

Abstract
The interleukin (IL)-1 family of cytokines is strongly associated with systemic sclerosis (SSc) and pulmonary involvement, but the molecular mechanisms are poorly understood. The aim of this study was to assess the role of IL-1α and IL-1β in pulmonary vascular and interstitial remodelling in a mouse model of SSc.
IL-1α and IL-1β were localised in lungs of SSc patients and in the fos-related antigen-2 (Fra-2) transgenic (TG) mouse model of SSc. Lung function, haemodynamic parameters and pulmonary inflammation were measured in Fra-2 TG mice with or without 8 weeks of treatment with the IL-1 receptor antagonist anakinra (25 mg·kg−1·day−1). Direct effects of IL-1 on pulmonary arterial smooth muscle cells (PASMCs) and parenchymal fibroblasts were investigated in vitro.
Fra-2 TG mice exhibited increased collagen deposition in the lung, restrictive lung function and enhanced muscularisation of the vasculature with concomitant pulmonary hypertension reminiscent of the changes in SSc patients. Immunoreactivity of IL-1α and IL-1β was increased in Fra-2 TG mice and in patients with SSc. IL-1 stimulation reduced collagen expression in PASMCs and parenchymal fibroblasts via distinct signalling pathways. Blocking IL-1 signalling in Fra-2 TG worsened pulmonary fibrosis and restriction, enhanced T-helper cell type 2 (Th2) inflammation, and increased the number of pro-fibrotic, alternatively activated macrophages.
Our data suggest that blocking IL-1 signalling as currently investigated in several clinical studies might aggravate pulmonary fibrosis in specific patient subsets due to Th2 skewing of immune responses and formation of alternatively activated pro-fibrogenic macrophages.
Short abstract
IL-1 dampens collagen production of lung structural cells and balances pro-fibrotic actions of the immune system. Blockade of IL-1 signalling in Fra-2 TG mice worsens lung function by increased Th2 inflammation and collagen production in the lung. http://bit.ly/2IVUGLX
Introduction
Systemic sclerosis (SSc) is a rare progressive fibrotic disorder that affects the skin and internal organs [1]. Lung involvement occurs in ∼50% of patients, and can manifest in both the parenchymal and vascular compartments as fibrosis and vascular remodelling, respectively. Vascular remodelling may result in the development of pulmonary hypertension, which together with pulmonary fibrosis is strongly associated with poor outcome [2]. Treatment options for SSc-associated pulmonary fibrosis are limited and disease management is mostly confined to nonselective immunosuppressants [3]. A recent study of the EUSTAR group highlights the necessity to further investigate and identify more effective treatment strategies [4].
Although autoimmunity plays an important role, the underlying molecular mechanisms of SSc-induced lung damage are poorly understood. Various cytokines and inflammatory mediators have been implicated in the development and progression of SSc [5]. The interleukin (IL)-1 family members IL-1α and IL-1β are strongly increased in the serum of SSc patients [6, 7]. IL-1β levels are also increased in the bronchoalveolar lavage fluid (BALF) of SSc patients and negatively correlate with lung function [6]. Furthermore, the IL-1 receptor (IL-1R) signalling pathway was associated with SSc susceptibility and SSc-related fibrosing alveolitis [8]. Mice lacking IL-1R, or its downstream signalling, are protected against pulmonary hypertension [9] and fibrosis [10]. This led to the hypothesis that IL-1 contributes to pulmonary fibrosis and vascular remodelling. Accordingly, the recombinant IL-1R antagonist anakinra ameliorated bleomycin-induced pulmonary fibrosis in mice [10] as well as hypoxia- or monocrotaline-induced pulmonary hypertension [9, 11] and was considered a potential therapeutic option for the treatment of SSc-related lung disease [12].
Mice overexpressing the activator protein (AP)-1 transcription factor family member fos-related antigen-2 (Fra-2) represent a murine model for SSc-related pulmonary fibrosis and hypertension [13, 14]. Fra-2 transgenic (TG) mice spontaneously develop pulmonary inflammation, as well as vascular and parenchymal remodelling, closely resembling the clinical manifestations of SSc patients with pulmonary involvement [13, 15]. However, the molecular pathomechanisms behind this phenotype remain unknown. In this study, we used the Fra-2 TG mouse model to investigate the role of IL-1 for pulmonary fibrosis and pulmonary hypertension, and to assess a possible therapeutic potential of targeting IL-1 for the treatment of pulmonary manifestations in SSc.
Material and methods
A detailed description of the methods is provided in the supplementary material.
Study population
Samples of SSc lungs were obtained from archival, anonymised lung tissue samples removed as part of patients’ medical care or post-mortem examination (Dept of Pathology, University of Pittsburgh Medical Center, Pittsburgh, PA, USA) or from SSc patients undergoing lung transplantation (Division of Thoracic Surgery, Medical University of Vienna, Vienna, Austria). Downsized nontransplanted donor lungs served as controls. The protocol and tissue usage was approved by the local authorities (Vienna: Institutional Ethics Committee (976/2010); Pittsburgh: Institutional Review Board (PRO1110204)). Patient consent was obtained before lung transplantation. Scleroderma patients met American College of Rheumatology diagnostic criteria or LeRoy and Medsger criteria for SSc [16, 17]. Patient characteristics are listed in supplementary table S1.
Animal experiments
Female Fra-2 TG mice and wild-type (WT) littermates were maintained under specific pathogen-free conditions in isolated ventilated cages with 12 h light/dark cycles. All animal experiments met European Union guidelines (2010/63/EU) and were approved by the local authorities (Austrian Ministry of Education, Science and Culture, Vienna, Austria). The characterisation of haemodynamic and lung function was performed in Fra-2 TG and WT mice at 20 weeks of age. 25 mg·kg−1 anakinra (Kineret; Swedish Orphan Biovitrum, Stockholm, Sweden) was given via daily intraperitoneal injection for a total of 8 weeks as previously described [18]. Control groups received injections with an equal volume of sterile saline solution. Mice were sacrificed at 18–19 weeks of age. Anakinra treatment was performed in two independent experiments with n=5–7 mice per group.
Statistics
Statistical analysis was performed in Prism version 5 (GraphPad, La Jolla, CA, USA). Data in figures are expressed as single data points with median, if not stated otherwise. Comparisons between two groups with equal variances were done with the unpaired t-test. Two groups with significantly different variances were compared using the Mann–Whitney test. Treatment effects of anakinra on WT and TG mice were analysed by two-way ANOVA with Bonferroni's post-test. Multigroup comparisons of cell culture protein and expression data were done using a Kruskal–Wallis test with Dunn's post-test for multiple comparisons. All statistical tests used for a specific dataset are indicated in the figure legends. p-values <0.05 were considered statistically significant.
Results
The Fra-2 TG mouse model is characterised by pulmonary hypertension and restrictive lung function
Overexpression of Fra-2 in mice leads to age-dependent remodelling in skin and lung (figure 1a) [13, 15]. Assessment of pulmonary vascular remodelling revealed increased muscularisation of pulmonary vessels (10–100 µm diameter; figure 1b and supplementary figure S1a) and elevated right ventricular systolic pressure (RVSP), indicating pulmonary arterial hypertension (figure 1c) in Fra-2 TG mice. Lung function of TG mice was impaired, with decreased inspiratory capacity and compliance (figure 1d). Collagen deposition in the lungs was significantly increased as determined by morphometric collagen quantification using Sirius red-stained sections of the left lung (figure 1e and supplementary figure S1b). Accordingly, there was an increase in collagen but not α-smooth muscle actin (α-SMA (Acta2)) expression at both the mRNA and protein level (figure 1f and g). Furthermore, the pro-fibrotic factor insulin-like growth factor 1 (Igf1) was highly upregulated (figure 1f). In Fra-2 TG mice, collagen 1 was detected around and in the wall of smaller distal vessels and in the parenchyma (figure 1h). α-SMA immunoreactivity was mostly seen in remodelled vessels, with further staining in fibrotic parenchymal regions (arrows), indicating the presence of myofibroblasts (figure 1h).
FIGURE 1.

fos-related antigen-2 (Fra-2)-overexpressing (transgenic (TG)) mice develop vascular remodelling and parenchymal fibrosis. WT: wild-type; RVSP: right ventricular systolic pressure; RV: right ventricle; LV: left ventricle; S: septum; IC: inspiratory capacity; Crs: compliance of the respiratory system; SR: Sirius red; Ct: cycle threshold; COL1: collagen 1; α-SMA: α-smooth muscle actin; α-TUB: α-tubulin. a) Representative images of whole lung slides stained with Masson's trichrome and magnifications of remodelled vessels in 20-week-old WT and Fra-2 TG mice. Scale bar: 20 µm. Collagen is stained in blue. b) Percentage of nonmuscularised (NM), partially muscularised (PM) and fully muscularised (FM) vessels <100 µm in diameter. n=10; mean±sd. c) RVSP as determined by right heart catheterisation and the Fulton index (RV/(LV+S)) in WT and Fra-2 TG mice. d) Lung function measurements (IC and Crs) of WT and Fra-2 TG mice. e) Morphometric quantification of collagen on Sirius red-stained WT and Fra-2 TG lung slides. f) Quantitative real-time PCR analysis of Col1a1, Col1a2, Col3a1, Acta2 and Igf1 expression in WT and Fra-2 TG mice. ΔCt values were normalised to the mean of the WT group (ΔΔCt). B2m (β2-microglobulin) and Hmbs (hydroxymethylbilane synthase) were used as reference genes. g) Western blot analysis of COL1 and α-SMA levels in WT and Fra-2 TG mice lung homogenates. α-TUB served as a loading control. h) Immunohistochemical staining of COL1 (brown) and α-SMA (red/pink). B: bronchi; V: vessel. Scale bar: 50 µm. *: p<0.05; **: p<0.01; ***: p<0.001.
Fra-2 overexpression leads to increased IL-1 levels through enhanced promoter binding
Inflammatory cytokines such as IL-1α and IL-1β are known to be associated with SSc [19] and idiopathic pulmonary arterial hypertension [9, 20], and might represent a common pathogenic link between parenchymal and vascular remodelling in SSc. In lung homogenates of Fra-2 TG mice, protein levels of both IL-1α and IL-1β were significantly elevated (figure 2a). In BALF, IL-1α levels were also increased, whereas IL-1β levels were unchanged (figure 2b). Accordingly, immunohistochemical staining against IL-1α was strongly increased in Fra-2 TG mice and located in the bronchial epithelium (black arrows), structural and inflammatory cells (white arrows) around vessels and in the parenchyma (figure 1c, left panel). IL-1β staining was not detectable in WT mice or in structural cells of Fra-2 TG mice and was only observed in inflammatory infiltrates (white arrows) (figure 2c, right panel).
FIGURE 2.

Increased interleukin (IL)-1α and IL-1β levels in the fos-related antigen-2 (Fra-2)-overexpressing (transgenic (TG)) mouse. BALF: bronchoalveolar lavage fluid; AP-1: activator protein-1. a, b) IL-1α and IL-1β protein levels in a) lung homogenate and b) BALF of 20-week-old WT and Fra-2 TG mice. c) Immunohistochemical staining of IL-1α and IL-1β on lung sections of Fra-2 TG and WT mice. B: bronchi, V: vessel; black arrows: bronchial epithelial cells; white arrows: structural and inflammatory cells. Scale bar: 20 µm. d) Schematic representation of the human IL1A and mouse Il1a promoter sequences including AP-1-binding sites. e, f) Electrophoretic mobility shift assay analysis performed on nuclear extracts of e) Fra-2-overexpressing primary human parenchymal fibroblasts or cells transfected with empty control plasmid (–) and f) lung homogenates from WT or Fra-2 TG mice. –NE: negative control of the binding reaction without nuclear extract. For loading and overexpression controls, see supplementary figure S1. **: p<0.01.
Promoter analysis of IL-1α and IL-1β gene loci revealed potential AP-1-binding sites in the promoter regions of human and mouse IL-1α genes (figure 2d), but not in the IL-1β genes. To further substantiate the direct effect of Fra-2 on the IL-1α promoter, we performed an electrophoretic mobility shift assay on nuclear extracts from primary human parenchymal fibroblasts overexpressing Fra-2 (figure 2e) and from lung homogenates of Fra-2 TG and WT mice (figure 2f). Equal loading of nuclear extracts was confirmed by Western blotting (supplementary figure S2). In both experimental setups, overexpression of Fra-2 led to increased binding to the AP-1 promoter region of IL-1α (figure 2e and f). Therefore, increased IL-1α levels might be a direct consequence of aberrant Fra-2 expression.
Cellular localisation of Fra-2 and IL-1 expression is altered in human SSc lungs
Analysis of Fra-2 localisation in donor and SSc lungs revealed that in donor lungs Fra-2-positive staining was mainly observed in alveolar macrophages (asterisks), whereas in SSc lungs it was also localised in remodelled vessels (white arrow) including endothelial cells, inflammatory cells (asterisks) and parenchymal cells (black arrows) (figure 3a). Furthermore, the staining intensity of Fra-2 was increased in SSc lungs, as already reported by others [13, 21]. A similar pattern of IL-1α staining was observed, with IL-1α localised to alveolar macrophages in donor lungs (asterisk), whereas in SSc lungs, IL-1α staining was also detected in remodelled vessels (white arrow), endothelial cells and generally in remodelled parenchymal regions (black arrows) (figure 3b). IL-1β was expressed in single inflammatory cells (asterisks), but not in other cell types of SSc lungs; no IL-1β staining could be detected in donor lungs (figure 3c).
FIGURE 3.

Localisation of fos-related antigen-2 (Fra-2), interleukin (IL)-1α and IL-1β in healthy donor and systemic sclerosis (SSc) lungs. Immunohistochemical staining (brown) of a) Fra-2, b) IL-1α and c) IL-1β. Left panels: overview; right panels: magnified insets. Asterisks: inflammatory cells/alveolar macrophages; black arrows: parenchymal cells; white arrows: endothelial cells. Scale bar: 100 µm (overview)/20 µm (insets). n=3–4; representative images are shown.
Blocking of IL-1 signalling in Fra-2 TG mice worsens lung function
To assess the role of IL-1 signalling in pulmonary remodelling in the Fra-2 TG mouse model, mice were treated for 8 weeks with the recombinant IL-1R antagonist anakinra to block IL-1R signalling (figure 4a). Treatment with anakinra did not alter the RVSP, Fulton index (figure 4b) or muscularisation of small pulmonary vessels (figure 4c), but further worsened the restrictive lung function in Fra-2 TG mice (figure 4d). Although collagen quantification on Sirius red-stained lung sections did not show detectable differences (figure 4e), the expression levels of Col1a2 and Col3a1 were increased upon anakinra treatment (figure 4f). The levels of the pro-fibrotic factor Igf1 [22] further increased upon anakinra treatment (figure 4g). Collagen protein levels in lung homogenates of anakinra-treated Fra-2 TG mice were slightly increased (p=0.059) compared with the untreated control group (figure 4h). Localisation of collagen deposition and α-SMA immunoreactivity was consistent with untreated control Fra-2 TG mice (figure 4i). Furthermore, anakinra led to more inflammatory cell infiltrates in the lungs of Fra-2 TG mice, evident by increased staining with the inflammatory cell marker CD45 (figure 4i).
FIGURE 4.

Blocking of interleukin (IL)-1 signalling worsens lung function and increases extracellular matrix production in fos-related antigen-2 (Fra-2)-overexpressing (transgenic (TG)) mice. RSVP: right ventricular systolic pressure; RV: right ventricle, LV: left ventricle; S: septum; ns: nonsignificant; IC: inspiratory capacity; Crs: compliance of the respiratory system; SR: Sirius red; Ct: cycle threshold; COL1: collagen 1; α-SMA: α-smooth muscle actin; α-TUB: α-tubulin; AU: arbitrary units; vWF: von Willebrand Factor. a) Overview of anakinra treatment: Fra-2 TG mice received 25 mg·kg−1 anakinra per day as intraperitoneal injections for 8 weeks and were sacrificed at the age of 18–19 weeks. b) RVSP as determined by right heart catheterisation and the Fulton index (RV/(LV+S)) of Fra-2 TG and WT mice with anakinra treatment or vehicle control (saline). #: p<0.05, significance of genotype effect determined by two-way ANOVA. c) Percentage of nonmuscularised, partially muscularised and fully muscularised vessels <100 µm in diameter. n=3 for WT and n=9 for TG; mean±sd. *: p<0.05, unpaired t-test. d) Lung function measurements (IC and Crs) of Fra-2 TG and WT mice with anakinra treatment or vehicle control (saline). #: p<0.05, significance of genotype effect determined by two-way ANOVA; ¶: p<0.05, significance of the difference between Fra-2 TG with and without anakinra treatment determined by two-way ANOVA with Bonferroni's post-test. e) Morphometric quantification of collagen on SR-stained lung slides from Fra-2 TG and WT mice with (TG+A) and without (TG) anakinra treatment. f, g) Quantitative real-time PCR analysis of f) Col1a1, Cola1a2, Col3a1 and Acta2 and g) Igf1 expression in Fra-2 TG mice. ΔCt values were normalised to the mean of the untreated Fra-2 TG group (ΔΔCt). B2m (β2-microglobulin) and Hmbs (hydroxymethylbilane synthase) were used as reference genes. *: p<0.05, unpaired t-test. h) Western blot analysis and quantification of COL1 and α-SMA levels in Fra-2 TG and TG+A mice lung homogenates. α-TUB served as a loading control. i) Collagen staining with SR (collagen in red), immunohistochemical double staining of vessels against α-SMA (violet) and endothelium marker vWF (light brown), and immunohistochemical staining against inflammatory cell marker CD45. B: bronchi. Scale bar: 100 µm (main)/20 µm (insets).
IL-1α and IL-1β suppress collagen 1 and α-SMA expression through distinct cell type-specific signalling pathways
To determine which mechanisms could lead to the deterioration of lung function in vivo, we addressed the potential effects of IL-1 in vitro, and investigated how IL-1α and IL-1β influence important resident lung cells regarding proliferation, α-SMA expression and collagen production. IL-1α and IL-1β stimulation of pulmonary arterial smooth muscle cells (PASMCs) and parenchymal fibroblasts led to decreased Col1a1 and Acta2 mRNA (figure 5a and b). Accordingly, collagen 1 and α-SMA protein levels were diminished (figure 5c and d), suggesting direct anti-fibrotic actions of IL-1α and IL-1β on resident lung cells. Stimulation with IL-1α or IL-1β did not alter cell proliferation after 24 h, but only after 48 h (figure 5e and f), pointing towards indirect effects of IL-1 on cell proliferation. Interestingly, PASMCs and parenchymal fibroblasts showed regulation of distinct and cell type-specific pathways upon IL-1α and IL-1β stimulation (supplementary figure S3).
FIGURE 5.

Interleukin (IL)-1 downregulates collagen 1 (COL1) and α-smooth muscle actin (α-SMA) expression in pulmonary arterial smooth muscle cells (PASMCs) and parenchymal fibroblasts. IL: interleukin; Ct: cycle threshold; α-TUB: α-tubulin; PDGF: platelet-derived growth factor; NC: negative control. a, b) Relative expression levels of Col1a1 and Acta2 after 4, 8 and 24 h of IL-1α (1 ng·mL−1) and IL-1β (10 ng·mL−1) stimulation in a) PASMCs and b) parenchymal fibroblasts (extracellular matrix production). Data from six independent experiments with cells isolated from different donor lungs are depicted. B2m (β2-microglobulin) and Hmbs (hydroxymethylbilane synthase) were used as reference genes. c, d) Representative Western blots of COL1 and α-SMA in c) PASMCs and d) parenchymal fibroblasts treated with IL-1α and IL-1β for 4, 8 or 24 h. e, f) Proliferation of e) PASMCs and f) parenchymal fibroblasts assessed by 3H-thymidine incorporation over 24 h upon stimulation with IL-1α, IL-1β and PDGF-BB. Data from 10 independent experiments on cells from five different donors; mean±sd. *: p<0.05, Kruskal–Wallis test with Dunn's post-test for multiple comparisons.
Anakinra treatment exacerbates T-helper cell type 2 inflammation in Fra-2 TG mice
Since we observed increased inflammatory infiltrates in Fra-2 TG mice treated with anakinra (figure 4i), a detailed analysis of inflammatory cell populations and mediators was performed. First, histological scoring revealed significantly more inflammatory infiltrates in the interstitium and around the bronchi, with eosinophils and lymphocytes as the most abundant cell populations (supplementary table S6). In the next step, inflammatory cells in the BALF of mice were quantified and analysed by flow cytometry. As previously described, Fra-2 TG have increased numbers of inflammatory cells in the BALF, predominately consisting of eosinophils, T-lymphocytes and B-lymphocytes (figure 6a) [23]. While anakinra treatment did not lead to changes in inflammatory cell numbers in WT mice, increased amounts of T-cells, B-cells and eosinophils in Fra-2 TG mice treated with anakinra were observed (figure 6a). Two-way ANOVA indicated an interaction of genotype and treatment for B-cells (p=0.0329) and eosinophils (p<0.0001). The number of total inflammatory cells as well as the number of eosinophils in the BALF negatively correlated with pulmonary compliance in Fra-2 TG mice (figure 6b), indicating that worsened lung function upon anakinra treatment is not only due to increased collagen production, but also increased inflammation in the lungs of Fra-2 TG mice.
FIGURE 6.

Blocking of interleukin (IL)-1 signalling increases inflammation in fos-related antigen-2 (Fra-2)-overexpressing (transgenic (TG)) mice. BALF: bronchoalveolar lavage fluid; WT: wild-type; Cst: quasi-static lung compliance; Ct: cycle threshold; RELMα: resistin-like molecule-α; COL1: collagen 1; DAPI: 4′,6-diamidino-2-phenylindole; qRT: quantitative real-time. a) Flow cytometric analysis of inflammatory cell populations in the BALF of Fra-2 TG and WT mice with anakinra or vehicle control (saline) treatment. ##: p<0.01; ###: p<0.001, significance of genotype effect determined by two-way ANOVA; ¶: p<0.05, significance of difference between Fra-2 TG with and without anakinra treatment determined by two-way ANOVA with Bonferroni's post-test; *: p<0.05, unpaired t-test. b) Correlation plots of Cst with BALF total cell count or BALF eosinophils. c–e) Relative expression levels determined by qRT-PCR of key inflammatory mediators in lung homogenates of Fra-2 TG mice upon anakinra treatment. ΔCt values were normalised to the mean of the untreated Fra-2 TG group (ΔΔCt). B2m (β2-microglobulin) and Hmbs (hydroxymethylbilane synthase) were used as reference genes. *: p<0.05, unpaired t-test. f) qRT-PCR analysis of markers of alternative macrophage polarisation. ΔCt values were normalised to the mean of the untreated control group (ΔΔCt). *: p<0.05, unpaired t-test. g) Immunohistochemical staining of CD206 (brown) on lung sections from Fra-2 TG mice with (TG+A) and without (TG) anakinra treatment. V: vessel. Scale bar: 100 µm (main)/20 µm (insets). f) Immunofluorescence staining of RELMα (red), CD68 (green), COL1 (white) and DAPI (nuclei; blue). Arrows: CD68+/RELMα+ double-positive cells. Scale bar: 25 µm.
As there was no effect of anakinra on the inflammatory profile in WT mice, we further investigated the expression levels of cytokines for T-helper cell type 1, 2 or 17 (Th1, Th2 or Th17) immune responses in Fra-2 TG mice after anakinra treatment (figure 6c–e). Expression of the Th1-inducing cytokine IL-12 was strongly decreased, whereas interferon-γ was unchanged (figure 6c). The Th17 cytokines, IL-17 and IL-22, were decreased or unaltered (figure 6d). Although IL-13 expression was unaffected by anakinra treatment, IL-4, a key mediator of Th2 immunity, was strongly increased (figure 6e). Taken together, these results indicate a shift towards a Th2-driven immune response after blockade of IL-1 signalling. Since IL-4 is a potent inducer of alternatively activated macrophages that are involved in wound healing and were shown to induce fibrogenic responses in fibroblasts [24], we analysed expression levels of alternatively activated macrophage markers. Arginase 1 (Arg1), chitinase-like 3 (Chil3) and resistin-like molecule-α (RELMα (Retnla)) were significantly overexpressed in Fra-2 TG mice after treatment with anakinra compared with vehicle-treated TG mice (figure 6f). Immunoreactivity of the pro-fibrotic macrophage marker CD206 was strongly increased in lung sections of Fra-2 TG mice upon treatment with anakinra (figure 6g). Triple immunofluorescence for CD68, RELMα and collagen 1 highlighted the accumulation of CD68+/RELMα+ double-positive macrophages around collagen-positive fibrotic areas (figure 6f). These data indicate that blocking of IL-1 signalling leads to increased numbers of pro-fibrotic macrophages in Fra-2 TG mice.
Discussion
Pulmonary complications are the leading cause of mortality in patients with SSc [2]. In the lung parenchyma these can manifest as pulmonary fibrosis and in the vasculature as pulmonary hypertension. We and others have shown that ectopic overexpression of the AP-1 transcription factor family member Fra-2 in the mouse leads to fibrotic changes of the skin and to pulmonary structural changes similar to those observed in SSc patients, such as vascular remodelling to the point of pulmonary arterial occlusion, aberrant extracellular matrix (ECM) deposition with fibrosis and inflammation [13, 15, 25]. These structural changes manifest in severe functional impairment, characterised by restrictive lung function and elevated pulmonary pressure, as can be seen in SSc patients, making Fra-2 TG mice a valuable tool to investigate pathophysiological aspects of lung involvement in SSc and to perform pre-clinical proof-of-concept studies.
To date it is uncertain which factors are responsible for pulmonary remodelling in SSc patients. Several studies showed an association of SSc with pro-inflammatory cytokines, such as IL-1 [5]. Both IL-1α and IL-1β levels are significantly increased in the serum of SSc [6, 7] and idiopathic pulmonary arterial hypertension [20] patients. IL-1α gene polymorphisms raising IL-1α protein expression were associated with increased susceptibility to SSc [26], while IL-1β polymorphisms have been associated with lung involvement in patients with SSc [27]. In this study, we describe a novel mechanism leading to increased IL-1α in SSc through direct Fra-2 promoter binding. In SSc patients, Fra-2 is overexpressed and accumulates in the nucleus [21], where it could bind to activate the IL1A promoter and lead to increased IL-1α expression. Accordingly, IL-1α immunoreactivity was increased in the parenchymal and vascular compartments of the lungs of SSc patients and Fra-2 TG mice, whereas IL-1β was mainly observed in inflammatory cells. Increased IL-1β might therefore be a secondary effect due to increased inflammation (in contrast to IL-1α, which is directly regulated by Fra-2).
Previously, it was shown that IL-1R signalling deficiency or blockade with the IL-1R antagonist anakinra protects from pulmonary hypertension and fibrosis [9–11]. Furthermore, a case study of a silicosis-induced pulmonary fibrosis patient treated with anakinra reported an improvement of respiratory symptoms and normalisation of inflammatory parameters [28]. We therefore hypothesised that increased IL-1 levels due to aberrant Fra-2 activity may be a driving factor for the pulmonary phenotype and that anakinra treatment ameliorates pulmonary hypertension and fibrosis. Unexpectedly, in vivo application of anakinra further deteriorated pulmonary function of Fra-2 TG mice in this study, and led to increased activation of the Th2/pro-fibrotic macrophage axis and thus collagen synthesis. As a transcription factor, Fra-2 orchestrates an array of inflammatory mediators and other target genes, beyond IL-1. The phenotype of Fra-2 TG mice is therefore not due to the upregulation of one single factor, but to dysregulation of several counteracting pathways. Here, we show that the detrimental effects of blocking IL-1 signalling may have two explanations: 1) a loss of direct anti-fibrotic effects of IL-1 on resident structural cells and 2) a further shift of inflammation towards a Th2-predominant inflammatory response.
Despite the clear association of IL-1 cytokines with SSc, it is still unknown if and how these cytokines contribute to tissue remodelling and ECM production. The direct effects of IL-1α and IL-1β on structural cells have been extensively studied, but revealed controversial results regarding their pro-fibrotic properties. Following IL-1 stimulation of fibroblasts, downregulation as well as upregulation of collagen have been observed [29–32]. Furthermore, both IL-1α and IL-1β were reported to inhibit transforming growth factor (TGF)-β-induced collagen production [30, 33]. We show that in pulmonary structural cells, such as PASMCs or parenchymal fibroblasts, both IL-1α and IL-1β dampen collagen expression without prior application of any pro-fibrotic stimuli, and that this effect is mediated via distinct signalling pathways in each cell type. Lack of IL-1 signalling could therefore be the cause of exaggerated collagen production by resident lung cells and decreased lung function in Fra-2 TG mice following anakinra treatment. However, IL-1 actions are very complex, and it cannot be excluded that IL-1 may play a role in the initial development of pulmonary fibrosis by induction of secondary mediators and pro-survival factors, such as IL-6. Increased PASMC and parenchymal fibroblast proliferation has been observed after prolonged treatment with IL-1α and IL-1β. However, the lack of proliferation after 24 h speaks against a direct proliferative effect of IL-1, and in combination with the suppression of collagen and α-SMA expression it points towards a direct anti-fibrotic activity of IL-1 on mesenchymal cells.
An additional confounding factor is the effect of IL-1 on inflammation in this mouse model. We previously reported that inflammation in Fra-2 TG mice is predominately Th2 driven with strong eosinophilia [23]. Upon treatment with anakinra, Fra-2 TG mice developed even higher eosinophilia (Th2 response), whereas no effects were observed in WT mice, indicating that the complex immunomodulatory actions of IL-1 strongly depend on the inflammatory microenvironment. The Th2 cytokine IL-4 was also increased in anakinra-treated Fra-2 TG mice. IL-4 has potent pro-fibrotic roles [34, 35] and its levels are significantly increased in the serum of SSc patients [36]. The number of eosinophils is also significantly increased in the BALF of SSc patients, and some studies show correlation with interstitial lung disease and skin fibrosis [37, 38]. In addition to the direct pro-fibrotic effects, IL-4 can lead to alternative activation of macrophages, as seen in our study. Alternatively activated macrophages are involved in the development of fibrosis [39] and increased numbers have been reported in the skin of patients with SSc [40] as well as in the Fra-2 TG mouse [41]. Successful treatment of SSc pathology was associated with decreased numbers of alternatively activated macrophages in Fra-2 TG mice [41].
The question how IL-1 signalling blockade can induce the shift towards a Th2-mediated immunity is difficult to answer. The effects of IL-1 on T-helper subset differentiation are complex [42]; even though Th1 cells do not express IL-1R, both Th2 and Th17 cells can respond to IL-1 [43]. For example, unrestrained IL-1 signalling (in IL-1R antagonist-deficient mice) leads to an increased Th17 response [44]. Regarding Th2 inflammatory responses, IL-1 was mostly reported to favour Th2 differentiation [45, 46]. However, it was also shown that increased IL-1β expression inhibits the production of thymic stromal lymphopoietin, a key Th2 cytokine implicated in atopic dermatitis and asthma [47], and that activated splenocytes isolated from IL-1R1-deficient mice, lacking IL-1 signalling, have increased production of IL-4 [48]. Together with our data, this suggests that in some circumstances IL-1 may act as a negative regulator of IL-4. Adding to this complexity, Th17 and Th2 responses are closely intertwined and can counteract each other [49]. Therefore, further investigations on the effect of IL-1 on T-helper subsets in the context of SSc are needed.
Of note, we cannot fully exclude that the observed effects are due to a blockade of IL-1R signalling. Although there are no such studies regarding the lung or inflammation, a few studies report IL-1R-independent effects of the IL-1R antagonist, e.g. in the hippocampus where it can act as an agonist mimicking IL-1β effects [50] or in the liver where it contributes to insulin resistance [51]. Despite their similarities, IL-1α and IL-1β have distinct expression patterns, functions and, as we show here, downstream signalling pathways: while IL-1α is constitutively expressed by a variety of cells under homeostasis, IL-1β is only induced during inflammation [52, 53]. General blockade of IL-1R1 might therefore mask or override beneficial effects of specific IL-1β neutralisation. Furthermore, our study is confined by the small human sample size and the fact that explanted end-stage lung specimens are used to investigate SSc pathomechanisms in human tissue. Drawing conclusions about disease development and progression might therefore be limited and one must rely on data collected from the animal model. Nevertheless, our findings may have important clinical implications, since IL-1 blockade belongs to the concepts that are addressed in clinical studies. For example, IL-1 blockade using rilonacept (IL-1-trap) was tested in a phase I/II clinical trial; however, no changes of skin pathology or indicators of IL-1 activity were observed [54]. Adverse effects as observed in this study indicate that the positive anti-inflammatory effects of blocking IL-1R1 signalling may be overridden by an increase in fibrosis/Th2 inflammation in patients treated with IL-1R1 antagonists/IL-1 blockers. For instance, in patients with rheumatoid arthritis, treatment with newer biological therapies such as IL-6 receptor or tumour necrosis factor-α blockade seems to worsen pre-existing interstitial lung disease [55]. The benefits of biological drugs must therefore be carefully considered against their risks, especially in patients prone to inflammation or infection or in patients with mixed Th1/Th2 inflammation, where a disturbed inflammatory balance might drive severe adverse effects.
In summary, we have shown that IL-1 has a complex modulatory role in the development of pulmonary fibrosis, acting on structural cells by dampening collagen production and furthermore balancing the pro-fibrotic and pro-inflammatory actions of the immune system. Blockade of IL-1 signalling in Fra-2 TG mice, characterised by pulmonary inflammation, vascular remodelling and fibrosis, worsens lung function by increased Th2 inflammation and collagen production in the lung. These data have important implications for the search of new intervention strategies targeting specific inflammatory pathways in SSc.
Supplementary material
Please note: supplementary material is not edited by the Editorial Office, and is uploaded as it has been supplied by the author.
Supplementary material ERJ-00154-2019.SUPPLEMENT (279.7KB, pdf)
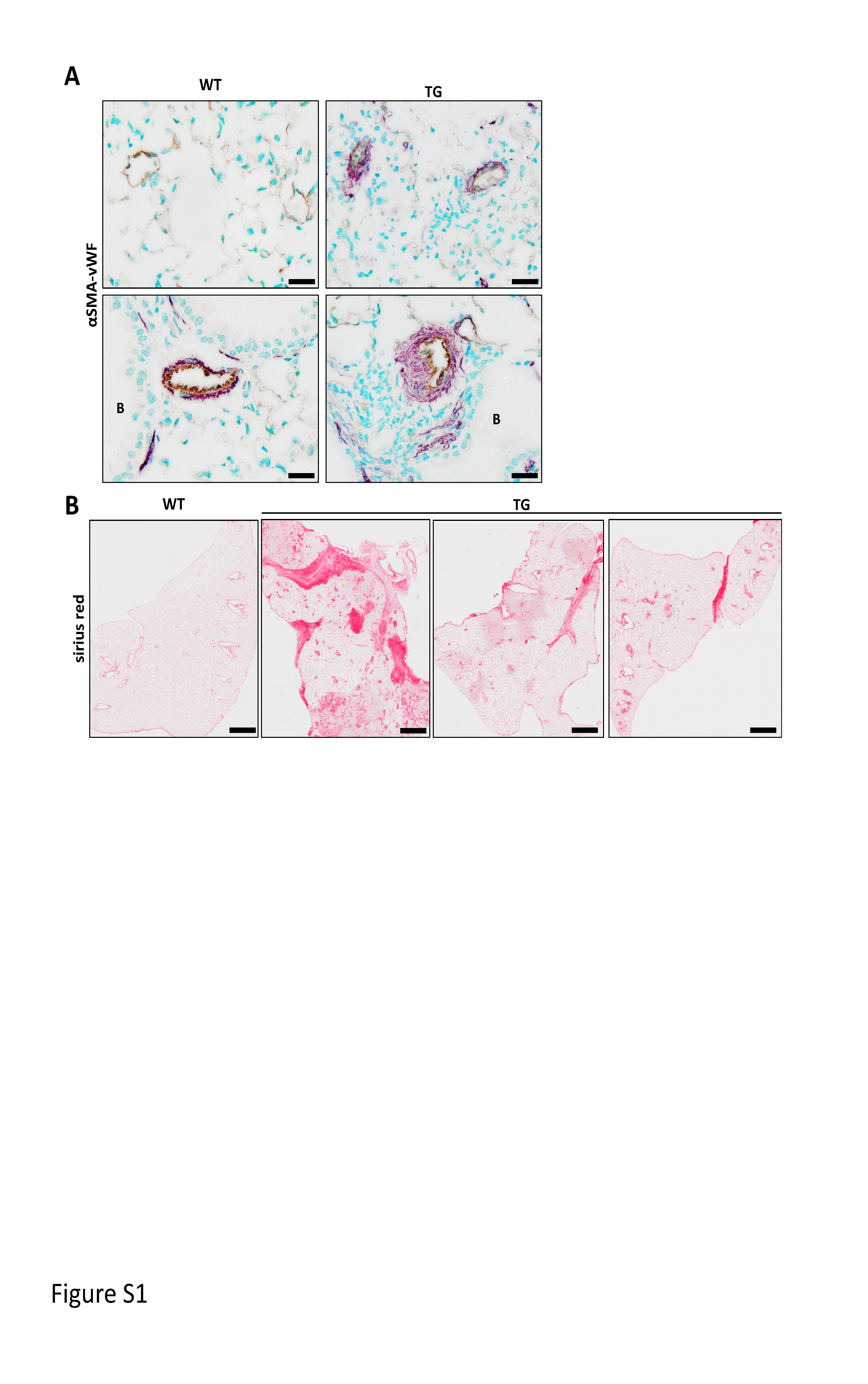
Supplementary figure S1. : Parenchymal and vascular remodelling in Fra2 overexpressing (TG). (A) Representative images of Sirius red stained whole lung sections from 3 different Fra-2 TG mice and one littermate control; scale bars 1 mm. (B) Representative images of immunohistochemical double staining against α-smooth muscle actin (α-sma, violet) and endothelium marker von Willebrand Factor (vWF, light brown) of wild-type (WT) or Fra-2 TG mice. Upper panel: small parenchymal vessels, lower panel: peribronchial arteries; scale bar: 20 μm; B: bronchi. ERJ-00154-2019.Figure_S1 (1.6MB, jpg)
{kind=link}
Supplementary figure S2. Loading control for electrophoretic mobility shift assays (presented in Figure 2E-F) of parenchymal fibroblasts (PF) transfected with Fra-2 overexpression plasmid (+) or empty control plasmid (-) and of lung homogenates from Fra-2 overexpressing (TG) or wild-type control (WT) mice. CE: cytoplasmic extract; NE: nuclear extract. ERJ-00154-2019.Figure_S2 (77.2KB, jpg)
{kind=link}
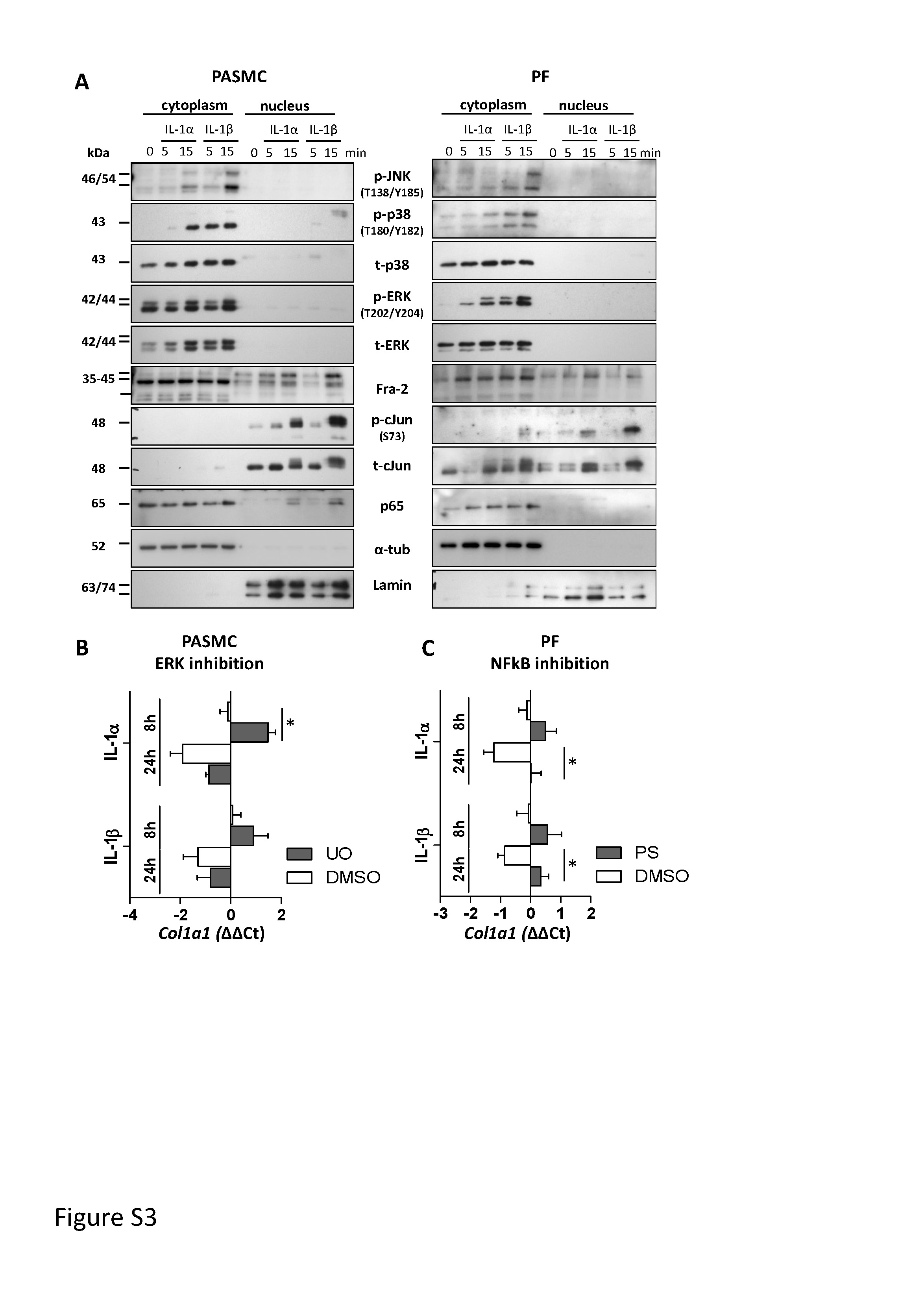
Supplementary figure S3. Signalling pathways regulated upon IL-1α and IL-1β stimulation. (A) Representative Western blot images for phosphorylated and total JNK, p38, ERK, cJun, Fra2 and p65 of cytoplasmic and nuclear extracts from pulmonary arterial smooth muscle cells (PASMC) and parenchymal fibroblasts (PF) after 5 and 15 minutes stimulation with IL-1α (1 ng/ml) or IL-1β (10 ng/ml). Lamin and α-tubulin served as a nuclear and cytoplasmic loading control. (B,C) Col1a1 expression upon IL-1α (1 ng/ml) and IL-1β (10 ng/ml) and inhibition of ERK signalling in PASMCs (B) and NFκB signalling in PFs (C). U0: ERK inhibitor U0126; PS: IκB inhibitor PS1145. DMSO: dimethysulfoxide – vehicle control. *p < 0.05. ERJ-00154-2019.Figure_S3 (538.8KB, jpg)
{kind=link}
Shareable PDF
Acknowledgements
We would like to thank Verena Braunschmid, Julia Schittl, Bettina Schrenk and Thomas Fuchs (Ludwig Boltzmann Institute for Lung Vascular Research, Graz, Austria) for technical support. We are grateful to Latifa Bakiri and Erwin Wagner (Spanish National Cancer Research Center, Madrid, Spain) for generously providing the CMV-hu-Fra-2 plasmid construct. We are grateful to the Research Institute for Molecular Pathology (Vienna, Austria) and Erwin Wagner for Fra-2 TG mice.
Footnotes
This article has supplementary material available from erj.ersjournals.com
Support statement: A. Birnhuber was funded by the Jubilee Foundation of the Austrian National Bank (grant 16187 to G. Kwapiszewska) and was trained within the frame of the PhD programme “Molecular Medicine” of the Medical University of Graz (Graz, Austria). A. Sahu-Osen was funded by the Austrian Science Fund (FWF): P27848-B28 (to G. Kwapiszewska). R. Eferl was supported by the Austrian Science Fund (FWF) Doktoratskolleg-plus grant “Inflammation and Immunity”: FWF grants P26908-B20 and P29222-B28. S. Crnkovic, V. Biasin, L. Brcic, N. Cikes, A. Olschewski and G. Kwapiszewska were recipients of Austrian Agency for International Cooperation in Education and Research (ÖAD) support (WTZ HR14/2012). Funding information for this article has been deposited with the Crossref Funder Registry.
Conflict of interest: A. Birnhuber reports grants from Jubilee Foundation of the Austrian National Bank, during the conduct of the study.
Conflict of interest: S. Crnkovic has nothing to disclose.
Conflict of interest: V. Biasin has nothing to disclose.
Conflict of interest: L.M. Marsh has nothing to disclose.
Conflict of interest: B. Odler has nothing to disclose.
Conflict of interest: A. Sahu-Osen has nothing to disclose.
Conflict of interest: E. Stacher-Priehse has nothing to disclose.
Conflict of interest: L. Brcic reports grants and personal fees from AstraZeneca, personal fees from Roche Austria, nonfinancial support from MSD Austria and Pfizer Austria, outside the submitted work.
Conflict of interest: F. Schneider has nothing to disclose.
Conflict of interest: N. Cikes has nothing to disclose.
Conflict of interest: B. Ghanim has nothing to disclose.
Conflict of interest: W. Klepetko has nothing to disclose.
Conflict of interest: W. Graninger has nothing to disclose.
Conflict of interest: Y. Allanore has nothing to disclose.
Conflict of interest: R. Eferl has nothing to disclose.
Conflict of interest: A. Olschewski has nothing to disclose.
Conflict of interest: H. Olschewski reports personal fees and nonfinancial support from Bayer, MSD, Pfizer and Novartis, grants, personal fees and nonfinancial support from Actelion, grants from Inventiva, personal fees from Bellerophon, outside the submitted work; and is a part-time employee of the Ludwig Boltzmann Institute for Lung Vascular Research, Graz, Austria.
Conflict of interest: G. Kwapiszewska reports grants from Jubilee Foundation of the Austrian National Bank (grant 16187), Austrian Science Fund (FWF) (P27848-B28), and Austrian Agency for International Cooperation in Education and Research (WTZ HR14/2012), during the conduct of the study.
References
- 1.Fan MH, Feghali-Bostwick CA, Silver RM. Update on scleroderma-associated interstitial lung disease. Curr Opin Rheumatol 2014; 26: 630–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Elhai M, Meune C, Boubaya M, et al. . Mapping and predicting mortality from systemic sclerosis. Ann Rheum Dis 2017; 76: 1897–1905. [DOI] [PubMed] [Google Scholar]
- 3.Giacomelli R, Liakouli V, Berardicurti O, et al. . Interstitial lung disease in systemic sclerosis: current and future treatment. Rheumatol Int 2017; 37: 853–863. [DOI] [PubMed] [Google Scholar]
- 4.Adler S, Huscher D, Siegert E, et al. . Systemic sclerosis associated interstitial lung disease – individualized immunosuppressive therapy and course of lung function: results of the EUSTAR group. Arthritis Res Ther 2018; 20: 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gu YS, Kong J, Cheema GS, et al. . The immunobiology of systemic sclerosis. Semin Arthritis Rheum 2008; 38: 132–160. [DOI] [PubMed] [Google Scholar]
- 6.Hussein MR, Hassan HI, Hofny ER, et al. . Alterations of mononuclear inflammatory cells, CD4/CD8+ T cells, interleukin 1beta, and tumour necrosis factor alpha in the bronchoalveolar lavage fluid, peripheral blood, and skin of patients with systemic sclerosis. J Clin Pathol 2005; 58: 178–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Maekawa T, Jinnin M, Ohtsuki M, et al. . Serum levels of interleukin-1alpha in patients with systemic sclerosis. J Dermatol 2013; 40: 98–101. [DOI] [PubMed] [Google Scholar]
- 8.Dieude P, Bouaziz M, Guedj M, et al. . Evidence of the contribution of the X chromosome to systemic sclerosis susceptibility: association with the functional IRAK1 196Phe/532Ser haplotype. Arthritis Rheum 2011; 63: 3979–3987. [DOI] [PubMed] [Google Scholar]
- 9.Parpaleix A, Amsellem V, Houssaini A, et al. . Role of interleukin-1 receptor 1/MyD88 signalling in the development and progression of pulmonary hypertension. Eur Respir J 2016; 48: 470–483. [DOI] [PubMed] [Google Scholar]
- 10.Gasse P, Mary C, Guenon I, et al. . IL-1R1/MyD88 signaling and the inflammasome are essential in pulmonary inflammation and fibrosis in mice. J Clin Invest 2007; 117: 3786–3799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Voelkel NF, Tuder RM, Bridges J, et al. . Interleukin-1 receptor antagonist treatment reduces pulmonary hypertension generated in rats by monocrotaline. Am J Respir Cell Mol Biol 1994; 11: 664–675. [DOI] [PubMed] [Google Scholar]
- 12.Dinarello CA, Simon A, van der Meer JW. Treating inflammation by blocking interleukin-1 in a broad spectrum of diseases. Nat Rev Drug Discov 2012; 11: 633–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Eferl R, Hasselblatt P, Rath M, et al. . Development of pulmonary fibrosis through a pathway involving the transcription factor Fra-2/AP-1. Proc Natl Acad Sci USA 2008; 105: 10525–10530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Maurer B, Busch N, Jungel A, et al. . Transcription factor fos-related antigen-2 induces progressive peripheral vasculopathy in mice closely resembling human systemic sclerosis. Circulation 2009; 120: 2367–2376. [DOI] [PubMed] [Google Scholar]
- 15.Biasin V, Marsh LM, Egemnazarov B, et al. . Meprin beta, a novel mediator of vascular remodelling underlying pulmonary hypertension. J Pathol 2014; 233: 7–17. [DOI] [PubMed] [Google Scholar]
- 16.Subcommittee for Scleroderma Criteria of the American Rheumatism Association Diagnostic and Therapeutic Criteria Committee. Preliminary criteria for the classification of systemic sclerosis (scleroderma). Arthritis Rheum 1980; 23: 581–590. [DOI] [PubMed] [Google Scholar]
- 17.LeRoy EC, Medsger TA Jr. Criteria for the classification of early systemic sclerosis. J Rheumatol 2001; 28: 1573–1576. [PubMed] [Google Scholar]
- 18.Petrasek J, Bala S, Csak T, et al. . IL-1 receptor antagonist ameliorates inflammasome-dependent alcoholic steatohepatitis in mice. J Clin Invest 2012; 122: 3476–3489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang L, Yan JW, Wang YJ, et al. . Association of interleukin 1 family with systemic sclerosis. Inflammation 2014; 37: 1213–1220. [DOI] [PubMed] [Google Scholar]
- 20.Humbert M, Monti G, Brenot F, et al. . Increased interleukin-1 and interleukin-6 serum concentrations in severe primary pulmonary hypertension. Am J Respir Crit Care Med 1995; 151: 1628–1631. [DOI] [PubMed] [Google Scholar]
- 21.Reich N, Maurer B, Akhmetshina A, et al. . The transcription factor Fra-2 regulates the production of extracellular matrix in systemic sclerosis. Arthritis Rheum 2010; 62: 280–290. [DOI] [PubMed] [Google Scholar]
- 22.Hung CF, Rohani MG, Lee SS, et al. . Role of IGF-1 pathway in lung fibroblast activation. Respir Res 2013; 14: 102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gungl A, Biasin V, Wilhelm J, et al. . Fra2 overexpression in mice leads to non-allergic asthma development in an IL-13 dependent manner. Front Immunol 2018; 9: 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Song E, Ouyang N, Horbelt M, et al. . Influence of alternatively and classically activated macrophages on fibrogenic activities of human fibroblasts. Cell Immunol 2000; 204: 19–28. [DOI] [PubMed] [Google Scholar]
- 25.Maurer B, Reich N, Juengel A, et al. . Fra-2 transgenic mice as a novel model of pulmonary hypertension associated with systemic sclerosis. Ann Rheum Dis 2012; 71: 1382–1387. [DOI] [PubMed] [Google Scholar]
- 26.Hutyrova B, Lukac J, Bosak V, et al. . Interleukin 1alpha single-nucleotide polymorphism associated with systemic sclerosis. J Rheumatol 2004; 31: 81–84. [PubMed] [Google Scholar]
- 27.Beretta L, Bertolotti F, Cappiello F, et al. . Interleukin-1 gene complex polymorphisms in systemic sclerosis patients with severe restrictive lung physiology. Hum Immunol 2007; 68: 603–609. [DOI] [PubMed] [Google Scholar]
- 28.Cavalli G, Fallanca F, Dinarello CA, et al. . Treating pulmonary silicosis by blocking interleukin 1. Am J Respir Crit Care Med 2015; 191: 596–598. [DOI] [PubMed] [Google Scholar]
- 29.Lawrance IC, Maxwell L, Doe W. Altered response of intestinal mucosal fibroblasts to profibrogenic cytokines in inflammatory bowel disease. Inflamm Bowel Dis 2001; 7: 226–236. [DOI] [PubMed] [Google Scholar]
- 30.Mia MM, Boersema M, Bank RA. Interleukin-1beta attenuates myofibroblast formation and extracellular matrix production in dermal and lung fibroblasts exposed to transforming growth factor-beta1. PLoS One 2014; 9: e91559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Siwik DA, Chang DL, Colucci WS. Interleukin-1beta and tumor necrosis factor-alpha decrease collagen synthesis and increase matrix metalloproteinase activity in cardiac fibroblasts in vitro. Circ Res 2000; 86: 1259–1265. [DOI] [PubMed] [Google Scholar]
- 32.Vesey DA, Cheung C, Cuttle L, et al. . Interleukin-1beta stimulates human renal fibroblast proliferation and matrix protein production by means of a transforming growth factor-beta-dependent mechanism. J Lab Clin Med 2002; 140: 342–350. [DOI] [PubMed] [Google Scholar]
- 33.van Nieuwenhoven FA, Hemmings KE, Porter KE, et al. . Combined effects of interleukin-1alpha and transforming growth factor-beta1 on modulation of human cardiac fibroblast function. Matrix Biol 2013; 32: 399–406. [DOI] [PubMed] [Google Scholar]
- 34.Huang XL, Wang YJ, Yan JW, et al. . Role of anti-inflammatory cytokines IL-4 and IL-13 in systemic sclerosis. Inflamm Res 2015; 64: 151–159. [DOI] [PubMed] [Google Scholar]
- 35.Wynn TA. Fibrotic disease and the TH1/TH2 paradigm. Nat Rev Immunol 2004; 4: 583–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hasegawa M, Fujimoto M, Kikuchi K, et al. . Elevated serum levels of interleukin 4 (IL-4), IL-10, and IL-13 in patients with systemic sclerosis. J Rheumatol 1997; 24: 328–332. [PubMed] [Google Scholar]
- 37.Ando K, Nakashita T, Kaneko N, et al. . Associations between peripheral blood eosinophil counts in patients with systemic sclerosis and disease severity. Springerplus 2016; 5: 1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hesselstrand R, Wildt M, Bozovic G, et al. . Biomarkers from bronchoalveolar lavage fluid in systemic sclerosis patients with interstitial lung disease relate to severity of lung fibrosis. Respir Med 2013; 107: 1079–1086. [DOI] [PubMed] [Google Scholar]
- 39.Wynn TA, Vannella KM. Macrophages in tissue repair, regeneration, and fibrosis. Immunity 2016; 44: 450–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Higashi-Kuwata N, Jinnin M, Makino T, et al. . Characterization of monocyte/macrophage subsets in the skin and peripheral blood derived from patients with systemic sclerosis. Arthritis Res Ther 2010; 12: R128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Huang J, Maier C, Zhang Y, et al. . Nintedanib inhibits macrophage activation and ameliorates vascular and fibrotic manifestations in the Fra2 mouse model of systemic sclerosis. Ann Rheum Dis 2017; 76: 1941–1948. [DOI] [PubMed] [Google Scholar]
- 42.Santarlasci V, Cosmi L, Maggi L, et al. . IL-1 and T helper immune responses. Front Immunol 2013; 4: 182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lichtman AH, Chin J, Schmidt JA, et al. . Role of interleukin 1 in the activation of T lymphocytes. Proc Natl Acad Sci USA 1988; 85: 9699–9703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Koenders MI, Devesa I, Marijnissen RJ, et al. . Interleukin-1 drives pathogenic Th17 cells during spontaneous arthritis in interleukin-1 receptor antagonist-deficient mice. Arthritis Rheum 2008; 58: 3461–3470. [DOI] [PubMed] [Google Scholar]
- 45.Helmby H, Grencis RK. Interleukin 1 plays a major role in the development of Th2-mediated immunity. Eur J Immunol 2004; 34: 3674–3681. [DOI] [PubMed] [Google Scholar]
- 46.Nakae S, Komiyama Y, Yokoyama H, et al. . IL-1 is required for allergen-specific Th2 cell activation and the development of airway hypersensitivity response. Int Immunol 2003; 15: 483–490. [DOI] [PubMed] [Google Scholar]
- 47.Schuepbach-Mallepell S, Philippe V, Bruggen MC, et al. . Antagonistic effect of the inflammasome on thymic stromal lymphopoietin expression in the skin. J Allergy Clin Immunol 2013; 132: 1348–1357. [DOI] [PubMed] [Google Scholar]
- 48.Satoskar AR, Okano M, Connaughton S, et al. . Enhanced Th2-like responses in IL-1 type 1 receptor-deficient mice. Eur J Immunol 1998; 28: 2066–2074. [DOI] [PubMed] [Google Scholar]
- 49.Choy DF, Hart KM, Borthwick LA, et al. . TH2 and TH17 inflammatory pathways are reciprocally regulated in asthma. Sci Transl Med 2015; 7: 301ra129. [DOI] [PubMed] [Google Scholar]
- 50.Loscher CE, Mills KH, Lynch MA. Interleukin-1 receptor antagonist exerts agonist activity in the hippocampus independent of the interleukin-1 type I receptor. J Neuroimmunol 2003; 137: 117–124. [DOI] [PubMed] [Google Scholar]
- 51.Franck N, Maris M, Nalbandian S, et al. . Knock-down of IL-1Ra in obese mice decreases liver inflammation and improves insulin sensitivity. PLoS One 2014; 9: e107487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bersudsky M, Luski L, Fishman D, et al. . Non-redundant properties of IL-1alpha and IL-1beta during acute colon inflammation in mice. Gut 2014; 63: 598–609. [DOI] [PubMed] [Google Scholar]
- 53.Dinarello CA. Interleukin-1 in the pathogenesis and treatment of inflammatory diseases. Blood 2011; 117: 3720–3732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mantero JC, Kishore N, Ziemek J, et al. . Randomised, double-blind, placebo-controlled trial of IL1-trap, rilonacept, in systemic sclerosis. A phase I/II biomarker trial. Clin Exp Rheumatol 2018; 36: Suppl. 113, 146–149. [PubMed] [Google Scholar]
- 55.Hadjinicolaou AV, Nisar MK, Bhagat S, et al. . Non-infectious pulmonary complications of newer biological agents for rheumatic diseases – a systematic literature review. Rheumatology 2011; 50: 2297–2305. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Please note: supplementary material is not edited by the Editorial Office, and is uploaded as it has been supplied by the author.
Supplementary material ERJ-00154-2019.SUPPLEMENT (279.7KB, pdf)
Supplementary figure S1. : Parenchymal and vascular remodelling in Fra2 overexpressing (TG). (A) Representative images of Sirius red stained whole lung sections from 3 different Fra-2 TG mice and one littermate control; scale bars 1 mm. (B) Representative images of immunohistochemical double staining against α-smooth muscle actin (α-sma, violet) and endothelium marker von Willebrand Factor (vWF, light brown) of wild-type (WT) or Fra-2 TG mice. Upper panel: small parenchymal vessels, lower panel: peribronchial arteries; scale bar: 20 μm; B: bronchi. ERJ-00154-2019.Figure_S1 (1.6MB, jpg)
Supplementary figure S2. Loading control for electrophoretic mobility shift assays (presented in Figure 2E-F) of parenchymal fibroblasts (PF) transfected with Fra-2 overexpression plasmid (+) or empty control plasmid (-) and of lung homogenates from Fra-2 overexpressing (TG) or wild-type control (WT) mice. CE: cytoplasmic extract; NE: nuclear extract. ERJ-00154-2019.Figure_S2 (77.2KB, jpg)
Supplementary figure S3. Signalling pathways regulated upon IL-1α and IL-1β stimulation. (A) Representative Western blot images for phosphorylated and total JNK, p38, ERK, cJun, Fra2 and p65 of cytoplasmic and nuclear extracts from pulmonary arterial smooth muscle cells (PASMC) and parenchymal fibroblasts (PF) after 5 and 15 minutes stimulation with IL-1α (1 ng/ml) or IL-1β (10 ng/ml). Lamin and α-tubulin served as a nuclear and cytoplasmic loading control. (B,C) Col1a1 expression upon IL-1α (1 ng/ml) and IL-1β (10 ng/ml) and inhibition of ERK signalling in PASMCs (B) and NFκB signalling in PFs (C). U0: ERK inhibitor U0126; PS: IκB inhibitor PS1145. DMSO: dimethysulfoxide – vehicle control. *p < 0.05. ERJ-00154-2019.Figure_S3 (538.8KB, jpg)
This one-page PDF can be shared freely online.
Shareable PDF ERJ-00154-2019.Shareable (479.3KB, pdf)