed that small substitutions on an aromatic ring in the S7 subpocket can deliver significant affinity gains and would suggest 35 as an attractive compound for further development although 35 currently lacks the requisite potency for a WDR5 tool compound.
E. Probing irreversible inhibition at C261
The proximity of the reported series to residue C261 suggested the potential to design irreversible inhibitors of WDR5. A compound possessing the scaffold of 29 and a reactive acrylamide headgroup (49) that could potentially form a covalent bond with the thiol side chain of C261 was synthesized. Compound 49 was incubated with a solution of WDR5 for 24 h at room temperature, prior to being crystallized. A high resolution (1.54 Å) structure was obtained; however, there was no evidence of covalent bonding to the protein. The cocrystal structure (Fig. 4) suggests that potentially the movement of the Cys261 side chain may be limited and that the hydrogen bond between the ligand and the amide of Cys261 is obstructing the required geometry to react with the thiol.
FIG. 4.

(a) Cocrystal structure of WDR5 bound to 49. Electron density maps (2mFo-DFc, 1 sigma) are displayed for 49 and residue C261. (b) Chemical structure of 49.
F. Imidazole-bearing peptidomimetics
Compound 1 was identified as a highly ligand efficient hit that binds the phenylalanine clamp of the WIN site, approximating the arginine headgroup of the WIN motif. The lack of net charge at physiological pH suggested this compound as a scaffold that might potentially reduce permeability issues associated with compounds possessing highly basic groups like guanidine or piperidine. Two peptidomimetics, with 1 replacing the arginine, were synthesized. Compounds 55 and 59 are analogs of the Ac-ARV-NH2 and Ac-α-MeAla-RV-NH2 tripeptides,21 respectively (Fig. 5). Binding was observed from both compounds to WDR5, and a cocrystal structure of 55 revealed that the ligand maintained the binding mode of the N-α-acetylated form of the H3 peptide ([3PSL],20 RMSD = 0.3 Å, for equivalent heavy atoms, Fig. 5). The hydroxymethyl imidazole thus appears to be a good replacement for the charged arginine residue in this context, and 59 may serve as a tool compound with more sensible physicochemical parameters than previous potent tripeptides. The high-resolution cocrystal structure of 55/WDR5 (1.50 Å) shows unambiguous density for the ligand, and the SPR sensorgrams of 55 and 59 fit well with a 1:1 binding model (supplementary Fig. S2), suggesting a single dominant binding partner. However, epimerization during synthesis resulted in an impure mixture of diastereomers for these two compounds (supplementary Scheme S1), and the proportion of each diastereomer is unclear. Binding constants of compounds 55 and 59 are thus difficult to derive. Instead, the (concentration independent) dissociation rates have been reported for 55 and 59 (kd = 0.067 s−1 and 0.025 s−1, respectively) to provide a more meaningful comparison to the analogous tripeptide Ac-ARV-NH2 (kd = 0.054 s−1). Considering the known SAR of the WIN site, it would be expected that D-amino acid-bearing peptidomimetics bind WDR5 with a markedly reduced affinity. However, if multiple diastereomers of 55/59 were indeed binding WDR5, it would not detract from the conclusion that hydroxymethyl imidazole-peptidomimetics have potential as WDR5 WIN-site inhibitors.
FIG. 5.

Cocrystal structures of WDR5 bound to 55 (yellow, WDR5 atoms in blue) overlaid with NαH3 peptide (purple, displayed Ac-ART-NH2, WDR5 atoms in grey) [3PSL].20 The drawn chemical structure (right) is as per the compound observed to bind in the displayed high-resolution structure (1.50 Å); however, the samples of 55 and 59 tested were a mixture of diastereomers. As the dissociation rate is concentration independent, this value is provided to allow a more meaningful comparison of binding.
III. CONCLUSION
Recent studies indicate WDR5 as a promising cancer target beyond MLL-r cancers,29,30 with the potential for WDR5 inhibitors to deliver clinical impact. However, high-affinity binding to WDR5 has not necessarily delivered compounds with useful levels of methyltransferase inhibition and lower cellular proliferation.25,28,33 Poor cellular permeability is presumably part of the explanation, but it is unlikely to fully account for the disconnect. This study identifies a highly ligand efficient motif with multiple hydrogen bonding interactions to the WDR5 protein. This could form the basis for the design of high-affinity drug-like molecules with enhanced responses in cellular assays through further optimization.
IV. METHODS
A. Protein expression and purification
WDR5 sequences (WDR523–334 and WDR532–334) were cloned into a modified pET43 (N-terminal hexahistidine-SUMO tags) vector. Transformed E. coli BL21-CodonPlus-RIL cells were grown overnight in TB media supplemented with 34 μg·ml−1 of chloramphenicol and 100 μg·ml−1 of ampicillin for selection. The overnight culture was subcultured into TB media (2 × 1 L) and grown at 310 K to an OD600 ∼ 0.8 with induction by 0.1 mM isopropyl β-D-1-thiogalactopyranoside (IPTG), carried out at 289 K for 20 h. The cultures were centrifuged at 4000 × g for 10 min, and the cell pellets were frozen at 253 K. For purification, cell pellets were thawed and suspended in 200 ml lysis buffer containing 50 mM Tris, pH 8.0, 150 mM NaCl, 10 mM imidazole, 2 mM MgCl2, 5 mM DTT, 6.25 units·ml−1 Benzonase, 0.5 mg·ml−1 lysozyme, 1 mM PMSF, and 4 EDTA-free cOmplete™ protease-inhibitor cocktail tablets (Roche). The suspension was lysed by three passes through an ice-cooled Avestin C5 cell crusher and then centrifuged at 50 000 × g for 20 min. The supernatant was filtered (5 μm) and purified via a native IMAC program on a Profinia™ (Bio-Rad), using a HiTrap IMAC FF (5 ml, GE Healthcare), being washed (10 column volumes per buffer) sequentially using buffers containing 10 mM and 20 mM imidazole (50 mM Tris, pH 8.0, 150 mM NaCl, 5 mM DTT), prior to elution with 250 mM imidazole. The eluate was further purified using gel filtration on a Superdex 200 26/60 (50 mM Tris, pH 8.0, 150 mM NaCl, 5 mM DTT). Ulp1 (SUMO) protease was added (1 U per 400 μg protein) to the sample overnight at 277 K. The sample was diluted 1:1 with 50 mM Tris, pH 8.0, 150 mM NaCl, 5 mM DTT, and 40 mM imidazole (final imidazole = 20 mM), added to the IMAC column, and washed with two column volumes of buffer containing 20 mM imidazole. The flowthrough was concentrated (Amicon Ultra, 10 kDa cut-off) and then purified using a Superdex 75 16/60 (50 mM Tris, pH 8.0, 150 mM NaCl, 5 mM DTT). Protein was concentrated, dispensed as aliquots, and snap frozen. Protein identity and purity were authenticated by SDS PAGE and mass spectrometry. ∼25 mg of purified protein was obtained per liter of culture.
B. Surface Plasmon Resonance (SPR)
SPR assay of small molecule binding used minimally biotinylated WDR523–334 protein captured on streptavidin-coated SPR chips. WDR5 (1 eq.) was mixed with freshly water-dissolved EZ-link Sulfo-NHS-LC-LC-biotin (0.8 eq., Thermofisher Scientific). The reaction mixture was incubated on ice for 2 h; unreacted biotin reagent was then removed from the mixture using size-exclusion chromatography on a Superdex 75 (10/300) column equilibrated in storage buffer (10 mM HEPES, 150 mM NaCl, 5 mM DTT).
SPR experiments were performed using a Biacore T200 or S200 biosensor with a CM5 chip (GE Healthcare). Streptavidin was coupled to the chip surface at 310 K in HBS-P+ running buffer (10 mM HEPES, pH 7.4, 150 mM NaCl, 0.05%[v/v] Tween-20) after a 10 min activation with a 1:1 mixture of NHS/EDC (N-hydroxysuccinimide/N-ethyl-N′-(3-diethylaminopropyl)carbodiimide). Protein was diluted to 100 μg·ml−1 at pH 4.5 (10 mM sodium acetate, 0.005%[v/v] Tween-20) and injected for 7 min. The surface was then blocked with 1 M ethanolamine, pH 8.0, for 7 min. Immobilization levels were typically 7000–13 000 RU, but as low as 1800 RU for compounds 55, 59, and Ac-ARV-NH2, where fitting uses a 1:1 kinetic model to determine dissociation rates. WDR5 capture was then performed over the streptavidin-coupled flow-cells at 293 K in HBS-P+ buffer with the addition of 5 mM DTT. Capture levels for fragment screening were ∼19 000 RU, while capture levels for subsequent testing were ∼9000 RU. The N-terminal domain of heat-shock protein 90 (Hsp90, UnprotKB P08238, residues 1–382) was captured (∼11 000 RU) during screening to provide an unrelated negative control surface, activity being monitored with intermittent ADP (adenosine diphosphate) injections. Biotin was subsequently injected over WDR5- and negative control protein-bound flow-cells to block remaining unbound streptavidin. SPR experiments were performed at 298 K in SPR running buffer containing 50 mM HEPES, 150 mM NaCl, 5 mM DTT, and 0.05%[v/v] Tween-20, 2%[v/v] DMSO. Initial fragments were screened at 100 μM. When performing dose-response testing, analytes were serially diluted (2- or 3-fold), injected for 30 s contact time, and allowed to dissociate for 30 s. The flow rate was 60 μl·min−1. Binding sensorgrams were processed, solvent-corrected, and double-referenced using Scrubber software (BioLogic Software, Australia). SPR binding analysis of most of the compounds investigated in this study revealed dissociation rates that were not sufficiently slow to allow global fitting to a kinetic binding model, for which the kd (dissociation rate constant) must typically be <0.5 s−1 for SPR instruments to be able to capture sufficient data points during the dissociation phase. Therefore, to determine binding affinities (KD values), responses at equilibrium for each analyte were fitted to a 1:1 steady-state affinity model available within Scrubber. For compounds where the top concentration tested was less than 2-fold above the KD, affinities were fit using a global Rmax against reference compounds. Sensorgrams and binding isotherms are displayed in supplementary Fig. S2.
C. Crystallization and x-ray structure determination
Crystallization experiments were performed at the CSIRO Collaborative Crystallization Centre . The sitting-drop vapor diffusion method was used at 281 K with droplets consisting of 150–200 nl of protein solution and reservoir solution and using a reservoir volume of 50 μl. Both constructs of WDR5 (WDR523–334 and WDR532–334) were typically used when attempting crystallization of WDR5 complexes. The protein solution was at 8.2 mg·ml−1 for the initial complex with 1 and at 5.1 mg·ml−1 for all subsequent complexes. Crystallization conditions are as follows: ammonium sulfate/ammonium fluoride, 0.024–0.237 M; sodium cacodylate/bis-tris chloride, 0.1 M, pH 5.62–7.06; PEG 3350/PEG 4000/PEG 8000/MPEG 5000, 22.1–30.5%[w/v]. Specific conditions for each complex are provided in supplementary Table S1. 1 mM of compound was added to the protein solution prior to dispensing into the plate. Compound 49 was added to the protein mix and left for 24 h at room temperature, prior to plating, to allow any potential reaction to take place that might otherwise be prevented at the low temperature (281 K) at which crystals were grown. Crystals were harvested from drops at 277 K and cryoprotected in 30%[v/v] ethylene glycol. Diffraction data were collected at the MX134 and MX235 beamlines at the Australian Synchrotron. Datasets were indexed using XDS36 or Dials37 and scaled using Aimless.38 Molecular replacement was performed using Phaser39 with 2H9L32 as a reference structure. Refinement was performed using Refmac40 and Coot41 with ligands' geometric restraints generated using Phenix eLBOW42 or PRODRG.43 A summary of data collection and refinement statistics is given in Table I, with full details in supplementary Table S1. Difference density maps for ligands are given in supplementary Fig. S1.
D. Compound synthesis and characterization
Unless otherwise stated, the following generalizations apply. 1H NMR spectra were recorded on a Bruker Ultrashield Plus (400 MHz) or a Bruker AVANCE III (400 MHz). The multiplicity of a signal is designated by the following abbreviations: s, singlet; d, doublet; t, triplet; q, quartet; dd, doublet of doublets; dt, doublet of triplets; tt, triplet of triplets; br, broad; and m, multiplet. All observed coupling constants, J, are reported in Hertz. Exchangeable protons are not always observed.
LCMS (liquid chromatography-mass spectrometry) data were generated using the conditions described below. Chlorine isotopes are reported as 35Cl, and bromine isotopes are reported as either 79Br or 81Br or both 79Br/81Br.
1. LCMS method A (LCMS-A)
Instrument: Agilent 6100 Series Single Quad LC/MS and Agilent 1200 Series HPLC. Pump: 1200 Series G1311A Quaternary pump. Autosampler: 1200 Series G1329A Thermostatted Autosampler. Detector: 1200 Series G1314B Variable Wavelength Detector. Column: Luna C8 (2) 5 μm 50 × 4.6 mm 100 Å. Column temperature: 30 °C. Injection volume: 5 μl. Solvent A: Water 0.1% Formic Acid. Solvent B: MeCN 0.1% Formic Acid. Gradient: 5%–100% solvent B over 10 min. Detection: 254 nm or 214 nm. MS Conditions: Ion Source: Quadrupole. Ion Mode: Multimode-ES. Drying gas temp: 300 °C. Vaporizer temperature: 200 °C. Capillary voltage (V): 2000 (positive). Capillary voltage (V): 4000 (negative). Scan range: 100–1000. Step size: 0.1 s. Acquisition time: 10 min.
2. LCMS method B (LCMS-B)
Instrument: Agilent 1260 Infinity Series UPLC/MS. Pump: 1260 Infinity G1312B Binary Pump. Autosampler: 1260 Infinity G1367E 1260 HiP ALS. Detector: 1290 Infinity G4212A 1290 DAD. Column: Poroshell 120 EC-C18 2.7 μm 50 × 3.0 mm. Column temperature: 35 °C. Injection volume: 1 μl. Solvent A: water 0.1% Formic Acid. Solvent B: MeCN 0.1% Formic Acid. Gradient: 5%–100% solvent B over 3.8 min. Detection: monitored at 254 nm and 214 nm. MS conditions: Ion Source: Quadrupole. Ion Mode: API-ES. Drying gas temp: 350 °C. Capillary voltage (V): 3000 (positive). Capillary voltage (V): 3000 (negative). Scan range: 100–1000. Step size: 0.1 s. Acquisition time: 5 min.
3. LCMS method C (LCMS-C)
Instrument: Agilent Technologies 1290 series. Pump type: binary pump. Detector type: Diode Array Detector. Column: Agilent Poroshell 120 EC-C18, 2.7 μm, 4.6 × 50 mm. Column temperature: 35 °C. Acquisition wavelengths: 214 nm and 254 nm. Mobile phase: A: 0.05% HCOOH in water (v/v) and B: 0.05% HCOOH in Acetonitrile (v/v). Run time: 5 min. Flow rate: 1.0 ml/min. MS model: Agilent G6120A, quadrupole LC/MS. Ion source: ES-API (+ve or −ve). TIC: 70 ∼ 1000 m/z. Fragmentor: 60. Drying gas flow: 10 l/min. Nebulizer pressure: 35 psi. Drying gas temperature: 350 °C. Vcap: 3000 V.
4. LCMS method D (LCMS-D)
Instrument: Agilent 6120 Series Single Quad LC/MS and Agilent 1200 Series HPLC. Pump: 1200 Series G1311A Quaternary pump. Autosampler: 1200 Series G1329A Thermostatted Autosampler. Detector: 1200 Series G1314B Variable Wavelength Detector. Column: Luna C8 (2) 5 μm 50 × 4.6 mm 100 Å. Column temperature: 30 °C. Solvent A: Water 0.1% Formic Acid. Solvent B: MeCN 0.1% Formic Acid. Gradient: 0%–95% solvent B over 10 min. Detection: 254 nm or 214 nm. MS conditions: Ion source: quadrupole. Ion mode: Multimode-ES & APCI. Drying gas temp: 250 °C. Vaporizer temperature: 200 °C. Capillary voltage (V): 4000 (positive). Capillary voltage (V): 4000 (negative). Scan range: 100–1000. Step size: 0.1 s. Acquisition time: 10 min.
Microwave irradiation was performed using a CEM Explorer SP Microwave Reactor.
Where necessary, anhydrous solvents were purchased from Sigma-Aldrich or dried using conventional methods. Solutions of inorganic acids or bases were made up as aqueous solutions unless stated otherwise.
Analytical thin-layer chromatography was performed on Merck silica gel 60 F254 aluminium-backed plates that were visualized using fluorescence quenching under UV light or a basic KMnO4 dip or Ninhydrin dip.
Preparative thin-layer chromatography (prep TLC) was performed using Tklst (China), grand grade: (HPTLC): 8 ± 2 μm > 80%; (TLC): 10–40 μm. Type: GF254. Compounds were visualized by UV (254 nm).
Flash chromatography was performed using a Biotage Isolera purification system using either Grace or RediSep® silica cartridges.
Column chromatography was performed using Tklst (China), grand grade, 100–200 meshes silica gel.
Cartridge details: SCX and SCX-2 cartridges: Manufacturer: Biotage. Product: ISOLUTE® SCX 1 g, (6 ml SPE Column unless otherwise stated); manufacturer: Biotage. Product: ISOLUTE® SCX-2 1 g (6 ml Column).
E. 4-(chloromethyl)-2-methyl-1H-imidazole hydrochloride 12
A solution of (2-methyl-1H-imidazol-4-yl)methanol hydrochloride (117 mg, 0.787 mmol) and thionyl chloride (1 ml) was stirred at room temperature overnight. Chloroform (1 ml) was added, and the mixture was concentrated in vacuo to give the title compound as a brown oil (144 mg, ∼80% purity, ∼88% yield). The material was used in the next step without further purification. 1H NMR (400 MHz, DMSO-d6) δ 7.59 (s, 1H), 4.84 (s, 2H), 2.56 (s, 3H). 1H NMR data matched the reported values.44
F. 4-methoxy-2-methyl-1H-imidazole hydrochloride 7
A solution of 4-(chloromethyl)-2-methyl-1H-imidazole hydrochloride 12 (34.5 mg, 0.207 mmol) and methanol (1 ml) was heated at reflux for 2 h and then irradiated in a microwave at 150 °C for 1 h. The mixture was returned to room temperature, concentrated in vacuo, dissolved in a minimum volume of MeOH, and loaded onto an SCX cartridge. The cartridge was washed with MeOH (10 ml) before the product was eluted with 7 M NH3 in MeOH (10 ml). The eluate was concentrated in vacuo and then further purified by column chromatography (4 g SiO2 cartridge, 1%–15% MeOH (with 10% 7 M NH3 in MeOH) in DCM) to give the title compound as a colorless solid (4.5 mg, 17%): 1H NMR (400 MHz, CDCl3) δ 6.87 (s, 1H), 4.38 (s, 2H), 3.37 (s, 3H), 2.40 (s, 3H).
G. General procedure for synthesis of 8–11
A solution of 4-(chloromethyl)-2-methyl-1H-imidazole hydrochloride 12 (30 mg, 0.180 mmol) and alcohol (1 ml) was irradiated in a microwave at 150 °C for 15 min. The solution was returned to room temperature and concentrated in vacuo. The residue was dissolved in a minimum volume of MeOH and loaded onto an SCX cartridge, washed with MeOH (10 ml), and then eluted with 7 M NH3 in MeOH (10 ml). The eluate was concentrated in vacuo and the residue was purified by column chromatography (4 g SiO2 cartridge, 5–15% MeOH (with 10% 7 M NH3 in MeOH) in DCM) to give the title compound (see below).
2-methyl-4-(propoxymethyl)-1H-imidazole 8: 6.8 mg, 24% yield. 1H NMR (400 MHz, chloroform-d) δ 6.88 (s, 1H), 5.79 (br s, 1H), 4.42 (s, 2H), 3.43 (t, J = 6.8 Hz, 2H), 2.43 (s, 3H), 1.68–1.52 (m, 2H), 0.90 (t, J = 7.4 Hz, 3H).
4-(isopropoxymethyl)-2-methyl-1H-imidazole 9: 8.4 mg, 30% yield. 1H NMR (400 MHz, chloroform-d) δ 6.84 (s, 1H), 4.43 (d, J = 0.7 Hz, 2H), 3.71 (hept, J = 6.1 Hz, 1H), 2.39 (s, 3H), 1.19 (d, J = 6.1 Hz, 6H).
2-methyl-4-((2,2,2-trifluoroethoxy)methyl)-1H-imidazole 10: 12.5 mg, 35% yield. 1H NMR (400 MHz, chloroform-d) δ 6.93 (s, 1H), 4.60 (s, 2H), 3.87 (q, J = 8.8 Hz, 2H), 2.42 (s, 3H). 19F NMR (376 MHz, chloroform-d) δ -73.83.
4-((benzyloxy)methyl)-2-methyl-1H-imidazole 11: 8.7 mg, 23% yield. 1H NMR (400 MHz, chloroform-d) δ 7.36 – 7.26 (m, 5H), 6.86 (s, 1H), 4.56 (s, 2H), 4.47 (d, J = 0.7 Hz, 2H), 2.38 (s, 3H).
H. General procedure for synthesis of 14 and 15
A suspension of dihydroxyacetone (50 mg, 0.55 mmol) and a commercially available amidine hydrochloride (0.555 mmol) in ammonium hydroxide 33%–35% (0.5 ml) was stirred in the microwave at 120 °C for 10 min. The reaction mixture was evaporated to dryness and loaded onto an SCX-2 cartridge (0.5 g). The cartridge was washed with MeOH (20 ml) before the product was eluted with 3.5 M NH3 in MeOH (10 ml). The basic fraction was concentrated under reduced pressure, and the resultant dark oil was triturated with acetone (∼10 ml). The oil was dried under high vacuum to yield the desired product.
(2-benzyl-1H-imidazol-4-yl)methanol 14: 1H NMR (400 MHz, Methanol-d4) δ 7.31 – 7.18 (m, 7H), 6.87 (s, 1H), 4.50 (d, J = 0.8 Hz, 2H), 4.01 (s, 2H).
(2-phenethyl-1H-imidazol-4-yl)methanol 15: 1H NMR (400 MHz, Methanol-d4) δ 7.27 – 7.21 (m, 2H), 7.18 – 7.13 (m, 3H), 6.83 (s, 1H), 4.50 (d, J = 0.8 Hz, 2H), 3.02 – 2.90 (m, 4H).
I. (2-(3-phenylpropyl)-1H-imidazol-4-yl)methanol 16
1. Step 1: 4-phenylbutanimidamide 60
Under an inert atmosphere, acetyl chloride (0.59 ml, 8.3 mmol) was added to a solution of 4-phenylbutyronitrile (0.150 g, 1.03 mmol) in anhydrous EtOH (1.21 ml) and the mixture was stirred at room temperature for 5 h. The volatiles were removed in vacuo, and the solid residue was stirred in 2 M NH3 in EtOH (2.5 ml) for 16 h at room temperature. The mixture was filtered, the filtrate was concentrated in vacuo, and the resultant residue was purified by column chromatography (Biotage Isolera, 12 g SiO2 cartridge, 0%–20% MeOH in DCM) to give the title compound as a white solid (0.081 g, 48%). The product had a weak chromophore and was unsuitable for analysis by LCMS. 1H NMR (400 MHz, DMSO) 8.80 (br s, 3H), 7.34 – 7.27 (m, 2H), 7.24 – 7.15 (m, 3H), 2.64 – 2.57 (m, 2H), 2.43 – 2.37 (m, 2H), 1.96 – 1.85 (m, 2H).
2. Step 2: (2-(3-phenylpropyl)-1H-imidazol-4-yl)methanol 16
A mixture of 4-phenylbutanimidamide 60 (0.081 g, 0.50 mmol) and dihydroxyacetone (0.045 g, 0.50 mmol) in 28%–30% ammonia solution (1 ml) was stirred in the microwave at 120 °C for 10 min. The solvent was evaporated in vacuo, and the residue was dissolved in MeOH and loaded onto an SCX-2 cartridge (500 mg). The cartridge was washed with 3 column volumes of MeOH before the desired product was eluted with 3 column volumes of 7 M NH3 in MeOH. The basic fraction was concentrated in vacuo and purified further by column chromatography (Biotage Isolera, 4 g SiO2 cartridge, 0%–20% MeOH (containing 10% 7 M NH3/MeOH solution) in DCM). Fractions containing the desired product were combined and purified further by mass directed preparative LCMS (5% –> 95% MeCN/0.1% TFA in H2O/0.1% TFA) to give the title compound as an orange oil (0.006 g, 6%). LCMS-B: rt 2.6 min; m/z 217.0 [M + H]+. The product had no significant chromophore at 254 nm or 214 nm. 1H NMR (400 MHz, DMSO) δ 13.89 (br s, 1H), 7.35 (s, 1H), 7.32 – 7.27 (m, 2H), 7.23 – 7.19 (m, 3H), 5.62 – 5.45 (m, 1H), 4.45 (d, J = 4.5 Hz, 2H), 2.87 (t, J = 7.7 Hz, 2H), 2.61 (t, J = 7.6 Hz, 2H), 2.01 (p, J = 7.7 Hz, 2H).
J. (2-(3-methoxy-3-phenylpropyl)-1H-imidazol-4-yl)methanol 17
1. Step 1: 4-methoxy-4-phenylbutanimidamide 61
Under an inert atmosphere, acetyl chloride (0.75 ml, 11 mmol) was added to a solution of 4-methoxy-4-phenylbutanenitrile (0.230 g, 1.31 mmol) in anhydrous EtOH (1.53 ml), and the mixture was stirred at room temperature for 5 h. The volatiles were removed in vacuo, and the solid residue was stirred in 2 M NH3 in EtOH (2.5 ml) for 16 h at room temperature. The mixture was filtered, and the filtrate was concentrated in vacuo to give the title compound as a white solid (0.219 g, 87%). The product had a weak chromophore and was unsuitable for analysis by LCMS. 1H NMR (400 MHz, DMSO) δ 9.11 – 8.47 (m, 3H), 7.41 – 7.36 (m, 2H), 7.33 – 7.27 (m, 3H), 4.16 (dd, J = 8.3, 4.6 Hz, 1H), 3.12 (s, 3H), 2.56 – 2.47 (m, peak obscured by solvent), 2.46 – 2.36 (m, 1H), 2.05 – 1.88 (m, 2H).
2. Step 2: (2-(3-methoxy-3-phenylpropyl)-1H-imidazol-4-yl)methanol 17
A mixture of 4-methoxy-4-phenylbutanimidamide 61 (0.219 g, 1.14 mmol) and dihydroxyacetone (0.103 g, 1.14 mmol) in 28%–30% ammonia solution (2 ml) was stirred in the microwave at 120 °C for 10 min. The solvent was evaporated in vacuo, and the residue was adsorbed onto silica and purified by column chromatography (Biotage Isolera, 12 g SiO2 cartridge, 0%–20% MeOH (+10% 7 M NH3 in MeOH) in DCM) to give an orange oil. The oil was triturated with Et2O before the solid was dissolved in MeOH and loaded onto an SCX-2 cartridge (500 mg). The cartridge was washed with 3 column volumes of MeOH before the desired product was eluted with 3 column volumes of 7 M NH3 in MeOH. The basic fraction was concentrated in vacuo to give the title compound as an orange oil (0.028 g, 10%). The product could not be characterized by LMCS due to the lack of a chromophore at 214 nm and 254 nm. 1H NMR (400 MHz, DMSO) δ 11.79 – 11.29 (m, 1H), 7.41 – 7.34 (m, 2H), 7.32 – 7.25 (m, 3H), 6.82 – 6.48 (m, 1H), 4.99 – 4.56 (m, 1H), 4.29 (s, 2H), 4.18 (dd, J = 7.4, 5.6 Hz, 1H), 3.12 (s, 3H), 2.63 – 2.51 (m, peaks obscured by solvent), 2.06 – 1.86 (m, 2H).
K. Synthesis of compounds 23 and 24
1. Intermediates
a. Step 1: 4-(((tert-butyldimethylsilyl)oxy)methyl)-1H-imidazole 18
TBSCl (3.38 g, 22.4 mmol) was added to a stirring solution of (1H-imidazol-4-yl)methanol (2.00 g, 20.4 mmol) and imidazole (2.78 g, 40.8 mmol) in DMF (N,N-dimethylformamide) (50 ml) under N2 at 0 °C. The mixture was allowed to slowly reach room temperature overnight. A saturated solution of NaHCO3 (50 ml) was added, and the mixture was stirred for 1 h. The mixture was extracted with EtOAc (3 × 100 ml), and the combined organic layers were washed with water (3 × 100 ml) and brine (100 ml), dried (Na2SO4), and concentrated in vacuo to give a yellow/orange oil. The material was purified by column chromatography (120 g SiO2 cartridge, 2%–10% methanol in DCM) to give the title compound as a colorless oil (3.0 g, 69%). 1H NMR (400 MHz, Chloroform-d) δ 7.59 (s, 1H), 6.94 (s, 1H), 4.73 (s, 2H), 0.91 (s, 9H), 0.08 (s, 6H). LCMS-B rt 2.89 min; (m/z) 213 [M + H]+.
b. Step 2: 4-(((tert-butyldimethylsilyl)oxy)methyl)-1-trityl-1H-imidazole 19
Trityl chloride (1.61 g, 5.78 mmol) was added to a solution of 4-(((tert-butyldimethylsilyl)oxy)methyl)-1H-imidazole 18 (1.12 g, 5.26 mmol) and triethylamine (1.47 ml, 10.5 mmol) in anhydrous DMF (20 ml) under a nitrogen atmosphere. The mixture was stirred for 2 h before water was added. The aqueous phase was extracted with DCM (3 × 100 ml), and the organics were combined and washed with brine (3 × 100 ml). The organic extract was dried (Na2SO4), and the solvent was removed in vacuo. Purification by column chromatography (Biotage Isolera, 40 g SiO2 cartridge, 0%–30% EtOAc in petroleum benzine 40–60 °C) gave the title compound as a white solid (1.93 g, 81%). 1H NMR (400 MHz, CDCl3) δ 7.37 (d, J = 1.5 Hz, 1H), 7.34 – 7.30 (m, 9H), 7.17 – 7.13 (m, 6H), 6.75 – 6.69 (m, 1H), 4.68 (d, J = 0.9 Hz, 2H), 0.84 (s, 9H), 0.03 (s, 6H). LCMS-B: rt 3.48 min; m/z 243.0 [Trt]+. Only trityl cation was observed in the mass spec.
c. Step 3: 4-(((tert-butyldimethylsilyl)oxy)methyl)-1-trityl-1H-imidazole-2-carbaldehyde 20
A solution of 4-(((tert-butyldimethylsilyl)oxy)methyl)-1-trityl-1H-imidazole 19 (0.211 g, 0.464 mmol) in anhydrous THF (5 ml) under an atmosphere of nitrogen was cooled to −40 °C. To this solution, n-butyllithium (1.54 M in hexanes, 0.60 ml, 0.93 mmol) was added, and the mixture was stirred at −40 °C for 30 min. Anhydrous DMF (0.14 ml, 1.9 mmol) was added, and the mixture was returned to room temperature and stirred for 30 min. A saturated aqueous solution of NH4Cl was added (∼20 ml), and the aqueous phase was extracted with EtOAc (3 × 20 ml). The organics were combined, washed with brine, and dried (Na2SO4), and the solvent was removed in vacuo. The crude solid was purified by column chromatography (Biotage Isolera, 24 g SiO2 cartridge, 0%–30% EtOAc in petroleum benzine 40–60 °C) to give the title compound as a white solid (0.190 g, 85%). LCMS-B: rt 4.02 min; m/z 243.0 [Trt]+. Only the trityl protecting group ion could be observed in the mass spectrum. 1H NMR (400 MHz, CDCl3) 9.13 (s, 1H), 7.35 – 7.31 (m, 9H), 7.15 – 7.11 (m, 6H), 6.93 (s, 1H), 4.77 – 4.74 (m, 2H), 0.82 (s, 9H), 0.03 (s, 6H).
d. Step 4: Ethyl (Z)-3-(4-(((tert-butyldimethylsilyl)oxy)methyl)-1-trityl-1H-imidazol-2-yl)acrylate and ethyl (E)-3-(4-(((tert-butyldimethylsilyl)oxy)methyl)-1-trityl-1H-imidazol-2-yl)acrylate 21
Sodium hydride (60% dispersion in mineral oil, 0.116 g, 2.90 mmol) was stirred in anhydrous THF (5 ml) at room temperature under nitrogen. The mixture was cooled to 0 °C before a solution of triethyl 2-phosphonoacetate (0.650 g, 2.90 mmol) in anhydrous THF (5 ml) was added. The mixture was returned to room temperature. A suspension of 4-(((tert-butyldimethylsilyl)oxy)methyl)-1-trityl-1H-imidazole-2-carbaldehyde 20 (0.700 g, 1.45 mmol) in anhydrous THF (10 ml) was then added, and the mixture was stirred at room temperature for 24 h. Water (∼50 ml) was added and THF was removed under reduced pressure. The aqueous layer was extracted with EtOAc (3 × 50 ml), the organics were combined, washed with brine, and dried (Na2SO4), and the solvent was removed in vacuo. The crude oil was purified by column chromatography (Biotage Isolera, 40 g SiO2 cartridge, 0%–30% EtOAc in petroleum benzine 40–60 °C) to give two components (in order of elution):
-
(a)
ethyl (E)-3-(4-(((tert-butyldimethylsilyl)oxy)methyl)-1-trityl-1H-imidazol-2-yl)acrylate 21 - 0.479 g, 60% yield. 1H NMR (400 MHz, CDCl3) δ 7.36 – 7.29 (m, 9H), 7.17 – 7.10 (m, 6H), 6.78 (s, 1H), 6.65 (d, J = 15.4 Hz, 1H), 6.49 (d, J = 15.5 Hz, 1H), 4.71 (s, 2H), 3.99 (q, J = 7.1 Hz, 2H), 1.12 (t, J = 7.1 Hz, 3H), 0.83 (s, 9H), 0.03 (s, 6H); LCMS-B rt 4.12 min; m/z 243.0 [Trt]+. Assigned as the (E) isomer on the basis of the alkenyl hydrogen signals: larger coupling constant and further downfield compared to the alkenyl hydrogens of the (Z)-isomer.
-
(b)
ethyl (Z)-3-(4-(((tert-butyldimethylsilyl)oxy)methyl)-1-trityl-1H-imidazol-2-yl)acrylate - 0.152 g, 19% yield. 1H NMR (400 MHz, CDCl3) δ 7.33 – 7.29 (m, 9H), 7.19 – 7.14 (m, 6H), 6.69 (s, 1H), 5.64 (d, J = 11.9 Hz, 1H), 5.37 (d, J = 11.9 Hz, 1H), 4.66 (s, 2H), 4.17 (q, J = 7.2 Hz, 2H), 1.24 (t, J = 7.1 Hz, 3H), 0.82 (s, 9H), 0.02 (s, 6H); LCMS-B: rt 3.69 min; m/z 243.0 [Trt]+. Assigned as the (Z) isomer: the alkenyl hydrogens have a smaller coupling constant and are further upfield compared to the alkenyl hydrogens of 21.
L. 3-(4-(hydroxymethyl)-1H-imidazol-2-yl)-N-methylpropanamide 23
1. Step 1: ethyl 3-(4-(((tert-butyldimethylsilyl)oxy)methyl)-1H-imidazol-2-yl)propanoate 22
A mixture of ethyl (E)-3-(4-(((tert-butyldimethylsilyl)oxy)methyl)-1-trityl-1H-imidazol-2-yl)acrylate 21 (0.479 g, 0.867 mmol) and Pd 10% on carbon (wetted with ca. 55% water, 0.075 g) in EtOAc was stirred under an atmosphere of hydrogen at room temperature for 21 h. The mixture was filtered through a plug of Celite, and the filtrate was concentrated under reduced pressure. The resultant residue was purified by column chromatography (Biotage Isolera, 24 g SiO2 cartridge, 0%–100% EtOAc in petroleum benzine 40–60 °C) to give the title compound as a colorless oil (0.144 g, 53% yield). 1H NMR (400 MHz, CDCl3) δ 6.80 – 6.75 (m, 1H), 4.66 (d, J = 0.9 Hz, 2H), 4.16 (q, J = 7.1 Hz, 2H), 3.04 – 2.99 (m, 2H), 2.75 – 2.70 (m, 2H), 1.26 (t, J = 7.1 Hz, 3H), 0.91 (s, 9H), 0.08 (s, 6H). LCMS-B RT 3.06 min; m/z 313.0 [M + H]+ (product had no absorption at 254 nm).
2. Step 2: 3-(4-(hydroxymethyl)-1H-imidazol-2-yl)-N-methylpropanamide 23
A solution of ethyl 3-(4-(((tert-butyldimethylsilyl)oxy)methyl)-1H-imidazol-2-yl)propanoate (22) (0.018 g, 0.058 mmol) in methylamine solution (33 wt. % in absolute ethanol, 5 ml) was stirred at 50 °C for 24 h. The volatiles were evaporated under reduced pressure, and the residue was dissolved in MeOH (2 ml). Hydrochloric acid (37%, 0.05 ml) was added, and the solution was stirred at 40 °C for 6 h. The volatiles were removed under reduced pressure, and the residue was taken up in a minimal amount of MeOH and loaded onto an SCX-2 cartridge (500 mg). The cartridge was washed with 5 column volumes of MeOH before the desired product was eluted with 7 N NH3 in MeOH. The basic fractions were combined, and the solvent was removed in vacuo to give the title compound as a pale yellow oil (0.011 g, >95% yield). 1H NMR (400 MHz, DMSO) δ 7.87 – 7.77 (m, 1H), 6.70 (s, 1H), 4.79 (s, 1H), 4.30 (s, 2H), 2.81 – 2.73 (m, 2H), 2.56 (d, J = 4.6 Hz, 3H), 2.47 – 2.40 (m, 2H), imidazole N-H not observed; LCMS-B rt 0.45 min; m/z 184.0 [M + H]+.
M. ethyl 3-(4-(hydroxymethyl)-1H-imidazol-2-yl)propanoate 24
A mixture of ethyl 3-(4-(((tert-butyldimethylsilyl)oxy)methyl)-1H-imidazol-2-yl)propanoate 22 (0.028 g, 0.090 mmol), HCl (37%, 0.02 ml), and EtOH was stirred at room temperature for 23 h. The volatiles were removed in vacuo, and the residue was taken up in MeOH and loaded onto an SCX-2 cartridge (500 mg). The cartridge was washed with 5 column volumes of MeOH, and then the product was eluted with 7 N NH3 in MeOH. The basic fractions were combined, and the solvent was removed under reduced pressure to give the title compound as a colorless oil (0.004 g, 23% yield): 1H NMR (400 MHz, MeOD) 6.86 (s, 1H), 4.51 – 4.46 (m, 2H), 4.11 (q, J = 7.1 Hz, 2H), 2.99 – 2.92 (m, 2H), 2.78 – 2.69 (m, 2H), 1.22 (t, J = 7.1 Hz, 3H); N-H and O-H not observed; the signal at 3.66 ppm indicates a small amount of methyl ester contamination; LCMS-B RT 0.46 min; m/z 199.0 [M + H]+; the product had no absorption at 254 nm and eluted with the solvent front.
N. (2-(4-aminobutyl)-1H-imidazol-5-yl)methanol bis trifluoroacetic acid 28
1. Step 1: 5-azidopentanenitrile 25
To a solution of 5-chloropentanenitrile (25.0 g, 213 mmol) in DMSO (200 ml) was added NaN3 (27.4 g, 425 mmol), and the mixture was heated at 50 °C overnight. The mixture was diluted with water (200 ml) and extracted with DCM (200 ml × 3). The combined organic extracts were washed with brine (200 ml), dried over Na2SO4, filtered, and concentrated under reduced pressure to give the crude title compound (38.3 g), which was used directly in the next step without further purification. LCMS-C: m/z 125.1 [M + H]+.
2. Step 2: ethyl 5-azidopentanimidate hydrochloride 26
A solution of 5-azidopentanenitrile 25 (20.2 g) in EtOH (200 ml) at 0 °C was bubbled with HCl gas for 3 h. The solvent was removed under reduced pressure to give the crude title compound (48.7 g), which was used directly in the next step without further purification. LCMS-C: m/z 171.1 [M + H]+.
3. Step 3: (2-(4-azidobutyl)-1H-imidazol-5-yl)methanol 27
A mixture of ethyl 5-azidopentanimidate hydrochloride 26 (2.4 g) and 1,3-dihydroxypropan-2-one (1.14 g, 12.7 mmol) in concentrated aqueous NH4OH (6 ml) and i-PrOH (6 ml) was heated at 90 °C under microwave irradiation for 1 h. This procedure was performed 20 times, and the reaction mixtures were combined and concentrated under reduced pressure. The residue was purified by silica gel chromatography (DCM/MeOH = 50:1 to 30:1 to 15:1) to give the title compound (13.8 g) as a yellow oil. LCMS-C: m/z 196.1 [M + H]+.
4. Step 4: (2-(4-aminobutyl)-1H-imidazol-5-yl)methanol bistrifluoroacetic acid 28
A mixture of (2-(4-azidobutyl)-1H-imidazol-5-yl)methanol 27 (100 mg, 0.51 mmol) and 10% Pd/C (20 mg) in MeOH (4 ml) was stirred at room temperature under an atmosphere of hydrogen overnight. The mixture was filtered, and the filtrate was concentrated under reduced pressure. The residue was purified by reverse phase chromatography (Biotage, C18 column, eluting with MeCN in water containing 0.1% TFA) to give the title compound (56 mg, 28%). LCMS-C: m/z 170.2 [M + H]+. 1H NMR (400 MHz, MeOD) δ 7.31 (s, 1H), 4.61 (s, 2H), 3.04 – 2.96 (m, 4H), 1.91 – 1.85 (m, 2H), 1.77 – 1.68 (m, 2H).
O. N-(4-(5-(hydroxymethyl)-1H-imidazol-2-yl)butyl)acetamide trifluoroacetic acid 29
To a solution of (2-(4-aminobutyl)-1H-imidazol-5-yl)methanol 28 (50 mg, 0.3 mmol) in DCM/MeOH (1 ml/1 ml) was added acetic anhydride (6.1 mg, 0.06 mmol), and the mixture was stirred at room temperature overnight. The solvent was removed under reduced pressure, and the residue was purified by reverse phase chromatography (Biotage, C18 column, eluting with MeCN in water containing 0.1% TFA) to give the title compound (25 mg, 40%). LCMS-C: m/z 212.1 [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ 14.2 (br s, 2H), 7.85 (t, J = 5.7 Hz, 1H), 7.38 (s, 1H), 5.63 (br s, 1H), 4.47 (s, 2H), 3.06 – 3.00 (m, 2H), 2.88 (t, J = 7.6 Hz, 2H), 1.78 (s, 3H), 1.72 – 1.64 (m, 2H), 1.41 – 1.33 (m, 2H).
P. N-(4-(5-(hydroxymethyl)-1H-imidazol-2-yl)butyl)benzamide 30
To a solution of (2-(4-aminobutyl)-1H-imidazol-5-yl)methanol 28 (40 mg, 0.24 mmol) and benzoyl chloride (15 mg, 0.11 mmol) in DCM/MeOH (1 ml/1 ml) was added Et3N (72 mg, 0.72 mmol), and the mixture was stirred at room temperature overnight. The mixture was concentrated under reduced pressure, and the residue was purified by prep-TLC (DCM/MeOH = 10:1) to give the title compound (5 mg, 8%). LCMS-C: m/z 274.0 [M + H]+. 1H NMR (400 MHz, MeOD) δ 7.85 – 7.79 (m, 2H), 7.53 (t, J = 7.3 Hz, 1H), 7.47 – 7.44 (m, 2H), 7.24 (s, 1H), 4.59 (s, 2H), 3.43 (t, J = 6.9 Hz, 2H), 2.99 (t, J = 7.7 Hz, 2H), 1.88 – 1.79 (m, 2H), 1.72 – 1.64 (m, 2H).
Q. N-(4-(5-(hydroxymethyl)-1H-imidazol-2-yl)butyl)-2-phenylacetamide trifluoroacetic acid 31
To a solution of (2-(4-aminobutyl)-1H-imidazol-5-yl)methanol 28 (30 mg, 0.18 mmol) in DCM (5 ml) and MeOH (1 ml) were added Et3N (75 μL, 0.54 mmol) and phenylacetyl chloride (10 μL, 0.07 mmol), and the mixture was stirred at room temperature overnight. The solvent was removed under reduced pressure, and the residue was diluted with water and washed with EtOAc. The aqueous layer was concentrated under reduced pressure, and the residue was purified by reverse phase chromatography (Biotage, C18 column, eluting with MeCN in water containing 0.1% TFA) to give the title compound (5 mg, 10%) as a yellow oil. LCMS-C: m/z 288.2 [M + H]+. 1H NMR (400 MHz, MeOD) δ 7.28 – 7.22 (m, 6H), 4.60 (s, 2H), 3.49 (s, 2H), 3.19 (t, J = 7.6 Hz, 2H), 2.93 (t, J = 7.6 Hz, 2H), 1.77 – 1.70 (m, 2H), 1.57 – 1.50 (m, 2H).
R. Methyl (4-(5-(hydroxymethyl)-1H-imidazol-2-yl)butyl)carbamate trifluoroacetic acid 32
To a solution of (2-(4-aminobutyl)-1H-imidazol-5-yl)methanol 28 (50 mg, 0.3 mmol) in DCM (3 ml) and MeOH (1 ml) were added Et3N (0.13 ml, 0.9 mmol) and methyl chloroformate (23 μl, 0.3 mmol), and the mixture was stirred at room temperature for 2 days. Water was added, and the mixture was washed with DCM. The aqueous layer was concentrated under reduced pressure, and the residue was purified by reverse phase chromatography (Biotage, C18 column, eluting with MeCN in water containing 0.1% TFA) to give the title compound (5 mg, 7%) as pale yellow oil. LCMS-C: m/z 228.2 [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ 14.1 (br s, 2H), 7.39 (s, 1H), 7.10 (t, J = 5.4 Hz, 1H), 5.59 (br s, 1H), 4.46 (s, 2H), 3.49 (s, 3H), 3.01 – 2.95 (m, 2H), 2.87 (t, J = 7.6 Hz, 2H), 1.75 – 1.59 (m, 2H), 1.41 – 1.32 (m, 2H).
S. Benzyl (4-(5-(hydroxymethyl)-1H-imidazol-2-yl)butyl)carbamate trifluoroacetic acid 35
1. Step 1: Benzyl (4-cyanobutyl)carbamate 33
A mixture of 5-azidopentanenitrile 25 (3.8 g, 31 mmol) and 10% Pd/C (400 mg) in MeOH (40 ml) was stirred at room temperature under an atmosphere of hydrogen overnight. The mixture was filtered, and the filtrate was concentrated under reduced pressure. The residue was dissolved in DCM (40 ml), Et3N (6.2 g, 61 mmol) and Cbz-Cl (6.3 g, 37 mmol) were added, and the mixture was stirred at room temperature overnight. The mixture was concentrated under reduced pressure, and the residue was purified by silica gel chromatography (DCM/MeOH = 10:1) to give the title compound (2.6 g, 37%) as a colorless oil. LCMS-C: m/z 233.1 [M + H]+.
2. Step 2: Benzyl (5-amino-5-iminopentyl)carbamate 34
To a solution of benzyl (4-cyanobutyl)carbamate 33 (460 mg, 1.98 mmol) in EtOH (5 ml) was added AcCl (1.2 g, 16 mmol) dropwise, and the mixture was heated at 30 °C overnight. The solvent was removed under reduced pressure, and the residue was taken up in EtOH and concentrated again under reduced pressure. The residue was dissolved in a 2 M NH3/MeOH solution (2 ml), and the mixture was stirred at room temperature overnight. The solvent was removed under reduced pressure, and the residue was purified by silica gel chromatography (DCM/MeOH = 1:0 to 7:1) to give the title compound (180 mg, 37%). LCMS-C: m/z 250.2 [M + H]+.
3. Step 3: Benzyl (4-(5-(hydroxymethyl)-1H-imidazol-2-yl)butyl)carbamate trifluoroacetic acid 35
A mixture of benzyl (5-amino-5-iminopentyl)carbamate 34 (400 mg, 1.70 mmol) and 1,3-dihydroxypropan-2-one (153 mg, 1.70 mmol) in concentrated aqueous NH4OH (4 ml) was heated at 70 °C in a sealed tube for 4 h. The mixture was filtered, and the filtrate was concentrated under reduced pressure. The residue was purified by reverse phase chromatography (Biotage, C18 column, eluting with MeCN in water containing 0.1% TFA) to give the title compound (100 mg, 20%). LCMS-C: M /z 304.1 [M + H]+. 1H NMR (400 MHz, MeOD) δ 7.35 – 7.26 (m, 6H), 5.05 (s, 2H), 4.59 (s, 2H), 3.15 (t, J = 6.9 Hz, 2H), 2.95 (t, J = 7.7 Hz, 2H), 1.82 – 1.74 (m, 2H), 1.57 – 1.50 (m, 2H).
T. Methyl benzyl(4-(5-(hydroxymethyl)-1H-imidazol-2-yl)butyl)carbamate trifluoroacetic acid 37
1. Step 1: (2-(4-(Benzylamino)butyl)-1H-imidazol-5-yl)methanol 36
To a solution of (2-(4-aminobutyl)-1H-imidazol-5-yl)methanol 28 (30 mg, 0.18 mmol), benzaldehyde (19 mg, 0.18 mmol), and AcOH (11 μl, 0.18 mmol) in MeOH (3 ml) was added NaCNBH3 (45 mg, 0.72 mmol), and the mixture was stirred at room temperature overnight. The mixture was concentrated under reduced pressure to give the title compound (47 mg, 100%), which was used in the next step without further purification. LCMS-C: m/z 260.1 [M + H]+.
2. Step 2: Methyl benzyl(4-(5-(hydroxymethyl)-1H-imidazol-2-yl)butyl)carbamate trifluoroacetic acid 37
To a solution of (2-(4-(benzylamino)butyl)-1H-imidazol-5-yl)methanol (47 mg, 0.18 mmol) in DCM (3 ml) at 0 °C were added Et3N (0.10 ml, 0.72 mmol) and methyl chloroformate (28 μl, 0.36 mmol), and the mixture was stirred at room temperature overnight. The mixture was concentrated under reduced pressure, and the residue was purified by prep. HPLC to give the title compound (4.5 mg, 8%). LCMS-C: m/z 318.2 [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ 14.0 – 13.8 (m, 2H), 7.40 (s, 1H), 7.35 – 7.21 (m, 5H), 5.58 (br s, 1H), 4.47 (s, 2H), 4.42 (s, 2H), 3.62 (s, 3H), 3.21 – 3.13 (m, 2H), 2.85 (t, J = 7.2 Hz, 2H), 1.67 – 1.60 (m, 2H), 1.49 – 1.40 (m, 2H).
U. Methyl (4-(4-(hydroxymethyl)-1H-imidazol-2-yl)butyl)((6-oxo-1,6-dihydropyridin-3-yl)methyl)carbamate bis trifluoroacetic acid 40
1. Step 1: tert-butyl 4-(((4-(5-(hydroxymethyl)-1H-imidazol-2-yl)butyl)amino)methyl)piperidine-1-carboxylate 38
To a solution of (2-(4-aminobutyl)-1H-imidazol-5-yl)methanol 28 (50 mg, 0.30 mmol), tert-butyl 4-formylpiperidine-1-carboxylate (63 mg, 0.30 mmol), and AcOH (18 mg, 0.30 mmol) in MeOH (10 ml) was added NaCNBH3 (75 mg, 1.20 mmol), and the mixture was stirred at room temperature overnight. The mixture was concentrated under reduced pressure to give the title compound (110 mg, 100%), which was used in the next step without further purification. LCMS-C: m/z 367.1 [M + H]+.
2. Step 2: tert-butyl 4-(((4-(5-(hydroxymethyl)-1H-imidazol-2-yl)butyl)(methoxycarbonyl)amino)methyl)piperidine-1-carboxylate 39
To a solution of tert-butyl 4-(((4-(5-(hydroxymethyl)-1H-imidazol-2-yl)butyl)amino)methyl)piperidine-1-carboxylate 38 (110 mg, 0.30 mmol) and Et3N (0.17 ml, 1.2 mmol) in DCM (5 ml) at 0 °C was added methyl chloroformate (56.7 mg, 0.6 mmol), and the mixture was allowed to warm to room temperature and stirred overnight. The mixture was concentrated under reduced pressure, and the residue was purified by prep-HPLC to give the title compound (16 mg, 12%). LCMS-C: m/z 425.1 [M + H]+.
3. Step 3: Methyl (4-(4-(hydroxymethyl)-1H-imidazol-2-yl)butyl)(piperidin-4-ylmethyl)carbamate bis trifluoroacetic acid 40
To a solution of tert-butyl 4-(((4-(5-(hydroxymethyl)-1H-imidazol-2-yl)butyl)(methoxycarbonyl)amino)methyl)piperidine-1-carboxylate 39 (16 mg, 0.038 mmol) in DCM (3 ml) at 0 °C was added TFA (0.2 ml), and the mixture was allowed to warm to room temperature and stirred for 4 h. The mixture was concentrated under reduced pressure to give the title compound (5 mg, 42%). LCMS-C: m/z 335.2 [M + H]+. 1H NMR (400 MHz, MeOD) δ 7.30 (s, 1H), 4.61 (s, 2H), 3.67 (s, 3H), 3.43 – 3.36 (m, 2H), 3.30 (2H obscured by solvent peak), 3.21 (d, J = 7.3 Hz, 2H), 3.02 – 2.90 (m, 4H), 2.05 – 1.93 (m, 1H), 1.91 – 1.81 (m, 2H), 1.80 – 1.72 (m, 2H), 1.65 – 1.57 (m, 2H), 1.48 – 1.36 (m, 2H).
V. Methyl (4-(4-(hydroxymethyl)-1H-imidazol-2-yl)butyl)((6-oxo-1,6-dihydropyridin-3-yl)methyl)carbamate trifluoroacetic acid 45
1. Step 1: 2-(4-azidobutyl)-5-(((tert-butyldiphenylsilyl)oxy)methyl)-1H-imidazole 41
To a solution of (2-(4-azidobutyl)-1H-imidazol-5-yl)methanol 27 (500 mg, 2.56 mmol) in DCM (10 ml) at 0 °C was added Et3N (1.1 ml, 7.68 mmol) followed by TBDPSCl (1.35 ml, 5.13 mmol), and the mixture was allowed to warm to room temperature and stirred for 2 days. The mixture was diluted with water and extracted with DCM, and the organic layer was washed with brine, dried over Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified by silica gel chromatography (Pet. Ether/EtOAc = 10:1 to 5:1 to 1:1) to give the title compound (420 mg, 38%). LCMS-C: m/z 434.2 [M + H]+. 1H NMR (400 MHz, MeOD) δ 7.68 – 7.66 (m, 4H), 7.46 – 7.36 (m, 6H), 6.62 (s, 1H), 4.64 (s, 2H), 2.70 (t, J = 7.6 Hz, 2H), 1.80 – 1.72 (m, 2H), 1.63 – 1.55 (m, 2H), 1.36 – 1.21 (m, 2H), 1.03 (s, 9H).
2. Step 2: 4-(5-(((tert-butyldiphenylsilyl)oxy)methyl)-1H-imidazol-2-yl)butan-1-amine 42
A mixture of 2-(4-azidobutyl)-5-(((tert-butyldiphenylsilyl)oxy)methyl)-1H-imidazole 41 (350 mg, 0.81 mmol) and 10% Pd/C (350 mg) in MeOH (5 ml) was stirred at room temperature under an atmosphere of hydrogen overnight. The mixture was filtered, and the filtrate was concentrated under reduced pressure. The residue was purified by silica gel chromatography (DCM/MeOH = 20:1 to 10:1 to 5:1) to give the title compound (310 mg, 94%) as a yellow oil. LCMS-C: m/z 408.3 [M + H]+. 1H NMR (400 MHz, CDCl3) δ 7.69 – 7.64 (m, 4H), 7.45 – 7.33 (m, 6H), 6.71 (s, 1H), 4.69 (s, 2H), 2.76 (t, J = 6.7 Hz, 2H), 2.68 (t, J = 7.4 Hz, 2H), 1.80 – 1.69 (m, 2H), 1.60 – 1.48 (m, 2H), 1.05 (s, 9H).
3. Step 3: 5-(((4-(4-(((tert-butyldiphenylsilyl)oxy)methyl)-1H-imidazol-2-yl)butyl)amino)methyl)pyridin-2(1H)-one 43
To a solution of 4-(5-(((tert-butyldiphenylsilyl)oxy)methyl)-1H-imidazol-2-yl)butan-1-amine 42 (120 mg, 0.29 mmol) and 6-hydroxynicotinaldehyde (37 mg, 0.29 mmol) in MeOH (10 ml) was added AcOH (0.15 ml), and the mixture was stirred at room temperature overnight. NaCNBH3 (73 mg, 1.2 mmol) was added, and the mixture was heated at 40 °C for 5 h. The mixture was diluted with water and extracted with DCM, and the organic extracts were washed with brine, dried over Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified by reverse phase chromatography (Biotage, C18 column, eluting with MeCN in water) to give the title compound (150 mg, 100%). LCMS-C: m/z 515.1 [M + H]+.
4. Step 4: Methyl (4-(4-(hydroxymethyl)-1H-imidazol-2-yl)butyl)((6-oxo-1,6-dihydropyridin-3-yl)methyl)carbamate trifluoroacetic acid 45
To a solution of 5-(((4-(4-(((tert-butyldiphenylsilyl)oxy)methyl)-1H-imidazol-2-yl)butyl)amino)methyl)pyridin-2(1H)-one 43 (50 mg, 0.13 mmol) in DCM (4 ml) were added Et3N (56 μl, 0.4 mmol) and methyl chloroformate (8 μl, 0.1 mmol), and the mixture was stirred at room temperature overnight. LCMS analysis revealed that only TBDPS deprotection had occurred. The mixture was extracted with ether and the organic extract was concentrated under reduced pressure. The residue was dissolved in DCM/MeOH (4 ml/0.5 ml) and treated with Et3N (100 μl, 0.52 mmol) and methyl chloroformate (10 μl, 0.13 mmol), and the mixture was stirred at room temperature overnight. MeOH (5 ml) and a 10% aqueous NaOH solution (0.5 ml) were added, and stirring was continued at room temperature overnight. The solvent was removed under reduced pressure, and the residue was purified by reverse phase chromatography (Biotage, C18 column, eluting with MeCN in water containing 0.1% TFA) to give the title compound (5 mg, 5%). LCMS-C: m/z 335.1 [M + H]+. 1H NMR (400 MHz, MeOD) δ 7.60 – 7.51 (m, 1H), 7.37 – 7.28 (m, 1H), 6.85 (s, 1H), 6.51 (d, J = 9.3 Hz, 1H), 4.50 (s, 2H), 4.24 (s, 2H), 3.71 (s, 3H), 3.24 (t, J = 7.4 Hz, 2H), 2.67 (t, J = 7.2 Hz, 2H), 1.69 – 1.61 (m, 2H), 1.54 – 1.45 (m, 2H).
W. Methyl (4-(4-(hydroxymethyl)-1H-imidazol-2-yl)butyl)((2-oxo-1,2-dihydropyridin-4-yl)methyl)carbamate hydrochloride 47
1. Step 1: 4-(((4-(4-(((tert-butyldiphenylsilyl)oxy)methyl)-1H-imidazol-2-yl)butyl)amino)methyl)pyridin-2(1H)-one 44
A solution of 4-(5-(((tert-butyldiphenylsilyl)oxy)methyl)-1H-imidazol-2-yl)butan-1-amine 42 (0.43 g, 1.1 mmol) and 2-hydroxyisonicotinaldehyde (0.20 g, 1.6 mmol) in MeOH (20 ml) was stirred at room temperature overnight. NaCNBH3 (0.13 g, 2.12 mmol) and AcOH (2 drops) were added, and stirring was continued at room temperature for 4 h. The mixture was diluted with water and extracted with DCM. The combined organic extracts were washed with brine, dried over Na2SO4, filtered, and concentrated under reduced pressure to give the crude title compound (0.6 g), which was used directly in the next step. LCMS-C: m/z 515.0 [M + H]+.
2. Step 2: Methyl (4-(4-(((tert-butyldiphenylsilyl)oxy)methyl)-1H-imidazol-2-yl)butyl)((2-oxo-1,2-dihydropyridin-4-yl)methyl)carbamate 46
To a solution of 4-(((4-(4-(((tert-butyldiphenylsilyl)oxy)methyl)-1H-imidazol-2-yl)butyl)amino)methyl)pyridin-2(1H)-one 44 (0.6 g, assumed 1.1 mmol) in DCM (10 ml) was added K2CO3 (0.29 g, 2.1 mmol) followed by a solution of methyl chloroformate (150 mg, 1.6 mmol) in DCM (2 ml) dropwise, and the mixture was stirred at room temperature for 2 h. The mixture was diluted with water and extracted with DCM, and the combined organic extracts were washed with brine, dried over Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified by prep-HPLC to give the title compound (45.2 mg, 7%). LCMS-C: m/z 573.0 [M + H]+.
3. Step 3: Methyl (4-(4-(hydroxymethyl)-1H-imidazol-2-yl)butyl)((2-oxo-1,2-dihydropyridin-4-yl)methyl)carbamate hydrochloride 47
A mixture of methyl (4-(4-(((tert-butyldiphenylsilyl)oxy)methyl)-1H-imidazol-2-yl)butyl)((2-oxo-1,2-dihydropyridin-4-yl)methyl)carbamate 46 (45 mg, 0.08 mmol) and a 4 M HCl in 1,4-dioxane solution (5 ml) was stirred at room temperature for 2 h. The solvent was removed under reduced pressure, and the residue was purified by prep-HPLC to give the title compound (3.2 mg, 12%). LCMS-C: m/z 335.2 [M + H]+. 1H NMR (400 MHz, MeOD) δ 7.40 – 7.31 (m, 2H), 6.46– 6.31 (m, 2H), 4.61 (s, 2H), 4.39 (s, 2H), 3.70 (s, 3H), 3.55 – 3.51 (m, 2H), 2.96 – 2.91 (m, 2H), 1.76 – 1.60 (m, 4H).
X. N-(4-(5-(hydroxymethyl)-1H-imidazol-2-yl)butyl)acrylamide 49
1. Step 1: N-(4-(5-(((tert-butyldiphenylsilyl)oxy)methyl)-1H-imidazol-2-yl)butyl)acrylamide 48
To a solution of 4-(5-(((tert-butyldiphenylsilyl)oxy)methyl)-1H-imidazol-2-yl)butan-1-amine 42 (400 mg, 0.98 mmol) in DCM (10 ml) at 0 °C was added a solution of NaOH (43 mg, 1.1 mmol) in water (1.0 ml) followed by a solution of acryloyl chloride (89 mg, 0.98 mmol) in DCM (2.0 ml) dropwise. The mixture was stirred at 0 °C for 2 h and then partitioned between DCM and water. The layers were separated, and the organic layer was dried over Na2SO4, filtered, and concentrated under reduced pressure to give the title compound (420 mg, 93%), which was used directly in the next step.
2. Step 2: N-(4-(5-(hydroxymethyl)-1H-imidazol-2-yl)butyl)acrylamide 49
To a solution of N-(4-(5-(((tert-butyldiphenylsilyl)oxy)methyl)-1H-imidazol-2-yl)butyl)acrylamide 48 (260 mg, 0.56 mmol) in THF (10 ml) at 0 °C was added TBAF (1 M solution in THF, 1.4 ml, 1.4 mmol), and the mixture was allowed to warm to room temperature and stirred for 2 h. The solvent was removed under reduced pressure, and the residue was purified by reverse phase chromatography (Biotage, C18 column, eluting with MeCN in water) to give the title compound (5.0 mg, 4%). LCMS-C: m/z 224.2 [M + H]+. 1H NMR (400 MHz, D2O) δ 7.23 (s, 1H), 6.27 – 6.11 (m, 2H), 5.72 (dd, J = 9.9, 1.6 Hz, 1H), 4.62 (s, 2H), 3.27 (t, J = 6.8 Hz, 2H), 2.97 (t, J = 7.5 Hz, 2H), 1.82 – 1.74 (m, 2H), 1.60 – 1.52 (m, 2H).
Y. Peptide-imidazole hybrids
1. 2-(2-acetamidopropanamido)-N-(1-amino-3-methyl-1-oxobutan-2-yl)-5-(4-(hydroxymethyl)-1H-imidazol-2-yl)pentanamide 55
a. Step 1: 4-(((tert-butyldimethylsilyl)oxy)methyl)-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-imidazole/5-(((tert-butyldimethylsilyl)oxy)methyl)-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-imidazole 50
Lithium hexamethyldisilazide (1 M in toluene) (2.6 ml, 2.6 mmol) was added to a stirring solution of 4-(((tert-butyldimethylsilyl)oxy)methyl)-1H-imidazole 18 (0.370 g, 1.74 mmol) in THF (8.7 ml) under N2 at 0 °C. The reaction mixture was stirred for 15 min before SEMCl (0.46 ml, 2.6 mmol) was added. The resulting solution was stirred for 4 h. A saturated solution of NaHCO3 (10 ml) was added, and the mixture was extracted with EtOAc (3 × 10 ml). The combined organic layers were washed with water (10 ml) and brine (10 ml), dried (Na2SO4), and concentrated in vacuo to give an orange oil. The material was purified by column chromatography (24 g SiO2 cartridge, 2%–10% methanol in DCM) to give the title compound as a colorless oil in an ∼2:1 mixture of regioisomers (0.464 g, 77%). LCMS-B rt 3.29 min (@ 214 nm); (m/z) 343 [M + H]+. Major regioisomer: 1H NMR (400 MHz, Chloroform-d) δ 7.53 (t, J = 0.8 Hz, 1H), 6.94 (d, J = 0.8 Hz, 1H), 5.35 (s, 2H), 4.71 (s, 2H), 3.54–3.44 (m, 2H), 0.95–0.83 (m, J = 0.6 Hz, 11H), 0.06 (d, J = 0.6 Hz, 6H), -0.02 (s, 9H). Minor regioisomer: 1H NMR (400 MHz, Chloroform-d) δ 7.52 – 7.50 (m, 1H), 6.96 – 6.94 (m, 1H), 5.22 (d, J = 0.6 Hz, 2H), 4.71 (s, 2H), 3.52 – 3.42 (m, 2H), 0.95–0.83 (m, 11H), 0.10 (d, J = 0.6 Hz, 6H), -0.02 (s, 9H).
b. Step 2: 4-(((tert-butyldimethylsilyl)oxy)methyl)-2-iodo-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-imidazole/5-(((tert-butyldimethylsilyl)oxy)methyl)-2-iodo-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-imidazole 51
n-BuLi (1.54 M in hexanes, 0.97 ml, 1.5 mmol) was added dropwise over 10 min to a stirring solution of 4-(((tert-Butyldimethylsilyl)oxy)methyl)-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-imidazole/5-(((tert-butyldimethylsilyl)oxy)methyl)-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-imidazole 50 (0.343 g, 1.00 mmol) in THF (10 ml) at −78 °C under N2. The resulting yellow solution was stirred at −78 °C for 1 h. A solution of iodine (0.381 g, 1.5 mmol) in THF (3 ml) was added dropwise until the orange/brown color persisted. The solution was stirred for 30 min. A saturated solution of sodium thiosulfate (10 ml) was added, and the aqueous was extracted with EtOAc (3 × 10 ml). The combined organic extracts were washed with water (10 ml) and brine (10 ml), dried (Na2SO4), and concentrated in vacuo. The material was purified by column chromatography (24 g SiO2 cartridge, 5%–100% EtOAc in petroleum benzine 40–60 °C) to give the title compound as a colorless oil in an ∼2:1 mixture of regioisomers (331 mg, 71%). LCMS-B: rt 3.97 min; m/z 468.8 [M + H]+. Major regioisomer: 1H NMR (400 MHz, Chloroform-d) δ 6.98 (s, 1H), 5.35 (s, 2H), 4.72 (d, J = 0.7 Hz, 2H), 3.58 – 3.50 (m, 2H), 0.95–0.87 (m, 11H), 0.07 (s, 6H), -0.00 (s, 9H). Minor regioisomer: 1H NMR (400 MHz, Chloroform-d) δ 7.06 (s, 1H), 5.19 (s, 2H), 4.69 (d, J = 0.7 Hz, 2H), 3.58 – 3.50 (m, 2H), 0.95–0.87 (m, 11H), 0.09 (s, 6H), −0.00 (s, 9H).
c. Step 3: 2-(2-acetamidopropanamido)-N-(1-amino-3-methyl-1-oxobutan-2-yl)-5-(4-(((tert-butyldimethylsilyl)oxy)methyl)-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-imidazol-2-yl)pent-4-ynamide/2-(2-acetamidopropanamido)-N-(1-amino-3-methyl-1-oxobutan-2-yl)-5-(5-(((tert-butyldimethylsilyl)oxy)methyl)-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-imidazol-2-yl)pent-4-ynamide 53
A suspension of 4-(((tert-Butyldimethylsilyl)oxy)methyl)-2-iodo-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-imidazole/5-(((tert-butyldimethylsilyl)oxy)methyl)-2-iodo-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-imidazole 51 (50.0 mg, 0.107 mmol), 2-(2-acetamidopropanamido)-N-(1-amino-3-methyl-1-oxobutan-2-yl)pent-4-ynamide 52 (38 mg, 0.117 mmol) and CuI (1.2 mg, 0.006 mmol) in Et3N (0.5 ml) and DMF (1 ml) was bubbled with N2 and sonicated. PdCl2(PPh3)2 (2.2 mg, 0.003 mmol) was added, and the suspension was stirred at room temperature until TLC indicated complete consumption of alkyne. A saturated solution of ammonium chloride (10 ml) was added, and the resulting suspension was extracted with EtOAc (3 × 30 ml). The organic extracts were combined, washed with 50% brine (3 × 20 ml), dried (Na2SO4), and concentrated in vacuo. The mixture was purified by column chromatography (12 g SiO2 cartridge, 0%–15% MeOH in DCM) to give the desired compound as an ∼2:1 mixture of regioisomers (5.7 mg, 8%). LCMS-B rt 3.51 min; m/z mass ion not detected. Major regioisomer: 1H NMR (400 MHz, Methanol-d4) δ 6.90 (s, 1H), 5.50 (s, 2H), 4.77 (s, 2H), 4.71 – 4.65 (m, 1H), 4.34 – 4.26 (m, 1H), 4.24 – 4.18 (m, 1H), 3.62 – 3.55 (m, 2H), 3.15 – 3.05 (m, 1H), 3.02 – 2.89 (m, 1H), 2.25 – 2.06 (m, 1H), 2.00 – 1.89 (m, 3H), 1.37 – 1.32 (m, 3H), 1.00 – 0.88 (m, 17H), 0.15 – 0.05 (m, 6H), 0.05 – -0.06 (m, 9H). Minor regioisomer: 1H NMR (400 MHz, Methanol-d4) δ 7.15 (s, 1H), 5.38 (d, J = 2.6 Hz, 2H), 4.60 (s, 2H), 4.71 – 4.65 (m, 1H), 4.34 – 4.26 (m, 1H), 4.24 – 4.18 (m, 1H), 3.62 – 3.55 (m, 2H), 3.15 – 3.05 (m, 1H), 3.02 – 2.89 (m, 1H), 2.25 – 2.06 (m, 1H), 2.00 – 1.89 (m, 3H), 1.37 – 1.27 (m, 5H), 1.00–0.88 (m, 15H), 0.15 – 0.05 (m, 6H), 0.05 – -0.06 (m, 9H).
d. Step 4: 2-(2-acetamidopropanamido)-N-(1-amino-3-methyl-1-oxobutan-2-yl)-5-(4-(((tert-butyldimethylsilyl)oxy)methyl)-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-imidazol-2-yl)pentanamide/2-(2-acetamidopropanamido)-N-(1-amino-3-methyl-1-oxobutan-2-yl)-5-(5-(((tert-butyldimethylsilyl)oxy)methyl)-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-imidazol-2-yl)pentanamide 54
A solution of 2-(2-acetamidopropanamido)-N-(1-amino-3-methyl-1-oxobutan-2-yl)-5-(4-(((tert-butyldimethylsilyl)oxy)methyl)-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-imidazol-2-yl)pent-4-ynamide/2-(2-acetamidopropanamido)-N-(1-amino-3-methyl-1-oxobutan-2-yl)-5-(5-(((tert-butyldimethylsilyl)oxy)methyl)-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-imidazol-2-yl)pent-4-ynamide 53 (5.2 mg, 0.008 mmol) in EtOH (1.0 ml) was evacuated and back filled with N2 three times. Pd/C (53% water wet, 3.2 mg) was added, and the flask was evacuated and back-filled with N2 three times and then evacuated and back-filled with H2 from a balloon. The reaction mixture was stirred at room temperature overnight and then evacuated and back filled with N2 three times. The suspension was diluted with EtOAc (10 ml) and filtered through a pad of Celite and washed with EtOAc (10 ml). The solution was concentrated in vacuo to give the desired product (4.3 mg, 82%) as a mixture of regioisomers. LCMS-B rt 3.47 min (no absorbance at 254/214 nm); m/z 669 [M + H]+. Major regioisomer: 1H NMR (400 MHz, Methanol-d4) δ 6.80 (s, 1H), 5.38 (s, 2H), 5.37 – 5.33 (m, 1H), 4.74 (s, 2H), 4.35 – 4.25 (m, 1H), 4.21 – 4.15 (m, 1H), 3.61 – 3.56 (m, 2H), 2.89–2.69 (m, 2H), 2.19 (t, J = 7.6 Hz, 2H), 2.08 – 2.00 (m, 1H), 1.98 – 1.94 (m, 3H), 1.85 (m, 2H), 1.81 – 1.71 (m, 1H), 1.66 – 1.56 (m, 1H), 1.39 – 1.27 (m, 3H obscured), 0.99 – 0.84 (m, 15H), 0.08 (dd, J = 2.8, 1.0 Hz, 6H), -0.00 (s, 9H). Minor regioisomer: 1H NMR (400 MHz, Methanol-d4) δ 7.00 (s, 1H), 5.38 – 5.33 (m, 1H), 5.28 (s, 2H), 4.61 (s, 2H), 4.35 – 4.25 (m, 1H), 4.21 – 4.15 (m, 1H), 3.61 – 3.56 (m, 2H), 2.89–2.69 (m, 2H), 2.19 (t, J = 7.6 Hz, 2H), 2.08 – 2.00 (m, 1H), 1.98 – 1.94 (m, 3H), 1.85 (m, 2H), 1.81 – 1.71 (m, 1H), 1.66 – 1.56 (m, 1H), 1.39 – 1.27 (m, 3H obscured), 0.99 – 0.84 (m, 15H), 0.08 (dd, J = 2.8, 1.0 Hz, 6H), -0.00 (s, 9H).
e. Step 5: 2-(2-acetamidopropanamido)-N-(1-amino-3-methyl-1-oxobutan-2-yl)-5-(4-(hydroxymethyl)-1H-imidazol-2-yl)pentanamide 55
TFA (0.5 ml) was added to a solution of 2-(2-Acetamidopropanamido)-N-(1-amino-3-methyl-1-oxobutan-2-yl)-5-(4-(((tert-butyldimethylsilyl)oxy)methyl)-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-imidazol-2-yl)pentanamide/2-(2-acetamidopropanamido)-N-(1-amino-3-methyl-1-oxobutan-2-yl)-5-(5-(((tert-butyldimethylsilyl)oxy)methyl)-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-imidazol-2-yl)pentanamide 54 (4.3 mg, 0.006 mmol) in DCM (0.5 ml) at room temperature. The solution was stirred overnight and then concentrated in vacuo. The residue was diluted in minimum MeOH and loaded onto an SCX cartridge (0.5 g, Silicycle), washed with MeOH (10 ml), and then eluted with 7 M NH3 in MeOH (3 × 5 ml). The residue was concentrated in vacuo to give the desired compound (1.1 mg, 40%) as a colorless oil. LCMS-B rt 0.54 min (no absorbance at 214/254 nm, rt based on +ve TIC chromatogram), m/z 425.0 [M + H]+. 1H NMR (400 MHz, Methanol-d4) δ 7.07 – 6.94 (m, 1H), 4.57 – 4.49 (m, 2H), 4.50 – 4.10 (m, 3H), 2.78 (m, 2H), 2.23 – 2.10 (m, 1H), 1.98 (dd, J = 7.8, 2.0 Hz, 3H), 1.92 – 1.64 (m, 4H), 1.40 – 1.31 (m, 3H), 1.01 – 0.80 (m, 6H). The product was isolated as a mixture of diastereomers.
Z. 2-(2-acetamido-2-methylpropanamido)-N-(1-amino-3-methyl-1-oxobutan-2-yl)-5-(4-(hydroxymethyl)-1H-imidazol-2-yl)pentanamide 59
1. Step 1: 2-(2-acetamido-2-methylpropanamido)-N-(1-amino-3-methyl-1-oxobutan-2-yl)-5-(4-(((tert-butyldimethylsilyl)oxy)methyl)-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-imidazol-2-yl)pent-4-ynamide/2-(2-acetamido-2-methylpropanamido)-N-(1-amino-3-methyl-1-oxobutan-2-yl)-5-(5-(((tert-butyldimethylsilyl)oxy)methyl)-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-imidazol-2-yl)pent-4-ynamide 57
A suspension of 4-(((tert-butyldimethylsilyl)oxy)methyl)-2-iodo-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-imidazole/5-(((tert-butyldimethylsilyl)oxy)methyl)-2-iodo-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-imidazole 51 (8.3 mg, 0.018 mmol) and 2-(2-acetamido-2-methylpropanamido)-N-(1-amino-3-methyl-1-oxobutan-2-yl)pent-4-ynamide 56 (6.0 mg, 0.018 mmol) in Et3N (0.5 ml) was degassed by three freeze (liq. N2)-pump-thaw cycles. CuI (0.4 mg, 1.8 μmol) and PdCl2(PPh3)2 (1.2 mg, 1.8 μmol) were added, and a freeze-pump-thaw cycle was repeated. The resulting suspension was stirred at room temperature for 4 days. Water (10 ml) was added, and the resulting suspension was extracted with EtOAc (3 × 30 ml). The organic extracts were combined, washed with water (30 ml) and brine (30 ml), dried (Na2SO4), and concentrated in vacuo. The mixture was purified by column chromatography (12 g SiO2 cartridge, 1%–15% MeOH in DCM) to give the title compound (1.2 mg, 10%). LCMS-A rt 6.17 min; m/z 679 [M + H]+, 677 [M-H]−.
2. Step 2: 2-(2-acetamido-2-methylpropanamido)-N-(1-amino-3-methyl-1-oxobutan-2-yl)-5-(4-(((tert-butyldimethylsilyl)oxy)methyl)-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-imidazol-2-yl)pentanamide/2-(2-acetamido-2-methylpropanamido)-N-(1-amino-3-methyl-1-oxobutan-2-yl)-5-(5-(((tert-butyldimethylsilyl)oxy)methyl)-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-imidazol-2-yl)pentanamide 58
A solution of 2-(2-acetamido-2-methylpropanamido)-N-(1-amino-3-methyl-1-oxobutan-2-yl)-5-(4-(((tert-butyldimethylsilyl)oxy)methyl)-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-imidazol-2-yl)pent-4-ynamide/2-(2-acetamido-2-methylpropanamido)-N-(1-amino-3-methyl-1-oxobutan-2-yl)-5-(5-(((tert-butyldimethylsilyl)oxy)methyl)-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-imidazol-2-yl)pent-4-ynamide 57 (1.1 mg, 1.7 μmol) in EtOH (1.0 ml) was evacuated and back filled with N2 three times. Pd/C (53% water wet, 1.2 mg) was added and the flask was evacuated and back-filled with N2 three times and then evacuated and back-filled with H2 from a balloon. The reaction mixture was stirred at room temperature overnight before being evacuated and back filled with N2 three times. The suspension was diluted with EtOAc (10 ml) and filtered through a pad of Celite that was then washed with EtOAc (10 ml). The solution was concentrated in vacuo to give the title compound that was used directly in the next step without further purification or characterization (assumed 100% conversion). LCMS-A rt 5.62 min (no absorbance at 214/254 nm, rt based on +ve TIC chromatogram), m/z 683 [M + H]+.
3. Step 3: 2-(2-acetamido-2-methylpropanamido)-N-(1-amino-3-methyl-1-oxobutan-2-yl)-5-(4-(hydroxymethyl)-1H-imidazol-2-yl)pentanamide 59
TFA (0.5 ml) was added to a solution of 2-(2-acetamido-2-methylpropanamido)-N-(1-amino-3-methyl-1-oxobutan-2-yl)-5-(4-(((tert-butyldimethylsilyl)oxy)methyl)-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-imidazol-2-yl)pentanamide/(S)-2-(2-acetamido-2-methylpropanamido)-N-(1-amino-3-methyl-1-oxobutan-2-yl)-5-(5-(((tert-butyldimethylsilyl)oxy)methyl)-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-imidazol-2-yl)pentanamide 58 (1.76 μmol) in DCM (0.5 ml) at room temperature. The solution was stirred overnight and then concentrated in vacuo. The residue was concentrated from MeOH in vacuo to give the title compound (0.6 mg, 68%) as a yellow oil. The compound was lypophilized from MeCN:H2O to give the title compound as an off-white oil (0.54 mg, 55% over 2 steps). LCMS-D rt 5.19 min, m/z 439.0 [M + H]+.
The tripeptide Ac-ARV-NH2 was purchased from Mimotopes (Victoria, Australia).
SUPPLEMENTARY MATERIAL
See the supplementary material for X-ray crystallography collection and refinement statistics, ligand difference density maps, SPR sensorgrams, and a scheme for the synthesis of compounds 55 and 59.
ACKNOWLEDGMENTS
We thank the Australian Synchrotron for beam time and the beamline scientists for their help. This research was undertaken in part using the MX2 beamline at the Australian Synchrotron and made use of the ACRF detector. We also thank the C3 Crystallization Centre for all crystallization experiments. This project received funding from the Australian Government through the Cooperative Research Center for Cancer Therapeutics (from the Department of Industry, Innovation and Science (DIIS) Cooperative Research Centres Program). The CRC for Cancer Therapeutics partially funds its work through the sale of discoveries to pharmaceutical and biotech companies.
Note: This article is part of the Special Issue, Transactions from the 68th Annual Meeting of the American Crystallographic Association: Shining a Light on Structure-Based Drug Design.
References
- 1. Guarnaccia D. A. and Tansey P. W., “ Moonlighting with WDR5: A cellular multitasker,” J. Clin. Med. 7, 21 (2018). 10.3390/jcm7020021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Dou Y., Milne T. A., Ruthenburg A. J., Lee S., Lee J. W., Verdine G. L., Allis C. D., and Roeder R. G., “ Regulation of MLL1 H3K4 methyltransferase activity by its core components,” Nat. Struct. Mol. Biol. 13, 713 (2006). 10.1038/nsmb1128 [DOI] [PubMed] [Google Scholar]
- 3. Cho Y.-W., Hong T., Hong S., Guo H., Yu H., Kim D., Guszczynski T., Dressler G. R., Copeland T. D., Kalkum M., and Ge K., “ PTIP associates with MLL3- and MLL4-containing histone H3 lysine 4 methyltransferase complex,” J. Biol. Chem. 282, 20395 (2007). 10.1074/jbc.M701574200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Steward M. M., Lee J.-S., O'Donovan A., Wyatt M., Bernstein B. E., and Shilatifard A., “ Molecular regulation of H3K4 trimethylation by ASH2L, a shared subunit of MLL complexes,” Nat. Struct. Mol. Biol. 13, 852 (2006). 10.1038/nsmb1131 [DOI] [PubMed] [Google Scholar]
- 5. Schuettengruber B., Martinez A.-M., Iovino N., and Cavalli G., “ Trithorax group proteins: Switching genes on and keeping them active,” Nat. Rev. Mol. Cell Biol. 12, 799 (2011). 10.1038/nrm3230 [DOI] [PubMed] [Google Scholar]
- 6. Thiel A. T., Blessington P., Zou T., Feather D., Wu X., Yan J., Zhang H., Liu Z., Ernst P., Koretzky G. A., and Hua X., “ MLL-AF9-induced leukemogenesis requires coexpression of the wild-type Mll allele,” Cancer Cell 17, 148 (2010). 10.1016/j.ccr.2009.12.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Muntean A. G. and Hess J. L., “ The pathogenesis of mixed-lineage leukemia,” Annu. Rev. Pathol. 7, 283 (2012). 10.1146/annurev-pathol-011811-132434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chen Y., Anastassiadis K., Kranz A., Stewart A. F., Arndt K., Waskow C., Yokoyama A., Jones K., Neff T., Lee Y., and Ernst P., “ MLL2, not MLL1, plays a major role in sustaining MLL-rearranged acute myeloid leukemia,” Cancer Cell 31, 755 (2017). 10.1016/j.ccell.2017.05.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Han Z., Guo L., Wang H., Shen Y., Deng X. W., and Chai J., “ Structural basis for the specific recognition of methylated histone H3 lysine 4 by the WD-40 protein WDR5,” Mol. Cell 22, 137 (2006). 10.1016/j.molcel.2006.03.018 [DOI] [PubMed] [Google Scholar]
- 10. Ruthenburg A. J., Wang W., Graybosch D. M., Li H., Allis C. D., Patel D. J., and Verdine G. L., “ Histone H3 recognition and presentation by the WDR5 module of the MLL1 complex,” Nat. Struct. Mol. Biol. 13, 704 (2006). 10.1038/nsmb1119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Couture J.-F., Collazo E., and Trievel R. C., “ Molecular recognition of histone H3 by the WD40 protein WDR5,” Nat. Struct. Mol. Biol. 13, 698 (2006). 10.1038/nsmb1116 [DOI] [PubMed] [Google Scholar]
- 12. Patel A., Vought V. E., Dharmarajan V., and Cosgrove M. S., “ A conserved arginine-containing motif crucial for the assembly and enzymatic activity of the mixed lineage leukemia protein-1 core complex,” J. Biol. Chem. 283, 32162 (2008). 10.1074/jbc.M806317200 [DOI] [PubMed] [Google Scholar]
- 13. Odho Z., Southall S. M., and Wilson J. R., “ Characterization of a novel WDR5-binding site that recruits RbBP5 through a conserved motif to enhance methylation of histone H3 lysine 4 by mixed lineage leukemia protein-1,” J. Biol. Chem. 285, 32967 (2010). 10.1074/jbc.M110.159921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Patel A., Dharmarajan V., and Cosgrove M. S., “ Structure of WDR5 bound to mixed lineage leukemia protein-1 peptide,” J. Biol. Chem. 283, 32158 (2008). 10.1074/jbc.C800164200 [DOI] [PubMed] [Google Scholar]
- 15. Dias J., Van Nguyen N., Georgiev P., Gaub A., Brettschneider J., Cusack S., Kadlec J., and Akhtar A., “ Structural analysis of the KANSL1/WDR5/KANSL2 complex reveals that WDR5 is required for efficient assembly and chromatin targeting of the NSL complex,” Genes Dev. 28, 929 (2014). 10.1101/gad.240200.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ee L.-S., McCannell K. N., Tang Y., Fernandes N., Hardy W. R., Green M. R., Chu F., and Fazzio T. G., “ An embryonic stem cell‐specific NuRD complex functions through interaction with WDR5,” Stem Cell Rep. 8, 1488 (2017). 10.1016/j.stemcr.2017.04.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wang Y.-L., Faiola F., Xu M., Pan S., and Martinez E., “ Human ATAC Is a GCN5/PCAF-containing acetylase complex with a novel NC2-like histone fold module that interacts with the TATA-binding protein,” J. Biol. Chem. 283, 33808 (2008). 10.1074/jbc.M806936200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Thomas L. R., Wang Q., Grieb B. C., Phan J., Foshage A. M., Sun Q., Olejniczak E. T., Clark T., Dey S., Lorey S., Alicie B., Howard G. C., Cawthon B., Ess K. C., Eischen C. M., Zhao Z., Fesik S. W., and Tansey W. P., “ Interaction with WDR5 promotes target gene recognition and tumorigenesis by MYC,” Mol. Cell 58, 440 (2015). 10.1016/j.molcel.2015.02.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Carugo A., Genovese G., Seth S., Nezi L., Rose J. L., Bossi D., Cicalese A., Shah P. K., Viale A., Pettazzoni P. F., Akdemir K. C., Bristow C. A., Robinson F. S., Tepper J., Sanchez N., Gupta S., Estecio M. R., Giuliani V., Dellino G. I., Riva L., Yao W., Di Francesco M. E., Green T., D'Alesio C., Corti D., Kang Y. A., Jones P., Wang H., Fleming J. B., Maitra A., Pelicci P. G., Chin L., DePinho R. A., Lanfrancone L., Heffernan T. P., and Draetta G. F., “ In vivo functional platform targeting patient-derived xenografts identifies WDR5-myc association as a critical determinant of pancreatic cancer,” Cell Rep. 16, 133 (2016). 10.1016/j.celrep.2016.05.063 [DOI] [PubMed] [Google Scholar]
- 20. Avdic V., Zhang P., Lanouette S., Voronova A., Skerjanc I., and Couture J.-F., “ Fine-tuning the stimulation of MLL1 methyltransferase activity by a histone H3-based peptide mimetic,” FASEB J. 25, 960 (2011). 10.1096/fj.10-171959 [DOI] [PubMed] [Google Scholar]
- 21. Karatas H., Townsend E. C., Cao F., Chen Y., Bernard D., Liu L., Lei M., Dou Y., and Wang S., “ High-affinity, small-molecule peptidomimetic inhibitors of MLL1/WDR5 protein–protein interaction,” J. Am. Chem. Soc. 135, 669 (2013). 10.1021/ja306028q [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Cao F., Townsend E. C., Karatas H., Xu J., Li L., Lee S., Liu L., Chen Y., Ouillette P., Zhu J., Hess J. L., Atadja P., Lei M., Qin Z. S., Malek S., Wang S., and Dou Y., “ Targeting MLL1 H3K4 methyltransferase activity in mixed-lineage leukemia,” Mol. Cell 53, 247 (2014). 10.1016/j.molcel.2013.12.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Senisterra G., Wu H., Allali-Hassani A., Wasney G. A., Barsyte-Lovejoy D., Dombrovski L., Dong A., Nguyen K. T., Smil D., Bolshan Y., Hajian T., He H., Seitova A., Chau I., Li F., Poda G., Couture J.-F., Brown P. J., Al-Awar R., Schapira M., Arrowsmith C. H., and Vedadi M., “ Small-molecule inhibition of MLL activity by disruption of its interaction with WDR5,” Biochem. J. 449, 151 (2013). 10.1042/BJ20121280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Grebien F., Vedadi M., Getlik M., Giambruno R., Grover A., Avellino R., Skucha A., Vittori S., Kuznetsova E., Smil D., Barsyte-Lovejoy D., Li F., Poda G., Schapira M., Wu H., Dong A., Senisterra G., Stukalov A., Huber K. V. M., Schönegger A., Marcellus R., Bilban M., Bock C., Brown P. J., Zuber J., Bennett K. L., Al-awar R., Delwel R., Nerlov C., Arrowsmith C. H., and Superti-Furga G., “ Pharmacological targeting of the Wdr5-MLL interaction in C/EBPα N-terminal leukemia,” Nat. Chem. Biol. 11, 571 (2015). 10.1038/nchembio.1859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Getlik M., Smil D., Zepeda-Velázquez C., Bolshan Y., Poda G., Wu H., Dong A., Kuznetsova E., Marcellus R., Senisterra G., Dombrovski L., Hajian T., Kiyota T., Schapira M., Arrowsmith C. H., Brown P. J., Vedadi M., and Al-awar R., “ Structure-based optimization of a small molecule antagonist of the interaction between WD repeat-containing protein 5 (WDR5) and mixed-lineage leukemia 1 (MLL1),” J. Med. Chem. 59, 2478 (2016). 10.1021/acs.jmedchem.5b01630 [DOI] [PubMed] [Google Scholar]
- 26. Zhu J., Sammons M. A., Donahue G., Dou Z., Vedadi M., Getlik M., Barsyte-Lovejoy D., Al-awar R., Katona B. W., Shilatifard A., Huang J., Hua X., Arrowsmith C. H., and Berger S. L., “ Gain-of-function p53 mutants co-opt chromatin pathways to drive cancer growth,” Nature 525, 206 (2015). 10.1038/nature15251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sun Y., Bell J. L., Carter D., Gherardi S., Poulos R. C., Milazzo G., Wong J. W. H., Al-Awar R., Tee A. E., Liu P. Y., Liu B., Atmadibrata B., Wong M., Trahair T., Zhao Q., Shohet J. M., Haupt Y., Schulte J. H., Brown P. J., Arrowsmith C. H., Vedadi M., MacKenzie K. L., Hüttelmaier S., Perini G., Marshall G. M., Braithwaite A., and Liu T., “ WDR5 supports an N-myc transcriptional complex that drives a protumorigenic gene expression signature in neuroblastoma,” Cancer Res. 75, 5143 (2015). 10.1158/0008-5472.CAN-15-0423 [DOI] [PubMed] [Google Scholar]
- 28. Wang F., Jeon K. O., Salovich J. M., Macdonald J. D., Alvarado J., Gogliotti R. D., Phan J., Olejniczak E. T., Sun Q., Wang S., Camper D., Yuh J. P., Shaw J. G., Sai J., Rossanese O. W., Tansey W. P., Stauffer S. R., and Fesik S. W., “ Discovery of potent 2-Aryl-6,7-dihydro-5H-pyrrolo [1,2-a]imidazoles as WDR5-WIN-site inhibitors using fragment-based methods and structure-based design,” J. Med. Chem. 61, 5623 (2018). 10.1021/acs.jmedchem.8b00375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Aho E. R., Wang J., Gogliotti R. D., Howard G. C., Phan J., Acharya P., Macdonald J. D., Cheng K., Lorey S. L., Lu B., Wenzel S., Foshage A. M., Alvarado J., Wang F., Shaw J. G., Zhao B., Weissmiller A. M., Thomas L. R., Vakoc C. R., Hall M. D., Hiebert S. W., Liu Q., Stauffer S. R., Fesik S. W., and Tansey W. P., “ Displacement of WDR5 from chromatin by a WIN site inhibitor with picomolar affinity,” Cell Rep. 26, 2916 (2019). 10.1016/j.celrep.2019.02.047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Aho E. R., Weissmiller A. M., Fesik S. W., and Tansey W. P., “ Targeting WDR5: A WINning anti-cancer strategy?,” Epigenet. Insights 12, 2516865719865282 (2019). 10.1177/2516865719865282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Francis C., Kenny P., Dolezal O., Saubern S., Kruger M., Savage P., Peat T., and Ryan J., “ Construction of the CSIRO fragment library,” Aust. J. Chem. 66, 1473 (2013). 10.1071/CH13325 [DOI] [Google Scholar]
- 32. Schuetz A., Allali‐Hassani A., Martín F., Loppnau P., Vedadi M., Bochkarev A., Plotnikov A. N., Arrowsmith C. H., and Min J., “ Structural basis for molecular recognition and presentation of histone H3 By WDR5,” EMBO J. 25, 4245 (2006). 10.1038/sj.emboj.7601316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Karatas H., Li Y., Liu L., Ji J., Lee S., Chen Y., Yang J., Huang L., Bernard D., Xu J., Townsend E. C., Cao F., Ran X., Li X., Wen B., Sun D., Stuckey J. A., Lei M., Dou Y., and Wang S., “ Discovery of a highly potent, cell-permeable macrocyclic peptidomimetic (MM-589) targeting the WD repeat domain 5 protein (WDR5)–mixed lineage leukemia (MLL) protein–protein interaction,” J. Med. Chem. 60, 4818 (2017). 10.1021/acs.jmedchem.6b01796 [DOI] [PubMed] [Google Scholar]
- 34. Cowieson N. P., Aragao D., Clift M., Ericsson D. J., Gee C., Harrop S. J., Mudie N., Panjikar S., Price J. R., Riboldi-Tunnicliffe A., Williamson R., and Caradoc-Davies T., “ MX1: A bending-magnet crystallography beamline serving both chemical and macromolecular crystallography communities at the Australian synchrotron,” J. Synchrotron Radiat. 22, 187 (2015). 10.1107/S1600577514021717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Aragao D., Aishima J., Cherukuvada H., Clarken R., Clift M., Cowieson N. P., Ericsson D. J., Gee C. L., Macedo S., Mudie N., Panjikar S., Price J. R., Riboldi-Tunnicliffe A., Rostan R., Williamson R., and Caradoc-Davies T. T., “ MX2: A high-flux undulator microfocus beamline serving both the chemical and macromolecular crystallography communities at the Australian Synchrotron,” J. Synchrotron Radiat. 25, 885 (2018). 10.1107/S1600577518003120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kabsch W., “ XDS,” Acta Crystallogr., Sect. D 66, 125 (2010). 10.1107/S0907444909047337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Winter G., Waterman D. G., Parkhurst J. M., Brewster A. S., Gildea R. J., Gerstel M., Fuentes-Montero L., Vollmar M., Michels-Clark T., Young I. D., Sauter N. K., and Evans G., “ DIALS: Implementation and evaluation of a new integration package,” Acta Crystallogr., Sect. D 74, 85 (2018). 10.1107/S2059798317017235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Evans P. R. and Murshudov G. N., “ How good are my data and what is the resolution?,” Acta Crystallogr., Sect. D 69, 1204 (2013). 10.1107/S0907444913000061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., and Read R. J., “ Phaser crystallographic software,” J. Appl. Cryst. 40, 658 (2007). 10.1107/S0021889807021206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Murshodov G., Vagin A., and Dodson E., “ Refinement of macromolecular structures by the maximum-likelihood method,” Acta Crystallogr., Sect. D 53, 240 (1997). 10.1107/S0907444996012255 [DOI] [PubMed] [Google Scholar]
- 41. Emsley P., Lohkamp B., Scott W. G., and Cowtan K., “ Features and development of Coot,” Acta Crystallogr., Sect. D 66, 486 (2010). 10.1107/S0907444910007493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Moriarty N. W., Grosse-Kunstleve R. W., and Adams P. D., “ electronic Ligand Builder and Optimization Workbench (eLBOW): A tool for ligand coordinate and restraint generation,” Acta Crystallogr., Sect. D 65, 1074 (2009). 10.1107/S0907444909029436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Schüttelkopf A. W. and van Aalten D. M. F., “ PRODRG: A tool for high-throughput crystallography of protein-ligand complexes,” Acta Crystallogr., Sect. D 60, 1355 (2004). 10.1107/S0907444904011679 [DOI] [PubMed] [Google Scholar]
- 44. Madsen C., Jensen A. A., Liljefors T., Kristiansen U., Nielsen B., Hansen C. P., Larsen M., Ebert B., Bang-Andersen B., Krogsgaard-Larsen P., and Frølund B., “ 5-Substituted imidazole-4-acetic acid analogues: Synthesis, modeling, and pharmacological characterization of a series of novel gamma-aminobutyric acid(C) receptor agonists,” J. Med. Chem. 50, 4147 (2007). 10.1021/jm070447j [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
See the supplementary material for X-ray crystallography collection and refinement statistics, ligand difference density maps, SPR sensorgrams, and a scheme for the synthesis of compounds 55 and 59.