

Abstract
The variability of human milk (HM) composition renders analysis of its components essential for optimal nutrition of preterm fed either with donor's or own mother's milk. To fulfil this requirement, various analytical instruments have been subjected to scientific and clinical evaluation. The objective of this study was to evaluate the suitability of a rapid method for the analysis of macronutrients in HM as compared with the analytical methods applied by cow's milk industry. Mature milk from 39 donors was analysed using an infrared human milk analyser (HMA) and compared with biochemical reference laboratory methods. The statistical analysis was based on the use of paired data tests. The use of an infrared HMA for the analysis of lipids, proteins and lactose in HM proved satisfactory as regards the rapidity, simplicity and the required sample volume. The instrument afforded good linearity and precision in application to all three nutrients. However, accuracy was not acceptable when compared with the reference methods, with overestimation of the lipid content and underestimation of the amount of proteins and lactose contents. The use of mid‐infrared HMA might become the standard for rapid analysis of HM once standardisation and rigorous and systematic calibration is provided.
Keywords: human milk, macronutrients, milk banking, quantitative methods, milk analysers, fortification
Introduction
The numerous benefits derived from the use of human milk (HM) as sole nutrient during the first months of life have been explicitly underscored by international health organisations and justify generalisation of its use in all newborn infants, particularly in the most vulnerable such as preterm and/or low‐birthweight infants [Henderson et al. 2007; World Health Organization (WHO) 2011; American Academy of Pediatrics 2012]. As a consequence, there has been a growing interest in developing human milk banks (HMBs) worldwide, with the objective of providing neonates whose mothers are not able to breastfeed with HM coming from donor mothers. In addition, the introduction of HMBs offers economic benefits as it reduces the incidence and/or severity of numerous diseases, particularly necrotizing enterocolitis (Arnold 2002; Italian Association of Human Milk Bank 2010; European Milk Bank Association 2012).
Routinely, donor milk is collected, subjected to chemical and microbiological control and pasteurisation, and stored until consumption. HMB is always consumed under medical prescription (Italian Association of Human Milk Bank 2010). HM composition is widely variable and may not always satisfy individual nutritional needs (Silvestre et al. 2001; Bokor et al. 2007; Bauer & Gerss 2011; Moltó‐Puigmartíet al. 2011). Preterm infants are at high risk of undernutrition. HM fortifiers added to HM in these patients enhance protein, energy and mineral provision, and improve growth and nitrogen retention (Arslanoglu et al. 2010). However, standard fortification is made assuming a fixed composition of HM. Information regarding the precise composition of HM would be of relevance to optimise fortification either with own mother's milk or with bank donor's milk (Arslanoglu et al. 2010).
In this scenario, generalisation of HMBs has led to an increasing demand for suitable methods capable of rapidly and reliably analysing large number of HM samples using only small aliquots. Conventional analytical methods used for quality control or research purposes do not meet these criteria because they are tedious, time‐consuming and require large milk volumes. In recent years, new spectrophotometric instruments similar to the ones used in the dairy industry for the analysis of cow's milk but technically adapted to the requirements of HMBs have been developed. Among the different options, infrared (IR) spectrophotometry has been shown to be very useful for the analysis of macronutrients in HM (Michaelsen et al. 1988; Sauer & Kim 2001; Corvaglia et al. 2008; Menjo et al. 2009; Casadio et al. 2010). The convenient handling characteristics of this technique offer an attractive alternative to classical analytical procedures. To add new information, we have launched a prospective study to compare the precision and suitability of the mid‐infrared (MIR) HMA (Miris AB®, Uppsala, Sweden) for routine quantification of fat, protein and lactose in HM with the biochemical methods routinely employed for the analysis of cow's milk as recommended by the International Dairy Federation (http://www.cd3wd.com/cd3wd_40/LSTOCK/004/X6537E/X6537E05.htm).
Key messages
-
•
Individualised fortification of HM favours the achievement of nutritional needs especially in preterm infants.
-
•
Traditional methods of analysis require large volumes of sample and are very tedious and time‐consuming.
-
•
On the contrary, MIR transmission spectroscopy HMAs only need a small volume of milk to perform analysis and are extremely rapid and accurate.
-
•
However, HMA needs to be periodically calibrated to avoid misreading that could endanger the quality of supplementation.
Material and methods
Milk donors and sample collection
Analysis of mature milk from 39 donors (HMB; Division of Neonatology, University & Polytechnic Hospital La Fe, Valencia, Spain) was performed in the Department of Pharmacy (University CEU Cardenal Herrera, Valencia, Spain). Milk donors delivered at term (range: 37–41 weeks; median: 39 weeks). Milk was expressed from both breasts using an electric pump (Lactina; Medela, Baar, Switzerland) into a sterile container until emptying was completed (c. 10–15 min in each breast). Extracted milk was poured into a labelled sterile container and kept frozen at −20°C until processed. For processing, the milk sample was allowed to thaw at room temperature. Before samples were analysed by HMA, they were warmed up to 40°C and homogenised (1.5 s per 1 mL of sample) with an ultrasonic vibrator (VCX 130; Sonics & Material, Newtown, CT, USA). Biochemical analysis did not require previous warming of the samples and were homogenised as described. Thereafter, a total of 44 mL was homogenised by manual agitation and further used for analytical purposes. Out of these, an aliquot of 2 mL was pipetted into a sterile recipient for HMA analysis, while the rest was fractioned into three aliquots: 20 mL for lipids, 2 mL for protein and 20 mL for lactose determination, respectively.
The study was approved by the Ethics and Scientific Committee of the Hospital Universitario y Politécnico La Fe and does not comprise ethical issues as only mothers who were producing exceeding amounts of milk for their own baby and willing to donate or already donating milk to the milk bank were approached to participate in the study.
Direct analysis with the HMA
HMA is based on semi‐solid MIR transmission spectroscopy. The chemical groups directly correlated with fat, protein and lactose are measured at different waveband areas through waveband filters. The wavebands are specific for each of the functional groups measured: carbonyl groups (5.7 mm) and carbon‐hydrogen groups (3.5 mm) for fat determination; amide groups (6.5 mm) for protein determination; and hydroxyl groups (9.6 mm) for lactose determination (Casadio et al. 2010).
The HMA was operated in ‘processed milk’ mode, indicated for frozen samples. The instrument has two built‐in modes –‘processed’ and ‘unprocessed’ for fresh and frozen milk, respectively. The analyser has a different calibration for fresh and frozen milk because milk fat globule size changes with storing, requiring homogenisation prior to its analysis. Manufacturer calibration is based on reference samples that have been homogenised with an ultrasonic vibrator. However, no negative control was used because the manufacturer does not provide with calibration standards and does not inform on how they should be performed.
Samples were allowed to thaw, warmed up to 40°C and homogenised (1.5 s per 1 mL of sample) with an ultrasonic vibrator (VCX 130; Sonics & Material).
Analysis with reference laboratory methods
The analytical assays employed in our studies follow the standard recommendations of the International Dairy Federation (http://www.cd3wd.com/cd3wd_40/LSTOCK/004/X6537E/X6537E05.htm).
Fat determination (Gerber method)
This is a volumetric determination of the lipid fraction using a specially calibrated butyrometer by centrifugation for 5 min at 2000 r.p.m. after aqueous fraction component (especially proteins) digestion with sulphuric acid. The sample volume required is 10 mL and the procedure lasts for 30 min (Kleyn et al. 2001).
Protein determination (Bradford method)
This a colorimetric technique that measures Coomasie blue protein complex absorbance using bovine sero‐albumin as the standard. A commercially available reactive was employed (Bio‐Rad Laboratories, Richmond, CA, USA). The sample volume required is 1 mL and the procedure lasts for 1–2 min (Bradford 1976).
Lactose determination (Chloramine‐T method)
After milk deproteinisation with tungstic acid and posterior filtration, lactose is quantified according to iodine production in lactose IK‐Chloramine‐T reaction based on the reducing characteristic of lactose using 0.04 M thiosulfate. The sample volume required is 10 mL and the procedure lasts for 120 min (Hinton & Macara 1927).
As initial step, we assessed precision and linearity of the different laboratory methods employed to analyse HM composition. Precision of a method refers to the degree of reproducibility of the specific method employed, considering that accidental mistakes may influence the results. Calculation is made in ‘n’ simultaneously obtained aliquots within the same working session (intra‐assay precision) and in aliquots obtained from the same sample in different working sessions (inter‐assay). Hence, precision between two different tests was calculated by performing an analysis of 16 aliquots of the same sample (8 aliquots per method). Precision was expressed as coefficient of variation (CV %) and was calculated as follows: CV % = 100 × standard deviation (SD)/mean value for ‘n’ determinations. The linearity of the methods for each nutrient was tested using various dilutions of a standard formula (Hero España SA®, Alcantarilla, Spain) due to the lack of certified HM samples. Dilutions employed assured that minimum and maximum concentration values for each of the analysed nutrients were comprised within the range present in HM. The artificial formula was diluted as follows: 6.2, 5, 3.8, 2.5 and 1.2 g per 100 mL of distilled water for fat; 2.5, 2, 1.5, 1 and 0.5 g per 100 mL of distilled water for proteins; and 8, 7, 6 and 5 g in 100 mL of distilled water for lactose. Each result was expressed as a correlation coefficient (r). Likewise, the between‐test precision of the HMA was evaluated based on determination in 20 aliquots of the same sample (10 aliquots in each test) and calculation of the mean percentage coefficient of variation between the two tests. The linearity of the HMA technique was evaluated based on dilutions of a sample of HM in proportions of 1:1, 1:2 and 1:3 for the analysis of fat and lactose as the HMA cannot be used in infant formulas or dairy products.
Differences between HMA and biochemical methods
Full analysis using biochemical methods requires >25 mL of sample volume, at least 2 h for completing the procedure and trained analyst, whereas HMA analysis of macronutrients can be performed using a substantially smaller sample volume (1 mL), less time (1 min) and minimum training. Moreover, MHA offers also the possibility of point‐of‐care analysis.
Statistical analysis
The Statgraphics plus 5.0 statistical package (Statpoint Technologies Inc., Warrenton, VA, USA) was used for the statistical analysis and MedCalc Software version 12.2.1(MedCalc Software, Broekstraat 52, 9030 Mariakerke, Belgium) for Bland–Altman test. The differences between the results obtained with the HMA technique and the traditional methods were individually analysed using the Bland–Altman test (Bland & Altman 1986), which allows the comparison of new measurement technique with an established one and verify if it agrees sufficiently as to replace the standard one. Results of the comparison were expressed using Bland–Altman plots, which allow the comparison of two measurements techniques. In this graphical method, the differences between the two techniques are plotted against the averages of the two techniques or, when one of the methods is a reference or ‘gold standard’ method, against that particular method (Krouwer 2008). Significant differences between methods were evaluated using linear correlation between both methods (r 2). Statistical significance was considered for P < 0.05.
Results
The Gerber (Kleyn et al. 2001), Bradford (Bradford 1976) and Chloramine‐T (Hinton & Macara 1927) methods proved to be valid for the determination of total lipids, protein and lactose in HM samples. Validation of the methods yielded the following precision values: 1.29% (Gerber), 6.91% (Bradford) and 2.15% (Chloramine‐T). Linearity at the considered concentrations was r 2 = 0.9996 for the Gerber method, r 2 = 0.9977 for the Bradford method and r 2 = 0.9928 for the Chloramine‐T, respectively. Accordingly, these methods were considered adequate for use as reference techniques vs. direct analysis with the HMA.
Conversely, evaluation of the HMA technique yielded a between‐test precision of 5.19%, 3.08% and 1.40% for fat, proteins and lactose, respectively. The study of linearity at the considered concentrations yielded the following linearity values expressed as correlation coefficients: r 2 = 0.9988 for fat, r 2 = 0.9822 for protein, and r 2 = 0.9975 for lactose, respectively.
1, 2, 3 show the differences between the biochemical methods employed for the determination of fat (Gerber), protein (Bradford) and lactose (Chloramine‐T) and the IR HMA using the Bland–Altman statistical method (Bland & Altman 1986; Krouwer 2008). Hence, Fig. 1 depicts the mean (−0.75) and ±1.96 SD (0.92–2.34) differences between the Gerber and HMA methods for fat analysis. All values are included within the confidence interval except for two. Differences do not show any statistically significant tendency. In Fig. 2, mean (0.6) and ±1.96 SD (0.07–1.12) differences for protein determination between Bradford and HMA methods for the determination of proteins are shown. Bradford method shows higher values than HMA; however, all values are within the confidence interval. Differences do not show any statistically significant tendency. Figure 3 represents mean (1.99) and ±1.96 SD (0.32–4.5) differences between Chloramine‐T and HMA methods for lactose analysis. Lactose values determined by the Chloramine‐T method are higher than those obtained using the HMA. All values but one are within the confidence interval. Graphic shows the tendency to enhance differences with greater lactose values.
Figure 1.

Bland–Altman plot for fat analysis. Difference between the values obtained with laboratory (Gerber; Kleyn et al. 2001) and HMA methods is represented in the Y‐axis. The results obtained using the Gerber method are represented in the X‐ axis.
Figure 2.
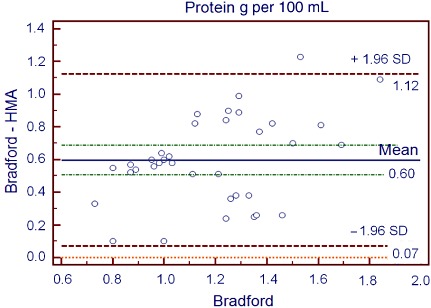
Bland–Altman plot for protein analysis. Difference between the values obtained with laboratory (Bio‐Rad; Bradford 1976) and HMA methods is represented in the Y‐axis. Bio‐Rad method results are represented in the X‐axis.
Figure 3.

Bland–Altman plot for lactose analysis. Difference between the values obtained with laboratory (Chloramine‐T; Hinton & Macara 1927) and HMA methods is represented in the Y‐axis. Chloramine‐T method results are represented in the X‐axis.
Table 1 reports the mean value and SD for macronutrients (fat, protein and lactose). As shown in Table 1, application of the Student's t‐test for paired data showed the differences between direct HMA analysis and analysis with the traditional methodology to be very significant (P < 0.01 and P < 0.001 for fat, proteins and lactose, respectively).
Table 1.
Comparison of lipid, protein and lactose concentrations in human milk (expressed in g per 100 mL) as determined with the infrared autoanalyser (HMA) and the conventional laboratory methods (LAB) using Student's t‐test for paired samples
| Component (g per 100 mL) | n | HMA | LAB | Significance |
|---|---|---|---|---|
| Lipids | 32 | 4.29 ± 1.43 | 3.59 ± 1.13 | <0.01 |
| Protein | 36 | 0.59 ± 0.29 | 1.19 ± 0.27 | <0.001 |
| Lactose | 32 | 4.9 ± 0.17 | 6.89 ± 0.91 | <0.001 |
Laboratory methods employed for the determination of lipids, protein and lactose were the Gerber, Bradford and Chloramine‐T techniques, respectively.
After confirming the existence of differences between the methods, we evaluated the existence of a linear relationship between them.
4, 5, 6 represent the scatter plots for fat, protein, and lactose evaluated by both traditional (represented as ‘biochemical’) and autoanalyser methods. Regression analysis for fat (Fig. 4) showed a highly significant correlation coefficient r 2 = 0.8155 (P < 0.01). In addition, regression analysis for proteins and lactose (5, 6) also showed a significant correlation (r 2 = 0.53244; P < 0.01 and r 2 = 0.4088; P < 0.002, respectively).
Figure 4.
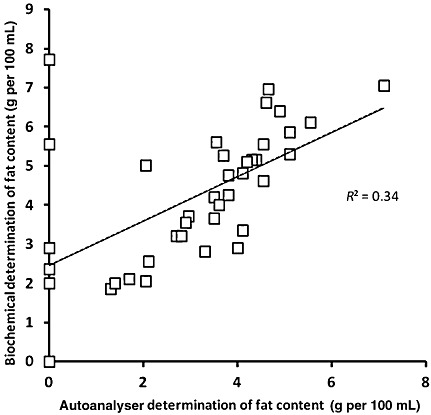
Scatter plot representing results obtained for lipid concentration using both methods (biochemical: Gerber technique and autoanalyser: HMA autoanalyser).
Figure 5.
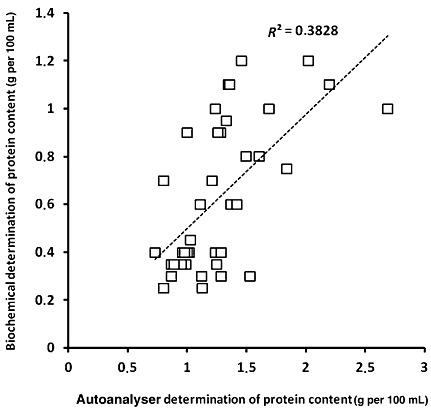
Scatter plot representing results obtained for protein concentration using both methods (biochemical: Bradford technique and autoanalyser: HMA autoanalyser).
Figure 6.
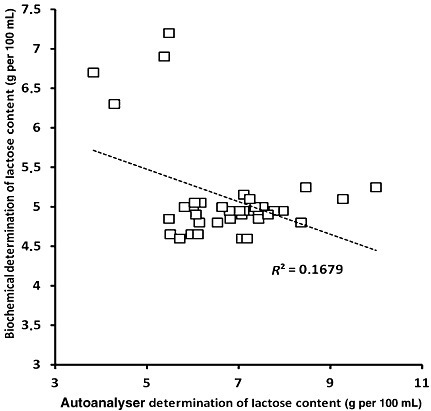
Scatter plot representing results obtained for lactose concentration using both methods (biochemical: Chloramine‐T technique and autoanalyser: HMA autoanalyser).
Discussion
The present study shows the presence of significant differences relative to macronutrient concentrations (fat, protein and lactose) in mature HM between routinely used biochemical assays and the IR HMA (Table 1). Hence, the Gerber method (Kleyn et al. 2001) for quantification of milk fat content consistently showed significantly lower concentrations than the HMA method. Both techniques showed the expected inter‐individual variations inherent to HM, and the concentrations obtained with both procedures were within the ranges published for lipid concentrations in HM (Lucas et al. 1978; Jensen & Clark 1984; Kleyn et al. 2001). Although the differences obtained were statistically significant, a good correlation was observed among the data, suggesting that the differences between the methods could be obviated by readjusting the calibration of the autoanalyser. Other authors have obtained similar results, comparing the autoanalyser with other biochemical methods (Folch and Röse Gottlieb methods, respectively) (Quigley et al. 2007). The protein concentrations obtained with both methods showed important differences on the order of 0.59 g per 100 mL, but with differences as high as 1.23 g per 100 mL in some of the samples. This striking difference is a relevant matter of concern because protein content of HM is a precluding condition to enrich HM. The analysis of these differences showed the Bradford method to yield significantly higher values than the HMA in all the samples analysed. Moreover, the linear correlation between the two techniques was low. As has been shown in the present study, the concentration of protein in HM is generally more stable than the concentration of lipids. In this sense, the SD was low and similar for both methods despite the fact that the mean concentration reported by the Bradford technique doubled that obtained with HMA. It is important to mention that the concentrations obtained with the HMA were far lower than expected, according to the mean protein concentrations in HM documented in the literature (Atkinson et al. 1980), while the Bradford method yielded values within the expected range. Therefore, in the absence of certified samples of HM and considering the analytical parameters obtained with both methods, it seems reasonable to recommend adjustment and calibration of the HMA before use for quantifying proteins. Other authors have conducted similar studies (Quigley et al. 2007), but using a different reference technique (the Kjeldahl method). Their results did not coincide with ours; however, significant differences were recorded with respect to the HMA, the latter technique overestimated the concentration of proteins in HM as opposed to our findings. Similarly as with the other components, lactose concentration in the samples showed significant differences according to whether the HMA or traditional Chloramine‐T technique was used. Hence, the HMA yielded a comparatively lesser concentration in all the analysed samples, with values of close to 5 g per 100 mL in all cases. The HMA therefore appears to underestimate the usually reported lactose concentration in HM (Andersson et al. 1981). In contrast, the conventional laboratory method yielded values similar to those expected for HM – a fact that reinforces its reliability. In addition to lactose, quantification with the Chloramine‐T method may also include other reducing sugars such as oligosaccharides with lactose terminals, which can be present in large and variable amounts in HM (Atkinson et al. 1980). Nevertheless, literature findings agree with our results. Specifically, the HMA recorded lesser lactose concentrations than the reference method; however, in the mentioned studies, the latter did not involve the Chloramine‐T method but an enzyme technique with a different analytical basis (Atkinson et al. 1980).
The advantages offered by the HMA are as follows: (1) requirement of a small sample volume of milk; (2) minimal training requirement; and (3) the HMA is easily transportable and needs only small volume of reactive. These advantages are supported by our findings and suggest that this method can easily be incorporated to HMBs for routine analysis of donor HM or mother's own milk and point‐of‐care analysis in the neonatal units. Using a volume sample <2 mL and in a running time of only 1 min, the HMA was able to determine the concentration of all three major nutrients (lipids, proteins and lactose) and compute the corresponding caloric value. In addition, the use of a single and easy‐to‐handle instrument considerably reduced the consumables needed for routine biochemical determinations and training of the technician. In comparison, the conventional laboratory techniques required a total volume of over 25 mL, with test duration of approximately 150 min and long‐lasting training of the technician in each of the methods.
In the selection of methods for the present study, we initially chose the Röse Gottlieb method recommended by the International Dairy Federation for total fat quantification (http://nmpf.org/washington_watch/labeling/international). However, the preliminary tests conducted in HM often caused formation of emulsions of milk and reagents. To avoid this inconvenience, the Gerber method was found more suitable (Kleyn et al. 2001). In addition, we used the Bradford (Bradford 1976) and Chloramine‐T (Hinton & Macara 1927) techniques for protein and lactose, respectively. A thorough evaluation of these biochemical assays revealed their suitability for analysis of HM. It should be mentioned here that for assessing the linearity of the HMA technique, we eliminated the 1:4 dilution as the sensitivity of the instrument was insufficient to quantify such low concentrations of fat and proteins, while the analysis of lactose in this aliquot lost the linearity observed in the dilutions of greater concentration.
Of relevance, both our results and those of other authors (Menjo et al. 2009; Casadio et al. 2010) have found significant differences between both traditional and HMA methods relative to the macronutrient concentrations. Hence, the Gerber method for quantification of milk fat content consistently showed significantly lower concentrations than the HMA method. Both techniques showed the expected inter‐individual variations inherent to HM, and the concentrations obtained with both procedures were within the ranges published for lipid concentrations in HM (Wojcik et al. 2009; Bauer & Gerss 2011; Moltó‐Puigmartíet al. 2011). Although the differences obtained were statistically significant, a good correlation was observed among the data, suggesting that the differences between the methods could be obviated by readjusting the calibration of the analyser. Other authors have obtained similar results, comparing the analyser with other biochemical methods (Folch and Röse Gottlieb methods, respectively) (Menjo et al. 2009; Casadio et al. 2010). The protein concentrations obtained with both methods showed important differences on the order of 0.59 g per 100 mL, but with differences as high as 1.23 g per 100 mL in some of the samples. This striking difference is a relevant matter of concern as protein content of HM is a precluding condition to enrich HM. The analysis of these differences showed the Bradford method to yield significantly higher values than the HMA autoanalyser in all the samples analysed. Moreover, the linear correlation between the two techniques was low. As has been supported in the present study, the concentration of proteins in HM is generally more stable than the concentration of lipids. In this sense, the SD was low and similar for both methods despite the fact that the mean concentration reported by the Bradford technique doubled that obtained with HMA. It is important to mention that the concentrations obtained with the HMA autoanalyser were far lower than expected, according to the mean protein concentrations in HM documented in the literature (Wojcik et al. 2009), while the Bradford method yielded values within the expected range. Therefore, in the absence of certified samples of HM and considering the analytical parameters obtained with both methods, it seems reasonable to recommend adjustment and calibration of the HMA before its use for quantification of milk protein content. Other authors have conducted similar studies but using a different reference technique (the Bradford method) (Casadio et al. 2010). Their results did not coincide with ours; however, although significant differences were recorded with respect to the HMA autoanalyser, the latter technique overestimated the concentration of proteins in HM as opposed to our findings (Casadio et al. 2010). Lactose concentration showed significant differences according to whether the HMA or traditional Chloramine‐T technique was used. Hence, the HMA autoanalyser yielded comparatively lesser concentration in all the analysed samples with values close to 5 g per 100 mL in all cases. The HMA therefore appears to underestimate the reported lactose concentration in HM (Kunz et al. 2000). In contrast, the conventional laboratory method yielded values coincident with those expected for HM – a fact that reinforces its reliability. In addition to lactose, quantification with the Chloramine‐T method may also include other reducing sugars such as oligosaccharides with lactose terminals, which can be present in large and variable amounts in HM (Kunz et al. 2000).
The principal strength of our study relies on the precision, linearity and reproducibility of the biochemical assays elected for determining milk macronutrients, which allowed critical comparison with results obtained with the IR HMA, rendering conclusions regarding the reliability of the latter valid. Limitations of the study include small sample size calculation and some missing observations that occurred due to lack of a sufficient milk volume to complete all the determinations in every sample.
We conclude that the use of autoanalysers such as the IR HMA appears to be more suitable for routine use in HMBs and neonatology units or milk banks. However, it should be underscored that routine calibration with a highly reliable standard is mandatory especially when used for fortification purposes in preterm infants. Although theoretical influence of such practice is apparently positive, further studies evaluating the benefits of incorporating routine milk analysis from milk banks and own mother's to preterm feeding protocols on nutritional outcome are needed.
Source of funding
This study was supported with grants RD008/0072/0022 from the Instituto Carlos III (Spanish Ministry of Science and Innovation) to MV and GE006‐10 from the Consellería de Sanitat i Consum (Autonomous Community of Valencia) to MG.
Conflicts of interest
The authors declare that they have no conflicts of interest.
Contributions
DS co‐designed the study, analysed the data and performed the initial draft of the manuscript; MF performed the biochemical analysis and performed the statistical analysis; MG performed the MIR spectroscopy analysis and helped with the design of the study and helped with the initial draft of the manuscript; ET recruited milk donors in the NICU, collected milk samples and performed MIR spectroscopy analysis; MV designed the study, reviewed the statistical analysis and figures, and wrote the final draft of the manuscript.
Acknowledgements
We would like to express our gratitude to Amparo Ramón, RN, and Rosario Sirvent, RN, for their contribution in obtaining and processing the milk sample.
References
- American Academy of Pediatrics ( 2012. ) Policy Statement. Breastfeeding and the use of human milk . Pediatrics 129 , e627 – e641 . [Google Scholar]
- Andersson G.H. , Atkinson D.S. & Bryan M.H. ( 1981. ) Energy and macronutrient content in human milk during early lactation from mothers living birth prematurely and a term . American Journal of Clinical Nutrition 34 , 258 – 265 . [DOI] [PubMed] [Google Scholar]
- Arnold L.D. ( 2002. ) The cost‐effectiveness of using banked donor milk in the neonatal intensive care unit: prevention of necrotizing enterocolitis . Journal of Human Lactation 18 , 172 – 177 . [DOI] [PubMed] [Google Scholar]
- Arslanoglu S. , Moro G. , Ziegler E.E. & the WAPM Working Group on Nutrition ( 2010. ) Optimization of human milk fortification for preterm infants: new concepts and recommendations . Journal of Perinatal Medicine 38 , 233 – 238 . [DOI] [PubMed] [Google Scholar]
- Atkinson S.A. , Bryan M.H. & Anderson G.H. ( 1980. ) Human milk: comparison of the nitrogen composition in milk from mothers of premature and full‐term infants . American Journal of Clinical Nutrition 33 , 811 – 815 . [DOI] [PubMed] [Google Scholar]
- Bauer J. & Gerss J. ( 2011. ) Longitudinal analysis of macronutrients and minerals in human milk produced by mothers of preterm infants . Clinical Nutrition 30 , 215 – 220 . [DOI] [PubMed] [Google Scholar]
- Bland J.M. & Altman D.G. ( 1986. ) Statistical methods for assessing agreement between two methods of clinical measurement . Lancet 1 , 307 – 310 . [PubMed] [Google Scholar]
- Bokor S. , Koletzko B. & Decsi T. ( 2007. ) Systematic review of fatty acid composition of human milk from mothers of preterm compared to full‐term infants . Advances Nutrition and Metabolism 51 , 550 – 556 . [DOI] [PubMed] [Google Scholar]
- Bradford M. ( 1976. ) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein‐dye binding . Analytical Biochemestry 72 , 248 – 254 . [DOI] [PubMed] [Google Scholar]
- Casadio Y.S. , Williams T.M. , Lai C.T. , Olsson S.E. , Hepworth A.R. & Hartman P.E. ( 2010. ) Evaluation of a mid‐infrared analyzer for the determination of the macronutrient composition of human milk . Journal of Human Lactation 26 , 376 – 383 . [DOI] [PubMed] [Google Scholar]
- Corvaglia L. , Battistini B. , Paoletti V. , Aceti A. , Capretti M.G. & Faldella G. ( 2008. ) Near‐Infrared reflectance analysis to evaluate the nitrogen and fat content of human milk in neonatal intensive care units . Archives Diseases of Children Fetal Neonatal Edition 93 , F372 – F375 . [DOI] [PubMed] [Google Scholar]
- European Milk Bank Association ( 2012. ) Available at: http://www.europeanmilkbanking.com (Accessed 1 April 2012 ).
- Henderson G. , Anthony M.Y. & McGuire W. ( 2007. ) Formula milk versus maternal breast milk for feeding preterm or low birth weight infants . Cochrane Database of Systematic Reviews ( 4 ), CD002972 . [DOI] [PubMed] [Google Scholar]
- Hinton C.L. & Macara T. ( 1927. ) The determination of aldose sugars by means of chloramine‐T, with special reference to the analysis of milk products . Analyst 52 , 668 – 688 . [Google Scholar]
- Italian Association of Human Milk Bank ( 2010. ) Guidelines for the establishment and operation of a donor human milk bank . Journal of Maternal Fetal and Neonatal Medicine 23 , 1 – 20 . [DOI] [PubMed] [Google Scholar]
- Jensen R.G. & Clark R.M. ( 1984. ) Methods of lipids analysis . Journal of Paediatric Gastroenterology and Nutrition 3 , 296 – 299 . [DOI] [PubMed] [Google Scholar]
- Kleyn D.H. , Lynch J.M. , Barbano D.M. , Bloom M.J. & Mitchel M.W. ( 2001. ) Determination of fat in raw and processed milks by the Gerber method: collaborative study . Journal of the Association of Analytical Communities 84 , 1499 – 1508 . [PubMed] [Google Scholar]
- Krouwer J.S. ( 2008. ) Why Bland‐Altman plots should use X, not (Y + X)/2 when X is a reference method . Statistics in Medicine 27 , 778 – 780 . [DOI] [PubMed] [Google Scholar]
- Kunz C. , Rudloff S. , Baier W. , Klein N. & Strobel S. ( 2000. ) Oligosaccharides in human milk: structural, functional and metabolic aspects . Annual Review of Nutrition 20 , 699 – 722 . [DOI] [PubMed] [Google Scholar]
- Lucas A. , Gibas J.A.H. , Lyster R.L. & Baum J.D. ( 1978. ) Creamatocrit: simple clinical technique for estimating fat concentration and energy value of human milk . British Medical Journal 1 , 1018 – 1020 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menjo A. , Mizuno K. , Murase M.Y. , Nishida Y. , Taki M. , Itabashi K. et al . ( 2009. ) Bedside analysis of human milk for adjustable nutrition strategy . Acta Paediatrica 98 , 380 – 384 . [DOI] [PubMed] [Google Scholar]
- Michaelsen K.F. , Pedersen S.B. , Skafte L. , Jaeger P. & Peitersen B. ( 1988. ) Infrared analysis for determining macronutrients in human milk . Journal of Pediatric Gastroenterology and Nutrition 2 , 229 – 235 . [DOI] [PubMed] [Google Scholar]
- Moltó‐Puigmartí C. , Castellote A.I. , Carbonell‐Estrany X. & López‐Sabater M.C. ( 2011. ) Differences in fat content and fatty acid proportions among colostrum, transitional, and mature milk from women delivering very preterm, preterm, and term infants . Clinical Nutrition 30 , 116 – 123 . [DOI] [PubMed] [Google Scholar]
- Quigley M. , Henderson G. , Anthony M.Y. & McGuire W. ( 2007. ) Formula milk versus donor breast milk for feeding preterm or low birth weight infants . Cochrane Database of Systematic Reviews ( 4 ), CD002971. DOI: 10.1002/14651858.CD002971.pub2 . [DOI] [PubMed] [Google Scholar]
- Sauer C.W. & Kim J.H. ( 2001. ) Human milk macronutrient analysis using point‐of‐care near infrared spectrophotometry . Journal of Perinatology 31 , 339 – 343 . [DOI] [PubMed] [Google Scholar]
- Silvestre D. , Martínez C. , Lagarda M.J. , Brines J. , Farré R. & Clemente G. ( 2001. ) Copper, iron and zinc contents in human milk during the first three months of lactation . Biology Trace Elements Research 80 , 1 – 11 . [DOI] [PubMed] [Google Scholar]
- Wojcik K.Y. , Rechtman D.J. , Lee M.L. , Montoya A. & Medo E.T. ( 2009. ) Macronutrient analysis of a nationwide sample of donor breast milk . Journal of the American Dietary Association 109 , 137 – 140 . [DOI] [PubMed] [Google Scholar]
- World Health Organization (WHO) ( 2011. ) Available at: http://www.who.int/topics/infantfeeding_recomendation/en/index.html (Accessed 12 December 2011 ).