

Abstract
Epidemiological and clinical observations have led to the hypothesis that the risk of developing some chronic diseases in adulthood is influenced not only by genetic and adult lifestyle factors, but also by environmental factors acting in early life. These factors act through the processes of developmental plasticity and possibly epigenetic modification, and can be distinguished from developmental disruption. The concept of predictive adaptation has been developed to explain the relationship between early life events and the risk of later disease. At its base, the model suggests that a mismatch between fetal expectation of its postnatal environment and actual postnatal environment contribute to later adult disease risk. This mismatch is exacerbated, in part, by the phenomenon of ‘maternal constraint’ on fetal growth, which implicitly provides an upper limit of postnatal nutritional environment that humans have adapted for and is now frequently exceeded. These experimental, clinical and conceptual considerations have important implications for prevention and intervention in the current epidemic of childhood obesity and adult metabolic and cardiovascular disorders.
Keywords: predictive adaptive response, evolution, metabolic, fetal growth, developmental plasticity
Introduction
The developmental origins of adult disease paradigm has its origins in the epidemiological associations made by Barker and colleagues between lower birth size and a greater risk of death in middle age from cardiovascular disease (Barker & Osmond, 1986) and Type 2 diabetes mellitus (Ravelli et al., 1998). To a large extent, the validity of the concept in the mind of many remains based on these epidemiological associations, but as this review will demonstrate, the model is also supported by extensive clinical and experimental data and has an increasingly solid conceptual and mechanistic basis.
While there have now been many papers showing inverse relationships between measures of birth size and later risks of insulin resistance or hypertension, few studies have actually incorporated disease manifestation into their reports. This muddled use of surrogate markers and biological end points has led to controversy and criticism within the epidemiological analyses. The confusion has arisen because the critics have focused on the association between indices of birth size (usually in weight) and surrogate measures (such as blood pressure), which only poorly reflect the later risk of disease, and have also been based on the misperception that the focus is about birth size. The developmental origins concept is not based on a causal role for birth size, but on the consequences of fetal responses to its environment. But because fetal growth and subsequent birth size can act as indicators of an adverse fetal environment, statistical relationships can be found. Only one estimate so far has allowed for any calculation of the possible importance of early life factors in the development of these diseases in later life (Barker et al., 2002), and it suggests a very major role for both fetal and childhood factors. Studies such as this, where long‐term disease risk in adult life is to be determined, require a stable cohort whose members can be followed from birth to their mature years (age 70+) to assess their disease risk. These are difficult studies to plan and maintain. Given the funding limitations and time constraints, it is more likely that we will have to continue relying on further estimates based on surrogate measures or on alternate approaches based on extrapolation from other clinical or experimental data.
The major interpretative problems have arisen from the focus on birth size. While it was fortuitous that a relationship was found between birth size and the later disease risk, the weight of data strongly suggest that size itself is not part of the causal pathway leading to disease. Instead, the consensus is that the pathway is one of early environmental cues inducing changes in functional development that, in turn, alter disease risk. Using fetal growth as a marker of an adverse intrauterine environment is a convenient but imprecise marker, namely because not all fetuses exposed to an abnormal fetal environment have altered growth, and not all altered fetal growth is a function of responses to environmental stimuli (reviewed in Harding, 2001).
Thus, the current working model is one whereby early life events, acting through the processes of developmental plasticity, alter development of the organism to such an extent that it affects its capacity to cope with the environment of adult life. Experimental data suggest that the period in which these early life events influence lifelong consequences can extend from conception (and possibly preconception) to infancy, depending on the organ system involved. It is this possible postnatal component that led to the change in nomenclature from fetal to developmental origins. However, there is ongoing controversy as to the relative importance of the fetal versus the postnatal components (Gluckman et al., 2005), and indeed it remains to be elucidated as to whether they are independent or interdependent influences.
This brief review considers these issues and places the underlying biology in the broader perspective of evolutionary and life history concepts. It will also demonstrate that there are robust experimental data that support the general model and indicate how this information may inform different strategies to improve lifelong health in various populations and contexts.
The epidemiology
The epidemiological data relating events of early life to heart disease and insulin resistance is extensively reviewed elsewhere (Osmond & Barker, 2000). However, a key point to note across the studies is that any association between birth size and disease risk is continuous across all birth sizes. In other words, that disease risk is not restricted solely to those born small. We will revisit this point later in our discussion.
As the relationship between causal factors and outcomes spans the human lifespan, prospective validation of the general model in humans is essentially impossible. There are, however, prospective data showing that children of lower birthweights develop insulin resistance in childhood (Hofman et al., 1997; Ong et al., 2004). Similarly, twins develop insulin resistance in childhood and this suggests that the constrained intrauterine environment of multiple pregnancy is sufficient to induce physiological changes in the fetus that lead to permanent change in metabolic regulation (Jefferies et al., 2004). More recently it has been demonstrated that infants born prematurely also develop insulin resistance in childhood (Hofman et al., 2004). The finding that there can be a lifelong impact of prematurity (24–34 weeks of gestation) suggests that programming can be induced by multiple forms of environmental cue acting in utero. Alterations in fetal growth or in maturation are two alternate, but not mutually exclusive, responses of the fetus to an adverse environment. While the early epidemiological data focused on birth size, such studies generally grouped all neonates born at >36 weeks gestation as ‘term infants’. It is indeed possible that subtle changes in maturation within this range may influence long‐term health risks.
There is an increasing body of knowledge suggesting parallel early life influences on the risks of developing osteoporosis (Dennison et al., 2001). In addition, a range of other diseases have been suggested as associated with early environmental insults as reflected in low birthweight, including mood disorders (Thompson et al., 2001), schizophrenia (Wahlbeck et al., 2001) and chronic lung disease (Lucas et al., 2004); however, the evidence supporting their association is weak. Interestingly, recent evidence suggests that larger babies have an increased risk of developing breast and other cancers in later life (Lahmann et al., 2004).
There is a body of knowledge pointing out the long‐term consequences of breastfeeding versus bottle‐feeding (Lucas, 1991). Lucas coined the term ‘programming’ to describe the long‐term effect of nutritional influences and it was later adopted by those studying the fetal origins of adult disease. In general, there is no reason to suppose that the phase of developmental plasticity in which cues may act is limited by parturition itself. Thus, it is not particularly surprising that nutritional manipulation in the neonatal period may have long‐term effects, which also include changes in metabolic control (Singhal et al., 2003, , 2004).
Catch‐up growth
A common observation of a number of epidemiological studies is that rapid childhood weight gain increases adult disease risk. This is best detailed in the Finnish cohort which has been followed to late middle age, and the pattern of growth can be linked directly to disease outcomes (2001a, 2003a). The data show that there is an interaction between birth size and weight gain in childhood in determining the risk of heart disease and diabetes. Risk of disease in adulthood is higher in those individuals born thin, but compounded if they experience a rapid weight gain and earlier adiposity rebound in childhood (2001a, 2003a, 2003b). Indeed, children who show rapid weight gain may be at risk regardless of birth size (Ong et al., 2000).
Experimental data
There are now a vast number of experimental studies in which manipulation of maternal status has long‐term effects on the offspring's physiology. Studies have been reported in the mouse, rat, guinea pig, sheep and pig (Bertram & Hanson, 2001), whereby maternal nutrition is manipulated by either reducing total nutritional intake in a balanced manner, reducing the protein intake specifically, or by exposing the mother to a high fat diet. In general, all result in relatively similar outcomes independent of whether the manipulations have produced lower birthweights. The offspring have disturbances of endothelial function (Brawley et al., 2003), disturbed metabolic homeostasis with insulin resistance (Petry et al., 2001), decreased feedback within the hypothalamic‐pituitary‐adrenal axis (Bloomfield et al., 2003a), hypertension and obesity (Vickers et al., 2000). Structural changes reported include disordered pancreatic islet formation (Petrik et al., 1999), reduced nephron number (Langley‐Evans et al., 1999) and reduced vascularization of several tissues (Bennis‐Taleb et al., 1999). As will be discussed below, it is not always clear, given the nature of the experimental approaches used, to what extent some of these findings represent developmental disruption as opposed to plastic responses.
The span of development in which induction of developmental responses can lead to long‐term consequences extends from conception to after birth. Brief periods of undernutrition limited just to the pre‐implantation period alone may have long‐term consequences (Kwong et al., 2000; Bloomfield et al., 2004); conversely, short exposures in the peripartum period may also programme the offspring (Khan et al., 2005).
A further feature of the experimental data is that they confirm the interaction between the prenatal and postnatal environments. In animals subject to prenatal maternal undernutrition, there is a synergistic effect of postnatal exposure to a high fat, postnatal diet leading to obesity, insulin resistance, leptin resistance and hypertension (Vickers et al., 2000). That is, fetal experiences change the physiological settings of the organism such that it responds to postnatal environmental challenges differently – we see this as the general model. Programming does not lead to disease directly; it changes the physiological set points of the organism so that it copes differently with postnatal environmental challenges.
Mechanisms
Developmental plasticity must be distinguished from developmental disruption (Bateson et al., 2004; Gluckman et al., 2004a). Developmental disruption is best demonstrated by teratogenesis but can be more subtle than gross malformations. For example, maternal iodine deficiency is associated with thyroid aplasia in the offspring, and clearly is of disruptive consequence. By contrast, developmental plasticity is a regulated phenomenon by which one genotype can give rise to a range of phenotypes. Developmental plasticity acts by allowing the developing conceptus to respond to environmental cues by choosing a trajectory of development that often has adaptive advantage. The processes of developmental plasticity involve the commitment of cells to specific lineages, tissue differentiation and growth, and are underpinned, in part, by epigenetic changes in gene expression.
It is now clear that environmental influences may effect gene expression in at least two distinct ways. They may act to transiently and reversibly affect gene expression – this is the normal process of gene promotion and repression. Alternatively, the environmental factor may lead to chemical modification of the DNA or chromatin such that it is fixed in an ‘on’ or ‘off’ position. This process is referred to as epigenetic modification and may involve either DNA methylation or histone acetylation (reviewed in Gibbs, 2003). DNA methylation occurs primarily on cytosine residues, particularly in promotor regions. In general, methylated genes are silenced and hypomethylated genes are induced. DNA methylation is the basis of genomic imprinting, but many non‐imprinted genes show variable methylation. Several studies now demonstrate that maternal nutritional manipulation may alter gene expression by alterations in DNA methylation in the offspring. In the offspring of rats subject to uteroplacental insufficiency, apoptosis in the kidney is increased and this is associated with altered methylation of the p53 gene (Pham et al., 2003). The agouti mouse mutant is an interesting model as it demonstrates that nutrition during the periconceptual period can permanently affect the degree of expression of a non‐imprinted gene (Wolff et al., 1998; Cooney et al., 2002). Weaver and colleagues have demonstrated in rats that in those mothers who show altered maternal grooming behaviours towards their newborns, their offspring develop changes in behaviour and hypothalamic‐pituitary‐adrenal axis function. Moreover, these changes are associated with a permanent change in the methylation status of the glucocorticoid gene promoter, and treatment with a drug that affects methylation can reverse the phenotype (Weaver et al., 2004).
We suspect that epigenetic change is the primary basis of the programming phenomenon and that the structural and regulatory effects on many organ systems are secondary to these environmentally induced changes in gene expression. The possibility that epigenetic change might also affect mitochondrial genes has been proposed as a mechanism (Petrie et al., 2002; McConnell & Petrie, 2004), but it is still unable to explain much of the experimental data.
Many specific changes have been identified to explain the development of obesity, insulin resistance and hypertension, but in general there appears to be a coordinated response of the offspring to its developmental exposures, which in turn suggest a regulated phenomenon. For example, the offspring of rats subject in pregnancy to maternal undernutrition develop central obesity, insulin resistance and hypertension, but they also develop hyperphagia and lethargy (2000, 2003). This combination of central and peripheral effects appears to be a suite of responses that would enable an individual to survive better in a nutritionally poor environment. This particular example, if extrapolated to the human, might explain why some individuals with insulin resistance appear resistant to therapeutic intervention using modified lifestyle approaches.
Attempts to relate specific outcomes to the specific timings of the environmental cues have not been particularly revealing. Following the Dutch winter famine, there was suggestion that the timing of the fetus’ exposure to maternal famine might have determined its propensity to diabetes or obesity in later life (Roseboom et al., 2001), as more data have accumulated this is less certain, and comparable observations in animals have been lacking. However, we have suggested elsewhere that there is a convergent phenotype, whereby an adverse intrauterine environment will lead to a common thrifty or ‘survival phenotype’ through different pathways, depending on the timing and nature of maternal cues (Gluckman & Hanson, 2004a).
Conceptual models
Two conceptual models were alternatively suggested as the basis of the relationship between birth size and later disease risk. The thrifty genotype model (Neel, 1962) argued that humans had selected for thrifty genes which allowed them to cope with their hunter‐gatherer past – these may have been particularly selected for in certain populations, such as the Pima Indians (Knowler et al., 1983). Modern exposure to high glycemic diets would expose this underlying genetic predisposition, manifest as an increased risk of insulin resistance and its complications. There have been a number of criticisms of the thrifty genotype explanation as a basis of the phenomenon, as it cannot explain the experimental data or the particular patterns of human disease in some populations. As an alternative, the thrifty phenotype model (Hales & Barker, 1992) was posited to explain the phenomenon. It stated that the fetus made adaptations in utero to survive maternal undernutrition or other environmental stresses, one possible consequence being a reduction in fetal growth of either the whole body or some organs. The organism would then be left to cope with the consequences of these in utero adaptations, thriving in a deprived environment, yet compromised in an enriched environment. The general model was widely accepted, but had limitations. While it could explain the consequences in individuals born small, it did not readily explain the continuous relationship across birth sizes seen in the epidemiological data, nor could it explain how programming could operate in a well nourished fetus – given that the continuous relationship implied programming could operate in the upper birth size range as well.
These limitations led in turn to a modification of this model in which we proposed that many of the adaptive responses made by the fetus were not made for immediate advantage, but rather in expectation of the future postnatal environment. This predictive adaptive response model has been widely reviewed elsewhere (Gluckman & Hanson, 2004a, 2004b, 2004c). Importantly, it suggests that the disease risk is a consequence of the degree of match or mismatch between the environment the organism is exposed to in the plastic (fetal and neonatal) phase, compared with the environment the organism is exposed to in the post‐plastic phase (Fig. 1). The greater the degree of mismatch, the greater the risk of disease. This model not only explains how the transition from an adverse in utero environment to an enriched postnatal one leads to disease risk, but allows the converse situation – how an enriched fetal environment followed by poor postnatal one can also induce risk (Eriksson et al., 2003b).
Figure 1.
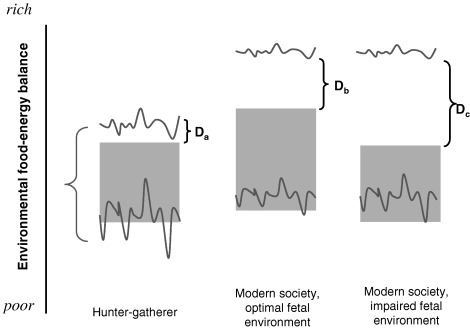
The predictive adaptive model is illustrated with respect to the human food‐energy environment. For each of three scenarios, variations in the actual postnatal environmental range experienced over time (between the red lines) and that predicted by the fetus (stippled region) are shown. The embryo/fetus/neonate sets its postnatal physiology for the predicted (stippled), rather than actual range. For the ancestral hunter‐gatherer (left panel), the predicted and actual postnatal environments are similar. The upper limit of the predicted range is below that of the actual range because of maternal constraint – this creates a small region (Da) where there is a risk of mismatch between the prenatal and postnatal environments, and hence of disease in later years (if longevity were ever great enough and the environment was consistently rich enough). The lower limit of the predicted range may or may not be below the actual range of postnatal nutrition seen – if it is, it creates a safety range of extreme conditions for which physiology can adapt. The middle panel illustrates the consequences of optimal fetal development in a modern society. The upper limit of the actual nutritional range is now shifted upward substantially. Maternal constraint continues to restrict the upper limit of predicted environmental range, creating a wider gulf between the predicted and actual postnatal environments (Db), which increases the probability of disease risk, particularly in middle age. The right panel shows the situation when the fetus is exposed to either extreme maternal constraint or maternal/placental disease. Predictive adaptive responses reduce the upper limit of the predicted postnatal environment even further, increasing the risk of mismatch (Dc) and disease. Irreversible plastic changes with immediate adaptive value in utero may also contribute to the narrowing of the postnatal range to which the fetus can adjust.
The model implies a non‐linear relationship between the environment the fetus (or neonate) predicts it will face and the actual environment it will be exposed to (Fig. 1). This non‐linear relationship is present, in part, because of maternal constraint; a term which describes the mechanisms that evolved such that the mammalian fetus would not outgrow the ability of its mother to deliver it naturally (Gluckman & Hanson, 2004d). This has been critically important to the monotocous species, such as the hominids, and particularly so once the upright posture was adopted and the pelvic canal narrowed. Thus, fetal growth is limited by the capacity of the uterus to allow for fetal growth, and of the placenta to transfer nutrients to the fetus. Maternal constraint is greater in first pregnancies, adolescent pregnancies and in mothers with shorter stature (Gluckman & Hanson, 2004d). These are physiological systems that must be distinguished from the pathophysiologic problems arising from maternal or placental disease. As maternal constraint is operative in all pregnancies, if nutrition is optimal in the mother, or excessive, fetal growth remains restricted to ensure it can exit the pelvic canal; thus, non‐linearity is created between the actual environment and that sensed by the fetus.
The effect is that maternal constraint determines a physiological ‘maximum’ for fetal nutrition which is optimal from the mother's perspective for fetal growth and development. In doing so, a limit is imposed on the nutritional information provided to the fetus, and through the processes of predictive adaptive responses, this dictates the maximum postnatal environment the fetus can predict and therefore cope with, without a risk to its fitness. Where constraint is greater or where the fetal environment is further compromised by maternal or placental disease, the maximal postnatal environment the fetus can cope with is reduced further. Thus, one would anticipate the emergence of lifestyle diseases at lower levels of nutrition in populations where maternal stunting is greater and birth size is consequently reduced. This may be the case in India (Yajnik, 2004).
The plastic phase and rebound growth
An issue that has more recently emerged is that of the role of rapid growth in the neonatal, infant and childhood periods. There is no doubt that prematurity can itself lead to long‐term consequences (Cutfield et al., 2004), including a greater risk of insulin resistance (Hofman et al., 2004). It remains unclear whether this is due to parallel effects of whatever induced prematurity also affecting the induction of longer‐term physiological changes, the effect of different timing of the perinatal glucocorticoid surge, or exposure of the premature infant to nutrition influences (i.e. high fat diet) that it ordinarily would not be exposed to had it still been in utero. Lucas and colleagues have suggested that the nature of infant feeding and rapid infant growth have long‐term sequelae (2003, 2004), in particular that adolescents born preterm who received enriched nutrition when born have later evidence of insulin resistance (Singhal et al., 2003). These studies are at risk of overinterpretation (Gluckman et al., 2005) as they are based on a mixed cohort of premature and term infants, many of whom were rather ill. However, it does seem likely that excessive or deficient nutritional exposures in any period during the phase of metabolic plasticity would have consequences, and this plastic period clearly does extend after birth for metabolic and neural systems.
As already discussed, there is evidence that rapid childhood growth and/or early adiposity rebound does have long‐term consequences in increasing disease risk (Ong et al., 2000; Eriksson et al., 2003a). The match/mismatch model would suggest that rapid childhood growth following normal fetal growth, albeit constrained, is fundamentally no different to that of normal childhood growth following impaired fetal growth. Both represent situations where the nutritional exposures in one phase of life – after plasticity is complete – are greater than that at an earlier phase. The matter is further complicated by the possibility that the two phases are linked. Rapid postnatal growth in the form of catch‐up growth is more likely in those born smaller. There is also the potential of parental overinvestment (i.e. overfeeding) in those born smaller, which may confound matters. In this discussion we have again focused on growth but the mismatch model may relate to some critical environmental factors that need not manifest as growth. Indeed, the same model has been shown to apply to fluid balance (Ross et al., 2005) and thermoregulation (Gluckman & Hanson, 2004a).
Intergenerational effects
There is a resurgence of interest in epigenetic inheritance – that is, non‐genomic transmission of environmentally induced information across generations. While largely ignored and misunderstood as a neo‐Lamarckian concept, there is ample empirical evidence in plant and animal biology that environmental factors can exert their influence over several generations. Often these are encompassed under the rubric ‘maternal effects’, whereby environmental impacts affecting the mother may be transmitted directly or indirectly to the next generation (For review, see Mousseau & Fox, 1998; Agrawal et al., 1999). In some ways, the developmental origins phenomenon is effectively a form of maternal effect, whereby the environmental impacts on the mother (F0 generation) are transmitted to her offspring (F1 generation), and possibly also to her grandchildren (F2). For instance, in rats (F0) administered glucocorticoids during pregnancy, the offspring (F1) have abnormalities of glucose homeostasis, which are then transmitted without further exposure to the F2 generation (Drake et al., 2005). Intriguingly, these effects are transmitted by both male and female F1 generation animals, thereby excluding mitochondrial mechanisms as the basis of the intergenerational transfer. Similarly, in rats exposed to a low protein diet during pregnancy, endothelial dysfunction is reported in the F2 generation (Torrens et al., 2002). Third generation effects have also been reported in humans regarding birth size and the propensity to insulin resistance. Following the Dutch famine, there was evidence that some grandchildren were born of low birthweight when their mothers were exposed to undernutrition, in utero, in the first trimester (Lumey, 1992).
These intergenerational effects have been extensively reviewed elsewhere (Drake & Walker, 2004), and several mechanisms may potentially underlie their function. First, intergenerational epigenomic effects may arise because of incomplete loss of methylation at meiosis, as reported in studies of the Agouti mouse mutant (Wolff et al., 1998). Second, there may be indirect effects mediated by changes in reproductive tract development during fetal life in the F1 generation, which then have a secondary effect on subsequent generations. For instance, Ibanez and colleagues have shown that adolescent girls born small have smaller uteri (Ibanez et al., 2000). Last, there may be indirect effects mediated by programming of maternal homeostasis in the F1 generation, which in turn affect her transmission of information to the next generation.
An anthropological and evolutionary perspective
Our fundamental hypothesis is that the developmental origins phenomenon arises because of a mismatch between the usual postnatal environment of the modern world and that which humans evolved to live in. We suggest that the mechanisms of maternal constraint have set limits of the future environment that can be predicted in fetal life. We propose that the processes of predictive adaptive responses are designed to match the prenatal and postnatal environments. As discussed below, this was an appropriate strategy before the development of agriculture. Thus, the fetus attempts to choose a developmental trajectory to maximize its chances for reproductive fitness in postnatal life.
Because one type of immediate adaptive response is to reduce fetal growth under constrained circumstances, it is the coexistence of these immediate and predictive adaptive responses that gave rise to the original relationships observed by Barker. Predictive adaptive responses operating in isolation and limited by maternal constraint can explain why children of normal birthweight who exhibit rapid weight gain have similar health outcomes as children of lower birthweight.
The question then becomes why did evolution allow these apparently deleterious responses to persist? These responses did not manifest as deleterious until middle age and after reproduction was complete, thus they would not have been selected against over the course of human evolution. Indeed, they have been actively selected for if they confer advantage up to and during the period of reproductive fitness. This is how predictive adaptive responses function. Provided that biology allows the prediction to be correct more often than wrong, then predictive adaptive responses confer advantage to a population. Individuals expressing such responses are better adapted, on average, to the environment in which they will live. Having the capacity for a thrifty phenotype in an environment that was likely to be thrifty would have been advantageous throughout 95% of the span of human existence that occurred between our evolution as a species and the invention of agriculture.
The processes of maternal constraint have evolved to allow monotocous species to survive pelvic delivery under virtually all circumstances. We speculate that maternal constraint had the additional advantage of matching fetal growth and the setting of the future physiological set points to the nutritional‐energy environments our pre‐agricultural ancestors were born into. Given the role and capacity of homeostatic processes to allow us to tolerate environmental variation, predictive adaptive responses set the range of environments we can live in healthily as adults. The gradual rise in maternal stature (and lessening maternal constraint) have together, until recently, allowed the predictive adaptations to match the changing environments as agriculture developed and food intakes changed. But since the industrial revolution, the marked change in the nature of food intakes, particularly in the last 50 years in the developed world and more recently in the developing world, have led to the probability of developmental mismatch rising dramatically. This and changed longevity are reflected in the changing ecology of human disease and the explosive appearance of lifestyle diseases, such as type 2 diabetes, in populations undergoing rapid nutritional transition.
Implications
Such data readily explain the current patterns of disease, why migrant populations have been at particular risk and why childhood obesity has become an increasing problem. Experimental data have suggested that early onset obesity, reduced muscle mass, lethargy, hyperphagia and a preference for a high fat diet are consequences of an impaired environment in early life changing our capacity to cope with a high fat diet after the plastic phase of development is complete (Vickers et al., 2000; Vickers et al., 2003). Indeed, lowered birthweight children or those exposed to impaired maternal nutrition do have a greater risk of obesity developing in childhood (Ravelli et al., 1999; Eriksson et al., 2001b). Demographic changes, such as having fewer children, may create additional considerations as first born children have been subject to greater maternal constraint.
The focus of the discussion has been on nutrition as nutritional signals are likely to be those of greatest relevance to the phenomenon. The shifts in maternal nutrition need not be extreme. Recent data show that fetus will adjust its physiology to subtle variations in maternal food intake well within the accepted normal ranges (Godfrey et al., 1996; Mitchell et al., 2004). Other research has shown the importance of maternal nutrition at the beginning of pregnancy, as it relates to the length of gestation and to the growth pattern of the fetus (Harding, 2001; Rao et al., 2001; Bloomfield et al., 2003b). This suggests that identifying the optimal nutritional regimens for women before and during pregnancy remains a critical knowledge gap.
One implication from the proposed non‐linear relationship between the predicted postnatal environment and the actual adult environment (Gluckman & Hanson, 2004c) is that interventions at different stages in the life cycle may need to be population‐ and context‐dependent. It may well be that while a focus on postnatal diet and exercise remains the most promising approach in the developed world, a focus on the health of women and their pregnancies would have greater impact in the developing world (Gluckman et al., 2004b). A key knowledge gap is to identify markers of inappropriate developmental trajectories chosen in utero. This is likely to involve the development of a range of tools to monitor epigenetic settings. From this, it may be possible to identify from birth infants whose postnatal nutritional management may need to be carefully managed to reduce their disease risk.
Acknowledgements
Mark A. Hanson is supported by the British Heart Foundation. Peter D. Gluckman is supported by the National Research Centre for Growth and Development (New Zealand).
References
- Agrawal A.A., Laforsch C. & Tollrian R. (1999) Transgenerational induction of defences in animals and plants. Nature, 401, 60–63. [Google Scholar]
- Barker D.J. & Osmond C. (1986) Infant mortality, childhood nutrition, and ischaemic heart disease in England and Wales. Lancet, 1, 1077–1081. [DOI] [PubMed] [Google Scholar]
- Barker D.J.P., Eriksson J.G., Forsén T. & Osmond C. (2002) Fetal origins of adult disease: strength of effects and biological basis. International Journal of Epidemiology, 31, 1235–1239. [DOI] [PubMed] [Google Scholar]
- Bateson P., Barker D., Clutton‐Brock T., Deb D., D’Udine B., Foley R.A. et al. (2004) Developmental plasticity and human health. Nature, 430, 419– 421. [DOI] [PubMed] [Google Scholar]
- Bennis‐Taleb N., Remacle C., Hoet J.J. & Reusens B. (1999) A low‐protein isocaloric diet during gestation affects brain development and alters permanently cerebral cortex blood vessels in rat offspring. Journal of Nutrition, 129, 1613–1619. [DOI] [PubMed] [Google Scholar]
- Bertram C.E. & Hanson M.A. (2001) Animal models and programming of the metabolic syndrome. British Medical Bulletin, 60, 103–121. [DOI] [PubMed] [Google Scholar]
- Bloomfield F.H., Oliver M.H., Giannoulias D., Gluckman P.D., Harding J.E. & Challis J.R.G. (2003a) Brief undernutrition in late‐gestation sheep programmes the hypothalamic‐pituitary adrenal axis in adult offspring. Endocrinology, 144, 2933–2940. [DOI] [PubMed] [Google Scholar]
- Bloomfield F., Oliver M., Hawkins P., Campbell M., Phillips D., Gluckman P. et al. (2003b) A periconceptional nutritional origin for noninfectious preterm birth. Science, 300, 606. [DOI] [PubMed] [Google Scholar]
- Bloomfield F.H., Oliver M.H., Hawkins P., Holloway A.C., Campbell M., Gluckman P.D. et al. (2004) Periconceptional undernutrition in sheep accelerates maturation of the fetal hypothalamic‐pituitary‐adrenal axis in late gestation. Endocrinology, 145, 4278– 4285. [DOI] [PubMed] [Google Scholar]
- Brawley L., Poston L. & Hanson M. (2003) Mechanisms underlying the programming of small artery dysfunction: review of the model using low protein diet in pregnancy in the rat. Archives of Physiology and Biochemistry, 111, 25–35. [DOI] [PubMed] [Google Scholar]
- Cooney C.A., Dave A.A. & Wolff G.L. (2002) Maternal methyl supplements in mice affect epigenetic variation and DNA methylation of offspring. Journal of Nutrition, 132, 2393S–2400S. [DOI] [PubMed] [Google Scholar]
- Cutfield W.S., Regan F.A., Jackson W.E., Jefferies C.A., Robinson E.M., Harris M. et al. (2004) The endocrine consequences for very low birth weight premature infants. Growth Hormone and IGF Research, 14, S130–S135. [DOI] [PubMed] [Google Scholar]
- Dennison E.M., Arden N.K., Keen R.W., Syddall H., Day I.N.M., Spector T.D. et al. (2001) Birthweight, vitamin D receptor genotype and the programming of osteoporosis. Paediatric and Perinatal Epidemiology, 15, 211–219. [DOI] [PubMed] [Google Scholar]
- Drake A.J. & Walker B.R. (2004) The intergenerational effects of fetal programming: non‐genomic mechanisms for the inheritance of low birth weight and cardiovascular risk. Journal of Endocrinology, 180, 1–16. [DOI] [PubMed] [Google Scholar]
- Drake A.J., Walker B.R. & Seckl J.R. (2005) Intergenerational consequences of fetal programming by in utero exposure to glucocorticoids in rats. American Journal of Physiology, 288, R34–R38. [DOI] [PubMed] [Google Scholar]
- Eriksson J.G., Forsen T., Tuomilehto J., Osmond C. & Barker D.J. (2001a) Early growth and coronary heart disease in later life: longitudinal study. British Medical Journal, 322, 949–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eriksson J., Forsén T., Tuomilehto J., Osmond C. & Barker D. (2001b) Size at birth, childhood growth and obesity in adult life. International Journal of Obesity, 25, 735–740. [DOI] [PubMed] [Google Scholar]
- Eriksson J.G., Forsen T., Tuomilehto J., Osmond C. & Barker D.J. (2003a) Early adiposity rebound in childhood and risk of Type 2 diabestes in adult life. Diabetologia, 46, 190–194. [DOI] [PubMed] [Google Scholar]
- Eriksson J.G., Forsen T.J., Osmond C. & Barker D.J.P. (2003b) Pathways of infant and childhood growth that lead to type 2 diabetes. Diabetes Care, 26, 3006–3010. [DOI] [PubMed] [Google Scholar]
- Gibbs W.W. (2003) The unseen genome: beyond DNA. Scientific American, 289, 106–113. [DOI] [PubMed] [Google Scholar]
- Gluckman P.D. & Hanson M.A. (2004a) The Fetal Matrix: Evolution, Development, and Disease. Cambridge University Press: Cambridge. [Google Scholar]
- Gluckman P.D. & Hanson M.A. (2004b) Living with the past: evolution, development and patterns of disease. Science, 305, 1773–1776. [DOI] [PubMed] [Google Scholar]
- Gluckman P.D. & Hanson M.A. (2004c) The developmental origins of the metabolic syndrome. Trends in Endocrinology and Metabolism, 15, 183–187. [DOI] [PubMed] [Google Scholar]
- Gluckman P.D. & Hanson M.A. (2004d) Maternal constraint of fetal growth and its consequences. Seminars in Fetal and Neonatal Medicine, 9, 419– 425. [DOI] [PubMed] [Google Scholar]
- Gluckman P.D., Cutfield W., Hofman P. & Hanson M.A. (2005) The fetal, neonatal and infant environments – the long‐term consequences for disease risk. Early Human Development, 81, 51– 59. [DOI] [PubMed] [Google Scholar]
- Gluckman P.D., Hanson M.A., Spencer H.G. & Bateson P. (2004a) Environmental influences during development and their later consequences for health and disease: implications for the interpretation of empirical studies. Proceedings of the Royal Society of London – Biology Sciences Series B. (in press). [DOI] [PMC free article] [PubMed]
- Gluckman P.D., Hanson M.A., Morton S.M. & Pinal C.S. (2004b) Life long echoes – a critical analysis of the developmental origins of adult disease model. Biology of the Neonate, 87, 127–139. [DOI] [PubMed] [Google Scholar]
- Godfrey K., Robinson S., Barker D.J., Osmond C. & Cox V. (1996) Maternal nutrition in early and late pregnancy in relation to placental and fetal growth. British Medical Journal, 312, 410– 414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hales C.N. & Barker D.J. (1992) Type 2 (non‐insulin‐dependent) diabetes mellitus: the thrifty phenotype hypothesis. Diabetologia, 35, 595–601. [DOI] [PubMed] [Google Scholar]
- Harding J.E. (2001) The nutritional basis of the fetal origins of adult disease. International Journal of Epidemiology, 30, 15–23. [DOI] [PubMed] [Google Scholar]
- Hofman P.L., Cutfield W.S., Robinson E.M., Bergman R.N., Menon R.K., Sperling M.A. et al. (1997) Insulin resistance in short children with intrauterine growth retardation. Journal of Clinical Endocrinology and Metabolism, 82, 402–406. [DOI] [PubMed] [Google Scholar]
- Hofman P.L., Regan F., Jackson W.E., Jefferies C., Knight D.B., Robinson E.M. et al. (2004) Premature birth and later insulin resistance. New England Journal of Medicine, 351, 2179–2186. [DOI] [PubMed] [Google Scholar]
- Ibanez L., Potau N., Enriquez G. & De Zegher F. (2000) Reduced uterine and ovarian size in adolescent girls born small for gestational age. Pediatric Research, 47, 575–577. [DOI] [PubMed] [Google Scholar]
- Jefferies C., Hofman P.L., Knoblauch H., Luft F.C., Robinson E.M. & Cutfield W.S. (2004) Insulin resistance in healthy prepubertal twins. Journal of Pediatrics, 144, 608–613. [DOI] [PubMed] [Google Scholar]
- Khan I.Y., Dekou V., Douglas G., Jensen R., Hanson M.A., Poston L. et al. (2005) A high fat diet during rat pregnancy or suckling induces cardiovascular dysfunction in adult offspring. American Journal of Physiology, 288, R127–R133. [DOI] [PubMed] [Google Scholar]
- Knowler W.C., Pettitt D.J., Bennett P.H. & Williams R.C. (1983) Diabetes mellitus in the Pima Indians: genetic and evolutionary considerations. American Journal of Physical Anthropology, 62, 107–114. [DOI] [PubMed] [Google Scholar]
- Kwong W.Y., Wild A.E., Roberts P., Willis A.C. & Fleming T.P. (2000) Maternal undernutrition during the preimplantation period of rat development causes blastocyst abnormalities and programming of postnatal hypertension. Development, 127, 4195– 4202. [DOI] [PubMed] [Google Scholar]
- Lahmann P.H., Gullberg B., Olsson H., Boeing H., Berglund G. & Lissner L. (2004) Birth weight is associated with postmenopausal breast cancer risk in Swedish women. British Journal of Cancer, 91, 1666–1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langley‐Evans S.C., Welham S.J. & Jackson A.A. (1999) Fetal exposure to a maternal low protein diet impairs nephrogenesis and promotes hypertension in the rat. Life Sciences, 64, 965–974. [DOI] [PubMed] [Google Scholar]
- Lucas A. (1991) Programming by early nutrition in man. Ciba Foundation Symposium, 156, 38–50. [PubMed] [Google Scholar]
- Lucas J.S., Inskip H.M., Godfrey K.M., Foreman C.T., Warner J.O., Gregson R.K. et al. (2004) Small size at birth and greater postnatal weight gain: relationships to diminished infant lung function. American Journal of Respiratory and Critical Care Medicine, 170, 534–540. [DOI] [PubMed] [Google Scholar]
- Lumey L.H. (1992) Decreased birthweights in infants after maternal in utero exposure to the Dutch famine of 1944–1945. Paediatric and Perinatal Epidemiology, 6, 240–253. [DOI] [PubMed] [Google Scholar]
- McConnell J.L. & Petrie L. (2004) Mitochondrial DNA turnover occurs during preimplantation development and can be modulated by environmental factors. Reproductive Biomedicine Online, 9, 418–424. [DOI] [PubMed] [Google Scholar]
- Mitchell E.A., Robinson E., Clark P.M., Becroft D.M.O., Glavish N., Pattison N.S. et al. (2004) Maternal nutritional risk factors for small for gestational age babies in a developed country: a case‐control study. Archives of Disease in Childhood – Fetal and Neonatal Edition, 89, F431–F435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mousseau T.A. & Fox C.W. (1998) The adaptive significance of matenal effects. Trends in Ecology and Evolution, 13, 403–407. [DOI] [PubMed] [Google Scholar]
- Neel J.V. (1962) Diabetes mellitus: a ‘thrifty’ genotype rendered detrimental by ‘progress’? American Journal of Human Genetics, 14, 353–362. [PMC free article] [PubMed] [Google Scholar]
- Ong K.K., Ahmed M.L., Emmett P.M., Preece M.A. & Dunger D.B. (2000) Association between postnatal catch‐up growth and obesity in childhood: prospective cohort study. British Medical Journal, 320, 967–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ong K.K., Petry C.J., Emmett P.M., Sandhu M.S., Kiess W., Hales C.N. et al. (2004) Insulin sensitivity and secretion in normal children related to size at birth, postnatal growth, and plasma insulin‐like growth factor‐1 levels. Diabetologia, 47, 1064–1070. [DOI] [PubMed] [Google Scholar]
- Osmond C. & Barker D.J.P. (2000) Fetal, infant, and childhood growth are predictors of coronary heart disease, diabetes, and hypertension in adult men and women. Environmental Health Perspectives, 108, 545–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrie L., Duthie S.J., Rees W.D. & McConnell J.M.L. (2002) Serum concentrations of homocysteine are elevated during early pregnancy in rodent models of fetal programming. British Journal of Nutrition, 88, 471– 477. [DOI] [PubMed] [Google Scholar]
- Petrik J., Reusens B., Arany E., Remacle C., Coelho C., Hoet J.J. et al. (1999) A low protein diet alters the balance of islet cell replication and apoptosis in the fetal and neonatal rat and is associated with a reduced pancreatic expression of insulin‐like growth factor‐II. Endocrinology, 140, 4861– 4873. [DOI] [PubMed] [Google Scholar]
- Petry C.J., Dorling M.W., Pawlak D.B., Ozanne S.E. & Hales C.N. (2001) Diabetes in old male offspring of rat dams fed a reduced protein diet. International Journal of Experimental Diabetes Research, 2, 139–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pham T.D., MacLennan N.K., Chiu C.T., Laksana G.S., Hsu J.L. & Lane R.H. (2003) Uteroplacental insufficiency increases apoptosis and alters p53 gene methylation in the full‐term IUGR rat kidney. American Journal of Physiology, 285, R962–R970. [DOI] [PubMed] [Google Scholar]
- Rao S., Yajnik C.S., Kanade A., Fall C.H.D., Margetts B.M., Jackson A.A. et al. (2001) Intake of micronutrient‐rich foods in rural Indian mothers is associated with the size of their babies at birth: Pune maternal nutrition study. Journal of Nutrition, 131, 1217–1224. [DOI] [PubMed] [Google Scholar]
- Ravelli A.C., Van Der Meulen J.H., Michels R.P., Osmond C., Barker D.J., Hales C.N. et al. (1998) Glucose tolerance in adults after prenatal exposure to famine. Lancet, 351, 173–177. [DOI] [PubMed] [Google Scholar]
- Ravelli A.C., Van Der Meulen J.H., Osmond C., Barker D.J. & Bleker O.P. (1999) Obesity at the age of 50 y in men and women exposed to famine prenatally. American Journal of Clinical Nutrition, 70, 811–816. [DOI] [PubMed] [Google Scholar]
- Roseboom T.J., Van Der Meulen J.H., Ravelli A.C., Osmond C., Barker D.J.P. & Bleker O.P. (2001) Effects of prenatal exposure to the Dutch famine on adult disease in later life: an overview. Molecular and Cellular Endocrinology, 185, 93–98. [DOI] [PubMed] [Google Scholar]
- Ross M.G., Desai M., Guerra C. & Wang S. (2005) Prenatal programming of hypernatremia and hypertension in neonatal lambs. American Journal of Physiology, 288, R25–R33. [DOI] [PubMed] [Google Scholar]
- Singhal A., Fewtrel M., Cole T.J. & Lucas A. (2003) Low nutrient intake and early growth for later insulin resistance in adolescents born preterm. Lancet, 361, 1089–1097. [DOI] [PubMed] [Google Scholar]
- Singhal A., Cole T.J., Fewtrell M. & Lucas A. (2004) Breastmilk feeding and lipoprotein profile in adolescents born preterm: follow‐up of a prospective randomised study. Lancet, 363, 1571–1578. [DOI] [PubMed] [Google Scholar]
- Thompson C., Syddall H., Rodin I., Osmond C. & Barker D.J.P. (2001) Birth weight and the risk of depressive disorder in late life. British Journal of Psychiatry, 179, 450–455. [DOI] [PubMed] [Google Scholar]
- Torrens C., Brawley L., Dance C.S., Itoh S., Poston L. & Hanson M.A. (2002) First evidence for transgenerational vascular programming in the rat protein restriction model. Journal of Physiology, 543, 41P–42P. [Google Scholar]
- Vickers M.H., Breier B.H., Cutfield W.S., Hofman P.L. & Gluckman P.D. (2000) Fetal origins of hyperphagia, obesity and hypertension and its postnatal amplification by hypercaloric nutrition. American Journal of Physiology, 279, E83–E87. [DOI] [PubMed] [Google Scholar]
- Vickers M., Breier B.H., McCarthy D. & Gluckman P. (2003) Sedentary behaviour during postnatal life is determined by the prenatal environment and exacerbated by postnatal hypercaloric nutrition. American Journal of Physiology, 285, R271–R273. [DOI] [PubMed] [Google Scholar]
- Wahlbeck K., Forsen T., Osmond C., Barker D.J.P. & Eriksson J.G. (2001) Association of schizophrenia with low maternal body mass index, small size at birth, and thinness during childhood. Archives of General Psychiatry, 58, 48–52. [DOI] [PubMed] [Google Scholar]
- Weaver I.C.G., Cervoni N., Champagne F.A., D’Alessio A.C., Sharma S., Seckl J.R. et al. (2004) Epigenetic programming by maternal behavior. Nature Neuroscience, 7, 847–854. [DOI] [PubMed] [Google Scholar]
- Wolff G.L., Kodell R.L., Moore S.R. & Cooney C.A. (1998) Maternal epigenetics and methyl supplements affect agouti gene expression in Avy/a mice. FASEB Journal, 12, 949–957. [PubMed] [Google Scholar]
- Yajnik C.S. (2004) Early life origins of insulin resistance and type 2 diabetes in India and other Asian countries. Journal of Nutrition, 134, 205–210. [DOI] [PubMed] [Google Scholar]