

Introduction
The regulatory mechanisms of vascular tone are well described, as are the changes that occur as a result of hypertension1, 2. In hypertension, pathological changes occur directly to the vascular endothelium, smooth muscle cells, adventitia, and perivascular adipose tissue (PVAT) that promote hyper-contractile and hypo-dilatory responses3. As a result, vascular dysfunction is defined as one of the hallmark characteristics of hypertension.
Uncontrolled activation of the immune system and low-grade systemic inflammation are common manifestations of hypertension4–7. The contribution of immune cells to hypertension-associated vascular dysfunction, via their infiltration into the vascular wall, has been confirmed by several laboratories7–10. Most recent evidence has led to a whole new realm of research by demonstrating that immune system receptors, expressed directly on cells of the vascular wall, are capable of providing immunoregulation of vascular function11. The main objective of this brief review is to discuss recent evidence on how immunoreceptor-mediated changes in the cells of the vasculature contribute to vascular pathophysiology in hypertension. First, we briefly describe the main characteristics of the arterial wall in hypertension and then present findings from experimental animal research to address the role of immunoreceptor-mediated vascular dysfunction in hypertension.
Arterial structure and function in hypertension
The endothelium is a dynamic regulator of vascular tone, hemostasis, vascular smooth muscle cell (VSMC) phenotype, and arterial wall inflammation. When functioning properly, the endothelium limits the development of hypertension through:
The secretion of vasodilatory, anticoagulant and anti-inflammatory factors (e.g., nitric oxide, prostacyclin, and endothelium-derived hyperpolarizing factor),
The formation of a barrier to the media layers of the artery essential in maintaining proper compartmentalization of the vascular and interstitial space,
The degradation of pro-contractile, pro-inflammatory, and pro-oxidative molecules via proteolytic enzymes and uptake transporters.
However, when these important homeostatic processes are perturbed, the endothelium is considered dysfunctional and can contribute to the development and/or maintenance of hypertension. Specifically, vascular endothelial cells can switch their production from primarily vasodilatory to vasoconstrictive factors (e.g., reactive oxygen species, thromboxane A2, endothelin)12, as well as change the predominant vasodilatory factor used to relax the artery (e.g., in hypertension, non-nitric oxide, non-prostacyclin endothelium-derived hyperpolarization increases its contribution to acetylcholine-induced relaxation because nitric oxide- and cyclooxygenase-dependent pathways become disarranged13). Furthermore, pro-inflammatory stimuli destabilize intercellular junctions between endothelial cells, causing the disruption of the barrier and increased junctional permeability. This facilitates transendothelial migration of immune cells, as well as solutes, into the intima, thus promoting vascular wall inflammation and interstitial edema14. Finally, the pro-contractile, pro-inflammatory, and pro-oxidative milieu is able to persist in hypertension due to endothelium-derived formation of these molecules, coupled with inefficient and ineffective clearance mechanisms. For example, clearance of low-density lipoprotein, prostaglandins, and endothelin-1 is reduced, generation of angiotensin II is amplified, and bradykinin is prematurely destroyed3, 15.
Vasoconstriction by VSMCs is the primary determinant of resistance to blood flow and thus, total peripheral resistance. These cells perform tonic and phasic contractions in response to receptor activation, mechano-sensing, and/or membrane depolarization16. For example, changes in load or length, sympathetic outflow from autonomic nervous system, and endocrine, paracrine, and/or autocrine factors can all independently regulate contractile responses of vascular smooth muscle. Nonetheless, regardless of the stimulus, VSMCs use myosin (thick filament) and actin (thin filament) to form cross bridges, initiate power strokes, and develop force. Calcium ions serve to initiate this myosin and actin interaction. Specifically, as the intracellular concentration of calcium increases, calcium combines with calmodulin, and this complex activates myosin light chain (MLC) kinase to phosphorylate the light chain of myosin. Contraction is maintained as cytosolic calcium is further increased from intracellular stores (sarcoplasmic reticulum), as well as entry from the extracellular space (receptor-operated calcium channels). Defects in the regulation of calcium and calcium signaling are well known to mediate hypertension-associated hyper-contractility and chronic increases in total peripheral resistance. These defects include: increased calcium entry, increased calcium storage, impaired intracellular buffering, and decreased calcium extrusion17.
A unique phenomenon observed in vascular smooth muscle is that the force and duration of contraction is far greater than that predicted by the actual intracellular calcium concentration18. In addition to MLC kinase, MLC phosphatase has a regulatory role in MLC phosphorylation. Activation of MLC phosphatase promotes smooth muscle relaxation via dephosphorylation of MLC. Inhibition of the enzymatic activity of MLC phosphatase, thereby causing the light chain of myosin to remain phosphorylated, promotes contraction via calcium-sensitization. This calcium-sensitizing effect has primarily been attributed to the activation of the small G protein, RhoA, and its downstream effector Rho kinase. RhoA cycles between an inactive GDP-bound and an active GTP-bound state in response to various stimuli. Upon activation, RhoA engages the enzyme Rho kinase, which phosphorylates myosin phosphatase, thereby inhibiting enzymatic activity and promoting phosphorylation of MLC. In hypertension, it has been observed that Rho kinase presents amplified activity and as such, pharmacological inhibition of Rho kinase induces relaxation in isolated arterial segments and lowers blood pressure in experimental models of hypertension19.
Chronic increases in total peripheral resistance, as observed in hypertension, can subsequently result in deleterious structural remodeling of the media layer of artery. Vascular remodeling can be classified as hypertrophic, eutrophic, or hypotrophic. Furthermore, remodeling can be inward (reduced luminal diameter) or outward (increased luminal diameter)20, 21. The most common remodeling in hypertension is inward remodeling, causing a reduction of the luminal diameter under passive conditions. Outward remodeling is generally seen during anti-hypertensive therapy and in conditions of increased flow21. Inward eutrophic remodeling occurs by repositioning VSMCs, which normalizes the circumferential stress of the artery exposed to increased blood pressure, and thus preserves wall cross-sectional area. It is suggested that the inward eutrophic remodeling precedes and prevents hypertrophy if the rearrangement effectively normalizes the circumferential wall stress22.
The adventitia is another determinant of vascular wall structure and consists of a heterogeneous population of cells amongst extracellular matrix (ECM; e.g., collagen, fibronectin, tenascin, osteopontin, and thrombospondin). Fibroblasts are considered the major cell type found in the adventitia, but resident progenitor cells, fibrocytes, immune cells (e.g., macrophage and dendritic cells), and adrenergic nerves also reside there23. While the production and deposition of ECM components is the major function of fibroblasts, they can also aid in the degradation of ECM components, and interact with endothelial cells, VSMCs, and immune cells via the secretion vasoactive molecules (e.g., growth factors, cytokines, chemokines, reactive oxygen species, and ATP) that could in turn modulate vascular function24. In response to hypertensive stimuli (e.g., hemodynamic stress, hypoxia), fibroblasts can become activated and differentiate toward a myofibroblast phenotype (it is also now recognized that a variety of other cells including epithelial, endothelial, and resident and circulating progenitor cells can also differentiate into myofibroblasts)23. Myofibroblasts secrete chemokines, cytokines and matricellular proteins involved in the recruitment of monocytes, lymphocytes and progenitor cells25. Over time, adhesion molecule expression promotes the retention of these cells within the adventitia, and some of these newly recruited cells can subsequently differentiate themselves into fibroblasts. This results in perpetual pro-inflammatory cycle and subsequently stiffening of small and large arteries26. Similar to medial layer remodeling, it has been proposed that in hypertension, adventitial remodeling occurs as a means to normalize circumferential wall stress27.
Similar to the adventitia, the PVAT is also composed of several different cell types, including adipocytes, vasculature cells (that circulate blood within PVAT), fibroblasts, immune cells, and possibly nerves28. Because of the close proximity between PVAT and the vascular wall, paracrine cross-talk can easily occur29. Healthy PVAT is known as having anti-contractile properties in most vascular beds30. These anti-contractile effects are caused by the release of vasodilatory factors, including adipokines (e.g., adiponectin and leptin), gaseous molecules (e.g., hydrogen sulfide and nitric oxide), and angiotensin 1–728. In hypertension, the anti-contractile effect of PVAT is not only lost, but it can also contribute to the hyper-contractile state31. This is caused by the production and release of contractile-inducing adipokines (e.g., chemerin), reactive oxygen species, angiotensin II, and cytokines28. Furthermore, PVAT can host infiltrating immune cells, and in hypertension this infiltration of activated immune cells is exacerbated32. These immune cells can influence vascular function directly through the release of cytokines, chemokines, and reactive oxygen species, and indirectly by causing an inflamed and dysfunctional PVAT.
Immune system activation and inflammation in hypertension
Although acute inflammation is an important defense mechanism against danger (e.g., infection) and facilitates wound healing, sustained, uncontrolled inflammation is associated with many chronic diseases, including hypertension. In fact, chronic, low grade, systemic inflammation has been proposed as a unifying etiological factor linking the three major organ systems responsible for the control of blood pressure- vasculature, kidneys, and brain. Nonetheless, the exact mechanisms that initiate this pathophysiological response, thereby contributing to further increases in blood pressure, are not well understood. While participation of T cells and other hematopoietic cells has been well described in the pathogenesis of hypertension4–7, what precisely activates T cells to mediate further increases in blood pressure has yet to be fully elucidated11. Furthermore, the signal(s) that instigates immune cell infiltration into organs important for the control of blood pressure has not been confirmed.
The innate immune system is the body’s early warning system that rapidly detects danger and damage33. The inflammatory milieu dictated by the innate immune system components directs the adaptive immune system and allows it time to mount a robust, antigen specific response. Of note, innate immune system components can be expressed on immune and non-immune (somatic) cells, including cells of the vasculature. Therefore, the tissues important for the control of blood pressure are likely the first to respond to perturbations in immunological homeostasis in hypertension, and as a result, alter their function34. This paradigm further extends the effects of uncontrolled immune system activation in the pathophysiology of hypertension. Danger signals that likely arise in hypertension include high salt, angiotensin II, misfolded proteins, and chronic stress. These factors could also cause small increases in peripheral vascular resistance, subsequently leading to ischemic- or pressure-induced cell injury and/or death, which could also contribute to the release of danger molecules and signals.
Pattern recognition receptors and Toll-like receptor 9 in hypertension
Important components of the innate immune system are sentinel pattern recognition receptors (PRRs). PRRs recognize evolutionarily conserved motifs; therefore, molecular patterns that range from microbe-associated molecular patterns (MAMPs) to damage-associated molecular patterns (DAMPs) can activate specific PRRs and distinct pro-inflammatory pathways35. Until recently, there was limited knowledge of the role of PRRs in the pathogenesis of cardiovascular diseases, including hypertension. As a result, various components of the PRRs, and in particular Toll-like receptors (TLRs), have become a significant research focus in the field of hypertension11, 36.
While we are interested in various TLRs, due to distinct reasons related to cardiovascular health and disease37–42, TLR9 has been of particular interest to us due to the elevated expression of mitochondrial DNA (mtDNA) in the circulation of different cardiovascular conditions, including hypertension42–44. Mitochondria carry hallmarks of their bacterial ancestry as an endosymbiont45, and one of these hallmarks is that its DNA consists of predominantly hypomethylated cytosine and guanine nucleotides (CpG); the specific motif that activates TLR9. Consistent with this notion, hypomethylated CpG dinucleotides are also a common characteristic of prokaryotic DNA, but not eukaryotic DNA. This specificity is important for preventing unintended TLR9 activation by self-nuclear DNA46.
To understand if TLR9 could be involved in the development and/or maintenance of hypertension, our initial experiments focused on whether TLR9 could alter vascular function using a pharmacological approach and standard myograph procedures42, 47. Specifically, we incubated isolated aortic segments from normotensive rats with TLR9 agonist, oligonucleotide (ODN)2395 (10 μmol/L) for 12 or 16 hours. TLR9 incubation increased aortic contractile responses to thromboxane mimetic U46619 (Figure 1A), and this response was independent of biological sex (Figure 1B). Subsequent studies involved experimentally manipulating TLR9 activity in vivo again testing aortic contractile responses. Specifically, we administered ODN2395 systemically to female normotensive rats (following the same protocol as reported in male rats42) and observed that TLR9 activation reduced nitric oxide bioavailability (Figure 2A, 2B, and 2C) and increased reactive oxygen species generation (Figure 2D and 2E) in aortic segments.
Figure 1.
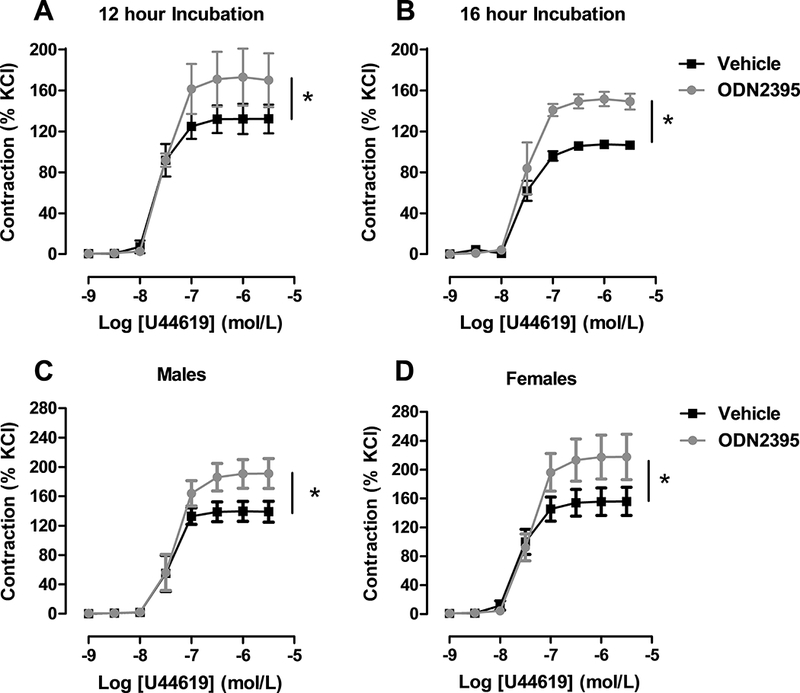
Isolated aortic segments from normotensive rats incubated with Toll-like receptor (TLR)9 agonist ODN2395 were more sensitive to thromboxane A2 mimetic U46619. Concentration-response curves to U46619 after incubation with vehicle or ODN2395 for (A) 12 (Emax, vehicle: 141±8 vs. ODN2395: 184±12) or (B) 16 hours (Emax, vehicle: 114±5 vs. ODN2395: 161±8). Concentration-response curves to U46619 in aorta from (C) male (Emax, vehicle: 139±7 vs. ODN2395: 190±9) and (D) female (Emax, vehicle: 154±7 vs. ODN2395: 216±12) rats after 12 hours incubation with vehicle or ODN2395. n=3–6. Non-linear regression analysis: *p<0.05 vs. vehicle.
Figure 2.
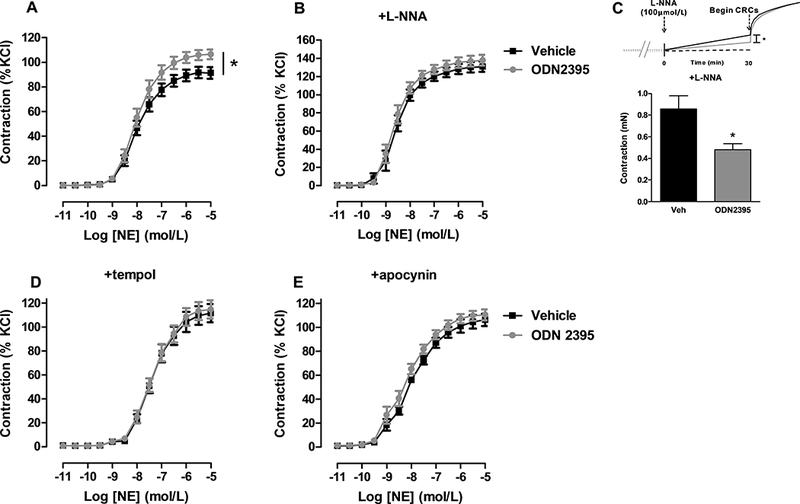
Isolated aortic segments from female normotensive rats treated systemically with Toll-like receptor (TLR)9 agonist ODN2395 are more sensitive to norepinephrine (NE) via decreased nitric oxide (NO) and increased reactive oxygen species (ROS). (A) Concentration-response curves to norepinephrine (NE) performed on aortic segments from normotensive rats treated with vehicle or ODN2395 (Emax, vehicle: 90±2 vs. ODN2395: 105±2). (B) Concentration-response curves to NE in the presence of NO synthase inhibitor L-NNA (100 μmol/L) (Emax, vehicle: 127±3 vs. ODN2395: 134±3). (C) Contractile tone after thirty minutes exposure to L-NNA (vehicle: 0.9±0.1 mN vs. ODN2395: 0.5±0.1 mN). Concentration-response curves to NE in the presence of reactive oxygen species inhibitors (C) tempol (1 mmol/L; Emax, vehicle: 111±4 vs. ODN2395: 115±4) and (D) apocynin (100 μmol/L; Emax, vehicle: 106±3 vs. ODN2395: 110±3). n=6–9. Non-linear regression analysis and student’s t-test: *p<0.05 vs. vehicle.
These data led to the hypothesis that TLR9 contributes to the etiology of hypertension11. Consequently, we subsequently inhibited TLR9 in spontaneously hypertensive rats (SHR) in vivo with the use of the ODN2088 (specific antagonist)42 and chloroquine (non-specific inhibitor)8, 47. Consistent with our initial observations, we observed that TLR9 inhibition in SHR was able to improve endothelial function of resistance and conduit arteries, and lower blood pressure.
Despite the clear contribution of TLR9 in hypertension-associated endothelial dysfunction, we wanted to understand if TLR9 activation on VSMCs caused defects in calcium homeostasis and thus enhanced contractile responses. Our curiosity for exploring this question was accentuated with the reports that TLR9 could impart an inhibitory influence on sarco/endoplasmic reticulum Ca2+-ATPase (SERCA2) activity, through a unique “non-canonical” signaling cascade specific for non-immune cells48, 49. Before activation TLR9 is localized to the endoplasmic reticulum (ER)50. Upon cellular recognition of its ligand in endosomes, canonical TLR9 signaling is initiated via its trafficking through the Golgi to endolysosomal compartments51, 52. TLR9-mediated inflammation is then mediated via adaptor protein, myeloid differentiation primary response gene 88 (MyD88)53. After proceeding through a complex, albeit TLR9-unique, intracellular signaling cascade, nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB), activator protein 1 (AP-1), and interferon regulatory factor (IRF)7 transcription factors augment pro-inflammatory gene transcription to promote a Th1 immune response (Figure 3A)53. In contrast, the recently reported “non-canonical” pathway for TLR9 was based on the rationale that Unc93b1, the chaperone-like protein that helps TLR9 traffic from the ER to endosome, is scantily expressed in non-immune cells48, 49. Therefore, endocytosed unmethylated CpG dinucleotides undergo retrograde transport from endosomes to the ER to activate TLR9. Uncleaved and ER-bound TLR9 subsequently decreases SERCA2 activity, leading to alterations in mitochondrial calcium handling and decreased ATP generation. Diminished ATP synthesis then stimulates the stress kinase 5’-AMP-activated protein kinase (AMPKα). Activation of AMPKα leads to stress tolerance, and thus this signaling cascade was appropriately named the TLR9 non-canonical “stress tolerance” signaling cascade (Figure 3B), and it was particularly unique to non-immune cells (cardiomyocytes and neurons specifically)48, 49.
Figure 3.
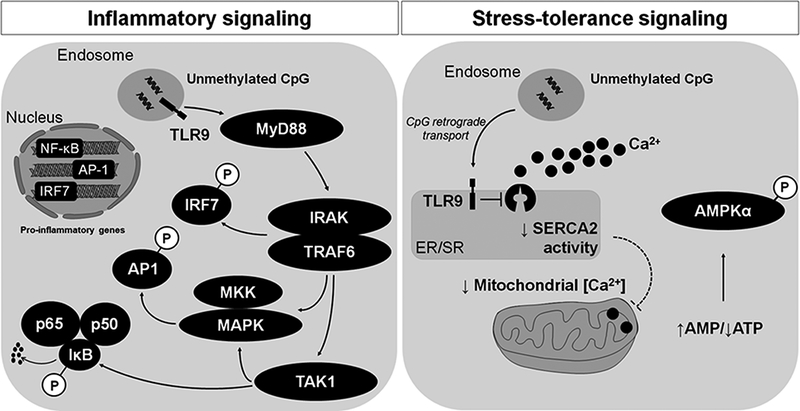
TLR9 has been proposed to signal through two independent cascades, (A) the canonical inflammatory pathway and (B) the non-canonical stress tolerance pathway. Signaling is initiated by TLR9 recognition of unmethylated CpG dinucleotides. In the canonical inflammatory pathway, this recognitions occurs after TLR9 trafficking to endolysosomes. In the non-canonical stress tolerance pathway, this recognition occurs after retrograde transport of unmethylated CpG dinucleotides to the endoplasmic reticulum where unstimulated TLR9 resides.
Our specific hypothesis was that in VSMCs, the non-canonical stress tolerance pathway for TLR9 activation inhibits SERCA2 contributing to enhanced contractile responses. Contrary to our hypothesis, and the previous reports in cardiomyocytes and neurons48, 49, we could not find any evidence of SERCA2 inhibition after TLR9 activation in VSMCs54. Nonetheless, consistent with our earlier work using ODN2395, we observed vascular dysfunction and it did involve calcium. Specifically we demonstrated that ODN2395 was able to induce calcium sensitization through Rho kinase and this process contributed to changes in the filamentous-to-globular actin ratio in VSMCs54. Therefore, we conclude that TLR9 is able to induce a dysfunction in the vasculature through endothelium- and smooth muscle-dependent mechanisms.
Unresolved questions regarding TLR9 in the vasculature include whether activation can contribute to fibrosis and collagen deposition in the adventitia layer of arteries from hypertensive animals, as well as if TLR9 on adipocytes contributes to the pro-contractile phenotypic transition of PVAT in hypertension. Our identification of Rho kinase activation downstream of TLR9 also raises the question as to whether cellular machinery generally reserved for vascular tone could also be involved in immune system activation. Calcium influx via STIM1 is important for T cell activation55 and hyper-contractility in SHR56. Thus, it is plausible that Rho kinase could modulate immune system regulation in hypertension through TLR9-independent mechanisms.
Our studies on TLR9 in experimental hypertension not only reveal mechanistic understanding of vascular dysfunction and immune system activation in this disease, they also propose novel therapeutic possibilities for patients with resistant hypertension and hypertension-associated pathologies (e.g., preeclampsia). While direct inhibition of immune system components such as PRRs or T cells is not clinically realistic, as this would immunocompromise an individual, targeted deletion of immunogenic factors such as DAMPs that contribute to unintended immune system activation in hypertension could be an intriguing option. For example, if we could degrade the elevated levels of mtDNA in the circulation of hypertensive patients42, 44, could we ameliorate the consequent vascular dysfunction and high blood pressure that would be a result of TLR9 activation? Preliminary studies performed by our group57 and others44 have suggested that mtDNA is a sensitive biomarker that can be exploited in human subjects. Nonetheless, whether its targeted removal can ameliorate the hypertensive phenotype has only been observed in genetically hypertensive rats thus far44.
Future directions surrounding uncontrolled immune system activation in hypertension
Novel mechanisms of inflammation and immune system hyperactivity in hypertension are constantly being revealed and being expanded upon. Therefore, this area of research is vast and has immeasurable potential for exploration. Here, we briefly propose some novel and alternative mechanisms that may mediate this pathophysiological response, including disturbances in the negative regulation of inflammation and the potential role for vascular resident immune cells and fibrocytes in hypertension-associated vascular dysfunction.
Negative regulation of immune system activity is an important physiological process named resolution of inflammation, and it is well known that disruptions to this process contribute to inflammatory disease states58. Resolution is an active phenomenon aimed at restoring tissue integrity and function via suppression of inflammatory processes once its objective (e.g. clearance of danger) is attained. In the context of immune cell-mediated resolution, chemokine expression decreases, neutrophils undergo apoptosis after performing their action at the inflamed site, and macrophages ingest apoptotic neutrophils59, 60. Clearance of apoptotic neutrophils prompts a switch from a pro- to an anti-inflammatory macrophage phenotype59. Although neutrophils and macrophages have traditionally been looked upon as dominant cell types during the resolution phase, additional cells such as myeloid-derived suppressor cells and T regulatory cells have more recently emerged as important players during resolution and may link innate and adaptive immune systems58. Furthermore, in line with earlier observations that arteries themselves can mediate pro-inflammatory responses in hypertension, it is possible that tissues themselves can also promote resolution via the production of pro-resolving factors. In fact, it is well established that the endothelium is fundamental to the resolution of injury through the initial expression of chemotactic cytokines and adhesion molecules that allow the recruitment and adherence of neutrophils and monocytes61, and subsequent secretion of anti-inflammatory cytokines62. Potentially novel mediators of resolution include metabolites of arachidonic acid and lipid mediators can promote and/or accelerate resolution of vascular inflammation directly in cells of the vasculature and indirectly via immune cell suppresion61, 63. These mediators include the lipoxins, resolvins, and protectins, which are derived from the omega-3 essential fatty acids eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA). Interestingly, a less known anti-inflammatory mechanism of action of aspirin is the acetylation of cyclooxygenase that not only redirects its catalytic activity from generating prostaglandins and thromboxane, but also promotes the production of lipoxin A4, an anti-inflammatory and pro-resolving agent64.
Another important discovery that has yet to be defined in the vasculature or in the context of hypertension is whether vascular resident immune cells can electrically couple with vascular cells and mediate reactivity. This hypothesis came to light with the recent discovery that tissue resident macrophages can electrically couple with surrounding cardiomyocytes and directly regulate cardiac rhythm65 and prune synaptic connections in the brain66. Evolutionarily, the presence of these cells in vital end organs may be to efficiently promote inflammation and subsequent regeneration post-insult. Nonetheless, several outstanding questions still remain, including:
Do immune cells reside in the healthy (non-diseased) vasculature and if so, how do vascular-resident immune cells affect homeostatic tissue function?
Do vascular-resident immune cells promote vascular dysfunction in hypertension via electrically coupling with endothelial cells and/or VSMCs?
Can vascular tissue resident immune cells be therapeutically targeted?
Interestingly, and pertinent to hypertension, a subset of non-circulatory and tissue resident T cells was recently identified in the livers of salmonella infected mice67. Therefore, we hypothesize that T cells could also permanently reside in the vasculature of hypertensive animals and humans and modulate function.
Finally, a unique cell type that has not been explored in the context of hypertension-associated vascular dysfunction is fibrocytes. Fibrocytes are a group of bone marrow-derived mesenchymal progenitor cells that can infiltrate end organs and differentiate into myofibroblasts that promote ECM deposition and fibrosis. Fibrocytes were first described as a circulating monocyte-derived cell capable of expressing a fibroblast phenotype68. Functionally, they migrate from the bone marrow to regions of tissue injury. These cells are unique in their expression of ECM proteins concomitantly with markers of hematopoietic and monocyte-macrophage lineage69. Fibrocytes contribute to the process of wound repair via several mechanisms including the production of cytokines, chemokines, and growth factors, serve as antigen presenting cells (APCs), and promotion of angiogenesis. Nonetheless, we postulate that uncontrolled fibrocyte-derived fibrosis is a novel immunovascular interaction that promotes deleterious vascular remodeling and stiffening in hypertension. It is already known that fibrocytes promote chronic disease via uncontrolled inflammation70 and circulating fibrocytes are associated with left ventricular hypertrophy in patients with hypertensive heart disease71.
Conclusion
In summary, revelation of the participation of TLRs in vascular dysfunction and hypertension proposes a mechanism by which the adaptive immune system can become activation and T cell infiltration into the vascular wall. Furthermore, our studies have revealed that innate immune system immunoreceptors, specifically TLR9 expressed on vascular cells, can mediate vascular function through their pro-inflammatory/pro-oxidative signaling pathways37–42, as well as their intersection with cellular machinery traditionally reserved for the contractile tone of the artery54. Nonetheless, as our future questions allude, TLRs41, 42 and T cells7, are only the beginning of our understanding of immunoregulation of vascular dysfunction in hypertension and many questions still remain (Figure 4).
Figure 4.
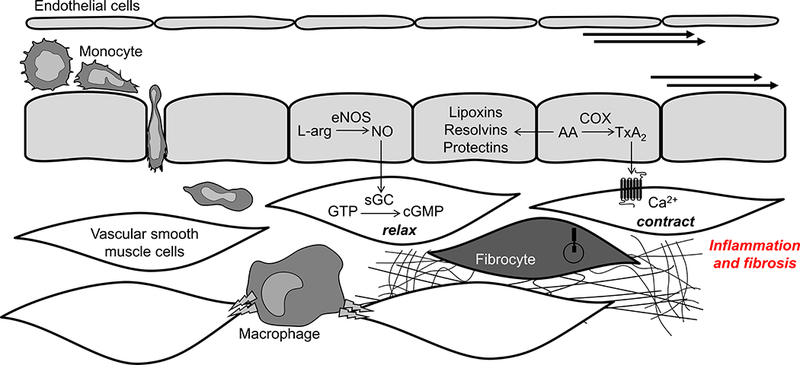
Vascular inflammation in hypertension: Outstanding questions and future directions. Dysregulated arachidonic acid metabolism in hypertension could impair the resolution of inflammation process via decreased production of endothelium-derived pro-resolving factors (e.g., lipoxins, resolvins, and protectins). Hypertension could stimulate the activity of vascular residing immune cells, such as macrophages or T cells, which could modulate vascular dysfunction by direct electrical coupling with vascular cells and/or production of pro-inflammatory cytokines directly into the vascular wall. Finally, newly identified components of hypertension-associated end organ injury, fibrocytes, could significantly contribute to vascular fibrosis and stiffening.
Abbreviations: AA, arachidonic acid; cGMP, cyclic guanosine monophosphate; COX, cyclooxygenase; eNOS, endothelial nitric oxide synthase; GTP, Guanosine-5’-triphosphate; L-arg, L-arginine; NO, nitric oxide; sGC, soluble guanylyl cyclase; TxA2; thromboxane A2
Acknowledgments
SOURCES OF FUNDING
This work was supported by the AHA (18POST34060003 and 13SDG17050056) and NIH (P01HL134604).
Footnotes
CONFLICTS OF INTEREST/DISCLOSURES
None.
REFERENCES
- 1.Bohr DF, Dominiczak AF, Webb RC. Pathophysiology of the vasculature in hypertension. Hypertension. 1991;18:III69–75. [DOI] [PubMed] [Google Scholar]
- 2.Webb RC, Bohr DF. Regulation of vascular tone, molecular mechanisms. Prog Cardiovasc Dis. 1981;24:213–242. [DOI] [PubMed] [Google Scholar]
- 3.Martinez-Quinones P, McCarthy CG, Watts SW, Klee NS, Komic A, Calmasini FB, Priviero F, Warner A, Chenghao Y, Wenceslau CF. Hypertension induced morphological and physiological changes in cells of the arterial wall. Am J Hypertens. 2018;31:1067–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.White FN, Grollman A. Autoimmune factors associated with infarction of the kidney. Nephron. 1964;1:93–102. [DOI] [PubMed] [Google Scholar]
- 5.Okuda T, Grollman A. Passive transfer of autoimmune induced hypertension in the rat by lymph node cells. Tex Rep Biol Med. 1967;25:257–264. [PubMed] [Google Scholar]
- 6.Crowley SD, Song YS, Lin EE, Griffiths R, Kim HS, Ruiz P. Lymphocyte responses exacerbate angiotensin II-dependent hypertension. Am J Physiol Regul Integr Comp Physiol. 2010;298:R1089–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Guzik TJ, Hoch NE, Brown KA, McCann LA, Rahman A, Dikalov S, Goronzy J, Weyand C, Harrison DG. Role of the T cell in the genesis of angiotensin II induced hypertension and vascular dysfunction. J Exp Med. 2007;204:2449–2460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McCarthy CG, Wenceslau CF, Goulopoulou S, Baban B, Matsumoto T, Webb RC. Chloroquine suppresses the development of hypertension in spontaneously hypertensive rats. Am J Hypertens. 2017;30:173–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wei Z, Spizzo I, Diep H, Drummond GR, Widdop RE, Vinh A. Differential phenotypes of tissue-infiltrating T cells during angiotensin II-induced hypertension in mice. PLoS One. 2014;9:e114895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mikolajczyk TP, Nosalski R, Szczepaniak P, Budzyn K, Osmenda G, Skiba D, Sagan A, Wu J, Vinh A, Marvar PJ, Guzik B, Podolec J, Drummond G, Lob HE, Harrison DG, Guzik TJ. Role of chemokine RANTES in the regulation of perivascular inflammation, T-cell accumulation, and vascular dysfunction in hypertension. FASEB J. 2016;30:1987–1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McCarthy CG, Goulopoulou S, Wenceslau CF, Spitler K, Matsumoto T, Webb RC. Toll-like receptors and damage-associated molecular patterns: Novel links between inflammation and hypertension. Am J Physiol Heart Circ Physiol. 2014;306:H184–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Versari D, Daghini E, Virdis A, Ghiadoni L, Taddei S. Endothelium-dependent contractions and endothelial dysfunction in human hypertension. Br J Pharmacol. 2009;157:527–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ozkor MA, Quyyumi AA. Endothelium-derived hyperpolarizing factor and vascular function. Cardiol Res Pract. 2011;2011:156146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chistiakov DA, Orekhov AN, Bobryshev YV. Endothelial barrier and its abnormalities in cardiovascular disease. Front Physiol. 2015;6:365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Eelen G, de Zeeuw P, Simons M, Carmeliet P. Endothelial cell metabolism in normal and diseased vasculature. Circ Res. 2015;116:1231–1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Webb RC. Smooth muscle contraction and relaxation. Adv Physiol Educ. 2003;27:201–206. [DOI] [PubMed] [Google Scholar]
- 17.Goulopoulou S, Webb RC. Symphony of vascular contraction: How smooth muscle cells lose harmony to signal increased vascular resistance in hypertension. Hypertension. 2014;63:e33–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.DeFeo TT, Morgan KG. Calcium-force relationships as detected with aequorin in two different vascular smooth muscles of the ferret. J Physiol. 1985;369:269–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Uehata M, Ishizaki T, Satoh H, Ono T, Kawahara T, Morishita T, Tamakawa H, Yamagami K, Inui J, Maekawa M, Narumiya S. Calcium sensitization of smooth muscle mediated by a rho-associated protein kinase in hypertension. Nature. 1997;389:990–994. [DOI] [PubMed] [Google Scholar]
- 20.Folkow B, Grimby G, Thulesius O. Adaptive structural changes of the vascular walls in hypertension and their relation to the control of the peripheral resistance. Acta Physiol Scand. 1958;44:255–272. [DOI] [PubMed] [Google Scholar]
- 21.Mulvany MJ. Vascular remodelling of resistance vessels: Can we define this? Cardiovasc Res. 1999;41:9–13. [DOI] [PubMed] [Google Scholar]
- 22.Castorena-Gonzalez JA, Staiculescu MC, Foote C, Martinez-Lemus LA. Mechanisms of the inward remodeling process in resistance vessels: Is the actin cytoskeleton involved? Microcirculation. 2014;21:219–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stenmark KR, Yeager ME, El Kasmi KC, Nozik-Grayck E, Gerasimovskaya EV, Li M, Riddle SR, Frid MG. The adventitia: Essential regulator of vascular wall structure and function. Annu Rev Physiol. 2013;75:23–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gutterman DD. Adventitia-dependent influences on vascular function. Am J Physiol. 1999;277:H1265–72. [DOI] [PubMed] [Google Scholar]
- 25.Buckley CD, Pilling D, Lord JM, Akbar AN, Scheel-Toellner D, Salmon M. Fibroblasts regulate the switch from acute resolving to chronic persistent inflammation. Trends Immunol. 2001;22:199–204. [DOI] [PubMed] [Google Scholar]
- 26.McGrath JC, Deighan C, Briones AM, Shafaroudi MM, McBride M, Adler J, Arribas SM, Vila E, Daly CJ. New aspects of vascular remodelling: The involvement of all vascular cell types. Exp Physiol. 2005;90:469–475. [DOI] [PubMed] [Google Scholar]
- 27.Schulze-Bauer CA, Regitnig P, Holzapfel GA. Mechanics of the human femoral adventitia including the high-pressure response. Am J Physiol Heart Circ Physiol. 2002;282:H2427–40. [DOI] [PubMed] [Google Scholar]
- 28.Szasz T, Webb RC. Perivascular adipose tissue: More than just structural support. Clin Sci (Lond). 2012;122:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rajsheker S, Manka D, Blomkalns AL, Chatterjee TK, Stoll LL, Weintraub NL. Crosstalk between perivascular adipose tissue and blood vessels. Curr Opin Pharmacol. 2010;10:191–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Soltis EE, Cassis LA. Influence of perivascular adipose tissue on rat aortic smooth muscle responsiveness. Clin Exp Hypertens A. 1991;13:277–296. [DOI] [PubMed] [Google Scholar]
- 31.Galvez B, de Castro J, Herold D, Dubrovska G, Arribas S, Gonzalez MC, Aranguez I, Luft FC, Ramos MP, Gollasch M, Fernandez Alfonso MS. Perivascular adipose tissue and mesenteric vascular function in spontaneously hypertensive rats. Arterioscler Thromb Vasc Biol. 2006;26:1297–1302. [DOI] [PubMed] [Google Scholar]
- 32.Guzik TJ, Skiba DS, Touyz RM, Harrison DG. The role of infiltrating immune cells in dysfunctional adipose tissue. Cardiovasc Res. 2017;113:1009–1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Matzinger P The danger model: A renewed sense of self. Science. 2002;296:301–305. [DOI] [PubMed] [Google Scholar]
- 34.Matzinger P, Kamala T. Tissue-based class control: The other side of tolerance. Nat Rev Immunol. 2011;11:221–230. [DOI] [PubMed] [Google Scholar]
- 35.Newton K, Dixit VM. Signaling in innate immunity and inflammation. Cold Spring Harb Perspect Biol. 2012;4: 10.1101/cshperspect.a006049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Singh MV, Chapleau MW, Harwani SC, Abboud FM. The immune system and hypertension. Immunol Res. 2014;59:243–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nunes KP, de Oliveira AA, Szasz T, Biancardi VC, Webb RC. Blockade of toll-like receptor 4 attenuates erectile dysfunction in diabetic rats. J Sex Med. 2018;15:1235–1245. [DOI] [PubMed] [Google Scholar]
- 38.Stallmann-Jorgensen I, Ogbi S, Szasz T, Webb RC. A toll-like receptor 1/2 agonist augments contractility in rat corpus cavernosum. J Sex Med. 2015;12:1722–1731. [DOI] [PubMed] [Google Scholar]
- 39.Szasz T, Wenceslau CF, Burgess B, Nunes KP, Webb RC. Toll-like receptor 4 activation contributes to diabetic bladder dysfunction in a murine model of type 1 diabetes. Diabetes. 2016;65:3754–3764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Thompson JA, Hardigan TA, Carrillo-Sepulveda MA, Mintz JD, Ergul A, Diamond MP, Webb RC. The contribution of toll-like receptors to placental inflammation in diet-induced maternal obesity. Placenta. 2015;36:1204–1206. [DOI] [PubMed] [Google Scholar]
- 41.Bomfim GF, Dos Santos RA, Oliveira MA, Giachini FR, Akamine EH, Tostes RC, Fortes ZB, Webb RC, Carvalho MH. Toll-like receptor 4 contributes to blood pressure regulation and vascular contraction in spontaneously hypertensive rats. Clin Sci (Lond). 2012;122:535–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.McCarthy CG, Wenceslau CF, Goulopoulou S, Ogbi S, Baban B, Sullivan JC, Matsumoto T, Webb RC. Circulating mitochondrial DNA and toll-like receptor 9 are associated with vascular dysfunction in spontaneously hypertensive rats. Cardiovasc Res. 2015;107:119–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Oka T, Hikoso S, Yamaguchi O, Taneike M, Takeda T, Tamai T, Oyabu J, Murakawa T, Nakayama H, Nishida K, Akira S, Yamamoto A, Komuro I, Otsu K. Mitochondrial DNA that escapes from autophagy causes inflammation and heart failure. Nature. 2012;485:251–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Veiko NN, Konorova IL, Neverova ME, Fidelina OV, Mkrtumova NA, Ershova ES, Kon’kova MS, Postnov AI. Delayed appearance of hypertension in spontaneously hypertensive rat (SHR) injected with CpG-rich DNA early in ontogenesis. Biomed Khim. 2010;56:686–699. [DOI] [PubMed] [Google Scholar]
- 45.Sagan L On the origin of mitosing cells. J Theor Biol. 1967;14:255–274. [DOI] [PubMed] [Google Scholar]
- 46.Stacey KJ, Young GR, Clark F, Sester DP, Roberts TL, Naik S, Sweet MJ, Hume DA. The molecular basis for the lack of immunostimulatory activity of vertebrate DNA. J Immunol. 2003;170:3614–3620. [DOI] [PubMed] [Google Scholar]
- 47.McCarthy CG, Wenceslau CF, Goulopoulou S, Ogbi S, Matsumoto T, Webb RC. Autoimmune therapeutic chloroquine lowers blood pressure and improves endothelial function in spontaneously hypertensive rats. Pharmacol Res. 2016;113:384–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shintani Y, Kapoor A, Kaneko M, Smolenski RT, D’Acquisto F, Coppen SR, Harada-Shoji N, Lee HJ, Thiemermann C, Takashima S, Yashiro K, Suzuki K. TLR9 mediates cellular protection by modulating energy metabolism in cardiomyocytes and neurons. Proc Natl Acad Sci U S A. 2013;110:5109–5114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shintani Y, Drexler HC, Kioka H, Terracciano CM, Coppen SR, Imamura H, Akao M, Nakai J, Wheeler AP, Higo S, Nakayama H, Takashima S, Yashiro K, Suzuki K. Toll-like receptor 9 protects non-immune cells from stress by modulating mitochondrial ATP synthesis through the inhibition of SERCA2. EMBO Rep. 2014;15:438–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Leifer CA, Kennedy MN, Mazzoni A, Lee C, Kruhlak MJ, Segal DM. TLR9 is localized in the endoplasmic reticulum prior to stimulation. J Immunol. 2004;173:1179–1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chockalingam A, Brooks JC, Cameron JL, Blum LK, Leifer CA. TLR9 traffics through the golgi complex to localize to endolysosomes and respond to CpG DNA. Immunol Cell Biol. 2009;87:209–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Latz E, Schoenemeyer A, Visintin A, Fitzgerald KA, Monks BG, Knetter CF, Lien E, Nilsen NJ, Espevik T, Golenbock DT. TLR9 signals after translocating from the ER to CpG DNA in the lysosome. Nat Immunol. 2004;5:190–198. [DOI] [PubMed] [Google Scholar]
- 53.Klinman DM. Immunotherapeutic uses of CpG oligodeoxynucleotides. Nat Rev Immunol. 2004;4:249–258. [DOI] [PubMed] [Google Scholar]
- 54.McCarthy CG, Wenceslau CF, Ogbi S, Szasz T, Webb RC. Toll-like receptor 9-dependent AMPKalpha activation occurs via TAK1 and contributes to RhoA/ROCK signaling and actin polymerization in vascular smooth muscle cells. J Pharmacol Exp Ther. 2018;365:60–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Oh-Hora M, Yamashita M, Hogan PG, Sharma S, Lamperti E, Chung W, Prakriya M, Feske S, Rao A. Dual functions for the endoplasmic reticulum calcium sensors STIM1 and STIM2 in T cell activation and tolerance. Nat Immunol. 2008;9:432–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Giachini FR, Chiao CW, Carneiro FS, Lima VV, Carneiro ZN, Dorrance AM, Tostes RC, Webb RC. Increased activation of stromal interaction molecule-1/orai-1 in aorta from hypertensive rats: A novel insight into vascular dysfunction. Hypertension. 2009;53:409–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Angiotensin II-mediated increases in damage-associated molecular patterns during acute mental stress: Erratum. Psychosom Med. 2018;80:590. [DOI] [PubMed] [Google Scholar]
- 58.Ortega-Gomez A, Perretti M, Soehnlein O. Resolution of inflammation: An integrated view. EMBO Mol Med. 2013;5:661–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Fadok VA, Bratton DL, Konowal A, Freed PW, Westcott JY, Henson PM. Macrophages that have ingested apoptotic cells in vitro inhibit proinflammatory cytokine production through autocrine/paracrine mechanisms involving TGF-beta, PGE2, and PAF. J Clin Invest. 1998;101:890–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Michlewska S, Dransfield I, Megson IL, Rossi AG. Macrophage phagocytosis of apoptotic neutrophils is critically regulated by the opposing actions of pro-inflammatory and anti-inflammatory agents: Key role for TNF-alpha. FASEB J. 2009;23:844–854. [DOI] [PubMed] [Google Scholar]
- 61.Serhan CN, Chiang N, Van Dyke TE. Resolving inflammation: Dual anti-inflammatory and pro-resolution lipid mediators. Nat Rev Immunol. 2008;8:349–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Shao Y, Cheng Z, Li X, Chernaya V, Wang H, Yang XF. Immunosuppressive/anti-inflammatory cytokines directly and indirectly inhibit endothelial dysfunction--a novel mechanism for maintaining vascular function. J Hematol Oncol. 2014;7:80-014-0080-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Romano M, Cianci E, Simiele F, Recchiuti A. Lipoxins and aspirin-triggered lipoxins in resolution of inflammation. Eur J Pharmacol. 2015;760:49–63. [DOI] [PubMed] [Google Scholar]
- 64.Claria J, Serhan CN. Aspirin triggers previously undescribed bioactive eicosanoids by human endothelial cell-leukocyte interactions. Proc Natl Acad Sci U S A. 1995;92:9475–9479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hulsmans M, Clauss S, Xiao L, Aguirre AD, King KR, Hanley A, Hucker WJ, Wulfers EM, Seemann G, Courties G, Iwamoto Y, Sun Y, Savol AJ, Sager HB, Lavine KJ, Fishbein GA, Capen DE, Da Silva N, Miquerol L, Wakimoto H, Seidman CE, Seidman JG, Sadreyev RI, Naxerova K, Mitchell RN, Brown D, Libby P, Weissleder R, Swirski FK, Kohl P, Vinegoni C, Milan DJ, Ellinor PT, Nahrendorf M. Macrophages facilitate electrical conduction in the heart. Cell. 2017;169:510–522. e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Paolicelli RC, Bolasco G, Pagani F, Maggi L, Scianni M, Panzanelli P, Giustetto M, Ferreira TA, Guiducci E, Dumas L, Ragozzino D, Gross CT. Synaptic pruning by microglia is necessary for normal brain development. Science. 2011;333:1456–1458. [DOI] [PubMed] [Google Scholar]
- 67.Benoun JM, Peres NG, Wang N, Pham OH, Rudisill VL, Fogassy ZN, Whitney PG, Fernandez-Ruiz D, Gebhardt T, Pham QM, Puddington L, Bedoui S, Strugnell RA, McSorley SJ. Optimal protection against salmonella infection requires noncirculating memory. Proc Natl Acad Sci U S A. 2018;115:10416–10421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bucala R, Spiegel LA, Chesney J, Hogan M, Cerami A. Circulating fibrocytes define a new leukocyte subpopulation that mediates tissue repair. Mol Med. 1994;1:71–81. [PMC free article] [PubMed] [Google Scholar]
- 69.Pilling D, Fan T, Huang D, Kaul B, Gomer RH. Identification of markers that distinguish monocyte-derived fibrocytes from monocytes, macrophages, and fibroblasts. PLoS One. 2009;4:e7475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Reilkoff RA, Bucala R, Herzog EL. Fibrocytes: Emerging effector cells in chronic inflammation. Nat Rev Immunol. 2011;11:427–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Keeley EC, Mehrad B, Janardhanan R, Salerno M, Hunter JR, Burdick MM, Field JJ, Strieter RM, Kramer CM. Elevated circulating fibrocyte levels in patients with hypertensive heart disease. J Hypertens. 2012;30:1856–1861. [DOI] [PMC free article] [PubMed] [Google Scholar]