

Abstract
Introduction
The presence or absence of cirrhosis in patients with chronic hepatitis C virus (HCV) infection influences the type and duration of antiviral therapy. Non-invasive markers, like serum aspartate aminotransferase (AST) to platelet ratio index (APRI), may help identify appropriate HCV treatment-naive patients for 8-week treatment with the pangenotypic regimen of glecaprevir/pibrentasvir.
Methods
This single-arm, open-label, international, prospective study (NCT03212521) evaluated the efficacy and safety of 8-week glecaprevir/pibrentasvir regimen in HCV treatment-naïve adults with chronic HCV genotypes 1–6 infection, APRI ≤ 1, and no prior evidence of cirrhosis. The primary and secondary outcomes were sustained virologic response at 12 weeks post-treatment (SVR12) by modified intent-to-treat (mITT) and intent-to-treat (ITT) analyses, respectively. Additional endpoints included virologic failures, treatment adherence, and genotype-specific SVR12 rates.
Results
Among the 230 patients enrolled, most were less than 65 years old (90%); 37% and 43% had a history of injection drug use or psychiatric disorders, respectively. SVR12 rates were 100% (222/222; 95% CI 98.3–100%) and 96.5% (222/230; 95% CI 94.2–98.9%) by mITT and ITT analyses, respectively. There were no virologic failures. ITT SVR12 rates were greater than 94% for all HCV genotypes. In patients with available data, treatment adherence was 99% (202/204). There were no grade 3 or higher laboratory abnormalities in alanine aminotransferase (ALT), aspartate aminotransferase (AST), and total bilirubin, and low rates of serious adverse events (2%).
Conclusions
Glecaprevir/pibrentasvir was highly efficacious and well tolerated in HCV treatment-naïve patients with APRI ≤ 1 and no prior evidence of cirrhosis.
Trial Registration
ClinicalTrials.gov number, NCT03212521.
Funding
AbbVie.
Plain Language Summary
Plain language summary available for this article.
Electronic supplementary material
The online version of this article (10.1007/s12325-019-01123-0) contains supplementary material, which is available to authorized users.
Keywords: Chronic hepatitis C, Direct acting antiviral, Glecaprevir/pibrentasvir, Infectious diseases, Simplification
Key Summary Points
| Why carry out this study? |
| The presence or absence of cirrhosis in patients with chronic hepatitis C virus (HCV) infection influences the type and duration of antiviral therapy. |
| Non-invasive markers, like serum aspartate aminotransferase (AST) to platelet ratio index (APRI), may help identify appropriate HCV treatment-naive patients for 8-week treatment with the pangenotypic regimen of glecaprevir/pibrentasvir in countries where 8-week G/P treatment in patients with compensated cirrhosis may take longer to get approved. |
| What was learned from the study? |
| Glecaprevir/pibrentasvir was highly efficacious and well tolerated in HCV treatment-naïve patients with APRI ≤ 1 and no prior evidence of cirrhosis. |
| Use of APRI may help HCV elimination efforts by simplifying care pathways and treatment scale-up in community-based settings with glecaprevir/pibrentasvir. |
Plain Language Summary
Elimination of chronic hepatitis C virus (HCV) infection is now achievable owing to powerful drug combinations, like glecaprevir/pibrentasvir (G/P), that get rid of all major types of the virus in patients; however, such elimination efforts led by the World Health Organization (WHO) depend on the availability of low-cost and scalable testing as well as access to the drugs. In order to determine if a simple and low-cost blood test called aspartate aminotransferase to platelet ratio index (APRI) can be used to select appropriate patients for shorter duration treatment, this study evaluated whether 8-week G/P is safe and eliminates the virus in patients with an APRI less than a predetermined threshold of 1, no prior HCV treatment experience, and no evidence of liver scarring or damage. We found that 8-week G/P in these selected patients was safe and eliminated the virus in over 96% of patients with no one failing to respond to treatment or losing their initial response; thus, this predetermined APRI threshold could be used in clinical practice as a simplified pretreatment assessment to select patients with chronic HCV, no prior HCV treatment experience, and no evidence of liver scarring or damage for the 8-week G/P regimen.
Introduction
Chronic hepatitis C virus (HCV) infection afflicts 71 million people worldwide [1]. Recently, the population with chronic HCV has shifted from an older population with cirrhosis to an increasingly younger HCV treatment-naïve population without cirrhosis particularly as a result of its spread among people who use drugs (PWUDs) [1, 2]. When left untreated, patients with chronic HCV are at increased risk for liver cirrhosis, and liver-related and all-cause mortality [3, 4]. In the past decade, novel direct acting antivirals (DAAs) have revolutionized the treatment of chronic HCV infection by yielding rates of over 90% sustained virologic response (SVR) in both treatment-naïve and experienced patients [5]. Achievement of SVR decreases long-term health risks associated with chronic HCV infection and improves patient quality of life, thereby delivering a cost-effective treatment for patients with chronic HCV infection [6–9]. However, despite the availability and benefit of DAAs, linkage to care following HCV diagnosis continues to be a gap in the HCV care cascade due, in part, to the need for specialists to identify the presence or absence of cirrhosis using either liver biopsy or transient elastography prior to initiating treatment [10–12].
Achievement of the World Health Organization (WHO) global target for HCV elimination by 2030 depends on access to low-cost and scalable testing as well as access to effective DAA treatment [1, 10]. Current treatment guidelines in the USA and Europe recommend both genotype (GT) and fibrosis or cirrhosis testing in order to determine the most suitable DAA regimen and treatment duration [11, 12]. Although liver biopsy and transient elastography have primarily been utilized for fibrosis or cirrhosis testing, non-invasive markers, including the aspartate aminotransferase to platelet ratio index (APRI), may be used to assess for cirrhosis prior to HCV treatment according to current WHO, European, Australian, and Canadian guidelines [11, 13–15]. APRI is determined from a blood test, thereby providing a low-cost, widely available, non-invasive method that has high negative predictive value (94%) for cirrhosis at an APRI cutoff of 1.0 compared to liver biopsy [16, 17].
Glecaprevir (a potent pangenotypic NS3/4A protease inhibitor identified by AbbVie and Enanta) plus pibrentasvir (a potent pangenotypic NS5A inhibitor), co-formulated as G/P, is an efficacious and safe DAA regimen approved for the treatment of patients with chronic HCV GT1–6 infection and compensated liver disease with or without cirrhosis, including those co-infected with HIV or with severe renal impairment [18–20]. G/P is approved for an 8-week treatment duration in patients with chronic HCV GT1–6 infection and without cirrhosis on the basis of data from its registrational trials, which demonstrated comparably high rates (at least 95%) of sustained virologic response at 12 weeks post-treatment (SVR12) with either 8 or 12 weeks of treatment [20, 21]. There was no significant difference observed in SVR12 rates between the 8- and 12-week durations in patients without cirrhosis regardless of baseline patient or disease characteristics analyzed, including patients who were HCV treatment-naïve and those with a pretreatment APRI < 1 or ≥ 1 [20]. Real-world evidence has been consistent with the G/P registrational trials, demonstrating that 8-week G/P is highly effective and well tolerated [22, 23]. However, a prospective study is necessary to determine if the use of non-invasive biomarker APRI can simplify the selection of patients for 8-week G/P treatment.
In the current study, we aim to determine whether a screening APRI ≤ 1 can be used to select appropriate patients for an 8-week G/P treatment duration by evaluating the efficacy and safety of 8-week G/P in HCV treatment-naïve patients with a screening APRI ≤ 1. Although 8-week treatment with G/P regimen has recently been approved for the treatment of treatment-naïve patients with GT1, 2, 4, 5, and 6 infections and with compensated cirrhosis in the USA and EU [24], we believe that APRI screening is useful in countries where 8-week G/P treatment in patients with compensated cirrhosis may take longer to get approved.
Methods
Study Design
This multicenter, open-label, single-arm, prospective phase III trial assessed the efficacy and safety of 8-week G/P regimen in treatment-naïve patients with APRI ≤ 1 at screening. Patients received three co-formulated tablets containing 100 mg glecaprevir and 40 mg pibrentasvir once daily with food for 8 weeks (total dose of 300 mg of glecaprevir and 120 mg pibrentasvir). Patients were enrolled in ten countries (Bulgaria, Canada, France, Germany, Poland, Puerto Rico, Russia, Spain, UK, and USA) across 43 sites. All patients provided written informed consent prior to screening. All authors had access to the study data, and reviewed and approved the final manuscript.
Patients
Patients were eligible if they were male or female, at least 18 years old at screening, and were positive for anti-HCV with plasma HCV RNA ≥ 1000 IU/mL for at least 6 months prior to and at screening. Women were eligible if they were not pregnant or breastfeeding, or were sterile or practicing one method of birth control. Patients were selected for the study if they had an APRI score ≤ 1 at screening and were HCV treatment-naïve. Patients with HCV GT1, 2, 3, 4, 5, or 6 infection were eligible for the study, including patients with mixed or indeterminate GT. Patients with human immunodeficiency virus type 1 (HIV-1) were eligible for the study if they were HIV treatment-naïve or on a stable, qualifying antiretroviral therapy (ART) regimen. Patients with drug or alcohol misuse could enroll unless they were considered an unsuitable candidate for the study by the site investigator as a result of recent misuse (within 6 months prior to study drug administration).
Patients were eligible unless they had evidence of cirrhosis in previous or current medical assessments; however, liver biopsy or transient elastography was not required to be performed for study eligibility. Patients were excluded if they met the following pre-defined laboratory values: platelet count < 150,000 cells/mm3, alanine aminotransferase (ALT) > 10× upper limit of normal (ULN), aspartate aminotransferase (AST) > 10× ULN, direct bilirubin > ULN, and/or albumin < lower limit of normal. Patients were required to be HBsAg and anti-HBc negative, or HBV DNA < lower level of quantification (LLOQ) with an isolated positive anti-HBc. Patients who had chronic kidney disease stage 4 or 5 (calculated creatinine clearance < 30 mL/min), previous organ transplantation, or a history of hepatocellular carcinoma (HCC) were excluded. Patients were required to discontinue prohibited medications or supplements at least 14 days or ten half-lives, whichever was longer, prior to the first G/P dose. Complete patient eligibility criteria are provided in the Supplementary Appendix.
Assessments
APRI was assessed at screening based on concurrent measures for AST and platelet count. The following formula was used to calculate APRI:
Real-time reverse transcriptase-polymerase chain reaction (RT-PCR) was used to quantify plasma HCV RNA for both baseline viral load and SVR12 assessments. HCV genotype was determined using the Versant® HCV Genotype Inno LiPA Assay, Version 2.0 or higher (Siemens Healthcare Diagnostics, Tarrytown, NY), and confirmed by phylogenetic analysis of viral sequences. SVR12 was assessed as HCV RNA < LLOQ 12 weeks after last G/P dose for all patients receiving at least one G/P dose in both intent-to-treat (ITT) and a modified ITT (mITT) that excluded patients not achieving SVR12 for reasons other than virologic failure (e.g., premature G/P discontinuation or missing HCV RNA data 12 weeks after last G/P dose). Treatment adherence was assessed comparing pills taken with expected pill count.
Safety was evaluated by physical examination, vital signs, electrocardiogram, clinical laboratory testing, and adverse events (AEs) monitoring throughout the duration of the study. All AEs were coded using the Medical Dictionary for Regulatory Activities (MedDRA) and were assessed for their relationship to G/P by study investigators.
Endpoints
Efficacy of 8-week G/P treatment was assessed using a fixed sequence testing procedure. The primary endpoint was the percentage of patients with SVR12 in the mITT population. If this primary endpoint was met, the secondary endpoint about the percentage of patients with SVR12 in the ITT population would be evaluated in the sequential testing. Non-sequential secondary efficacy endpoints were the percentage of patients with on-treatment virologic failure or post-treatment relapse in the ITT population. Additional analysis assessed treatment adherence, defined as use of at least 80% and at most 120% of tablets taken relative to the expected total number of tablets to be taken. Safety was evaluated by the number and percentage of patients with treatment-emergent AEs and laboratory abnormalities, and through characterization of reported AEs.
Statistical Analysis
The study enrolled 230 patients with chronic HCV GT1–6 infection, including 35 with GT3 infection, in order to achieve 90% and 83% power to demonstrate efficacy compared with pre-specified thresholds for the mITT and ITT populations, respectively. The pre-specified thresholds were set on the basis of historical SVR12 rates from the G/P registrational trials [21]. The number and percentage of patients in both mITT and ITT population achieving SVR12 were summarized with two-sided 95% confidence intervals (CI) calculated using the normal approximation to the binomial distribution. If the number of SVR12 non-responders was less than five, then Wilson’s score method would be used to calculate the confidence interval. The primary efficacy endpoint in the mITT population was met if the lower bound of the 95% CI was greater than the pre-specified threshold of 92.4% based on the historical rate observed in the G/P registrational studies in treatment-naïve patients without cirrhosis (98.4%) minus 6%. The secondary efficacy endpoint of SVR12 in the ITT population was met if the lower bound of the 95% CI was greater than 91.4% based on the mITT threshold minus an expected 1% rate of non-virologic failure.
Compliance with Ethics Guidelines
The trial was conducted in accordance with Good Clinical Practice and the Declaration of Helsinki, and was approved at all sites by their independent ethics committee or institutional review board prior to enrollment. The master ethics committee of this study is the Quorum Institutional Review Board. A complete list of institutional ethics committees or institutional review boards is provided in Supplementary Table S3.
Results
Baseline Characteristics
Between August 7, 2017 and August 13, 2018, 230 treatment-naïve patients with chronic HCV genotypes 1–6 infection and APRI ≤ 1 were enrolled (Fig. 1). Table 1 summarizes the complete baseline demographics and disease characteristics for patients in the mITT and ITT populations. Overall, in the ITT population, patients (n, %) were predominantly Caucasian (207, 90%) and less than 65 years old (207, 90%), and most patients had GT1 infection (151, 66%) and a screening APRI ≤ 0.5 (140, 61%). There were 87 (38%) patients with a history of injection drug use (most of which were from more than 12 months ago) and 99 (43%) patients with a history of a psychiatric disorder, including 45 (20%) with a history of depression or bipolar disorder. Although all 230 patients were HCV treatment-naïve, 31% (71/230) of patients presented with a key baseline NS5A polymorphism. At baseline, a blinded FibroTest assessment yielded a median value of 0.26 (range 0.02–0.87).
Fig. 1.
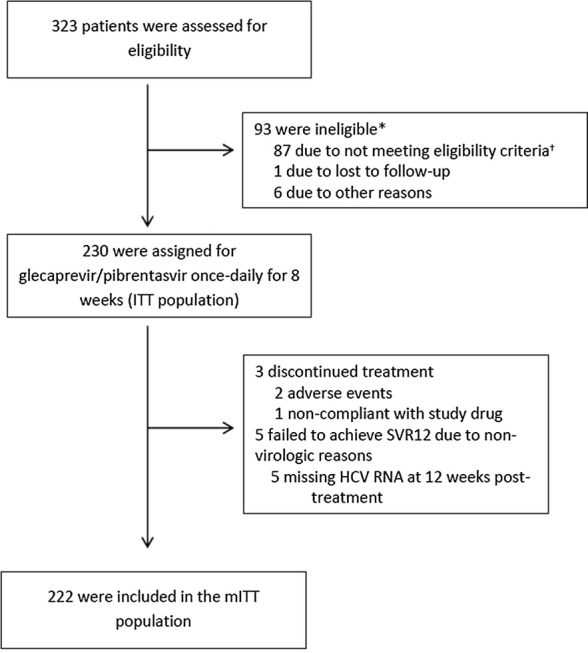
Trial profile. *Patients who were screening failures are counted under each reason given for screen fail; therefore, the sum of the counts given for the reasons for screen fail may be greater than the overall number of screen failures. †Majority of patients who did not meet the eligibility criteria was not due to failure to meet APRI score of ≤ 1 at the time of screening
Table 1.
Baseline demographics and patient characteristics
| Characteristic | mITT population N = 222 | ITT population N = 230 |
|---|---|---|
| Male, n (%) | 111 (50) | 117 (51) |
| Race, n (%) | ||
| White | 202 (91) | 207 (90) |
| Black or African American | 10 (5) | 13 (6) |
| Asian | 10 (5) | 10 (4) |
| Hispanic or Latino ethnic origin, n (%) | 23 (10) | 25 (11) |
| Age, median (range), years | 48 (19–82) | 48 (19–82) |
| Age ≥ 65 years old, n (%) | 23 (10) | 23 (10) |
| BMI, median (range), kg/m2 | 25.3 (16.9–55.6) | 25.2 (16.9–55.6) |
| Baseline HCV RNA level, median (range), log10 IU/mL | 6.3 (2.2–7.7) | 6.3 (2.2–7.7) |
| Baseline HCV RNA ≥ 1 million IU/mL, n (%) | 141 (64) | 146 (63) |
| HCV genotype, n (%) | ||
| GT1 | 145 (65) | 151 (66) |
| GT1a | 77 (35) | 82 (36) |
| GT1b | 67 (30) | 68 (30) |
| GT1i | 1 (< 1) | 1 (< 1) |
| GT2 | 33 (15) | 33 (14) |
| GT3 | 33 (15) | 35 (15) |
| GT4 | 9 (4) | 9 (4) |
| GT6 | 2 (< 1) | 2 (< 1) |
| Key baseline polymorphisms, n (%)a | ||
| Any NS3 polymorphism | 1 (< 1) | 1 (< 1) |
| Any NS5A polymorphism | 68 (31) | 71 (31) |
| Screening APRI, median (range) | 0.41 (0.13–1.0) | 0.41 (0.13–1.0) |
| Screening APRI, n (%) | ||
| ≤ 0.5 | 136 (61) | 140 (61) |
| 0.5–0.7 | 54 (24) | 57 (25) |
| 0.71–1.0 | 32 (14) | 33 (14) |
| Blinded fibrotest, median (range)b | 0.26 (0.02–0.87) | 0.26 (0.02–0.87) |
| FIB-4, median (range) | 1.04 (0.26–4.09) | 1.05 (0.26–4.09) |
| Platelet count, median (range), count/109/L | 243 (126–462) | 243 (126–483) |
| HIV co-infection, n (%)c | 8 (4) | 10 (4) |
| CD4+ T cell countd, median (range), cells/mm3 | 762 (444–1199) | 692 (444–1199) |
| History of injection drug use, n (%) | 83 (37) | 87 (38) |
| Within the last 12 months | 5 (2) | 6 (3) |
| More than 12 months ago | 78 (35) | 81 (35) |
| On stable opiate substitution, n (%) | 19 (9) | 19 (8) |
| History of diabetes, n (%) | 7 (3) | 9 (4) |
| History of depression or bipolar disorder | 43 (19) | 45 (20) |
BMI body mass index, HCV hepatitis C virus, GT genotype, APRI aspartate aminotransferase to platelet ratio, HIV human immunodeficiency virus
aIncludes any baseline resistance-associated variants in NS3 (155, 156, and 168) or NS5A (24, 28, 30, 31, 58, 92, and 93) at a 15% detection threshold. No patients had both an NS3 and an NS5A key resistance-associated variant
bPerformed at baseline, blinded to the investigators and therefore not used for patient eligibility. A value > 0.80 has a high positive predictive value (PPV) for cirrhosis [16]
cAll patients with HIV co-infection were antiretroviral therapy-naïve
dOnly assessed in the 10 HIV/HCV co-infected patients
Efficacy Outcomes
For the primary efficacy endpoint, overall SVR12 rate by mITT analysis was 100% (222/222; 95% CI 98.3–100%) with no patients experiencing virologic failure (Fig. 2). The primary efficacy endpoint for the study was met since the lower bound of the 95% CI (98.3%) was greater than the pre-specified threshold of 92.4%. All patients with baseline NS3 or NS5A polymorphisms achieved SVR12 (70/70; 100%).
Fig. 2.

Efficacy of 8-week G/P regimen in HCV treatment-naïve patients with APRI ≤ 1. G/P efficacy, defined as SVR12, is reported overall using modified intent-to-treat (mITT; blue) and intent-to-treat (ITT; green) analyses. Bar graphs show mean with 95% confidence intervals and include reasons for non-response. Dotted lines indicate threshold above which lower bound of mITT and ITT analysis must be greater than in order to meet primary and secondary endpoint, respectively. *Includes two patients who prematurely discontinued because of a serious AE (see Table 2). †All five patients missing SVR12 data had no detectable HCV RNA at the end of treatment
For the sequential secondary efficacy endpoint, overall SVR12 rate in the ITT population was 96.5% (222/230, 95% CI 94.2–98.9%). The secondary efficacy endpoint was met since the lower bound of the 95% CI (94.2%) was greater than the threshold of 91.4%. Eight (3%) patients did not achieve SVR12 because of non-virologic failure reasons, specifically three (1%) due to premature G/P discontinuations and five (2%) due to missing HCV RNA data 12 weeks after last dose of G/P. Overall, four (2%) patients prematurely discontinued G/P; one achieved SVR12 despite discontinuing G/P treatment at day 12 after becoming pregnant during treatment. Among the patients failing to achieve SVR12, two patients discontinued because of adverse events at days 8 and 15, respectively, while one patient discontinued at day 29 because of non-compliance. All five patients missing SVR12 data had no detectable HCV RNA at their last treatment visit (four at post-treatment week 4 and one at end of treatment).
Additional endpoints included adherence to G/P treatment and SVR12 by HCV genotype. Among all patients with available data at all treatment visits, 99% (202/204) of patients were adherent to treatment. One patient who was non-adherent achieved SVR12, while the other patient did not achieve SVR12 after prematurely discontinuing G/P at day 29 because of non-adherence as mentioned above. SVR12 rates by HCV genotype are reported in Fig. 3 using both mITT and ITT analyses. High SVR12 rates were observed in all HCV genotypes.
Fig. 3.

Efficacy by genotype for 8-week G/P regimen in HCV treatment-naïve patients with APRI ≤ 1. G/P efficacy, defined as SVR12, is reported by HCV genotype using modified intent-to-treat (mITT; blue) and intent-to-treat (ITT; green) analyses. Bar graphs show mean with 95% confidence intervals. *One GT1 patient with subtype GT1i achieved SVR12
Safety Outcomes
Overall, 124/230 (54%) patients experienced an AE, of which eight (3%) patients had a grade 3 or higher AE (Table 2). The most common AEs occurring in at least 5% of patients were headache (13%) and fatigue (7%). Among the four (2%) patients who experienced a serious AE (listed in Supplementary Table 1), two (1%) patients experienced serious AEs of angioedema that led to premature G/P discontinuation on days 8 and 15, respectively. After G/P was discontinued, both cases of angioedema resolved within 7 and 3 days, respectively. Both patients were Black or African American, former drug users, taking an angiotensin-converting enzyme (ACE) inhibitor (lisinopril), and had HIV co-infection (see Supplementary Table 2 for more information).
Table 2.
Adverse events and laboratory abnormalities
| Event, n (%) | Eight-week G/P treatment, N = 230 |
|---|---|
| Any AE | 124 (54) |
| Grade ≥ 3 AE | 8 (3) |
| Serious AE | 4 (2) |
| DAA-relateda serious AE | 2 (< 1)b |
| AE leading to premature G/P discontinuation | 2 (< 1)b |
| AEs occurring in ≥ 5% of all patients by preferred term | |
| Headache | 29 (13) |
| Fatigue | 17 (7) |
| Laboratory abnormalities (grade ≥ 3) | |
| ALT > 5 × ULNc | 0 |
| AST > 5 × ULN | 0 |
| Total bilirubin > 3 × ULN | 0 |
| Deaths | 0 |
G/P glecaprevir/pibrentasvir, AE adverse event, DAA direct acting antiviral, ALT alanine aminotransferase, ULN upper limit of normal, AST aspartate aminotransferase
aAs assessed by the investigator
bTwo patients experienced a serious AE of angioedema leading to premature G/P discontinuation on days 8 and 15, respectively
cPost-nadir increase in grade to grade ≥ 3
There were no laboratory abnormalities in ALT, AST, or total bilirubin. There were no events consistent with hepatic decompensation, hepatic failure, or drug-induced liver injury.
Discussion
Using APRI ≤ 1 as a selection tool for an 8-week G/P regimen yielded high SVR12 rates and no virologic failures among treatment-naïve patients with chronic HCV GT1–6 infection and no prior evidence of cirrhosis. This finding suggests that simplification of pretreatment testing is feasible specifically amongst the growing population of patients with chronic HCV infection that are being evaluated and treated in the community-based setting, many of whom are younger, HCV treatment-naïve, and have less severe liver disease [1, 2]. In this emerging population that includes high proportions of PWUDs and patients with psychiatric disorders, treatment adherence is a concern that has persisted despite clinical trial data demonstrating high adherence [25, 26]. Similar to previous findings, 99% of patients in our study were adherent despite the inclusion of patients with histories of injection drug use and psychiatric disorders [25, 27, 28]. Overall, the current data are consistent with previous findings from the G/P registrational trials since efficacy remained high regardless of HCV genotype and presence of baseline NS3 or NS5A polymorphisms [20].
The 8-week G/P regimen was safe and well tolerated in these HCV treatment-naïve patients with APRI ≤ 1 and no prior evidence of cirrhosis, consistent with previous data reported with G/P. The most common AEs were headache and fatigue, occurring in a comparably low proportion of the patient population [19, 20]. Rates of AEs leading to premature discontinuation (1%) and serious AEs (2%) were also similarly low in this study compared with prior integrated analysis of G/P safety [19, 20]. While there was only one non-serious case of angioedema among the 2369 patients within the G/P registrational trials, two serious cases of angioedema were reported in this study, both leading to premature G/P discontinuation. Both cases, however, were attributed to concomitant use of an ACE inhibitor (lisinopril). While there is no clinically significant pharmacokinetic interaction between G/P and lisinopril, there is a clear link between ACE inhibitors, like lisinopril, and angioedema, especially among African Americans [21, 29]. Consistent with the low rates of laboratory abnormalities observed in the registrational trials, no grade 3 or higher laboratory abnormalities in ALT, AST, or total bilirubin were observed [19]. Overall, the favorable safety profile of the 8-week G/P regimen in HCV treatment-naïve patients with APRI ≤ 1 and no prior evidence of cirrhosis demonstrated in this study is consistent with safety data from the registrational trials and post-marketing real-world evidence, supporting the safety of the 8-week regimen in this patient population [20, 22, 23].
Since 8 weeks of this pangenotypic DAA regimen was both efficacious and safe in HCV treatment-naïve patients with APRI ≤ 1 and no prior evidence of cirrhosis, this study suggests that pretreatment testing can be further simplified when using a pangenotypic therapy such as G/P. First, as recommended by the European Association for the Study of the Liver (EASL) and WHO guidelines, genotyping is not necessary when using G/P in all treatment-naïve patients owing to its high efficacy across all genotypes [11, 13]. Second, on the basis of our findings, the well-studied, widely available, low-cost blood test for APRI can be used to determine G/P treatment duration, thereby removing the need for a specialist to perform more invasive and costly screening tests for cirrhosis prior to treatment initiation [16]. Using this simplified screening approach, treatment-naive patients can rapidly initiate treatment with G/P in community-based settings by triaging to primary care providers, while more invasive cirrhosis testing and follow-up in more well-resourced or specialized settings can be used as a second-line test in HCV treatment-naïve patients with APRI > 1. This approach could reduce the need for liver biopsy or transient elastography especially given the growing population of younger treatment-naïve patients with chronic HCV infection that are less likely to be cirrhotic. Patients with prior HCV treatment experience will still require both genotyping and more comprehensive cirrhosis testing to determine G/P treatment duration. Thus, 8-week G/P regimen for these treatment-naïve patients with APRI ≤ 1 and no prior evidence of cirrhosis provides a simplified and shortened treatment program which may improve health benefits and save costs for healthcare systems [30]. Further simplification may be possible on the basis of preliminary results from EXPEDITION-8 that show high SVR12 rates with 8-week G/P treatment in patients with chronic HCV GT1, 2, 4, 5, or 6 infection and compensated cirrhosis; however, 8-week G/P treatment is currently not a recommended regimen for patients with prior evidence of cirrhosis [31].
There are limitations to this study inherent to its design. This was a single-arm, open-label trial without a placebo or active control; however, the use of objective measures for efficacy (SVR12) and the comparison with a pre-specified threshold considering historical reference SVR12 rates from G/P registrational trials mitigates this concern. Given that these data are from a controlled clinical trial, further real-world data using this approach for simplification of pretreatment testing is necessary to validate its use in clinical practice.
Conclusion
These data support the use of APRI ≤ 1 in clinical practice as a simplified pretreatment assessment to select treatment-naïve patients with chronic HCV GT1–6 infection and no prior evidence of cirrhosis for the 8-week G/P regimen. Use of this approach could aid in HCV elimination efforts by simplifying care pathways and treatment scale-up in community-based settings by non-specialist providers for HCV treatment-naïve patients without prior evidence of cirrhosis.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgements
AbbVie and authors thank all the trial investigators and the patients who participated in this clinical trial.
Funding
This work and the Rapid Service and Open Access Fees were supported by AbbVie. AbbVie sponsored the study (NCT03212521), contributed to its design, data collection, analysis, and interpretation of the data, and participated in the writing, review, and approval of the manuscript.
Medical Writing and/or Editorial Assistance
Medical writing support was provided by Daniel O’Brien, Ph.D., and Salil Sharma, Ph.D., both of AbbVie.
Authorship
All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this article, take responsibility for the integrity of the work as a whole, and have given their approval for this version to be published.
Author Contributions
Ana Gabriela Pires dos Santos designed the study. Zhenyi Xue led study analyses. Daniel O’Brien did the literature search and provided medical writing support. All authors contributed to the implementation, conduct, data interpretation, writing, and review of this work. All authors approved the final version of the manuscript and the decision to submit to the journal.
Prior Presentation
This work was presented at AASLD (2018) The Liver Meeting.
Disclosures
Robert Fontana: Research support from BMS, Gilead, AbbVie. Consulting: Alynam. Stuart McPherson: Consultancy/speakers fees-Abbvie, Allergan, BMS, Gilead, MSD, Novartis. Sabela Lens: Advisor: Janssen, Gilead, AbbVie. Magdy Elkhashab: Grant support from AbbVie, Advisor for AbbVie. Victor Ankoma-Sey: investigator in an AbbVie-sponsored clinical trial. Mark Bondin is an employee of AbbVie, Inc. and may hold stock or stock options. Ana Gabriela Pires dos Santos is an employee of AbbVie, Inc. and may hold stock or stock options. Roger Trinh is an employee of AbbVie, Inc. and may hold stock or stock options. Zhenyi Xue is an employee of AbbVie, Inc. and may hold stock or stock options. Ariel Porcalla is an employee of AbbVie, Inc. and may hold stock or stock options. Stefan Zeuzem: Consultancies for AbbVie, BMS, Gilead, Janssen, Merck.
Compliance with Ethics Guidelines
The trial was conducted in accordance with Good Clinical Practice and the Declaration of Helsinki, and was approved at all sites by their independent ethics committee or institutional review board prior to enrollment. The master ethics committee of this study is the Quorum Institutional Review Board. A complete list of institutional ethics committees or institutional review boards is provided in Supplementary Table S3.
Data Availability
AbbVie is committed to responsible data sharing regarding the clinical trials we sponsor. This includes access to anonymized, individual and trial-level data (analysis data sets), as well as other information (e.g., protocols and Clinical Study Reports), as long as the trials are not part of an ongoing or planned regulatory submission. This includes requests for clinical trial data for unlicensed products and indications. This clinical trial data can be requested by any qualified researchers who engage in rigorous, independent scientific research, and will be provided following review and approval of a research proposal and Statistical Analysis Plan (SAP) and execution of a Data Sharing Agreement (DSA). Data requests can be submitted at any time and the data will be accessible for 12 months, with possible extensions considered. For more information on the process, or to submit a request, visit the following link: https://www.abbvie.com/our-science/clinical-trials/clinical-trials-data-and-information-sharing/data-and-information-sharing-with-qualified-researchers.html.
Abbreviations
- AE
Adverse event
- ALT
Alanine aminotransferase
- APRI
Aspartate aminotransferase to platelet ratio index
- ART
Antiretroviral therapy
- AST
Aspartate aminotransferase
- DAA
Direct acting antiviral
- G/P
Glecaprevir/pibrentasvir
- GT
Genotype
- HBV
Hepatitis B virus
- HCC
Hepatocellular carcinoma
- HCV
Hepatitis C virus
- HIV
Human immunodeficiency virus
- ITT
Intent-to-treat
- LLOQ
Lower limit of quantification
- MedDRA
Medical Dictionary for Regulatory Activities
- mITT
Modified intent-to-treat
- PWUD
People who use drugs
- RT-PCR
Real-time polymerase chain reaction
- SVR
Sustained virologic response
- SVR12
Sustained virologic response at 12 weeks post-treatment
- ULN
Upper limit of normal
- WHO
World Health Organization
Footnotes
Enhanced Digital Features
To view enhanced digital features for this article go to 10.6084/m9.figshare.9924842.
References
- 1.World Health Organization . Global hepatitis report. Geneva: World Health Organization; 2017. [Google Scholar]
- 2.Huppe D, Serfert Y, Buggisch P, et al. Hepatitis C therapy in Germany: results from the german hepatitis C registry 4 years after approval of the new direct antiviral substances (DAAs) Wiesbaden: Viszeralmedizin; 2018. [Google Scholar]
- 3.Alazawi W, Cunningham M, Dearden J, Foster GR. Systematic review: outcome of compensated cirrhosis due to chronic hepatitis C infection. Aliment Pharmacol Ther. 2010;32(3):344–355. doi: 10.1111/j.1365-2036.2010.04370.x. [DOI] [PubMed] [Google Scholar]
- 4.Xu F, Moorman AC, Tong X, et al. All-Cause mortality and progression risks to hepatic decompensation and hepatocellular carcinoma in patients infected with hepatitis C virus. Clin Infect Dis. 2016;62(3):289–297. doi: 10.1093/cid/civ860. [DOI] [PubMed] [Google Scholar]
- 5.Falade-Nwulia O, Suarez-Cuervo C, Nelson DR, Fried MW, Segal JB, Sulkowski MS. Oral direct-acting agent therapy for hepatitis C virus infection: a systematic review. Ann Intern Med. 2017;166(9):637–648. doi: 10.7326/M16-2575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Messori A, Badiani B, Trippoli S. Achieving sustained virological response in hepatitis c reduces the long-term risk of hepatocellular carcinoma: an updated meta-analysis employing relative and absolute outcome measures. Clin Drug Investig. 2015;35(12):843–850. doi: 10.1007/s40261-015-0338-y. [DOI] [PubMed] [Google Scholar]
- 7.Liu Z, Wei X, Chen T, Huang C, Liu H, Wang Y. Characterization of fibrosis changes in chronic hepatitis C patients after virological cure: a systematic review with meta-analysis. J Gastroenterol Hepatol. 2017;32(3):548–557. doi: 10.1111/jgh.13500. [DOI] [PubMed] [Google Scholar]
- 8.Younossi Z, Henry L. Systematic review: patient-reported outcomes in chronic hepatitis C—the impact of liver disease and new treatment regimens. Aliment Pharmacol Ther. 2015;41(6):497–520. doi: 10.1111/apt.13090. [DOI] [PubMed] [Google Scholar]
- 9.Cipriano LE, Goldhaber-Fiebert JD. Population health and cost-effectiveness implications of a “treat all” recommendation for HCV: a review of the model-based evidence. MDM Policy Pract. 2018;3(1):2381468318776634. doi: 10.1177/2381468318776634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Calvaruso V, Petta S, Craxi A. Is global elimination of HCV realistic? Liver Int. 2018;38(Suppl 1):40–46. doi: 10.1111/liv.13668. [DOI] [PubMed] [Google Scholar]
- 11.EASL European Association for the Study of the Liver recommendations on treatment of hepatitis C 2018. J Hepatol. 2018;69:461–511. doi: 10.1016/j.jhep.2018.03.026. [DOI] [PubMed] [Google Scholar]
- 12.AASLD. HCV guidance: recommendations for testing, managing, and treating hepatitis C. 2018. https://www.hcvguidelines.org/. Accessed 27 Nov 2018.
- 13.World Health Organization . Guidelines for the screening care and treatment of persons with chronic hepatitis C infection. Geneva: World Health Organization; 2018. [Google Scholar]
- 14.Australian recommendations for the management of hepatitis C virus infection: a consensus statement (September 2018). 2018. https://www.hepatologyassociation.com.au/. Accessed 22 Oct 2018.
- 15.Shah H, Bilodeau M, Burak KW, et al. The management of chronic hepatitis C: 2018 guideline update from the Canadian Association for the Study of the Liver. CMAJ. 2018;190(22):E677–E687. doi: 10.1503/cmaj.170453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chou R, Wasson N. Blood tests to diagnose fibrosis or cirrhosis in patients with chronic hepatitis C virus infection. Ann Intern Med. 2013;158(11):807–820. doi: 10.7326/0003-4819-158-11-201306040-00005. [DOI] [PubMed] [Google Scholar]
- 17.Asselah T, Lens S, Zadeikis N, et al. Analysis of AST to platelet ratio index (APRI) for determining eligibility for 8 weeks of glecaprevir/pibrentasvir. J Viral Hepat. 2018;25(S2):19–20. [Google Scholar]
- 18.Rockstroh JK, Lacombe K, Trinh R, et al. Efficacy and safety of glecaprevir/pibrentasvir in patients co-infected with hepatitis C virus and human immunodeficiency virus-1: the EXPEDITION-2 study. Clin Infect Dis. 2018;67(7):1010–1017. doi: 10.1093/cid/ciy220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gane Edward, Poordad Fred, Zadeikis Neddie, Valdes Joaquin, Lin Chih-Wei, Liu Wei, Asatryan Armen, Wang Stanley, Stedman Catherine, Greenbloom Susan, Nguyen Tuan, Elkhashab Magdy, Wörns Marcus-Alexander, Tran Albert, Mulkay Jean-Pierre, Setze Carolyn, Yu Yao, Pilot-Matias Tami, Porcalla Ariel, Mensa Federico J. Safety and Pharmacokinetics of Glecaprevir/Pibrentasvir in Adults With Chronic Genotype 1–6 Hepatitis C Virus Infections and Compensated Liver Disease. Clinical Infectious Diseases. 2019;69(10):1657–1664. doi: 10.1093/cid/ciz022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Puoti M, Foster GR, Wang S, et al. High SVR12 with 8-week and 12-week glecaprevir/pibrentasvir therapy: an integrated analysis of HCV genotype 1–6 patients without cirrhosis. J Hepatol. 2018;69(2):293–300. doi: 10.1016/j.jhep.2018.03.007. [DOI] [PubMed] [Google Scholar]
- 21.MAVYRET (glecaprevir and pibrentasvir) [package insert]. North Chicago: Approved on August 2017. https://www.rxabbvie.com/pdf/mavyret_pi.pdf.
- 22.D’Ambrosio R, Pasulo L, Puoti M, et al. Real-life effectiveness and safety of glecaprevir/pibrentasvir in 723 patients with chronic hepatitis C. J Hepatol. 201970(3):379–87. [DOI] [PubMed]
- 23.Wiegand J, Naumann U, Stoehr A, et al. Glecaprevir/pibrentasvir for the treatment of patients with chronic hepatitis c virus infection: updated real-world data from the german hepatitis C-registry. Hepatology. 2018;68(S1):364A. [Google Scholar]
- 24.MAVIRET (SmPC); AbbVie 2019/MAVYRET (US package insert); AbbVie 2019.
- 25.Grebely J, Hajarizadeh B, Dore GJ. Direct-acting antiviral agents for HCV infection affecting people who inject drugs. Nat Rev Gastroenterol Hepatol. 2017;14(11):641–651. doi: 10.1038/nrgastro.2017.106. [DOI] [PubMed] [Google Scholar]
- 26.Back D, Belperio P, Bondin M, et al. Integrated efficacy and safety of glecaprevir/pibrentasvir in patients with psychiatric disorders. J Hepatol. 2018;68(S1):S280–S281. doi: 10.1016/S0168-8278(18)30778-5. [DOI] [Google Scholar]
- 27.Brown AS, Welzel TM, Conway B, et al. Adherence to pangenotypic glecaprevir/pibrentasvir treatment and SVR12 in HCV-infected patients: an integrated analysis of the phase 2/3 clinical trial program. J Hepatol. 2017;66(S1):114A–115A. [Google Scholar]
- 28.Foster GR, Grebely J, Sherman KE, et al. Safety and efficacy of glecaprevir/pibrentasvir in patients with chronic hepatitis C genotypes 1–6 and recent drug use. Hepatology. 2017;66(S1):636A–637A.
- 29.Kamil RJ, Jerschow E, Loftus PA, et al. Case-control study evaluating competing risk factors for angioedema in a high-risk population. Laryngoscope. 2016;126(8):1823–1830. doi: 10.1002/lary.25821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Feld JJ, Sanchez Gonzalez Y, Pires dos Santos AG, Ethgen O. Clinical benefits, economic savings and faster time to HCV elimination with a simplified 8-week treatment and monitoring program in chronic F0-F3 naive patients in the US. Hepatology. 2018;68(S1):408A–409A. [Google Scholar]
- 31.Brown RS, Jr, Hezode C, Wang S, et al. Preliminary efficacy and safety of 8-week glecaprevir/pibrentasvir in patients with HCV genotypes 1–6 infection and compensated cirrhosis: the EXPEDITION-8 study. Hepatology. 2018;68(S1):425A–426A. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
AbbVie is committed to responsible data sharing regarding the clinical trials we sponsor. This includes access to anonymized, individual and trial-level data (analysis data sets), as well as other information (e.g., protocols and Clinical Study Reports), as long as the trials are not part of an ongoing or planned regulatory submission. This includes requests for clinical trial data for unlicensed products and indications. This clinical trial data can be requested by any qualified researchers who engage in rigorous, independent scientific research, and will be provided following review and approval of a research proposal and Statistical Analysis Plan (SAP) and execution of a Data Sharing Agreement (DSA). Data requests can be submitted at any time and the data will be accessible for 12 months, with possible extensions considered. For more information on the process, or to submit a request, visit the following link: https://www.abbvie.com/our-science/clinical-trials/clinical-trials-data-and-information-sharing/data-and-information-sharing-with-qualified-researchers.html.