

Background
Glucose transporter type 1 deficiency syndrome (GLUT1-DS) is caused by heterozygous mutations in the SLC2A1 gene. Glucose transport is impaired across the blood–brain barrier and into astrocytes. This eventually results in cerebral energy deficiency.1 Typically, GLUT1-DS is associated with developmental delay, permanent motor disorders and paroxysmal manifestations including epileptic and non-epileptic paroxysmal episodes.1 2 The phenotypic spectrum is however much wider: exercise-induced paroxysmal dyskinesia may sometimes be the main or sole manifestation of the disease.3 4
Ketogenic diet is the standard of care in GLUT1-DS,1 5 providing ketone bodies as an alternate source of energy to the brain. Other alternative treatments are needed as many patients have difficulties following the heavy constraints of this diet. Triheptanoin (UX007; Ultragenyx Pharmaceuticals, Novato, USA) is a medium odd-chain triglyceride containing three 7-carbon fatty acids with anaplerotic properties.6–8 Unlike even-chain fatty acids that can only generate acetyl-CoA, triheptanoin provides indeed both acetyl-CoA and propionyl-CoA, two key carbon sources for the Krebs cycle.9 10
We recently showed that triheptanoin dramatically reduced by 90% the number of non-epileptic paroxysmal manifestations over 2 months in GLUT1-DS and improved patient’s brain energy profile.9 Here, we wished to evaluate the long-term clinical efficacy of triheptanoin in children and adults with GLUT1-DS.
Methods
We extended our study protocol (NCT02014883) based on the striking short-term response with triheptanoin.9 All participants and/or their legal guardians signed a new informed consent. Among the six patients previously reported who completed the initial study,9 five patients (P1, P3–P6) opted to participate in the study extension. Patient P2, who prior to the study failed being on a ketogenic diet after a few months, declined the extension due to her strong appetite for glucose. Patient’s characteristics were the same as in the initial trial.9 Triheptanoin was reintroduced after the withdrawal phase from the initial study (figure 1A). The study was extended for 3 years, divided into four phases: (1) resumption short-term (6 months); (2) resumption mid-term (6 months); (3) second year of follow-up (1 year); and (4) third year of follow-up (1 year).
Figure 1.
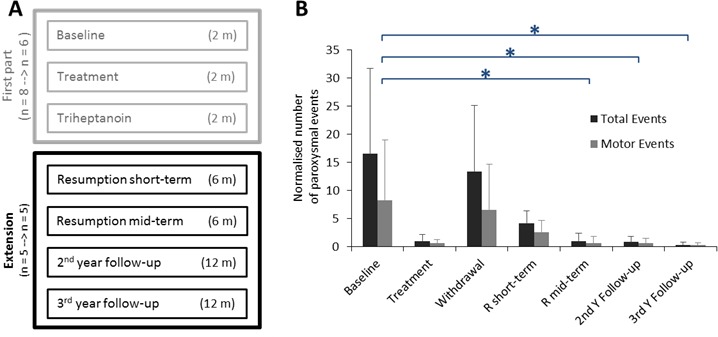
(A) Flow chart. (B) Normalised number of paroxysmal events in patients with glucose transporter type 1 deficiency syndrome (GLUT1-DS) during the different phases of the study. Total and motor numbers of paroxysmal events are expressed in mean per month. Error bars represent SEM. *P<0.05 (Dunn’s tests). m, number of months; n, number of patients.
Patient’s diet was isocaloric, but restricted in fast sugars, and triheptanoin, distributed during meals, represented about 30% of patient’s calorie intake.9 10 Dietary and therapeutic compliance was assessed by a trained dietitian at each visit.
During each study phase, the patients and/or their caregivers filled a comprehensive diary to record all motor and non-motor paroxysmal events, as well as their approximate duration in minutes.9 At each visit, diaries were comprehensively reviewed by the evaluating physician. The primary endpoint was the total number of paroxysmal events normalised over a month. Secondary endpoints were their mean duration, the number of motor events and the clinical Global Impression Severity Scale (CGI-S, range 0–7) completed by the patient at each visit. Compliance was verified with dosage of triheptanoin-derived metabolites in plasma (acylcarnitines and C5-keto acids) collected after an overnight fast.
For clinical parameters, Friedman tests were used to test the global hypothesis that study phases were equal. If significant, Dunn’s multiple comparison tests were applied for pairwise phase comparisons with an α of 0.05.
Results
Compared with baseline, triheptanoin reduced the number of monthly total paroxysmal events during the resumption mid-term phase (p=0.048) as well as during the second and third year of follow-up (p=0.036 and 0.004, respectively) (figure 1B, online supplementary table e1). This therapeutic response was similar to what we previously demonstrated during the initial treatment phase with triheptanoin.9 Furthermore, triheptanoin significantly reduced the number of monthly motor paroxysmal events during the second and third year of follow-up (p=0.028 and 0.016, respectively) (figure 1B, online supplementary table e1). Notably, compared with baseline, the number of paroxysmal events decreased during the resumption short-term phase of the study but that decrease did not reach significance (figure 1B).
jnnp-2018-320283supp001.pdf (115.8KB, pdf)
Triheptanoin tended to reduce the mean duration of total paroxysmal events, particularly during the second and third year of follow-up (online supplementary table e1). The same trend of improvement on triheptanoin was observed on the CGI-S score: on average, patients evolved from moderately ill at baseline to mildly ill after 2 or 3 years of treatment with triheptanoin (online supplementary table e1).
Plasma propionylcarnitine and C5-keto acid levels measured at the end of each period were higher after triheptanoin treatment (data not shown), reflecting proper metabolism of triheptanoin.
Although our study was not designed to evaluate cognitive performances, we observed an improvement of cognitive performance during the study extension. Compared with before baseline, P1 improved her total IQ (63 to >72), verbal IQ (62 to >72) and performance IQ (67 to >75) after 2 years of treatment with triheptanoin, and P3 improved his school performance.
Discussion
In five patients with GLUT1-DS, treatment with triheptanoin led to a dramatic (97%) and sustained reduction of motor and non-motor paroxysmal events over 3 years. The magnitude and duration of this response confirm the therapeutic benefit of triheptanoin and eliminate a placebo effect.
Proper compliance is key to obtaining good results. It means compliance to the prescribed triheptanoin dosage and to a low-sugar diet. Indeed, when triheptanoin was reintroduced, we observed that paroxysmal events were reduced compared with baseline but without reaching statistical significance, suggesting that the magnitude of the effect was less pronounced than in the initial study. We realised that the remaining events were associated with increased intake of fast sugars due to relaxed habits during the withdrawal phase. Renewed explanation and closer monitoring of sugar intake led to a significant reduction of paroxysmal events during the subsequent study phases. Over the last year of treatment, patients have become almost free of events. As for any dietary intervention, proper diet instructions and monitoring are mandatory for triheptanoin to provide its best efficacy. This is a major lesson for future trials using triheptanoin in GLUT1-DS or other diseases. Continued treatment with triheptanoin is currently provided to our patients through a temporary use of authorisation, but a study on a larger patient group should be encouraged.
Footnotes
Contributors: EH, AM, FM and ER drafted/revised the manuscript for content. FM designed the study. EH, DG, MPL, MA, MB and FM acquired data. EH and FM analysed/interpreted data. EH and MD performed the statistical analysis.
Funding: Ultragenyx Pharmaceutical provided the investigational drug triheptanoin and funding for the study. The research leading to these results has also received funding from the programme 'Investissements d’avenir' ANR-10-IAIHU-06.
Competing interests: FM holds a patent on the use of triheptanoin in GLUT1-DS (WO2014093901).
Patient consent for publication: Not required.
Ethics approval: The extension study was sponsored by INSERM and approved by the local ethical committee (ID RCB: 2013-A01300-45).
Provenance and peer review: Not commissioned; externally peer reviewed.
References
- 1. De Vivo DC, Trifiletti RR, Jacobson RI, et al. Defective glucose transport across the blood-brain barrier as a cause of persistent hypoglycorrhachia, seizures, and developmental delay. N Engl J Med 1991;325:703–9. 10.1056/NEJM199109053251006 [DOI] [PubMed] [Google Scholar]
- 2. Leen WG, Klepper J, Verbeek MM, et al. Glucose transporter-1 deficiency syndrome: the expanding clinical and genetic spectrum of a treatable disorder. Brain 2010;133:655–70. 10.1093/brain/awp336 [DOI] [PubMed] [Google Scholar]
- 3. Alter AS, Engelstad K, Hinton VJ, et al. Long-term clinical course of GLUT1 deficiency syndrome. J Child Neurol 2015;30:160–9. 10.1177/0883073814531822 [DOI] [PubMed] [Google Scholar]
- 4. Pons R, Collins A, Rotstein M, et al. The spectrum of movement disorders in GLUT-1 deficiency. Mov Disord 2010;25:275–81. 10.1002/mds.22808 [DOI] [PubMed] [Google Scholar]
- 5. Klepper J, Leiendecker B. GLUT1 deficiency syndrome and novel ketogenic diets. J Child Neurol 2013;28:1045–8. 10.1177/0883073813487600 [DOI] [PubMed] [Google Scholar]
- 6. Mochel F, DeLonlay P, Touati G, et al. Pyruvate carboxylase deficiency: clinical and biochemical response to anaplerotic diet therapy. Mol Genet Metab 2005;84:305–12. 10.1016/j.ymgme.2004.09.007 [DOI] [PubMed] [Google Scholar]
- 7. Adanyeguh IM, Rinaldi D, Henry P-G, et al. Triheptanoin improves brain energy metabolism in patients with Huntington disease. Neurology 2015;84:490–5. 10.1212/WNL.0000000000001214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Pascual JM, Liu P, Mao D, et al. Triheptanoin for glucose transporter type I deficiency (G1D): modulation of human ictogenesis, cerebral metabolic rate, and cognitive indices by a food supplement. JAMA Neurol 2014;71:1255–65. 10.1001/jamaneurol.2014.1584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mochel F, Hainque E, Gras D, et al. Triheptanoin dramatically reduces paroxysmal motor disorder in patients with GLUT1 deficiency. J Neurol Neurosurg Psychiatry 2016;87:550–3. 10.1136/jnnp-2015-311475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Mochel F. Triheptanoin for the treatment of brain energy deficit: a 14-year experience. J Neurosci Res 2017;95:2236–43. 10.1002/jnr.24111 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
jnnp-2018-320283supp001.pdf (115.8KB, pdf)