

Abstract
The importance of physiological supply of folate is well recognized in human health; the crucial roles of folate in one‐carbon metabolism for physiological DNA synthesis and cell division, as well as in the conversion of homocysteine (Hcy) to methionine, and subsequently, to S‐adenosylmethionine, have been convincingly demonstrated. Improved folate status may reduce the risk of macrocytic anaemia, cardiovascular diseases, neuropsychiatric disorders and adverse pregnancy outcomes. Inadequate folate status results in a decrease in the methylation cycle and in increased blood levels of the neurotoxic Hcy. The aim of this review is to provide insight into the influence of folate status on pregnancy health outcomes, and to consider increasing evidence of a link between the extent of genome/epigenome damage and elevated risk for adverse obstetrical endpoints.
Pregnant women are at risk for folate insufficiency because of the increased need for folate for rapid fetal growth, placental development and enlargement of the uterus. Inadequate folate status may cause fetal malformations, impaired fetal growth, pre‐term delivery and maternal anaemia. Even some diseases of the placenta may arise from folate deficiencies. Fetal growth seems to be vulnerable to maternal folate status during the periconception period, because it has the potential to affect both the closure of the neural tube and several epigenetic mechanisms within the placenta and the fetus. Mainly on the basis of the well recognized link between maternal folate status and fetal neural tube defects, women are advised to receive folic acid supplement during the periconceptional period. Because an adequate folate supply seems to play an important role in the implantation and development of the placenta and in improving endothelial function, folic acid supplementation in the late first trimester or early second trimester might also be beneficial.
Keywords: nutrigenomics, polymorphism, pregnancy, EURRECA, folate/folic acid, one‐carbon metabolism
1. General considerations on folate
1.1 Absorption, bioavailability and metabolism
Folate was first isolated in 1941 from four tons of spinach leaves, and finally synthesized in 1946 (Mitchell et al. 1941; Hall & Solehdin 1998). Folate is the generic term for all related compounds that exhibit vitamin activity similar to folic acid (pteroylmonoglutamic acid). Folic acid, i.e. the synthetic, fully oxidized form of folate, consists of three parts: a pteridine ring, p‐aminobenzoic acid and one molecule of L‐glutamic acid (Fig. 1). The naturally‐occurring folates usually have a polyglutamate side chain. Humans are able to synthesize the pteridine ring but unable to link it with the other compounds (Birn 2006). Consequently, human health depends on exogenous sources of preformed folate, which consists of not only dietary folate, but also folate synthesized by the normal microflora of the large intestine (Dudeja et al. 2001). Natural folates are highly unstable to exposure to oxygen, light and heating, and therefore a considerable amount of them can be degraded during storage and food processing. Unfortunately, the highly bioavailable and chemically stable folic acid does not occur in nature in significant quantities; nevertheless, it is the most common form of folate used in supplements and in fortified foods (Gregory 1997).
Figure 1.
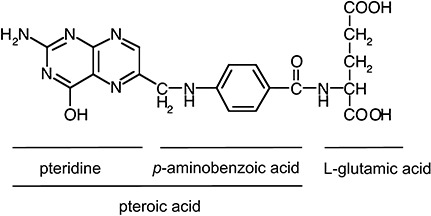
Structure of folic acid. Adapted from Shane 2008.
Our present knowledge on folate bioavailability is far from complete and the results of absorption studies are especially controversial. However, some metabolic details have been studied and the results of these studies suggest that deconjugation of polyglutamyl folates, food matrix, nutrient status of the body and food processing may be the major modifying factors contributing to the bioavailability of folates (McNulty & Pentieva 2004; Melse‐Boonstra et al. 2004). A common polymorphism (677C→T) in a key folate enzyme, methylenetetrahydrofolate reductase (MTHFR) also affects the bioavailability, which leads to increased requirement for folate (Robitaille et al. 2009). Dietary folate equivalents (DFEs) are the units which are used to account for differences in the absorption of natural food folates and the more bioavailable synthetic folic acid. When folic acid is consumed without food, its bioavailability is nearly 100%. When folic acid is consumed with food, its estimated bioavailability is approximately 85%. In contrast, the bioavailability of food folates is about 50%. DFE is defined as the quantity of food folate plus 1.7 times (85/50) the quantity of folic acid in the diet (Suitor & Bailey 2000). Good dietary sources of natural folate include a wide variety of fruits, dark green leafy vegetables, legumes and inner organs of animals (Table 1). Dietary polygutamyl folates are unable to cross the cell membranes. Prior to absorption, they have to be enzymatically deconjugated to monoglutamyl form by the membrane‐bound, zinc dependent γ‐glutamyl‐carboxypeptidase in the enterocytes. Folic acid and reduced monoglutamyl folates are absorbed in the proximal small intestine by saturable, carrier‐mediated, pH‐ and energy‐sensitive transporters having similar affinity for both oxidized and reduced folate forms (Shane 2008). Most absorbed folate is converted to 5‐methyl‐tetrahydrofolate (5‐MTHF) during passage through the enterocyte. 5‐MTHF is the predominant folate form in serum, it can be present either in free form, or bound to high‐affinity folate binding protein, or loosely associated with other serum proteins including albumin (Birn 2006). However, if high doses of folic acid are consumed, they may appear in the peripheral circulation untransformed in the postprandial state after supplementation (Kelly et al. 1997). 5‐MTHF transported into cells has to be converted into tetrahydrofolate (THF) via methionine synthase (MS) before it can participate into other folate‐dependent reactions (Akoglu et al. 2008). The active forms of folate, folylpolyglutamates which are produced by folylpoly‐γ‐glutamate synthetase (FPGS), use THF as a substrate (Suh et al. 2001). The polyglutamyl chain is required for the retention of the folate in the cell; it may also enhance the binding of the folate cofactor to folate‐requiring enzymes and allow channelling between the active sites without dissociating from the surface of the enzyme (Brody 1999).
Table 1.
Folate content of common foods
| Food item | Measure | Folate content (µg DFEs) |
|---|---|---|
| Beef liver | 1 slice (85 g) | 185 |
| Lentils, cooked | ½ cup | 180 |
| Chickpeas, cooked | ½ cup | 140 |
| Beans (black, kidney, navy), cooked | ½ cup | 115–130 |
| Spinach, cooked | ½ cup | 100 |
| Asparagus, cooked | 5 spears | 100 |
| Greens (mustard, turnip), cooked | ½ cup | 85–90 |
| Orange juice, ready to drink | 1 cup | 80 |
| Strawberries, fresh | 8 medium | 80 |
| Brussels sprout, cooked | ½ cup | 80 |
| Broccoli, cooked | ½ cup | 50 |
| Tomato juice | 1 cup | 50 |
| Peanut, dry roasted | 1 oz | 40 |
| Orange | 1 medium | 40 |
| Grapes | 1 cup | 40 |
| Cauliflower, cooked | ½ cup | 35 |
| Cantaloupe, fresh | ¼medium | 40 |
| Egg | 1 large | 25 |
| Banana | 1 medium | 20 |
| Grapefruit | ½ medium | 15 |
| Milk, fluid | 1 cup | 10–15 |
| Tomato, raw | ½ cup | 10 |
| Cucumber, raw | ½ cup | 5 |
| Potatoes, french fries | 10 strips | 5 |
DFEs, dietary folate equivalents; excellent folate sources: 100–200 µg DFE per serving; good folate sources: 50–100 µg DFE per serving; moderate folate sources: 25–49 µg DFE per serving; fair to poor folate sources: <25 µg DFE per serving; adapted from Suitor & Bailey (2000).
Under normal conditions of dietary intake and status, folate exhibits slow turnover in the human body, with a half‐life in excess of 100 days (Gregory 1997). In addition to urinary excretion, folate is secreted by the liver into the bile. However, most of this folate is reabsorbed following enterohepatic recirculation; the relatively high amount of folate in the faeces arises from gut bacteria (Shane 2008).
Reactions requiring folate are collectively referred to as one‐carbon metabolism; 5,10‐methylene‐tetrahydrofolate plays a central role in these cycles (Fig. 2). These synthetic cycles are located in the cytosol; however, the large mitochondrial folate pools also provide one‐carbon precursors into the cytosol. One‐carbon metabolism includes synthesis of purine and pyrimidine precursors of DNA and RNA synthesis, interconversion of serine and glycine and conversion of homocysteine (Hcy) to methionine, which is subsequently converted to S‐adenosylmethionine (SAM). SAM is methyl donor in more than 100 chemical reactions including methylation of DNA, RNA, cellular membrane phospholipids, neurotransmitters, hormones and myelin (Bailey & Gregory 1999).
Figure 2.
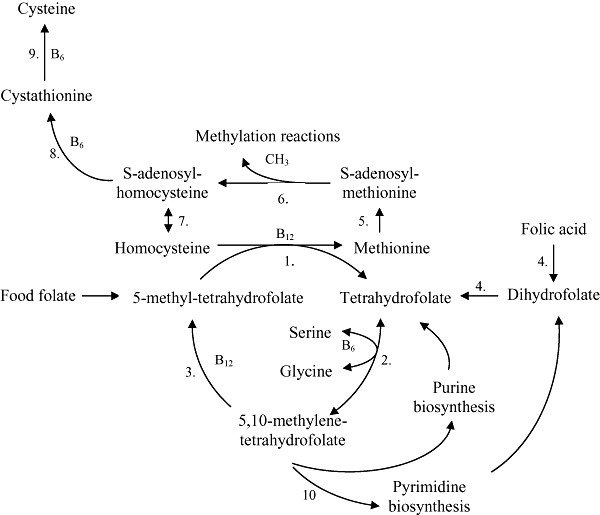
Overview of one‐carbon metabolism. 1. methionine synthase (MS); 2. serine hydroxymethyltransferase (SHMT); 3. methylenetetrahydrofolate reductase (MTHFR); 4. dihydrofolate reductase (DHFR); 5. methionine adenosyltransferase (MAT); 6. methyltransferases (MT); 7. S‐adenosylhomocysteine hydrolase; 8. cystathionine β‐synthase (CBS); 9. cystathionine γ‐lyase (CHT); 10. thymidylate synthase (TS).
Key messages
-
•
Pregnant women may be at risk for folate insufficiency in view of the increased physiological need for folate.
-
•
Women are generally advised to take 0.4 mg/day folic acid when planning a pregnancy or 4 mg/day when previous pregnancy affected by NTD is in the obstetrical history.
-
•
Further research on the role of the effect of folic acid supplementation on placental function needs to be conducted.
1.2 Hyperhomocysteinemia
Inadequate folate status results in reduction of circulating folate concentrations. The sign of decreased activity in the methylation cycle may be the elevation in plasma total Hcy (tHcy), i.e. hyperhomocysteinemia (HHCY) (Tamura & Picciano 2006). Hcy, the demethylated derivative of methionine, is metabolized via two pathways (Fig. 2). Hcy can be either remethylated to methionine by means of MS belonging to the family of enzymes with methyltransferase (MT) activity, requiring 5‐MTHF as methyl donor and vitamin B12 as a cofactor. A further route of Hcy‐elimination is the trans‐sulfuration pathway, where the conversion from Hcy to cysteine takes place in two enzymatic steps involving vitamin B6 as a cofactor (Goddijn‐Wessel et al. 1996; Castro et al. 2006). Several trials demonstrated that daily supplementation with folic acid effectively lowered plasma Hcy concentrations (Homocysteine Lowering Trialists' Collaboration 1998; van Oort et al. 2003; Lamers et al. 2004). Even if elevated Hcy levels are usually related to inadequacy of folate supply, they may also point to deficiency of enzymes involved in folate‐homocysteine metabolism due to inherited conditions, i.e. polymorphisms of MTHFR gene 677 C→T and 1298 A→C; methionine synthase gene (MTR) 2756 A→G and methionine synthase reductase gene (MTRR) 66 A→G (Guéant et al. 2003; Dhillon et al. 2009). For example, a point mutation at 677 C→T of the MTHFR gene causes the replacement of cytosine with thymine, and leads to a reduction in MTHFR activity (de Bree et al. 2003). Insufficient production of 5‐MTHF leads to an increase in Hcy levels, as the methyl group of 5‐MTHF is no longer available to a sufficient extent for the re‐methylation of Hcy into methionine. Thereby, subjects with the 677 C→T mutation need more dietary folate to achieve an adequate plasma folate levels (de Bree et al. 2003). However, plasma Hcy cannot be considered a highly specific biomarker of folate status. Other determinants of circulating Hcy are vitamin B6‐, choline‐ and vitamin B12 deficiencies due to deregulation of their metabolism or low dietary availability, or the combination of these two factors (Locksmith & Duff 1998; Steen et al. 1998; Obeid & Herrmann 2005; Castro et al. 2006; Sadananda Adiga et al. 2009; Zeisel 2009).
HHCY is suggested to be implicated in the aetiology of cardiovascular diseases, occlusive vascular diseases, demyelination, neuropathy, depression and cognitive impairment (Eskes 2001; Refsum et al. 2006; Forges et al. 2007; Dodds et al. 2008). The triggering mechanisms are not fully elucidated. However, cell‐culture studies, studies in experimentally induced HHCY in animals, and clinical studies in hyperhomocysteinemic humans suggest that high levels of tHcy may promote vascular inflammation through oxidative stress (Beaudin & Stover 2007). Both increased accumulation of reactive oxygen species (ROS) and involvement of the redox‐sensitive signalling events may be involved in increased stress (Papatheodorou & Weiss 2007). Elevated tHcy stimulates proinflammatory pathways in vascular cells and results in leukocyte recruitment to the arterial wall, in the infiltration of leukocytes into the arterial wall and in the differentiation of monocytes into cholesterol‐scavenging macrophages. Furthermore, tHcy stimulates the proliferation of vascular smooth muscle cells and subsequent production of extracellular matrix (Eskes 2001; Papatheodorou & Weiss 2007), and may be cytotoxic (Beaudin & Stover 2007).
Despite what might be assumed from cell culture and in‐vitro studies as well as from observational studies, there is actually no reliable clinical evidence that elevated plasma tHcy is the causal factor of all these disease. Several large randomized controlled trials have been designed to assess the Hcy lowering effect of folic acid and other B‐vitamin supplementation. Although the vitamin supplementation was effective in reducing Hcy levels, it failed to reduce the risk of cardiovascular disease events (Ntaios et al. 2009) and cognitive decline in Alzheimer's disease (Aisen et al. 2008). In this context, a recent review on the public significance of elevated Hcy concludes that ‘This multitude of relationships between elevated plasma homocysteine and diseases that afflict the elderly, pregnant women, and the embryo points to the existence of a common denominator which may be responsible for these diseases. Whether this denominator is homocysteine itself, or homocysteine is merely a marker, remains to be determined’ (Selhub 2008).
1.3 Potential risks of high folate supply
Elevated intake of synthetic folic acid (but not that of naturally‐occurring reduced folates) may be associated with some adverse effects as well. The tolerable upper intake level (UL) of folic acid is 1 mg for adults, which was developed to avoid masking vitamin B12 deficiency. This 1 mg UL can be also applied to pregnant and lactating women; the UL for children increases with increasing age (Scientific Committee on Food 2000).
1.3.1 Folate and vitamin B12 interrelationship
Vitamin B12 deficiency rarely occurs in infants under 4 months of age, because vitamin B12 is concentrated and stored in the liver of the fetus. However, later on breastfed infants of vitamin B12 deficient mothers may be vulnerable to vitamin B12 deficiency (Rasmussen et al. 2001; Black 2008). Malabsorption of vitamin B12 becomes more frequent with increasing age. Prevalence of vitamin B12 deficiency in the elderly ranges from 5% to 46%, depending on the population and the different criteria of deficiency (Cuskelly et al. 2007).
Vitamin B12 deficiency has many consequences, including HHCY, megaloblastic anaemia and neuropathy (Stabler & Allen 2004). Methionine synthase activity is decreased in vitamin B12 deficiency, which causes impairment of the methylation pathway, resulting in Hcy accumulation and decreased production of SAM, which is the most important methyl donor in biological reactions. The interrupted methylation process causes disturbed neurotransmitter metabolism and myelin damage. Subacute combined degeneration of the spinal cord is a specific type of neuropathy in vitamin B12 deficiency. Another symptom of vitamin B12 deficiency is megaloblastic anaemia due to delayed DNA synthesis (Smulders et al. 2006).
The interrelationship between folate and vitamin B12 is probably best explained by the methyl trap hypothesis. The conversion of 5,10‐MTHF to 5‐MTHF, catalyzed by methylenetetrahydrofolate reductase (MTHFR), is irreversible. The only way for 5‐MTHF to participate in folate‐dependent reactions is through the vitamin B12‐dependent methionine synthase (Fig. 2). In vitamin B12 deficient cells, the availability of folate is normal; however, folate is trapped as 5‐MTHF that cannot participate in methylation cycle and DNA biosynthesis; this situation is termed as folate pseudodeficiency. In addition, monoglutamate 5‐MTHF is a poor substrate for polyglutamation by FPGS, therefore it is not retained in the cell (Smulders et al. 2006). Intramuscularly given vitamin B12 reactivates methionine synthase, restarts methylation cycle and DNA synthesis, and therefore restarts myelination and depresses the anaemia. High dose folic acid supplementation is effective in treating the anaemia but not the neuropathy. When folic acid enters the cell, it has to be converted via dihydrofolate to tetrahydrofolate, thus it can initiate DNA biosynthesis. However, folic acid supplementation cannot restart the methylation cycle, which is blocked by the vitamin B12‐dependent methionine synthase, thus the neuropathy remains. Hence, folic acid treatment may mask the haematologic changes caused by vitamin B12 deficiency and may lead to delayed diagnosis and treatment of neurological symptoms (Scott 1992; Cuskelly et al. 2007).
1.3.2 Adverse effects on zinc absorption
Zinc is one of the most important and ubiquitous trace elements in biologic systems. It plays critical role in gene expression, protein synthesis, cell division, growth and immune defence. In zinc deficiency, several disturbances may occur, including impairments of immunologic, neurologic and gastrointestinal functions. Infants, children, pregnant and lactating women and elderly people may be particularly at risk of zinc deficiency because of their increased zinc requirements (Hotz & Brown 2004).
The suggestion that folic acid supplementation might have an adverse effect on zinc status was first raised in the 1980s. Supplementation with 0.4 mg/day folic acid was found to influence zinc balance in men: faecal zinc excretion increased, while urinary zinc excretion decreased in the treatment groups compared with controls (Milne et al. 1984). In another study, significant association was found between the rate of fetomaternal complications and the combination of low maternal plasma zinc and high maternal plasma folate concentrations (Mukherjee et al. 1984). Furthermore, significantly decreased zinc absorption was found when pregnant women received oral iron‐folic acid supplements; or when non‐pregnant subjects received folic acid supplements alone (Simmer et al. 1987). In contrast, a number of studies failed to detect any adverse effect of folic acid supplementation on zinc absorption (Kauwell et al. 1995; Hansen et al. 2001; Green et al. 2003). Some studies suggest that folic acid and zinc may form insoluble complexes in the acidic pH of the stomach. These complexes may dissolve at the higher pH of the duodenum, however, various pathological conditions, in that the pH of the duodenum remains below 6.0, prevent the dissolution of zinc‐folate complexes and disturb zinc absorption (Ghishan et al. 1986). To sum up, although several absorption and supplementation studies investigated the effects of folic acid on zinc absorption, the results are highly controversial. Basic differences in study designs and methodology, as well as lack of sensitive and reliable biomarker of zinc status, may be the major reasons of the inconsistent outcomes (Butterworth & Tamura 1989; Zimmermann & Shane 1993; Hansen et al. 2001).
1.3.3 Carcinogenicity
Folate is involved in DNA methylation, therefore it plays an important role in the regulation of gene expression, maintenance of DNA stability and integrity. Low folate status may be a potential risk factor for cancer development, via the mechanisms of DNA strand breaks, impaired DNA repair, increased mutation rate, global DNA hypomethylation and genomic instability (Sauer et al. 2009). Adequate folate status is crucial for physiological DNA synthesis and cell division. However, recent observational and intervention studies suggest that elevated folate status may also increase the risk of breast cancer (Charles et al. 2004), prostate cancer (Figueiredo et al. 2009) and colon cancer (Lonn et al. 2006). The timing and the dose of folate intervention appears to be responsible for this dual modulatory role of folate in carcinogenesis. Whereas folic acid supplementation suppresses the development of tumours in normal tissues, it may accelerate the development and progression of already growing neoplasms (Kim 2006; Ulrich & Potter 2006).
2. The physiological role of folate in pregnancy
In contrast to the paucity of data on the potential adverse effects of very high doses of folic acid supplementation, the benefits of improved folate status are clear: reduced risk of macrocytic anaemia, cardiovascular diseases, neuropsychiatric disorders, neural tube defects and some other adverse pregnancy outcomes.
The aim of this review is to provide insight into the relevance of folate in pregnancy by discussing and scientifically evaluating its role in pregnancy health outcomes. The importance of optimal folate supply in pregnancy has been well recognized by the medical community. Mainly on the basis of the link between maternal folate status and fetal neural tube defects (NTDs) (MRC Vitamin Study Research Group 1991; Czeizel & Dudás 1992; Pitkin 2007), in most countries women are advised to use a folic acid supplement, in addition to a healthy diet, during the periconceptional period.
Pregnancy is associated with increases in the rates of cellular proliferation and one‐carbon metabolism. Folate is involved in one‐carbon transfer reactions, which are fundamental for maintenance and repair of the genome, regulation of gene expression, amino acid metabolism and neurotransmitter synthesis (Djukic 2007). Moreover, the derivate SAM is the most important methyl donor in the human body for DNA methylation, it is also involved in epigenetic mechanisms modifying gene expression (Maloney & Rees 2005). Consequently, folate is essential for fetal growth and development as well as for maternal health (Reynolds 2006).
During gestation folate requirements are 5‐ to 10‐fold higher than in the non‐pregnant condition (Antony 2007) because of rapid fetal growth, placenta development, uterine enlargement and expansion of blood volume (Tamura & Picciano 2006; Kim et al. 2009). Indeed, pregnant women may be at risk for folate insufficiency in view of the increased physiological need for folate.
Even the timing of adequate maternal folate supply may play a crucial role in health of the offspring, because developing organ systems may respond with permanent adaptations to the suboptimal availability of nutrients during critical periods of rapid development (Finnell et al. 2004; Maloney & Rees 2005). Fetal growth seems to be vulnerable to maternal folate deficiency during the periconception period, because folate has the potential to affect epigenetic mechanisms both in the placenta and in the fetus (McMillen et al. 2008; Cetin et al. 2009). Low dietary folate intake depletes folate metabolites and decreases SAM concentrations, resulting in hypomethylation of DNA. The inhibition by SAM of MTHFR is minimized resulting in the irreversible conversion of 5,10‐MTHF to 5‐MTHF, favouring an increase in the uracil incorporation into DNA (Fenech 2001). The interaction of nutrients with the epigenetic system may lead to variations associated with chromatin remodeling and regulation of gene expression and these variations may lead to the developmental programming origin of pathological consequences in adulthood (Finnell et al. 2004; Junien 2006). DNA can be methylated at cytosine bases that are followed by a guanosine (CpG islands) (Strathdee et al. 2004). Once CpG islands in genes are methylated, the methylation is reproduced every time the gene is copied. Thus, effects of methylation may persist throughout life: changes in dietary availability of methyl groups may induce stable changes in gene methylation, altering thereby gene expression and the resulting phenotype (Zeisel 2009).
Experiments in mice and rats demonstrated that methyl‐supplements during pregnancy affected phenotypic modifications in the offspring (Wolff et al. 1998; Cooney et al. 2002; Prasolova et al. 2006; Torrens et al. 2006; Maret & Sandstead 2008). The extent to which periconceptional supply of folate, vitamin B12 and methionine affected epigenetic alterations to DNA methylation and adult health‐related phenotype in the offspring was investigated by Sinclair et al. (2007). In mature female sheep they found that a methyl‐deficient diet around the time of conception resulted in a modification in methylation status of 4% of the 1400 gene‐associated CpG islands within ovarian follicles of ewes. This apparently slight modification led to the adult offspring being heavier and fatter, insulin‐resistant and hypertonic. These findings indicate that in the periconceptional period specific dietary inputs to the methionine cycles may affect a significant part of genome in the offspring. Despite its biologically plausibility, the postulated role of inadequate folate status as a determinant of long‐term epigenetic control through methylation of DNA has not yet been proven. Most of the research is based on animals and studies have shown associations but actual causation has not been demonstrated.
3. Inadequate folate status in pregnancy
Inadequate folate status and intake in pregnancy may affect both the fetus and the mother: fetal malformations, impaired fetal growth, pre‐term delivery and maternal anaemia may all be consequences of sub‐optimal availability of folate (1999, 2004; Tamura & Picciano 2006). Folate deficiency is postulated to lead to megaloblastosis and cell death (particularly in highly proliferative somatic cells) resulting in fetal adverse consequences (Antony 2007). Another factor may be an altered Hcy metabolism, i.e. a decrease in the methylation cycle leading to HHCY (Eskes 2001). HHCY is suggested as a risk factor for deep venous thrombosis, pre‐eclampsia, spontaneous abortion, intrauterine death, placental diseases (Forges et al. 2007; Dodds et al. 2008; Kim et al. 2009) and congenital birth defects (Locksmith & Duff 1998; Tamura & Picciano 2006).
Inadequate folate status and the related diseases may develop as a consequence of low maternal dietary intake or materno‐fetal folate transport defects or fetal biochemical disorders or a combination of these factors (Dhillon et al. 2009). Genetic conditions such as variations of specific genes encoding either folate uptake and transport proteins (Piedrahita et al. 1999; Ananth et al. 2008) or enzymes involved in the Hcy metabolism pathway or one‐carbon metabolism may also modify folate status in pregnancy (Nurk et al. 2004; Mtiraoui et al. 2006; Beaudin & Stover 2007; Deng et al. 2008). Placental uptake of folate from the maternal circulation is critical for adequate folate supply to the developing fetus. Though absence of in vivo human studies hampers complete clarification of the mechanisms involved, it was shown by using an ex vivo placental cotyledon perfusion model that maternal‐to‐fetal folate transfer was mediated by placental folate receptors (Antony 2007). The initial uptake step in placental folate transfer seems to involve endocytosis mediated by the folate receptor α (FR‐α) at the microvillous membrane of the syncytiotrophoblast (Bisseling et al. 2004). FR‐α faces maternal circulation and binds the circulating maternal 5‐MHTF with high affinity. Once bound, this folate‐receptor‐bound folate is destined for fetal transport and is preferentially displaced by incoming folate. The resulting intervillous blood folate concentration, which is three times higher than that of the maternal blood, allows for folate to be transferred to the fetal circulation along a downhill concentration gradient. On the basolateral side of the syncytiotrophoblast, folate efflux into the fetal circulation is facilitated by the reduced folate carrier. Recently, a heterogeneous nuclear ribonucleoprotein (E1) was identified that specifically interacts with folate‐receptor RNA and triggers its synthesis (Antony 2007). Cell and animal studies showed that folate deficiency resulting in HHCY leads to homocysteinylation of E1, and consequent up‐regulation of folate receptors, indicating thereby Hcy as a key modulator of this process (Antony 2007).
3.1 Placenta‐related disorders
Some evidence indicates the moderate extent of connection between pathological changes in the placenta (abruption or infarction), pre‐eclampsia and spontaneous abortion, on the one hand, and folate levels on the other hand (Ray & Laskin 1999; Steegers‐Theunissen et al. 2004; Forges et al. 2007). Moreover, significant inverse dose‐response relationship between serum folate concentrations and risk of spontaneous recurrent early pregnancy losses has been observed (Nelen et al. 2000), suggesting a protective effect by high serum folate concentrations. This connection was even stronger when Hcy levels were taken into account (Nelen et al. 2000; Vollset et al. 2000). A prospective cohort study including 2119 women showed that subjects with increased tHcy in early pregnancy were at higher risk of pregnancy loss or pre‐eclampsia, but not of giving birth to small for gestational age (SGA) infants or developing gestational hypertension. These observations indicate a potential role in high tHcy levels in abnormalities of the placental vasculature (Dodds et al. 2008). Similarly, a systematic review revealed significantly higher prevalence of HHCY among women with placenta abruption/infarction and pre‐eclampsia than among women without these symptoms (Ray & Laskin 1999). HHCY has been shown to provoke vascular inflammation, to decrease the bioavailability of nitric oxide, an important endothelium vasodilatator, and to be associated with the production of ROS (Forges et al. 2007). Consequently, folate‐deficiency or HHCY may cause endothelial dysfunction. This theory might be supported by the observation that elevated tHcy concentrations were associated with an increased risk of diseases, such as atherosclerotic, thromboembolic and neurodegenerative disorders (Diaz‐Arrastia 2000; Kuo et al. 2005; Folstein et al. 2007; Forges et al. 2007).
In contrast, a recent systematic review on the role of tHcy in pre‐eclampsia evidenced that homocysteine concentrations are slightly increased in normotensive pregnancies that later develop pre‐eclampsia and are considerably increased once pre‐eclampsia is established. Moreover, no dose‐response relationship between homocysteine concentration and severity of pre‐eclampsia have been observed (Mignini et al. 2005). In this context, even results regarding a strong association between 677 C→T mutation and placental abruption and early pregnancy loss are inconsistent (Eskes 2001; Forges et al. 2007). However, a prospective cohort study, which included 2951 pregnancies and was aimed at evaluating the relationship between folic acid supplementation and MTHFR genotype with the risk of pre‐eclampsia, showed that supplementation of folic acid in a dose of ≥1.0 mg in the second trimester was associated with reduced risk of pre‐eclampsia (Wen et al. 2008).
Taken together, these studies seem to suggest that an adequate cellular folate supply may not only play a role in the implantation and development of the placenta but may also improve endothelial function at both placental and systemic levels. Moreover, folic acid supplementation in the late first trimester or early second trimester might be beneficial both in suppressing genetic effects on pre‐eclampsia and in preventing early pregnancy loss. Nevertheless, precaution needs to be taken when relying on homocysteine for adverse pregnancy outcomes. Even if many causes and pathological mechanisms have been proposed, some obstetric disorders still remain ‘diseases of theories’ as their cause or pathophysiology are actually unclear (Mignini et al. 2006), due to heterogeneity of data, size of samples and overall bias in experimental design and analysis of data.
3.2 Fetal growth
In the beginning of the previous century, the definitions of birthweight were highly inconsistent and a clear distinction was only made after the 1970s. A low birthweight (LBW) infant is an infant with a birthweight of <2500 g regardless of gestational age. A SGA infant is an infant whose birthweight and/or length below the 10th percentile for that gestational age. The terms SGA and intrauterine growth retardation (IUGR) are usually used interchangeably (Wilcox 2001).
Pre‐term delivery and LBW are among the major risk factors of infant morbidity including bronchopulmonary dysplasia, respiratory distress syndrome, necrotizing enterocolitis and intraventricular haemorrhage. Moreover, perinatal mortality is more than 6–10 times higher in intrauterine growth retarded infants than in infants with normal intrauterine growth (Scholl & Johnson 2000). LBW influences not only infant health, but various aspects of later development as well. Several investigators indicated that LBW is associated with a higher risk of stroke, hypertension, coronary heart disease and type 2 diabetes in adulthood. The hypothesis of developmental origins of adult disease is based on the assumption that inadequate nutrition decreases the rate of cell division, and the consequent redistribution of blood flow and the metabolic and endocrine changes cause permanent adaptations in tissues and influence organ development in the fetus (Barker 1998; Le Clair et al. 2009).
Folate has a fundamental role in nucleic acid synthesis and cell division. Increased folate intake is required for the growth of the uterus and the placenta, as well as for the increase of blood volume and the growth of the fetus during pregnancy (Rondo & Tomkins 2000). If dietary folate supply is insufficient, maternal folate depletion may increase the prevalence of IUGR and pre‐term delivery (Smits & Essed 2001). Moreover, low folate status is associated with elevated serum Hcy level, which may increase the risk of placental vascular insufficiency, disturbing thereby the delivery of nutrients and oxygen to the fetus (Le Clair et al. 2009). Placental dysfunction and damage caused by HHCY has been associated with IUGR (Lindblad et al. 2005); furthermore a twofold increase in the risk of LBW and pre‐term delivery has been reported in those women who consumed low folate intake (<240 µg/day) during pregnancy (Scholl et al. 1996). Whereas observational studies unequivocally suggest that low folate status increases the risk of LBW and pre‐term delivery, and good folate status decreases the risk of these adverse health outcomes, supplementation studies show highly contradictory results (Charles et al. 2005). The controversy may originate from variable degree of compliance as well as from genetic polymorphisms and different environmental factors (Scholl & Johnson 2000).
3.3 Fetal malformations
Birth defects are structural malformations that may affect different organs. The most common types of birth defect are congenital heart defects (CHDs), followed by NTDs. NTDs consist of abnormality of the spine (spina bifida) and brain (anencephaly); anencephaly is inconsistent with life and spina bifida may lead to extremely severe clinical consequences that seriously impair quality of life (Padmanabhan 2006). Craniofacial malformations, which include cleft lip with or without cleft palate and cleft palate only, are also common (Zhu et al. 2009).
It is widely recognized that the interplay between genetic and environmental factors contributes to the aetiology of structural birth defects (Finnell et al. 2004; Zhu et al. 2009). Folic acid exerts a pivotal role in promoting normal embryonic development: folic acid supplementation is critically important both pre‐ and post‐conceptionally in protecting against NTDs (Molloy et al. 2009). Despite the consistent scientific evidence on the association between folate status and/or impaired folate metabolism with fetal birth defects, biological mechanisms involved in embryonic folate utilization are not yet well understood (Beaudin & Stover 2007). Sub‐optimal maternal folate status appears to impose biochemical stress to the embryo via inducing disturbances of the methionine one‐carbon metabolism and resulting in abnormal closure of the neural tube (Zhang et al. 2008).
Women are generally advised to take 0.4 mg/day folic acid when planning a pregnancy or 4 mg/day when previous NTD pregnancy is in the obstetrical history (de Bree et al. 1997; Geisel 2003; Pitkin 2007). The risk reduction in NTDs amounts up to 70% with 4 mg/day dose of supplementation (MRC Vitamin Study Research Group 1991), whereas a near 100% reduction of NTDs in addition to significant reductions of CHDs was achieved by periconceptional supplementation of a multi‐vitamin product containing 0.8 mg of folic acid (Czeizel & Dudás 1992; Czeizel et al. 2004). The folic acid dose of 0.8 mg daily was demonstrated to be the optimal dosage for lowering Hcy levels as well (Wald et al. 2001). HHCY has been postulated as a mechanism involved in NTDs, because an abnormality in Hcy metabolism was reported in women who gave birth to children with NTDs (Mills et al. 1995). These observations suggest that an increased tHcy level might be the actual causal factor in the aetiology of NTDs (Beaudin & Stover 2009). Homocysteinylation of placental folate receptor 1 seems to trigger an autoimmune response leading presumably to the block of folic acid transport (Taparia et al. 2007). This indicates that folate transport may be the critical step in ensuring normal embryonic development (Beaudin & Stover 2009; Blom 2009), as demonstrated in knockout mice, whose pups had significant morphological abnormalities depending on folinic acid supply provided to the dams (Taparia et al. 2007). The incidence of NTDs seems to correlate also with the level of vitamin B12 (Molloy et al. 2009). Women with low folate supply in combination with low blood vitamin B12 levels had a drastically increased risk of NTDs (Padmanabhan 2006). Low B12 concentrations seem to be associated not only with NTDs (Steen et al. 1998; Ray & Laskin 1999; Ray & Blom 2003) but with CHDs as well (Verkleij‐Hagoort et al. 2008).
Neural crest cells are involved in the embryogenesis of the neural tube, lip and palate, and cardiovascular system. Since the migration and differentiation of neural crest cells is influenced by Hcy, vitamin B12 seems to contribute to the embryogenesis of the heart in the first weeks after conception (Verkleij‐Hagoort et al. 2008). Additionally, it was observed in a case‐control study that periconceptional intake of thiamine, niacin and vitamin B6 seems to contribute to the prevention of orofacial cleft defects (Krapels et al. 2004).
In summary, methyl group donators seem to exert a profound effect on reproductive outcome through epigenetic mechanisms (Zeisel 2009). Though genetic factors (MTRR 66 A→G and the MTHFR 677C→T) are certainly also involved (Finnell et al. 2004; Padmanabhan 2006; Blom 2009), epigenetic mechanisms may significantly contribute to the complex aetiology of congenital malformations (Beaudin & Stover 2007; Blom 2009). On the basis of conclusive evidence showing that folic acid protects against NTDs, folic acid supplementation should be recommended.
3.4 Maternal megaloblastic anaemia
Anaemia is one of the most frequent haematological disorders. The major symptoms are fatigue, headache, poor concentration, tachycardia, heart failure, dyspnoea and pallor. Anaemia affects about 40% of all pregnant women and about 50% of all children worldwide (McLean et al. 2007). In these population groups, severe anaemia is associated with increased prevalence of morbidity and mortality. Pre‐term delivery, spontaneous abortion and LBW are the potential adverse outcomes of severe maternal anaemia.
Megaloblastic anaemia is the typical symptom of folate deficiency, which is the second most common anaemia after iron deficiency anaemia (Sifakis & Pharmakides 2000). Folate insufficiency leads to delayed synthesis and fragmentation of DNA and to reduced extent of cell division. Megaloblastic cells contain double amount of DNA than normal cells, because they are not able to enter into the mitosis phase (Shane 2008). Reduced division of erythrocytes results in macrocytic cells with raised mean corpuscular volume. Vitamin B12 deficiency also causes megaloblastic anaemia; in this case not the insufficient 5‐MTHF level, but the inactive vitamin B12‐dependent methionine synthase is responsible for the interrupted methylation cycle. Megaloblastic changes occur especially in the rapidly dividing cells of the bone marrow, and cause not only anaemia, but neutropenia and thrombocytopenia as well (Scott 2007). Megaloblastic anaemia appears typically in the latest phase of pregnancy, usually not sooner than the 36th week of gestation. Whereas the prevalence of maternal megaloblastic anaemia is quite low in developed countries due to the recommended folate supplementation, its prevalence may be more than 50% in the developing world (Wickramasinghe 2006).
4. Conclusion
Folate plays a fundamental role in several intracellular processes resulting in cell growth, and consequently, it may seriously influence the overall health outcome of pregnancy. Folic acid supplementation starting before pregnancy should be firmly recommended, as conclusive evidence exist in relation to the protective role folic acid exerts against NTDs, and public health efforts should be undertaken to ensure that the diet of all women who may bear children contains an adequate amount of folic acid. Considering the involvement of folate in several obstetric outcomes, appropriate folate levels seem to be important not only in the periconceptional period but also throughout the entire duration of pregnancy.
Source of funding
The work reported herein has been carried out within the EURRECA Network of Excellence (http://www.eurreca.org) which is financially supported by the Commission of the European Communities, specific Research, Technology and Development (RTD) Programme Quality of Life and Management of Living Resources, within the Sixth Framework Programme, contract no. 036196. This report does not necessarily reflect the Commission's views or its future policy in this area.
Conflict of interest
The authors have declared no conflict of interest.
References
- Aisen P.S., Schneider L.S., Sano M., Diaz‐Arrastia R., Van Dyck C.H., Weiner M.F., et al (2008) High‐dose B vitamin supplementation and cognitive decline in Alzheimer disease: a randomized controlled trial. Journal of the American Medical Association 300, 1774–1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akoglu B., Schrott M., Bolouri H., Jaffari A., Kutschera E., Caspary W.F., et al (2008) The folic acid metabolite L‐5‐methyltetrahydrofolate effectively reduces total serum homocysteine level in orthotopic liver transplant recipients: a double‐blind placebo‐controlled study. European Journal of Clinical Nutrition 62, 796–801. [DOI] [PubMed] [Google Scholar]
- Ananth C.V., Peltier M.R., Moore D.F., Kinzler W.L., Leclerc D. & Rozen R.R. for the New Jersey‐Placental Abruption Study Investigators (2008) Reduced folate carrier 80A→G polymorphism, plasma folate, and risk of placental abruption. Human Genetics 124, 137–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antony A.C. (2007) In utero physiology: role of folic acid in nutrient delivery and fetal development. American Journal of Clinical Nutrition 85, 598S–603S. [DOI] [PubMed] [Google Scholar]
- Bailey L.B. & Gregory J.F. 3rd (1999) Folate metabolism and requirements. Journal of Nutrition 129, 779–782. [DOI] [PubMed] [Google Scholar]
- Barker D.J. (1998) In utero programming of chronic disease. Clinical Science 95, 115–128. [PubMed] [Google Scholar]
- Beaudin A.E. & Stover P.J. (2007) Folate‐mediated one‐carbon metabolism and neural tube defects: balancing genome synthesis and gene expression. Birth Defects Research 81, 183–203. [DOI] [PubMed] [Google Scholar]
- Beaudin A.E. & Stover P.J. (2009) Insights into metabolic mechanisms underlying folate‐responsive neural tube defects: a minireview. Birth Defects Research 85, 274–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birn H. (2006) The kidney in vitamin B12 and folate homeostasis: characterization of receptors for tubular uptake of vitamins and carrier proteins. American Journal of Physiology – Renal Physiology 291, F22–F36. [DOI] [PubMed] [Google Scholar]
- Bisseling T.M., Steegers E.A., Van Den Heuvel J.J., Siero H.L., Van De Water F.M., Walker A.J., et al (2004) Placental folate transport and binding are not impaired in pregnancies complicated by fetal growth restriction. Placenta 25, 588–593. [DOI] [PubMed] [Google Scholar]
- Black M.M. (2008) Effects of vitamin B12 and folate deficiency on brain development in children. Food and Nutrition Bulletin 29 (Suppl.), S126–S131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blom H.J. (2009) Folic acid, methylation and neural tube closure in humans. Birth Defects Research 85, 295–302. [DOI] [PubMed] [Google Scholar]
- De Bree A., Van Dusseldorp M., Brouwer I.A., Van Het Hof K.H. & Steegers‐Theunissen R.P. (1997) Folate intake in Europe: recommended, actual and desired intake. European Journal of Clinical Nutrition 51, 643–660. [DOI] [PubMed] [Google Scholar]
- De Bree A., Verschuren W.M., Bjørke‐Monsen A.L., Van Der Put N.M., Heil S.G., Trijbels F.J. et al (2003) Effect of the methylenetetrahydrofolate reductase 677C→T mutation on the relations among folate intake and plasma folate and homocysteine concentrations in a general population sample. American Journal of Clinical Nutrition 77, 687–693. [DOI] [PubMed] [Google Scholar]
- Brody T. (1999) Nutritional biochemistry, 2nd edn. Academic Press: San Diego. [Google Scholar]
- Butterworth C.E. Jr & Tamura T. (1989) Folic acid safety and toxicity: a brief review. American Journal of Clinical Nutrition 50, 353–358. [DOI] [PubMed] [Google Scholar]
- Castro R., Rivera I., Blom H.J., Jakobs C. & Tavares de Almeida I. (2006) Homocysteine metabolism, hyperhomocysteinaemia and vascular disease: an overview. Journal of Inherited Metabolic Disease 29, 3–20. [DOI] [PubMed] [Google Scholar]
- Cetin I., Berti C. & Calabrese S. (2009) Role of micronutrients in the periconceptional period. Human Reproduction Update 16, 80–95. [DOI] [PubMed] [Google Scholar]
- Charles D., Ness A.R., Campbell D., Smith G.D. & Hall M.H. (2004) Taking folate in pregnancy and risk of maternal breast cancer. BMJ 329, 1375–1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charles D.H.M., Ness A.R., Campbell D., Smith G.D., Whitley E. & Hall M.H. (2005) Folic acid supplements in pregnancy and birth outcome: re‐analysis of a large randomised controlled trial and update of Cochrane review. Paediatric and Perinatal Epidemiology 19, 112–124. [DOI] [PubMed] [Google Scholar]
- Cooney C.A., Dave A.A. & Wolff G.L. (2002) Maternal methyl supplements in mice affect epigenetic variation and DNA methylation of offspring. Journal of Nutrition 132, 2393S–2400S. [DOI] [PubMed] [Google Scholar]
- Cuskelly G.J., Mooney K.M. & Young I.S. (2007) Folate and vitamin B12: friendly or enemy nutrients for the elderly. Proceedings of the Nutrition Society 66, 548–558. [DOI] [PubMed] [Google Scholar]
- Czeizel A.E. & Dudás I. (1992) Prevention of the first occurrence of neural‐tube defects by periconceptional vitamin supplementation. New England Journal of Medicine 327, 1832–1835. [DOI] [PubMed] [Google Scholar]
- Czeizel A.E., Tímár L. & Sárközi A. (1999) Dose‐dependent effect of folic acid on the prevention of orofacial clefts. Pediatrics 104, e66. [DOI] [PubMed] [Google Scholar]
- Czeizel A.E., Dobó M. & Vargha P. (2004) Hungarian cohort‐controlled trial of periconceptional multivitamin supplementation shows a reduction in certain congenital abnormalities. Birth Defects Research 70, 853–861. [DOI] [PubMed] [Google Scholar]
- Deng L., Elmore C.L., Lawrance A.K., Matthews R.G. & Rozen R. (2008) Methionine synthase reductase deficiency results in adverse reproductive outcomes and congenital heart defects in mice. Molecular Genetics and Metabolism 94, 336–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhillon V., Thomas P. & Fenech M. (2009) Effect of common polymorphisms in folate uptake and metabolism genes on frequency of micronucleated lymphocytes in a South Australian cohort. Mutation Research 665, 1–6. [DOI] [PubMed] [Google Scholar]
- Diaz‐Arrastia R. (2000) Homocysteine and neurologic disease. Archives of Neurology 57, 1422–1427. [DOI] [PubMed] [Google Scholar]
- Djukic A. (2007) Folate‐responsive neurologic diseases. Pediatric Neurology 37, 387–397. [DOI] [PubMed] [Google Scholar]
- Dodds L., Fell D.B., Dooley K.C., Armson B.A., Allen A.C., Nassar B.A., et al (2008) Effect of homocysteine concentration in early pregnancy on gestational hypertensive disorders and other pregnancy outcomes. Clinical Chemistry 54, 326–334. [DOI] [PubMed] [Google Scholar]
- Dudeja P.K., Kode A., Alnounou M., Tyagi S., Torania S., Subramanian V.S., et al (2001) Mechanism of folate transport across the human colonic basolateral membrane. American Journal of Physiology – Gastrointestinal and Liver Physiology 281, G54–G60. [DOI] [PubMed] [Google Scholar]
- Eskes T.K. (2001) Clotting disorders and placental abruption: homocysteine‐a new risk factor. European Journal of Obstetrics & Gynecology and Reproductive Biology 95, 206–212. [DOI] [PubMed] [Google Scholar]
- Fenech M. (2001) The role of folic acid and Vitamin B12 in genomic stability of human cells. Mutation Research 475, 57–67. [DOI] [PubMed] [Google Scholar]
- Figueiredo J.C., Grau M.V., Haile R.W., Sandler R.S., Summers R.W., Bresalier R.S., et al (2009) Folic acid and risk of prostate cancer: results from a randomized clinical trial. Journal of the National Cancer Institute 101, 432–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finnell R.H., Shaw G.M., Lammer E.J., Brandl K.L., Carmichael S.L. & Rosenquist T.H. (2004) Gene–nutrient interactions: importance of folates and retinoids during early embryogenesis. Toxicology and Applied Pharmacology 198, 75–85. [DOI] [PubMed] [Google Scholar]
- Folstein M., Liu T., Peter I., Buel J., Arsenault L., Scott T., et al (2007) The homocysteine hypothesis of depression. American Journal of Psychiatry 164, 861–867. [DOI] [PubMed] [Google Scholar]
- Forges T., Monnier‐Barbarino P., Alberto J.M., Guéant‐Rodriguez R.M., Daval J.L. & Guéant J.L. (2007) Impact of folate and homocysteine metabolism on human reproductive health. Human Reproduction Update 13, 225–238. [DOI] [PubMed] [Google Scholar]
- Geisel J. (2003) Folic acid and neural tube defects in pregnancy. A review. Journal of Perinatal & Neonatal Nursing 17, 268–279. [DOI] [PubMed] [Google Scholar]
- Ghishan F.K., Said H.M., Wilson P.C., Murrell J.E. & Greene H.L. (1986) Intestinal transport of zinc and folic acid: a mutual inhibitory effect. American Journal of Clinical Nutrition 43, 258–262. [DOI] [PubMed] [Google Scholar]
- Goddijn‐Wessel T.A., Wouters M.G., Van De Molen E.F., Spuijbroek M.D., Steegers‐Theunissen R.P., Blom H.J., et al (1996) Hyperhomocysteinemia: a risk factor for placental abruption or infarction. European Journal of Obstetrics, Gynecology, and Reproductive Biology 66, 23–29. [DOI] [PubMed] [Google Scholar]
- Green T.J., Skeaff C.M., Whiting S.J. & Gibson R.S. (2003) Effect of folic acid supplementation on plasma zinc concentrations of young women. Nutrition 19, 522–523. [DOI] [PubMed] [Google Scholar]
- Gregory J.F. 3rd (1997) Bioavailability of folate. European Journal of Clinical Nutrition 51, S54–S59. [PubMed] [Google Scholar]
- Guéant J.L., Guéant R.M., Anello G., Bosco P., Brunaud L., Romano C., et al (2003) Genetic determinants of folate and vitamin B12 metabolism: a common pathway in neural tube defect and down syndrome? Clinical Chemistry and Laboratory Medicine 41, 1473–1477. [DOI] [PubMed] [Google Scholar]
- Hall J.G. & Solehdin F. (1998) Folate and its various ramifications. Advances in Pediatrics 45, 1–35. [PubMed] [Google Scholar]
- Hansen M., Samman S., Madsen L.T., Jensen M., Sørensen S.S. & Sandström B. (2001) Folic acid enrichment of bread does not appear to affect zinc absorption in young women. American Journal of Clinical Nutrition 74, 125–129. [DOI] [PubMed] [Google Scholar]
- Homocysteine Lowering Trialists' Collaboration (1998) Lowering blood homocysteine with folic acid based supplements: metaanalysis of randomised trials. BMJ 316, 894–898. [PMC free article] [PubMed] [Google Scholar]
- Hotz C. & Brown K.H. (2004) International Zinc Nutrition Consultative Group (IZiNCG). Technical Document #1. Assessment of the risk of zinc deficiency in populations and options for its control. Food and Nutrition Bulletin 25, S91–S203. [PubMed] [Google Scholar]
- Junien C. (2006) Impact of diets and nutrients/drugs on early epigenetic programming. Journal of Inherited Metabolic Disease 29, 359–365. [DOI] [PubMed] [Google Scholar]
- Kauwell G.P., Bailey L.B., Gregory J.F. 3rd, Bowling D.W. & Cousins R.J. (1995) Zinc status is not adversely affected by folic acid supplementation and zinc intake does not impair folate utilization in human subjects. Journal of Nutrition 125, 66–72. [DOI] [PubMed] [Google Scholar]
- Kelly P., McPartlin J., Goggins M., Weir D.G. & Scott J.M. (1997) Unmetabolized folic acid in serum: acute studies in subjects consuming fortified food and supplements. American Journal of Clinical Nutrition 65, 1790–1795. [DOI] [PubMed] [Google Scholar]
- Kim Y.I. (2006) Does a high folate intake increase the risk of breast cancer? Nutrition Reviews 64, 468–475. [DOI] [PubMed] [Google Scholar]
- Kim J.M., Hong K., Lee J.H., Lee S. & Chang N. (2009) Effect of folate deficiency on placental DNA methylation in hyperhomocysteinemic rats. Journal of Nutritional Biochemistry 20, 172–176. [DOI] [PubMed] [Google Scholar]
- Krapels I.P., Van Rooij I.A., Ocké M.C., Van Cleef B.A., Kuijpers‐Jagtman A.M. & Steegers‐Theunissen R.P. (2004) Maternal dietary B vitamin intake, other than folate, and the association with orofacial cleft in the offspring. European Journal of Nutrition 43, 7–14. [DOI] [PubMed] [Google Scholar]
- Kuo H.K., Sorond F.A., Chen J.H., Hashmi A., Milberg W.P. & Lipsitz L.A. (2005) The role of hom.ocysteine in multisystem age‐related problems: a systematic review. Journal of Gerontology: Medical Sciences 60A, 1190–1201. [DOI] [PubMed] [Google Scholar]
- Lamers Y., Prinz‐Langenohl R., Moser R. & Pietrzik K. (2004) Supplementation with [6S]‐5‐methyltetrahydrofolate or folic acid equally reduces plasma total homocysteine concentrations in healthy women. American Journal of Clinical Nutrition 79, 473–478. [DOI] [PubMed] [Google Scholar]
- Le Clair C., Abbi T., Sandhu H. & Tappia P.S. (2009) Impact of maternal undernutrition on diabetes and cardiovascular disease risk in adult offspring. Canadian Journal of Physiology and Pharmacology 87, 161–179. [DOI] [PubMed] [Google Scholar]
- Lindblad B., Zaman S., Malik A., Martin H., Ekström A.M., Amu S., et al (2005) Folate, vitamin B12, and homocysteine levels in South Asian women with growth‐retarded fetuses. Acta Obstetricia et Gynecologica Scandinavica 84, 1055–1061. [DOI] [PubMed] [Google Scholar]
- Locksmith G.J. & Duff P. (1998) Preventing neural tube defects: the importance of periconceptional folic acid supplements. Obstetrics & Gynecology 91, 1027–1034. [DOI] [PubMed] [Google Scholar]
- Lonn E., Yusuf S., Arnold M.J., Sheridan P., Pogue J., Micks M., et al (2006) Homocysteine lowering with folic acid and B vitamins in vascular disease. New England Journal of Medicine 354, 1567–1577. [DOI] [PubMed] [Google Scholar]
- McLean E., Egli I., Cogswell M., Benoist B. & Wojdyla D. (2007) Worldwide prevalence of anemia in preschool aged children, pregnant women and non‐pregnant women of reproductive age In: Nutritional Anemia (eds Kraemer K. & Zimmermann M.B.), pp. 1–12. Sight and Life Press: Basel. [Google Scholar]
- McMillen I.C., MacLaughlin S.M., Muhlhausler B.S., Gentili S., Duffield J.L. & Morrison J.L. (2008) Developmental origins of adult health and disease: the role of periconceptional and foetal nutrition. Basic & Clinical Pharmacology & Toxicology 102, 82–89. [DOI] [PubMed] [Google Scholar]
- McNulty H. & Pentieva K. (2004) Folate bioavailability. The Proceedings of the Nutrition Society 63, 529–536. [DOI] [PubMed] [Google Scholar]
- Maloney C.A. & Rees W.D. (2005) Gene‐nutrient interactions during fetal development. Reproduction 130, 401–410. [DOI] [PubMed] [Google Scholar]
- Maret W. & Sandstead H.H. (2008) Possible roles of zinc nutriture in the fetal origins of disease. Experimental Gerontology 43, 378–381. [DOI] [PubMed] [Google Scholar]
- Melse‐Boonstra A., Verhoef P. & West C. (2004) Quantifying folate bioavailability: a critical appraisal of methods. Current Opinion in Clinical Nutrition & Metabolic Care 7, 539–545. [DOI] [PubMed] [Google Scholar]
- Mignini L., Latthe P.M., Villar J., Kilby M.D., Carroli G. & Khan K.S. (2005) Mapping the theories of preeclampsia: the role of homocysteine. Obstetrics & Gynecology 105, 411–425. [DOI] [PubMed] [Google Scholar]
- Mignini L., Villar J. & Khan K.S. (2006) Mapping the theories of preeclampsia: the need for systematic reviews of mechanisms of the disease. American Journal of Obstetrics and Gynecology 194, 317–321. [DOI] [PubMed] [Google Scholar]
- Mills J.L., McPartlin J.M., Kirke P.N., Lee J.L., Conley M.R. & Weir D.G. (1995) Homocysteine metabolism in pregnancies complicated by neural‐tube defects. Lancet 345, 149–151. [DOI] [PubMed] [Google Scholar]
- Milne D.B., Canfield W.K., Mahalko J.R. & Sandstead H.H. (1984) Effect of oral folic acid supplements on zinc, copper, and iron absorption and excretion. American Journal of Clinical Nutrition 39, 535–539. [DOI] [PubMed] [Google Scholar]
- Mitchell H.K., Snell E.E. & Williams R.J. (1941) The concentration of ‘folic acid’. Journal of the American Chemical Society 63, 2284–2285. [DOI] [PubMed] [Google Scholar]
- Molloy A.M., Brody L.C., Mills J.L., Scott J.M. & Kirke P.N. (2009) The search for genetic polymorphisms in the homocysteine/folate pathway that contribute to the etiology of human neural tube defects. Birth Defects Research 85, 285–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MRC Vitamin Study Research Group (1991) Prevention of neural tube defects: results of the Medical Research Council vitamin study. Lancet 338, 131–137. [PubMed] [Google Scholar]
- Mtiraoui N., Zammiti W., Ghazouani L., Jmili Braham N., Saidi S., Finan R.R., et al (2006) Methylenetetrahydrofolate reductase C677T and A1298C polymorphism and changes in homocysteine concentrations in women with idiopathic recurrent pregnancy losses. Reproduction 131, 395–401. [DOI] [PubMed] [Google Scholar]
- Mukherjee M.D., Sandstead H.H., Ratnaparkhi M.V., Johnson L.K., Milne D.B. & Stelling H.P. (1984) Maternal zinc, iron, folic acid, and protein nutriture and outcome of human pregnancy. American Journal of Clinical Nutrition 40, 496–507. [DOI] [PubMed] [Google Scholar]
- Nelen W.L., Blom H.J., Steegers E.A., Den Heijer M., Thomas C.M. & Eskes T.K. (2000) Homocysteine and folate levels as risk factors for recurrent early pregnancy loss. Obstetrics & Gynecology 95, 519–524. [DOI] [PubMed] [Google Scholar]
- Ntaios G., Savopoulos C., Grekas D. & Hatzitolios A. (2009) The controversial role of B‐vitamins in cardiovascular risk: An update. Archives of Cardiovascular Disease 102, 847–854. [DOI] [PubMed] [Google Scholar]
- Nurk E., Tell G.S., Refsum H., Ueland P.M. & Vollset S.E. (2004) Associations between maternal MTHFR polymorphisms and adverse outcomes of pregnancy: the Hordoland Homocysteine Study. American Journal of Medicine 117, 26–31. [DOI] [PubMed] [Google Scholar]
- Obeid R. & Herrmann W. (2005) Homocysteine, folic acid and vitamin B12 in relation to pre‐ and postnatal health aspects. Clinical Chemistry and Laboratory Medicine 43, 1052–1057. [DOI] [PubMed] [Google Scholar]
- Van Oort F.V., Melse‐Boonstra A., Brouwer I.A., Clarke R., West C.E., Katan M.B., et al (2003) Folic acid and reduction of plasma homocysteine concentrations in older adults: a dose‐response study. American Journal of Clinical Nutrition 77, 1318–1323. [DOI] [PubMed] [Google Scholar]
- Padmanabhan R. (2006) Etiology, pathogenesis and prevention of neural tube defects. Congenital Anomalies 46, 55–67. [DOI] [PubMed] [Google Scholar]
- Papatheodorou L. & Weiss N. (2007) Vascular oxidant stress and inflammation in hyperhomocysteinemia. Antioxidants & Redox Signaling 9, 1941–1958. [DOI] [PubMed] [Google Scholar]
- Piedrahita J.A., Oetama B., Bennett G.D., Van Waes J., Kamen B.A., Richardson J., et al (1999) Mice lacking the folic acid‐binding protein Folbp1 are defective in early embryonic development. Nature Genetics 23, 228–232. [DOI] [PubMed] [Google Scholar]
- Pitkin R.M. (2007) Folate and neural tube defects. American Journal of Clinical Nutrition 85, 285S–288S. [DOI] [PubMed] [Google Scholar]
- Prasolova L.A., Trut L.N., Oskina I.N., Gulevich R.G., Plyusnina I.Z., Vsevolodov E.B., et al (2006) The effect of methyl supplements during pregnancy on the phenotypic modification of offspring agouti coat color in rats. Russian Journal of Genetics 42, 67–71. [PubMed] [Google Scholar]
- Rasmussen S.A., Fernhoff P.M. & Scanlon K.S. (2001) Vitamin B12 deficiency in children and adolescents. Journal of Pediatrics 138, 10–17. [DOI] [PubMed] [Google Scholar]
- Ray J.G. & Blom H.J. (2003) Vitamin B12 insufficiency and the risk of fetal neural tube defects. QJM 96, 289–295. [DOI] [PubMed] [Google Scholar]
- Ray J.G. & Laskin C.A. (1999) Folic acid and homocyst(e)ine metabolic defects and the risk of placental abruption, pre‐eclampsia and spontaneous pregnancy loss: a systematic review. Placenta 20, 519–529. [DOI] [PubMed] [Google Scholar]
- Refsum H., Nurk E., Smith A.D., Ueland P.M., Gjesdal C.G. & Bjelland I. (2006) The Hordeland homocysteine Study: a community‐based study of homocysteine, its determinants, and associations with disease. Journal of Nutrition 136, S1731–S1740. [DOI] [PubMed] [Google Scholar]
- Reynolds E. (2006) Vitamin B12, folic acid, and the nervous system. Lancet Neurology 5, 949–960. [DOI] [PubMed] [Google Scholar]
- Robitaille J., Hamner H.C., Cogswell M.E. & Yang Q. (2009) Does the MTHFR 677C → T variant affect the Recommended Dietary Allowance for folate in the US population? American Journal of Clinical Nutrition 89, 1269–1273. [DOI] [PubMed] [Google Scholar]
- Rondo P.H. & Tomkins A.M. (2000) Folate and intrauterine growth retardation. Annals of Tropical Paediatrics 20, 253–258. [DOI] [PubMed] [Google Scholar]
- Sadananda Adiga M.N., Chandy S., Ramaswamy G., Appaji L., Kumari A.B. & Krishnamoorthy L. (2009) Association between plasma homocysteine and riboflavin status in acute lymphoblastic leukemia in children. Indian Journal of Clinical Biochemistry 24, 257–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauer J., Mason J.B. & Choi S.W. (2009) Too much folate: a risk factor for cancer and cardiovascular disease? Current Opinion in Clinical Nutrition and Metabolic Care 12, 30–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholl T.O. & Johnson W.G. (2000) Folic acid: influence on the outcome of pregnancy. American Journal of Clinical Nutrition 71 (Suppl.), 1295S–1303S. [DOI] [PubMed] [Google Scholar]
- Scholl T.O., Hediger M.L., Schall J.I., Khoo C.S. & Fischer R.L. (1996) Dietary and serum folate: their influence on the outcome of pregnancy. American Journal of Clinical Nutrition 63, 520–525. [DOI] [PubMed] [Google Scholar]
- Scientific Committee on Food (2000) Opinion of the Scientific Committee on Food on the tolerable upper intake level of folate Report ID. SCF/CS/NUT/UPPLEV/18 Final. European Commission, health & consumer protection directorate‐general, Brussels.
- Scott J.M. (1992) Folate‐vitamin B12 interrelationships in the central nervous system. Proceedings of the Nutrition Society 51, 219–224. [DOI] [PubMed] [Google Scholar]
- Scott J.M. (2007) Nutritional anemia: B‐vitamins In Nutritional Anemia (eds Kraemer K. & Zimmermann M.B.), pp. 111–132. Sight and Life Press: Basel. [Google Scholar]
- Selhub J. (2008) Public health significance of elevated homocysteine. Food and Nutrition Bulletin 29, S116–S125. [DOI] [PubMed] [Google Scholar]
- Shane B. (2008) Folate and vitamin B12 metabolism: overview and interaction with riboflavin, vitamin B6, and polymorphisms. Food and Nutrition Bulletin 29, S5–16. [DOI] [PubMed] [Google Scholar]
- Sifakis S. & Pharmakides G. (2000) Anemia in pregnancy. Annals of the New York Academy of Sciences 900, 125–136. [DOI] [PubMed] [Google Scholar]
- Simmer K., Iles C.A., James C. & Thompson R.P. (1987) Are iron‐folate supplements harmful? American Journal of Clinical Nutrition 45, 122–125. [DOI] [PubMed] [Google Scholar]
- Sinclair K.D., Allegrucci C., Sinh R., Gardner D.S., Sebastian S., Bispham J., et al (2007) DNA methylation, insulin resistance, and blood pressure in offspring determined by maternal periconceptional B vitamin and methionine status. Proceedings of the National Academy of Sciences of the United States of America 104, 19351–19356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smits L.J. & Essed G.G. (2001) Short interpregnancy intervals and unfavourable pregnancy outcome: role of folate depletion. Lancet 358, 2074–2077. [DOI] [PubMed] [Google Scholar]
- Smulders Y.M., Smith D.E., Kok R.M., Teerlink T., Swinkels D.W., Stehouwer C.D., et al (2006) Cellular folate vitamer distribution during and after correction of vitamin B12 deficiency: a case for the methylfolate trap. British Journal of Haematology 132, 623–629. [DOI] [PubMed] [Google Scholar]
- Stabler S.P. & Allen R.H. (2004) Vitamin B12 deficiency as a worldwide problem. Annual Review of Nutrition 24, 299–326. [DOI] [PubMed] [Google Scholar]
- Steegers‐Theunissen R.P., Van Iersel C.A., Petronella P.G., Nelen W.L. & Steegers E.A. (2004) Hyperhomocysteinemia, pregnancy complications, and the timing of investigation. Obstetrics & Gynecology 104, 336–343. [DOI] [PubMed] [Google Scholar]
- Steen M.T., Bodie A.M., Fisher A.J., MacMahon W., Saxe D., Sullivan K.M., et al (1998) Neural‐tube defects are associated with low concentrations of cobalamin (Vitamin B12) in amniotic fluid. Prenatal Diagnosis 18, 545–555. [PubMed] [Google Scholar]
- Strathdee G., Simand A. & Brown R. (2004) Control of gene expression by CpG island methylation in normal cells. Biochemical Society Transactions 32, 913–915. [DOI] [PubMed] [Google Scholar]
- Suh J.R., Herbig A.K. & Stover P.J. (2001) New perspectives on folate catabolism. Annual Reviev of Nutrition 21, 255–282. [DOI] [PubMed] [Google Scholar]
- Suitor C.W. & Bailey L.B. (2000) Dietary folate equivalents: interpretation and application. Journal of the American Dietetic Association 100, 88–94. [DOI] [PubMed] [Google Scholar]
- Tamura T. & Picciano M.F. (2006) Folate and hyman reproduction. American Journal of Clinical Nutrition 83, 993–1016. [DOI] [PubMed] [Google Scholar]
- Taparia S., Gelineau‐van Waes J., Rosenquist T.H. & Finnell R.H. (2007) Importance of folate‐homocysteine homeostasis during early embryonic development. Clinical Chemistry and Laboratory Medicine 45, 1717–1727. [DOI] [PubMed] [Google Scholar]
- Torrens C., Brawley L., Anthony F.W., Dance C.S., Dunn R., Jackson A.A., et al (2006) Folate supplementation during pregnancy improves offspring cardiovascular dysfunction induced by protein restriction. Hypertension 47, 982–987. [DOI] [PubMed] [Google Scholar]
- Ulrich C.M. & Potter J.D. (2006) Folate supplementation: too much of a good thing? Cancer Epidemiology, Biomarkers & Prevention 15, 189–193. [DOI] [PubMed] [Google Scholar]
- Verkleij‐Hagoort A.C., Van Driel L.M., Lindemans J., Isaacs A., Steegers E.A., Helbing W.A., et al (2008) Genetic and lifestyle factors related to the periconception vitamin B12 status and congenital heart defects: a Dutch case – control study. Molecular Genetics and Metabolism 94, 112–119. [DOI] [PubMed] [Google Scholar]
- Vollset S.E., Refsum H., Irgens L.M., Emblem B.M., Tverdal A., Gjessing H.K., et al (2000) Plasma total homocysteine, pregnancy complications, and adverse pregnancy outcomes: the Hordaland Homocysteine Study. American Journal of Clinical Nutrition 71, 962–968. [DOI] [PubMed] [Google Scholar]
- Wald D.S., Bishop L., Wald N.J., Law M., Hennessy E., Weir D., et al (2001) Randomized trial of folic acid supplementation and serum homocysteine levels. Archives of Internal Medicine 161, 695–700. [DOI] [PubMed] [Google Scholar]
- Wen S.W., Chen X.K., Rodger M., White R.R., Yang Q., Smith G.N., et al (2008) Folic acid supplementation in early second trimester and the risk of preeclampsia. American Journal of Obstetrics and Gynecology 198, 45.e1–45.e7. [DOI] [PubMed] [Google Scholar]
- Wickramasinghe S.N. (2006) Diagnosis of megaloblastic anaemias. Blood Reviews 20, 299–318. [DOI] [PubMed] [Google Scholar]
- Wilcox A.J. (2001) On the importance‐and the unimportance‐of birthweight. International Journal of Epidemiology 30, 1233–1241. [DOI] [PubMed] [Google Scholar]
- Wolff G.L., Kodell R.L., Moore S.R. & Cooney C.A. (1998) Maternal epigenetics and methyl supplements affect agouti gene expression in Avy/a mice. FASEB Journal 12, 949–957. [PubMed] [Google Scholar]
- Zeisel S.H. (2009) Importance of methyl donors during reproduction. American Journal of Clinical Nutrition 89, 673S–677S. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H.Y., Luo G.A., Liang Q.L., Wang Y., Yang H.H., Wang Y.M., et al (2008) Neural tube defects and disturbed maternal folate‐ and homocysteine‐mediated one‐carbon metabolism. Experimental Neurology 212, 515–521. [DOI] [PubMed] [Google Scholar]
- Zhu H., Kartiko S. & Finnell R.H. (2009) Importance of gene–environment interactions in the etiology of selected birth defects. Clinical Genetics 75, 409–423. [DOI] [PubMed] [Google Scholar]
- Zimmermann M.B. & Shane B. (1993) Supplemental folic acid. American Journal of Clinical Nutrition 58, 127–128. [DOI] [PubMed] [Google Scholar]