

Summary
Definitive haematopoiesis in the fetal liver supports self-renewal and differentiation of haematopoietic stem cells/multipotent progenitors (HSC/MPPs) but remains poorly defined in humans. Using single cell transcriptome profiling of ~140,000 liver and ~74,000 skin, kidney and yolk sac cells, we identify the repertoire of human blood and immune cells during development. We infer differentiation trajectories from HSC/MPPs and evaluate the impact of tissue microenvironment on blood and immune cell development. We reveal physiological erythropoiesis in fetal skin and the presence of mast cells, NK and ILC precursors in the yolk sac. We demonstrate a shift in fetal liver haematopoietic composition during gestation away from being erythroid-predominant, accompanied by a parallel change in HSC/MPP differentiation potential, which we functionally validate. Our integrated map of fetal liver haematopoiesis provides a blueprint for the study of paediatric blood and immune disorders, and a valuable reference for harnessing the therapeutic potential of HSC/MPPs.
Keywords: human development, haematopoiesis, immunology, single cell RNA-sequencing, liver, skin, kidney, yolk-sac
Introduction
The blood and immune systems develop during early embryogenesis. Our understanding of this process derives from murine and in vitro model systems as human fetal tissue is scarce. While haematopoietic development is conserved across vertebrates1, important differences between mouse and human have been noted2,3. Comprehensive interrogation of human tissue to understand the molecular and cellular landscape of early hematopoiesis has implications beyond life in utero, providing a blueprint for understanding immunodeficiencies, childhood leukemias and anaemias and generating insights into HSC/MPP propagation to inform stem-cell technologies.
The earliest blood and immune cells originate outside the embryo, arising from the yolk-sac between 2-3 post-conception weeks (PCW). At 3-4 PCW, intra-embryonic progenitors from the aorta-gonad-mesonephros (AGM) develop4. Yolk-sac and AGM progenitors colonise fetal tissues such as the liver, which remains the major organ of haematopoiesis until the mid-second trimester. Fetal bone marrow (BM) is colonised around 11 PCW and becomes the dominant site of haematopoiesis after 20 PCW in human4. Yolk sac-, AGM-, fetal liver- and BM-derived immune cells seed peripheral tissues including non-lymphoid tissues (NLT), where they undergo specific maturation programs which are both intrinsically determined and extrinsically nurtured by the tissue microenvironment5,6. Systematic, comprehensive analysis of multiple blood and immune lineages during human development has not previously been attempted.
In this study, we used single cell transcriptomics to map the molecular states of human fetal liver cells between 7-17 PCW, when the liver represents the predominant site of human fetal haematopoiesis. We integrate imaging mass cytometry, flow cytometry and cellular morphology to validate the transcriptome-based cellular profiles. We construct the functional organisation of the developing immune network through comparative analysis of immune cells in fetal liver with those in yolk sac, and skin and kidney as representative NLT.
Results
Single cell transcriptome of fetal liver
To investigate blood and immune cell development in the fetal liver, we generated single cell suspensions from embryonic and fetal livers between 6 and 17 PCW. We FACS-isolated CD45+ and CD45- cells using adjoining gates for comprehensive capture (Figure 1a and Extended Data 9a) for single cell RNA-sequencing (scRNA-seq) (both 10x Genomics platform Smart-seq2) (Figure 1, Extended Data 4d, and Supplementary Table 1). To allow parallel evaluation of blood and immune cell topography in NLT and the yolk sac during early development (Figure 1a) we profiled skin, kidney and yolk sac cells by FACS-isolation and 10x Genomics platform.
Figure 1. Single cell transcriptome map of fetal liver.

a, Schematic of tissue processing and cell isolation for scRNA-seq profiling of fetal liver, skin and kidney across four developmental stages (7-8, 9-11, 12-14, and 15-17 post conception weeks (PCW)), and yolk sac from 4-7 PCW. SS2, Smart-seq2. b, UMAP visualisation of fetal liver cells from 10x using 3’ chemistry. Colours indicate cell state. HSC/MPP, haematopoietic stem cell/multipotent progenitor; ILC, innate lymphoid cell; NK, natural killer cell; Neut-myeloid, neutrophil-myeloid; DC, dendritic cell; pDC, plasmacytoid DC; Mono-mac, monocyte-macrophage; EI, erythroblastic island; Early L/TL, Early lymphoid/T lymphocyte; MEMP, megakaryocyte-erythroid-mast cell progenitor. Statistical significance of cell frequency change by stage shown in parentheses (negative binomial regression with bootstrap correction for sort gates; * p < 0.05, *** p < 0.001, and **** p < 0.0001 as per SI Table 8) with up/down arrows to indicate positive/negative coefficient of change, respectively. c, Liver composition by developmental stage as the mean percentage of each population per stage corrected by CD45+/CD45- sort fraction. Colours indicate cell states as shown in b.
In total, 138,575 (n = 14) liver (an additional 1,206 cells were profiled using Smart-seq2), 54,690 (n = 7) skin, 9,643 kidney (n = 3) and 10,071 yolk sac (n = 3) cells passed quality control (QC) and doublet exclusion (Extended Data 1a-b, and Supplementary Table 2). We performed graph-based Louvain clustering and derived differentially expressed genes to annotate cell clusters. To minimize technical batch-effect while preserving biological variation due to gestational stage, we divided liver samples into four gestational stage categories and performed data integration between samples using Harmony (Extended Data 1c-e).
27 major cell states are present in the fetal liver (Figure 1b, Extended Data 1f). VCAM1+ erythroblastic island (EI) macrophages were validated as a distinct cell state as a result of their interactions with erythroid cells (Extended Data 4a-f). We applied a descriptive nomenclature based on gene expression profiles. All cell states are found throughout the developmental period studied, but frequency varied by gestation stage (Figure 1c and Extended Data 1e). Neutrophils, basophils and eosinophils are not detected, consistent with reports of granulocytes emerging during fetal BM haematopoiesis7. Early stage samples show erythroid lineage bias, with later lymphoid and myeloid lineages representation, as previously shown5 (Figure 1c and Extended Data 1g).
Our fetal liver dataset can be explored using an interactive web portal through the following weblink: https://developmentcellatlas.ncl.ac.uk//datasets/hca_liver/. We provide comprehensive expression profiles of genes known to cause primary immunodeficiencies8 to aid future molecular phenotyping of these disorders (Extended Data 8).
Validation of genes and cell states
We manually selected 48 genes from the 4,471 differentially expressed genes between all clusters (log fold change >0.5) (Figure 2a). The predictive power of these 48 genes to determine cell states by the Random Forest classifier was on average 89% for precision and recall (Extended Data 2a). We designed a FACS panel for prospective cell isolation using genes encoding surface proteins (Figure 2a, Extended Data 9b), permitting validation of 19 cell types by mini-bulk transcriptome profiling (6 of the 19 cell types also by scRNA-seq) using Smart-seq2 (Extended Data 2b-d). Cytospins from FACS-isolated cells are morphologically consistent with their designated cell type, including hypogranularity of embryonic/fetal mast cells9 and early erythroid cells resemblance to the previously reported ‘early erythroid progenitors’ (Figure 2b)10.
Figure 2. Multi-modal and spatial validation of cell types.

a, Median scaled ln-normalised gene expression of 48 selected differentially expressed genes for the liver cell states from 1b visualised by dot; asterisk (*) indicates markers used for FACS-isolation of cells. Gene expression frequency (% cells within cell type expressing) indicated by spot size and expression level by colour intensity. Neut-my, Neutrophil-myeloid; prec., precursor; mono, monocyte. b, Representative Giemsa-stained cytospins showing morphology of populations isolated by FACS based on differentially expressed genes with * in a. Scale bar, 10μm.
Next, we evaluated the spatial distribution of erythroid, mast cell, myeloid and lymphoid lineages using imaging mass cytometry (Extended Data 2e). The liver architecture evolves considerably between 8 and 15 PCW. Organization of hepatocyte aggregates increases, though hepatic lobules around a central vein and portal triad are not clearly visible. Haematopoietic islands are present in sinusoids and surrounding hepatocyte aggregates. Sinusoidal CD68+ macrophages are surrounded by GlycophorinA (GYPA)+ erythroid cells (Extended Data 2e). CD1c+ DCs and CD79a/CD20+ cells from the B-lineage are sparsely distributed (Extended Data 2e). Cell proportions approximate our scRNA-seq profile for haematopoietic cells but not hepatocytes (Extended Data 2e and Figure 1b), in keeping with the fragility of hepatocytes following ex vivo isolation and their high expression of mitochondrial genes11. By validating our single cell transcriptome dataset with multiple modalities, we provide an integrated map of haematopoietic cells in the fetal liver.
Fetal liver and NLT haematopoiesis
Next, we inferred trajectories of haematopoietic development. By force directed graph (FDG), we identify three connections to a central HSC/MPP node featuring erythroid-megakaryocyte-mast cells, B cell and innate/T-lymphoid cells and myeloid cells (Figure 3a and Supplementary Video 1). Partition based approximate graph abstraction (PAGA) also supports the presence of a shared megakaryocyte-erythroid-mast cell progenitor (MEMP) downstream of HSC/MPP (Extended Data 3a). Genes dynamically modulated in the specification of erythroid, megakaryocyte and mast cell lineages are distinct: TAL1 and KLF1 in erythroid; F11R, PBX1 and MEIS1 in megakaryocyte; and HES1 in mast cell differentiation (Extended Data 3b)12–15. We explored supporting factors for fetal liver erythropoiesis using CellPhoneDB16 to predict specific/enriched receptor-ligand interactions between erythroblasts and VCAM1+ EI macrophages (Extended Data 4a). We identify statistically significant interactions for VCAM1, ITGB1, ITGA4, SIGLEC1, ICAM4 and SPN, molecules known to be important in haematopoiesis (Extended Data 4a)17,18. The presence of VCAM1 on EI macrophages and ITGA4 on early/mid erythroid cells is confirmed by immunohistochemical analysis on serial fetal liver sections (Extended Data 4b). VCAM1+ EI macrophage interaction with erythroblasts is also observed using Imaging Flow Cytometry ImageStream analysis (Extended Data 4d) and may explain their combined erythroblast and macrophage transcriptome (Figure 2a), which has also been described in mouse central EI macrophages (Extended Data 4g)19.
Figure 3. Fetal liver and NLT haematopoiesis.

a, Force-directed graph (FDG) visualisation of all haematopoietic cells from 1b. b, Dot plot showing the median scaled ln-normalised expression of globin genes encoding haemoglobin subunits; HBZ and HBE1 (Gower 1), HBE1 and HBA1 (Gower 2) and HBA1 and HBG2 (fetal) in liver, skin, and yolk sac erythroid lineages (MEMP, early, mid and late erythroids). Gene expression frequency (% cells within cell type expressing) indicated by spot size and expression level by colour intensity.
Comparing across haematopoietic tissues, mast cells are also present in yolk sac (Extended Data 1b). Erythroblasts show expression of haemoglobin genes, and a temporal shift from Gower 1 and 2 subunit expression (HBZ, HBE1) to fetal haemoglobin subunit expression (HBA1 and HBG2) between yolk sac and liver (Figure 3b).
Megakaryocytes, erythroid cells, mast cells and MEMP, are present in NLT, but HSC/MPPs are absent (Extended Data 1a, Extended Data 3c-e). We compared the highly expressed and differentially expressed genes of corresponding cell types in fetal liver, skin and kidney (Extended Data 3d). Mast cells, megakaryocytes and cells of the erythroid lineages show high connectivity (PAGA scores) across all four tissues (Extended Data 3e). Local maturation of progenitors in NLT or influx of cells at various differentiation stages are two possibilities. Erythroid cells are absent in kidney, suggesting restricted differentiation of the MEMP lineage in certain sites (Extended Data 1a). Immunohistochemical analysis of serial skin sections show nucleated GYPA+ cells inside and outside of CD34+ blood vessels, in keeping with local differentiation of MEMPs (Extended Data 3f). Light sheet fluorescence microscopy supported this finding (Extended Data 3g, Supplementary Video 2). The proliferative capacity of MEMP in NLT is confirmed using MKI67 and cell cycle gene expression (Extended Data 3h). Skin MEMP express some early erythroblast genes including MYL4 (Extended Data 3d)20, suggesting that these may act as erythroid progenitors in situ in the skin. These findings demonstrate that during early development, the skin in physiological state can contribute to erythropoiesis and supplement fetal liver erythroid output.
Lymphoid lineages in fetal liver and NLT
Previous studies have reported the presence of T and B lymphocytes21, NK cells22, and ILCs23 in the human fetal liver. We observe two lymphoid branches; an NK/T/ILC lineage and a B-lineage (Figure 4a and Extended Data 5a). The ‘early lymphoid/T lymphocyte’ cluster varies by gestational stage, with cells expressing GATA3, KLRB1, CD3D, CD7 and JCHAIN at 7-8PCW before T cells emerge from the thymus (Figure 4a and Extended Data 5b-c). Early in gestation, this cluster may contain the fetal liver early thymocyte progenitor, which is capable of generating αβT cells upon co-culture with thymic epithelial cells24,25. At the 12-14 PCW stage, cells express TRDC and TRAC, but no GZMB or PRF1, the cytoplasmic granular products characteristic of mature CD8+ T cells. TRDC expression is absent at the 15-17 PCW stage (Extended Data 5b-d). These findings are in keeping with the seeding of fetal liver by γδT cells and αβT cells sequentially following their exit from thymus after 12 PCW26 and are consistent with previous reports of T cell identification only after 18 PCW25,27.
Figure 4. Lymphoid lineages in fetal liver and NLT.

a, FDG visualisation of fetal liver HSC/MPP and lymphoid cell types from 1b showing changes over four developmental stages. b, FDG visualisation of fetal liver and corresponding skin, kidney and yolk sac lymphoid cells.
NK cells (expressing NCAM1, CD7, IL2RB and CD3E) and ILC precursors (expressing KIT, KLRB1, IL7R, RORC) share a common origin in the lymphoid branch by PAGA and diffusion map analyses (Figure 2a, 4a, Extended Data 5a and 5i, and Supplementary Table 3). This is in keeping with existing literature of a shared progenitor for NK and ILCs in human and mouse28,29.
The B-lineage cells are a continuum of differentiation states, from primitive ‘pre pro-B’ clusters expressing CD34, SPINK2, and IGLL1 to ‘pro-B’ and ‘pre-B’ clusters with increasing expression of B cell transcripts MS4A1, CD79B, DNTT and HLA-DRA and reducing expression of JCHAIN and LTB30 (Figure 2a, 4a and Supplementary Table 3). ‘Pro/pre-B’ cell clusters have high nuclear to cytoplasmic ratio, immature chromatin and nucleoli (Extended Data 5e). Differentially regulated genes in the HSC/MPPs to B cell transition include SPIB, SP100 and CTSS (Extended Data 5e). Pre-B cells are detected between 7-8 PCW, but mature B cells only after 9 PCW (Figure 1b-c and 4a). We observe a decline in HSC/MPP expression of NFKBIA, an inhibitor of NF-κB, and an increase in Kupffer cell TNFSF13B (BAFF) expression with gestation (Extended Data 5f). NF-κB and BAFF are known B cell survival and differentiation factors31. The cell-intrinsic versus tissue-microenvironment factors controlling B cell differentiation in the fetal liver require further investigation.
Comparing liver with yolk sac and NLT, pro-B, pre-B and B cells are present in NLT but HSC/MPPs and pre pro-B cells are absent (Figure 4b and Extended Data 5g). NK cell precursors, NK cells and ILC in NLT share a transcriptional signature with their liver counterparts, however tissue-specific expression of chemokine (XCLI, CXCL8) and cytotoxic granule genes (GNLY) suggest maturation and tissue adaptation in the skin and kidney (Extended Data 5h). ILC precursors in NLT lack the full characteristic markers and TFs of their mature progenies; ILC1, ILC2 and ILC3 (Extended Data 5i). NK cells and ILC precursors are present in yolk sac (Figure 4b). Together, these findings suggest that NLTs are seeded by NK and ILC precursors from fetal liver and potentially yolk sac, which differentiate in situ and acquire tissue-related gene expression profiles.
Tissue signatures in myeloid cells
In mice, fate-mapping studies have demonstrated that tissue macrophages are seeded by yolk sac and fetal liver progenitors32,33, while dendritic cells (DCs) originate from BM-derived HSC/MPPs through a monocyte-independent lineage34. We observe myeloid progenitors, monocytes, macrophages, DC1 and DC2 in fetal liver and NLT as early as 7 PCW (Figure 1b, 5a-b, Extended Data 1a, 6a).
Figure 5. Tissue signatures in developing myeloid cells.

a, FDG visualisation of HSC/MPP, myeloid progenitors, monocytes and macrophages from fetal liver, decidua/placenta and yolk sac. Mac, Macrophage; Monocyte prec., Monocyte precursor; Neut-myeloid prog., Neutrophil-myeloid progenitor. b, PAGA connectivity scores of the populations shown in a.
Myeloid lineages stem from HSC/MPP via three intermediates: a neutrophil-myeloid progenitor expressing CD34, SPINK2, AZU1, PRTN3, ELANE, MPO and LYZ, monocyte- and DC-precursors (Figure 2a, Figure 3a and Supplementary Table 3). DC1 differentiate from neutrophil-myeloid progenitors and DC-precursors link closely to DC2. DC and monocyte differentiation involve dynamic regulation of CLEC11A, BATF3 and ID2 while monocyte differentiation involves S100A8/A9, FCGR1A/2A and S100A12 (Extended Data 6b). Plasmacytoid DC (pDC) precursors branch from both early myeloid precursors and pre pro-B cells in keeping with recent reports of their mixed lymphoid and myeloid origin in mice (Extended Data 6a)35,36. We compared monocytes, macrophages and their putative precursors between placenta, yolk sac and fetal liver (Figure 5a-b and Extended Data 6c). The macrophage transcriptome profile is highly tissue specific, showing connectivity to other macrophage subtypes within their tissue of residence and obscuring potential ontogenic relationships (Figure 5a-b and Extended Data 6c-e).
Monocytes, macrophages, pDC, DC1 and DC2 are present in both skin and kidney (Extended Data 1a). NLT monocytes and DCs correlate strongly to fetal liver counterparts but macrophages are more tissue-specific, with VCAM1 expression in liver and F13A1 in skin, as is observed in adult tissue macrophages37,38 (Extended Data 6e). Tissue specific gene expression patterns conferring functional specialization are discernible for DCs, for example S100A4 in skin DC1 (a molecule involved in T cell activation)39 and AOAH in liver DC2 (an enzyme involved in lipopolysaccharide response modulation)40 (Extended Data 6e). DC activation despite the sterile fetal environment suggests an active role for fetal DCs in mediating tolerance as previously reported41.
HSC/MPP differentiation potential by age
Our observation of an HSC/MPP cell state from which the earliest lineage-committed cells radiate is in keeping with recent observations from scRNA-seq analysis in post-natal mice and humans35,42,43 (Figure 6a). At the base of this cluster by FDG visualization is a population expressing CLEC9A, HLA-DRA and highest levels of primitive genes including MLLT3 consistent with a multipotent long-term repopulating (LT)-HSC (Figure 6a-b)44. Using differentially expressed genes and supervised analysis, HSC/MPP clusters with gene expression intermediate between LT-HSC and early progenitors similar to human lymphoid-primed short-term (ST)-HSCs44, the mouse erythroid-biased MMP2, and myeloid-biased MMP345,46 are identifiable, demonstrating early transcriptome priming along all differentiation branches within the MPP pool (Figure 6a-b).
Figure 6. HSC/MPP differentiation potential by gestation.

a, FDG visualisation of liver HSC/MPP and early haematopoietic progenitor populations from Figure 1b. b, Violin plots showing ln-normalised median gene expression of statistically significant, dynamically variable genes that are up or downregulated during HSC/MPP transition to neutrophil-myeloid progenitors, MEMP and pre pro-B cells from fetal liver. Bar and ‘ns’ indicate not significant. H/M, HSC/MPP. c, Stacked barplot of all different types of colonies generated by single ‘HSC pool’ gate cells in an assay with MS5 stroma. * p < 0.05, *** p < 0.001, individual samples shown in Extended Data 7c. The colour of the stat bar corresponds to the type of colony tested vs all others. My, Myeloid; Ery, Erythroid, Meg, Megakaryocyte. d, Percentage of colonies generated by single ‘HSC pool’ gate cells containing erythroid cells (defined as the sum of Ery, Ery/Meg, Ery/Meg/My, Ery/My, Ery/NK and Ery/NK/My colonies shown in c), *** p < 0.001. My, Myeloid; Ery, Erythroid; Meg, Megakaryocyte. e, Percentage of colonies containing B cells following culture in B/NK optimized conditions from 10 cells from ‘HSC pool’ gate (** p < 0.01). f, Mean +/- s.d. percentage of cells in the G0 phase of the cell cycle assessed using Ki67 and DAPI flow cytometry analysis (* p = 0.0136). g, ln-normalised median expression of selected genes in yolk sac progenitors, cord blood HSC and adult bone marrow HSC with significant differential expression compared to fetal liver HSC/MPP, visualised by violin plots (**** p < 0.0001).
We hypothesized that the cellular composition of the developing fetal liver resulted from local modulation of HSC/MPP potential. To test this, we FACS-isolated single cells from the CD34+CD38+, CD34+CD38-CD45RA- and CD34+CD38-CD45RA+ FACS gates and profiled them by both plate-based single cell transcriptomics (Smart-seq2) and single cell clonal differentiation assays44,47 (Extended Data 7a). A support vector machine trained on the fetal liver dataset identified enrichment of HSC/MPPs (~85%) in the CD34+CD38- gate, with the majority of cells in the CD34+CD38-CD45RA+ MLP gate also classified as HSC/MPP (Extended Data 7b). This is in agreement with reported transcriptional similarity between HSC/MPP and MLP78 and our identification of lymphoid priming in the HSC/MPP compartment. Single cell culture from CD34+CD38-CD45RA- HSC/MPPs yield both uni- and multipotential colonies (Figure 6c-e and Extended Data 7c-g). There is a significant reduction in trilineage colonies with gestational age (Extended Data 7f). Colonies containing erythroid cells significantly decrease while those containing NK cells and B cells increase with gestational age (Figure 6c-e, Extended Data 7c-g). HSC/MPP from <9 PCW embryonic livers generate almost no B cells (Figure 6e), in keeping with the paucity of B cells at this stage (Figure 1c, 4a and Extended Data 1g). These findings support the hypothesis of differential HSC/MPP intrinsic potential by gestational stage and mirror our observation of early erythroid predominance and greater lymphoid representation at later stages (Figure 1c and Figure 4a).
Comparing HSC/MPPs and early progenitors across haematopoietic tissues during development, higher MKI67 expression and cell cycle staining suggest enhanced proliferative potential of yolk sac and fetal liver progenitors/HSC/MPPs48–50 (Extended Data 7h). The fraction of fetal liver HSC/MPP in G0increases with gestational age (Figure 6f, Extended Data 7i), indicating a progressive shift to quiescence during fetal life. Fetal liver HSC/MPP have higher expression of genes encoding a heat shock protein (HSPA1A), potentially for maintenance of genome and proteome integrity, and lower levels of MHC-I (HLA-B) suggesting reduced antigen presenting potential compared to cord blood and adult BM HSC/MPPs (Figure 6g).
Collectively, our findings demonstrate that intrinsic changes in HSC/MPP numbers, proliferation and differentiation potential occur over the first and second developmental trimesters. These changes are likely to be pivotal for fetal liver haematopoiesis to adapt to the needs of the developing fetus; first the establishment of an effective oxygen transport system and subsequently the development of a complete blood and immune system.
Discussion
Development of the human immune system in utero has remained poorly understood. Using single cell transcriptome profiling, we resolve the cellular heterogeneity and abstract dynamic temporal information on blood and immune development in fetal liver, yolk sac, skin and kidney. Large scale scRNA-seq studies during human development must account for technical batch effects without compromising detection of biological variations over gestation. Our approach highlights key insights; physiological erythropoiesis in fetal skin, establishment of DC network as early as 7 PCW, potential dual myeloid and lymphoid origin of pDCs, seeding of mast cells, NK and ILCs from the yolk sac and tissue adaptation of NKs, ILCs, DCs and macrophages during development. Our findings reveal modulation of HSC/MPP intrinsic differentiation potential over gestation age suggesting this as an additional functional mechanism to regulate haematopoietic output of the fetal liver throughout the first and second trimesters.
In summary, our comprehensive fetal liver atlas provides a foundational resource for understanding fetal liver haematopoiesis and the developing immune system. Our reference dataset will be invaluable for studies on paediatric blood and immune disorders and exploiting HSC/MPPs for therapy. Our approach using single cell transcriptomics to study human development provides a framework that can be applied to study any temporal processes across the human lifespan.
Materials and methods
Tissue Acquisition
Human fetal tissues were obtained from the MRC/Wellcome Trust-funded Human Developmental Biology Resource (HDBR; http://www.hdbr.org)51 with appropriate written consent and approval from the Newcastle and North Tyneside NHS Health Authority Joint Ethics Committee (08/H0906/21+5). HDBR is regulated by the UK Human Tissue Authority (HTA; www.hta.gov.uk) and operates in accordance with the relevant HTA Codes of Practice.
Embryos and fetal specimens used for light sheet fluorescence microscopy were obtained with written informed consent from the parents (Gynecology Hospital Jeanne de Flandres, Lille, France) with the approval of the local ethics committee (protocol N°PFS16-002). Tissues were made available in accordance with the French bylaw (Good practice concerning the conservation, transformation and transportation of human tissue to be used therapeutically, published on December 29, 1998). Permission to utilize human tissues was obtained from the French agency for biomedical research (Agence de la Biome decine, Saint-Denis La Plaine, France).
Tissue Processing
All tissues were processed immediately after isolation using the same protocol. Tissue was transferred to a sterile 10mm2 tissue culture dish and cut into <1mm3 segments before being transferred to a 50mL conical tube. Yolk sac content was aspirated for analysis prior to yolk sac digestion. Tissue was digested with 1.6mg/mL collagenase type IV (Worthington) in RPMI (Sigma-Aldrich) supplemented with 10%(v/v) heat-inactivated fetal bovine serum (Gibco), 100U/mL penicillin (Sigma-Aldrich), 0.1mg/mL streptomycin (Sigma-Aldrich), and 2mM L-Glutamine (Sigma-Aldrich) for 30 minutes at 37°C with intermittent shaking. Digested tissue was passed through a 100μm filter, and cells collected by centrifugation (500g for 5 minutes at 4°C). Cells were treated with 1X RBC lysis buffer (eBioscience) for 5 minutes at room temperature and washed once with flow buffer (PBS containing 5%(v/v) FBS and 2mM EDTA) prior to counting.
Fetal developmental stage assignment and chromosomal assessment
Embryos up to 8 post conception weeks (PCW) were staged using the Carnegie staging method52. After 8 PCW, developmental age was estimated from measurements of foot length and heel to knee length and compared against a standard growth chart53. A piece of skin, or where this was not possible, chorionic villi tissue was collected from every sample for Quantitative Fluorescence-Polymerase Chain Reaction analysis using markers for the sex chromosomes and the following autosomes 13, 15, 16, 18, 21, 22, which are the most commonly seen chromosomal abnormalities. All samples were karyotypically normal.
Flow cytometry and FACS for scRNA-seq
Antibody panels were designed to allow enrichment of cell fractions for sequencing and cell type validation. Antibodies used for FACS isolation are listed in Supplementary Table 14. An antibody cocktail was prepared fresh by adding 3μL of each antibody in 50μL Brilliant Stain Buffer (BD) per tissue. Cells (<10x106) were resuspended in 50-100μL flow buffer and an equal volume of antibody mix was added to cells from each tissue. Cells were stained for 30 minutes on ice, washed with flow buffer and resuspended at 10x106 cells/mL. DAPI (Sigma-Aldrich) was added to a final concentration of 3μM immediately prior to sorting. Flow sorting was performed on a BD FACSAria™ Fusion instrument using DIVAv8, and data analysed using FlowJo (v10.4.1, BD). Cells were gated to exclude dead cells and doublets, and then isolated for scRNA-seq analysis (10x or Smart-seq2). For 10x, cells were sorted into chilled FACS tubes coated with FBS and prefilled with 500μL sterile PBS. For Smart-seq2, single cells were index-sorted into 96-well lo-bind plates (Eppendorf) containing 10μL lysis buffer (TCL (Qiagen) + 1% (v/v) β-mercaptoethanol) per well. B cells were also investigated by flow cytometry as per Roy et. al.54.
Cytospins and mini bulk RNA-seq validation
Fetal liver cells were immunostained with two separate panels (see Supplementary Table 15 for antibody details). Cells were stained for 30 minutes on ice followed by DAPI staining. FACS was performed on a BD FACSAria™ Fusion instrument, and data analysed using FlowJo (v10.4.1). Cells were isolated into chilled FACS tubes coated with FBS and prefilled with 500μL sterile PBS for cytospin (500 – 2000 cells), or into 1.5mL microfuge tubes containing 20μL lysis buffer (100 cells). Giemsa staining (Sigma-Aldrich) was used to determine the morphology of sorted cells on cytospins. Slides were viewed using a Zeiss AxioImager microscope, images taken of 4 fields from n = 3 samples using the 100x objective, and viewed using Zen (v2.3). Raw images supplied in Supplementary Images.
HSC/MPP Culture
MS5 in log-phase growth (DSMZ, Germany, passage 6-10, or imported from Prof. Katsuhiko Itoh at Kyoto University; all tested mycoplasma-free, not authenticated) were seeded into 96-well flat-bottom plates (Nunclon delta surface; Thermo) at a density of 3000 cells per well 24 hours prior to sorting. Medium was Myelocult H5100 (Stem Cell Technologies) supplemented with 100U/mL Penicillin and 0.1mg/mL Streptomycin (Sigma-Aldrich). On the day of sorting, media were replaced with Stem Pro-34 SFM media (Life Technologies) supplemented with 100U/mL Penicillin and 0.1mg/mL Streptomycin, 2mM L-glutamine (Sigma-Aldrich), stem cell factor 100ng/ml (Miltenyi), Flt3 20ng/ml (Miltenyi), TPO 100ng/ml (Miltenyi), EPO 3ng/ml (Eprex), IL-6 50ng/ml (Miltenyi), IL-3 10ng/ml (Miltenyi), IL-11 50ng/ml (Miltenyi), GM-CSF 20ng/ml (Miltenyi), IL-2 10ng/ml (Miltenyi), IL-7 20ng/ml (Miltenyi) and Lipids (hLDL) 50ng/ml (Life Technologies)44.
Frozen fetal liver cells were thawed and stained with 10μl/106 cells of antibody cocktail (see Supplementary Table 16-17 for antibody details) for 30 minutes on ice. Three populations of HSC/MPPs and progenitors were isolated from fetal liver suspension. Populations were identified from the DAPI-, doublet-excluded gate as CD3/CD16/CD11c/CD14/CD19/CD56-, CD34+ cells (see Supplementary Table 16 for antibody details). The HSC/MPP pool and MLP were found within the 20% of cells with lowest CD38 expression: HSC/MPP pool were CD90+/- and CD45RA- whilst MLP were CD90-CD45RA+. Progenitors with the highest 20% of CD38 expression were sorted for comparison. Single cells were sorted using a BD FACSAria™ Fusion, and sorted directly onto MS5 or medium for culture, or into 96-well lo-bind plates containing 10μl/well lysis buffer for Smart-seq2 scRNAseq (Supplementary Table 12). Single-cell-derived colonies analysis was performed as described by the Laurenti Lab44. In brief, colonies were harvested into 96 U-bottom plates using a plate filter to prevent the carryover of MS5 cells. Cells were stained with 50μl/well of antibody cocktail (Supplementary Table 16-17 for antibody details), incubated for 20 minutes in the dark at room temperature and then washed with 100μl/well of PBS + 3% FBS. The type (lineage composition) and the size of the colonies formed were assessed by high-throughput flow cytometry (BD FACS Symphony). Colony output was determined using the gating strategy shown in Extended Data 9c. A single cell was defined as giving rise to a colony if the sum of cells detected in the CD45+ 5+ and GYPA+ gates was ≥ 30 cells. Erythroid colonies were identified as CD45−GYPA+ ≥ 30 cells, Megakaryocyte colonies as CD41+ ≥ 30 cells, Myeloid colonies as [(CD45+CD14+) + (CD45+CD15+)] ≥ 30 cells, NK colonies as CD45+CD56+ ≥ 30 cells. All high-throughput screening flow cytometry data was recorded in a blinded way, and correlation between the colony phenotype and originating population was only performed at the final stage. Two-tailed Fisher’s exact test, performed in Prism (v8.1.2, GraphPad Software), were applied to the numbers of colonies of each type by stage to determine statistical significance in lineage differentiation potential with development. For differentiation of B cells from HSC/MPP pool, 10 cells were sorted directly on MS5 stroma in the same conditions as Doulatov et al.47 (see Supplementary Table 17 for antibody details). An example of gating strategy is shown in Extended Figure 8d. For Ki67 staining, cells were stained using antibody panel in Supplementary Table 18, CD34+ cells were sorted, fixed/permeabilised using the BD Cytofix/Cytoperm kit according to the manufacturer’s instructions and then stained overnight for Ki67-FITC followed by DAPI as a DNA dye.
ImageStream analysis of fetal liver cell suspensions
Frozen fetal liver cells were thawed and stained with the antibody cocktail (see Supplementary Table 19 for antibody details) for 30 minutes on ice. Cells were washed with flow buffer and resuspended at the same cell density employed for cell sorting experiments (10x106 cells/mL). DAPI (Sigma-Aldrich) was added to a final concentration of 3μM immediately prior to acquisition in order to identify and exclude dead cells from the experiment. Samples were acquired on a fully calibrated ImageStream X MKII system (Luminex Corporation, USA) using 488nm, 561nm, 405nm and 642nm excitation lasers and the 60x magnification collection optic. Laser powers were set in order to maximise signal resolution but minimise any saturation of the CCD camera with bright-field (BF) images collected in channels 1 and 9. A minimum of 50,000 cell events were collected per sample. In order to calculate spectral compensation, single stained antibody capture beads were acquired with the bright-field illumination turned off. Spectral compensation and data analysis were performed using the IDEAS analysis software (v6.2.64, Luminex Corp). Briefly, dead cells were first excluded based on DAPI positivity. Overt doublets and debris were excluded from the live cell population using the aspect ratio and area of the BF image in channel 1. Single cells had an intermediate area value and an aspect ratio between 1 and 0.6 and were gated as such. Overt doublets present as having an increased area and a lower aspect ratio value and were also gated as such. In all cases the position and boundaries of a gate was checked for appropriateness using the underlying imagery. The key cell types within the sample were then identified and gated using the total integrated (post-compensation) fluorescence signals from each labelled antibody in the panel in an analogous fashion to conventional flow cytometry data. Each major cell type was then interrogated using the associated multi-spectral images for true single cell identity or for the presence of “pernicious doublets” (cells with either significant debris attached or large cells with much smaller cells attached). The same phenotypic analysis was extended to the cells in the overt doublet gate. Further analysis was performed in FlowJo (v10.4.1). Kruskal-Wallis with Dunn’s post hoc, performed in Prism (v8.1.2, GraphPad Software), were applied to the proportion of each cell type found within the doublet gate to determine statistical significance in doublet rates across fetal liver cell types.
Library Preparation and Sequencing
For the droplet-encapsulation scRNA-seq experiments, 7,000 live, single, CD45+ or CD45- FACS-isolated cells were loaded onto each channel of a Chromium chip before encapsulation on the Chromium Controller (10x Genomics, Pleasanton, CA, USA). Single cell sequencing libraries were generated using the Single Cell 3' v2 and V(D)J Reagent Kits (for T cell receptor repertoire analysis) as per the manufacturer’s protocol. Libraries were sequenced using an Illumina HiSeq 4000 using v4 SBS chemistry to achieve a minimum depth of 50,000 raw reads per cell. The libraries were sequenced using the following parameters: Read1: 26 cycles, i7: 8 cycles, i5: 0 cycles; Read2: 98 cycles to generate 75bp paired end reads.
For the plate-based scRNA-seq experiments, a slightly modified Smart-seq2 protocol was used as previously described55. After cDNA generation, libraries were prepared (384 cells per library) using the Illumina Nextera XT kit (Illumina Inc, San Diego, CA, USA). Index v2 sets A, B, C and D were used per library to barcode each cell before multiplexing. Each library was sequenced to achieve a minimum depth of 1-2 million raw reads per cell using an Illumina HiSeq 4000 using v4 SBS chemistry to generate 75bp paired end reads.
For the mini bulk RNA-seq experiments, each cell lysate was transferred into a 96-well lo-bind plate (Eppendorf) then processed using the same modified Smart-seq2 protocol as described above. After cDNA generation, libraries were prepared using the Illumina NexteraXT kit with Index v2 set A to barcode each mini bulk library before multiplexing. All libraries were sequenced on one lane of an Illumina HiSeq 4000 using v4 SBS chemistry to generate 75bp paired end reads and aiming to achieve a minimum depth of 10 million reads per library.
Immunohistochemistry
Formalin fixed, paraffin embedded blocks of fetal livers aged 6 PCW, 8 PCW, 10 PCW and 13 PCW were obtained from the HDBR. Each was sectioned at 4μm thickness onto APES-coated slides. Sections were dewaxed for 5 minutes in Xylene (Fisher Chemical) then rehydrated through graded ethanol (99%, 95% and 70%; Fisher Chemical) and washed in running water. Sections were treated with hydrogen peroxide block (1%v/v in water; Sigma) for 10 minutes and rinsed in tap water prior to antigen retrieval. Citrate antigen retrieval was used for all sections. Citrate buffer, pH6 was used with pressure heating for antigen retrieval, and then slides placed in TBS, pH7.6 for 5 minutes prior to staining. Staining was done using the Vector Immpress Kit (Vector Laboratories). Sections were blot dried and blocked sequentially with 2.5% normal horse serum, avidin (Vector Laboratories) and then biotin (Vector Laboratories) for 10 minutes each and blot dried in between. Sections were incubated for 60 minutes with primary antibody diluted in TBS pH7.6 (see Supplementary Table 20 for antibody details). Slides were washed twice in TBS pH7.6 for 5 minutes each before incubation for 30 minutes with the secondary antibody supplied with the kit. Slides were washed twice in TBS pH7.6 for 5 minutes each, and developed using peroxidase chromogen DAB. Sections were counterstained in Mayer’s Haematoxylin for 30 seconds, washed and put in scots tap water for 30 seconds. Slides were dehydrated through graded ethanol (70% to 99%) and then placed in Xylene prior to mounting with DPX (Sigma-Aldrich). Sections were imaged on a Nikon Eclipse 80i microscope using NIS-Elements Fv4.
Alignment, quantification and quality control of scRNA-seq data
Droplet-based (10x) sequencing data was aligned and quantified using the Cell Ranger Single-Cell Software Suite (version 2.0.2, 10x Genomics Inc) using the GRCh38 human reference genome (official Cell Ranger reference, version 1.2.0). Smart-seq2 sequencing data was aligned with STAR (version 2.5.1b), using the STAR index and annotation from the same reference as the 10x data. Gene-specific read counts were calculated using htseq-count (version 0.10.0). Cells with fewer than 200 detected genes and for which the total mitochondrial gene expression exceeded 20% were removed. Genes that were expressed in fewer than 3 cells were also removed. We detected on average ~3,000 genes per cell with the 10x Genomics platform and ~6,000 genes with the Smart-seq2 protocol.
Doublet detection
Doublets were detected with an approach adapted from Pijuan-Sala et al.56. In the first step of the process, each 10X lane was processed independently. Scrublet57 was run, obtaining per-cell doublet scores. The standard Seurat-inspired Scanpy processing pipeline was performed up to the clustering stage, using default parameters. Each cluster was subsequently separately clustered again, yielding an over clustered manifold, and each of the resulting clusters had its Scrublet scores replaced by the median of the observed values. The resulting scores were assessed for statistical significance, with p-values computed using a right-tailed test from a normal distribution centred on the score median and a MAD-derived standard deviation estimate. The MAD was computed from above-median values to circumvent zero-truncation. The p-values were FDR-corrected with the Benjamini-Hochberg procedure, and a significance threshold of 0.1 was imposed. In the second step of the process, all 10X lanes for a single tissue were pooled together and the Seurat-inspired Scanpy processing was repeated, with the addition of Harmony58 with a theta of 3 for batch correction between the lanes before the neighbour graph identification step. The joint manifold was clustered, and the frequency of identified doublets was computed. The same statistical framework as in the first step was used to identify clusters significantly enriched in doublets, which were subsequently flagged as doublets in their entirety and removed.
Clustering and annotation
Downstream analysis included data normalisation (NormalizeData, LogNormalize method, scaling factor 10000), data feature scaling (ScaleData), variable gene detection (FindVariableGenes), PCA (RunPCA, from variable genes) and Louvain graph-based clustering (FindClusters, data dimensionality reduction using PCA, clustering resolution (res.30)) performed using the R package Seurat (version 2.3.4). Cluster cell identity was assigned by manual annotation using known marker genes and computed differentially expressed genes (DEGs) using FindAllMarkers function in Seurat package (one-tailed Wilcoxon rank sum test, p-values adjusted for multiple testing using the Bonferroni correction; Supplementary Table 3). For computing DEGs all genes were probed provided they were expressed in at least 25% of cells in either of the two populations compared and the expression difference on a natural log scale was at least 0.25. Manual annotation was performed iteratively, which included validating proposed cell labels with known markers and further investigating clusters whose gene signatures indicated additional diversity. Number of each cell type per sample, annotations per cell, and nGene and nUMI per cell type are reported in Supplementary Tables 4, 5, 8 and 9, respectively.
Clustering and cell type assignment for fetal liver data was assessed using two additional clustering methods (not shown): Agglomerative clustering (with Ward linkage and Euclidean affinity) and Gaussian mixture (AgglomerativeClustering class from cluster module and GaussianMixture from mixture module in sklearn version 0.19.1 Python 3.6.3). Consensus agreement between the 3 clustering methods was measured by Rand index and adjusted mutual information implemented in the metrics module in sklearn package. The Rand Index scores were 0.89 and 0.85 for Agglomerative and Gaussian Mixture clustering methods respectively.
After annotation was completed, a cell type classifier was built by training an SVM on labelled fetal liver scRNA-seq data with grid search for parameter optimization based on training data. 70% of the data was used for training and the other 30% for test. The SVM was previously compared in terms of accuracy and recall with a random forest and logistic regression classifiers trained on the same data. Out of the 3 classifiers the SVM was chosen due to showing a mean accuracy and weighted mean recall of 95%. Random forest showed 89% for both precision and recall (Supplementary Table 11). The SVM classifier was used for automatic annotation of the Smart-seq2 and mini bulk RNA sequencing data sets to allow identification of biologically meaningful clusters and DEG computation.
Data generated from fetal skin, kidney and yolk sac was pre-processed, normalised, clustered and manually annotated, in parallel with, and using the same pipeline as, the liver data. Annotation by cell type for skin and kidney, and yolk sac are reported in Supplementary Tables 6 and 7, respectively. Skin and kidney data were combined using the MergeSeurat function. Clusters characterised by differentially expressed immune gene markers were extracted from the NLT dataset for subsequent comparative analysis with liver-derived immune populations. Human cord blood and adult bone marrow datasets were downloaded from Human Cell Atlas data portal (https://preview.data.humancellatlas.org/). These were processed using the same approach as described above, followed by manual annotation. Decidua and placental data from Vento-Tormo et al.16, were downloaded from ArrayExpress record E-MTAB-6701 (https://www.ebi.ac.uk/arrayexpress/experiments/E-MTAB-6701/).
Data integration
We used Harmony data integration58 to correct for batch effect between sample identities. The average kBET rejection rate statistically significantly improved from 0.735 to 0.471 (Supplementary Table 13) following Harmony data integration (p-value 3.83e-3 in Kolmogorov–Smirnov test and p-value 8.8e-6 in Wilcoxon signed-rank test). The manifold was subjected to re-clustering using Harmony adjusted PCs with parameters as mentioned above in “Clustering and annotation”. Cell type classifications were then ascertained through re-annotation of the clusters derived from Harmony adjusted PCs to produce the final annotation.
Changes in cell proportions over development
Comparison of cell proportions across gestational stages was assessed by modelling cell number data with negative binomial regression based on Poisson-gamma mixture distribution. Cell numbers were corrected for CD45-/CD45+ FACS sorted ratio (Supplementary Table 1) prior to applying negative binomial regression modelling. Modelling was achieved using the glm.nb function in the R MASS package. Modelled cell number data were studied for regression coefficient significance (variable coefficient p-value ≤ 0.05) to the response variable of gestational age with the corresponding z-score and p-values taken (Supplementary Table 10)
Dimensionality reduction and trajectory analysis
Dimensionality reduction methods included tSNE (Seurat, computed from the first 20 PCs, Barnes-Hut fast computation), UMAP (Python UMAP package, 5 nearest neighbours, correlation metric, minimum distance 0.3, computed from the first 20 PCs), FDG (ForceAtlas2 class from fa2 Python Package, Barnes-Hut implementation for faster computation with theta 0.8, 2000 iterations) and partition-based approximate graph abstraction (PAGA) (paga in scanpy Python package version 1.2.2). Development trajectories were inferred by comparing FDG, PAGA and diffusion map plots. Inferred trajectory analysis included computing diffusion map (scanpy tl.diffmap with 20 components), pseudotime (scanpy tl.dpt setting the earliest known cell type as root) and variable genes across pseudotime. Order of cells in pseudotime was statistically significant using Kruskal-Wallis test (p < 1x10-7).
Comparisons of trajectories across stage were performed by subsetting liver dataset by stage using SubsetData function, computing dimensional reduction coordinates, batch correcting by sample using Harmony, and plotting PAGA and FDG by stage. Cell type comparisons across tissue involved subsetting for cell types of interest using SubsetData function, merging cross-tissue datasets using MergeSeurat function, and processing data using the same approach as for the liver and NLT datasets. Harmony batch correction was then performed by tissue type, with results presented as combined UMAPs, FDGs and PAGA score heatmaps.
Dynamically expressed genes across pseudotime
Genes that vary across pseudotime were calculated using DifferentialGeneTest function in Monocle in R (version 2.6.4) and a cut-off of adjusted p-value < 0.001 applied. This was applied on the entire pseudotime range and also on the pseudotime intervals specific to each cell type in order to avoid limitation to the genes characterised by monotonic changes across the inferred trajectory. Expression of pseudotime variable genes were min-max normalised prior to visualization and annotated based on each gene’s involvement in relevant cell-specific functional modules or hallmark functional pathways from MSigDB v6.2, a curated molecular signature database59. Peak expression for each gene over pseudotime was calculated and grouped into ‘Early’, ‘Mid’ or ‘Late’ categories. For visualisation purposes, the resulting gene lists were minimised by ordering them from those present in the most selected functional pathways to least, as well as ensuring coverage across pseudotime. These genes were manually compared against current literature to determine if they have known functional or cell type associations. The top 20-25 genes in each list were displayed using the ggplot2 package. Transcription factors were marked within the dataset based on AnimalTFDB transcription factor prediction database60. The full pseudotime gene list is available in the interactive files accompanying diffusion maps.
Mouse data generated by Li et al.19 used for comparison of erythroid-associated genes expressed in mouse Kupffer cells vs EI macrophages shown in Extended Data 4g is available at GEO with accession code GSE127980.
Visualisation by animated force-directed graph representation
The FDG animation was created using an in-house modified version of the ForceAtlas2 class in fa2 Python package by saving all the intermediate states (published version only outputs the final state and discards all intermediates). The FDG coordinates at each iteration were plotted and the resulting graphs were assembled in a mp4 video format using VideoWriter in cv2 (version 3.3.1) Python package.
Differential gene extraction and validation
Differential gene validation was done using a random forest classifier (RandomForestClassifier class in ensemble module of sklearn Python package v0.19.1, with 500 estimators, min_sample_split of 5, class weights set to the “balanced” policy and all other parameters set to default). The Random Forest algorithm was chosen as it resembled the FACS gating hierarchy. 70% of the data was used for training and 30% for test. Parameter tuning was performed on training data using grid search. To determine whether tissue-related transcriptome variations were present in equivalent immune populations between liver, skin and kidney, each equivalent population was taken in turn and grouped according to its tissue of origin. Seurat FindMarkers function was then applied in a pair-wise manner between each tissue subset to produce a cell type-specific list of genes marking each tissue subset. These were investigated in turn for biological relevance, with representative genes displayed using VlnPlot function of Seurat.
DEGs from B cell pseudotime were studied for significant expression change across stage and differentiation state using a one-way ANOVA with Tukey’s multiple comparison test. DEGs displaying significant variance in ln-normalised expression were further studied for correlation to DEGs identified within all other cell-types across stage. B cell pseudotime DEGs with significantly correlated trends of expression to DEGs within other cell-types across stage (p-value <0.05, Two-tailed Pearson’s R at 95% CI) were plotted in Prism (v8.1.2, GraphPad Software). All graphs presented in the manuscript were plotted using ggplot2 R package, Seurat implementation of ggplot2, matplotlib Python package, Prism (v.8.1.2, GraphPad Software) or FlowJo (v10.4.1). Spot plots are shown throughout the manuscript, displaying scaled expression of ln-normalised counts.
Primary immunodeficiency (PID) gene list curation
Disease and genetic deficiency information was extracted from Picard et al.8 and manually annotated to include HGNC symbol names for each disease-associated genetic defect for subsequent correlation with the liver dataset. Diseases implicated in PID were divided according to the International Union of Immunological Societies (IUIS) major categories and screened across the liver scRNA-seq dataset. 315 unique genes were identified in the dataset from the 354 inborn errors of immunity highlighted in the article. For each disease category a dot plot was generated using Seurat DotPlot function and ordered by highest expression across each gene and across each cell type, highlighting those cell types in each disease category which express the highest number of genes associated with a genetic defect.
CellPhoneDB analysis
CellPhoneDB v2.0 (www.cellphonedb.org)61 was used for the receptor-ligand analysis in Extended Data 4a. Significant (p < 0.05) receptor-ligand interactions between VCAM1+ Erythroblastic Island macrophages and the two erythroid (early and mid) populations were displayed.
Whole genome sequencing and fetal cell identification
To identify maternal cells present in our data we combined the information from fetal whole genome DNA sequencing with the single cell RNA-seq data. For each sample we measured the allele frequency in the fetal DNA of SNPs from the 1000 genomes project62 falling within exons with a population allele frequency in excess of 1%. We then consider only those SNPs which are homozygous in the fetal DNA for follow up in the scRNA-seq data. A SNP was considered to be homozygous if its allele frequency in the fetal DNA was less than 0.2 or greater than 0.8 and had an FDR adjusted p-value of less than 0.01 under a binomial test for the null hypothesis that the allele frequency in the DNA was in the range [0.3,0.7].
The allele frequency of each of these SNPs with population allele frequency > 1% that are known to be homozygous in the fetal DNA was then measured in each cell in the scRNA-seq data. Any deviations from homozygosity in the RNA-seq data must be a consequence of either sequencing errors, RNA editing, or the genotype of the cell differing from the fetal DNA. For each cell, we calculated the total fraction of reads at the SNPs (selected as described above) that differ from the fetal genotype. We then assume that the genome-wide rate of deviations due to sequencing errors and RNA editing is less than or equal to 2%. For maternal cells, the expected genome wide rate of deviation at these SNPs is equal to half the mean of the population allele frequency at the interrogated SNPs. Finally, for each cell we calculated the posterior probability of the cell being fetal or maternal assuming a binomial distribution with rate 2% for a fetal cell and half the mean of the population allele frequency for the maternal cell and assign a cell as: maternal/fetal if either posterior probability exceeds 99%, ambiguous otherwise. We validated this method using samples for which both the fetal and maternal DNA were available.
‘Hyperion’ Imaging mass cytometry (IMC)
Antibodies were conjugated to metals using the Fluidigm MaxPar conjugation kits and the associated method with following modifications; the lanthanides were used at 1.5 mM and washed for a shorter duration (4x 5 minutes) in W buffer in prior to elution. Ultrapure MilliQ water was used throughout for any dilutions and washes. 4 μm Formalin-fixed paraffin-embedded sections obtained from 8 and 15 PCW fetal liver tissue blocks were incubated at 60 °C for 1 hour then dewaxed in Xylene (Fisher). After rehydration through graded alcohols (Fisher) and a 5 minute wash in water, the sections were subjected to Heat-Induced Epitope Retrieval with Citrate buffer (pH- 6.0). Sections were then washed in water and PBS (Gibco) and blocked with 3% BSA (Sigma-Aldrich) for 45 minutes. A mixture of 8 metal-conjugated antibodies diluted in 0.5% BSA (see Supplementary Table 21 for antibody details, was added to the sections for overnight incubation at 4 °C in a humidified chamber. Slides were washed twice in 0.2% Triton X-100 diluted in PBS for 8 minutes and then twice in PBS for 8 minutes.
To counterstain nucleated cells, sections were incubated with 312.5nm (193 Ir) Intercalator-Ir (Fluidigm) for 30 minutes at room temperature. Slides were then washed in water for 5 minutes, and allowed to air-dry at room temperature prior to imaging on the Hyperion imaging mass cytometer. Using expected target cell frequencies from previous fluorescence flow cytometry data, Region of Interest (ROI) size was set to 2.8mm by 3.8mm. The ablation energy was set at 2 db with a laser frequency of 200Hz. Each session of ablation generated a .MCD image file containing information for every panorama and ROI measured whereby each 1μm piece of tissue liberated by the laser was analysed for ionic content on a per channel basis by Time of Flight. Single cell segmentation and feature extraction was performed using CellProfiler (v3.1.5). Nuclei were identified using the “IdentifyPrimaryObjects” module where the input images were the sum of the DNA stained Iridium channels (191 and 193) constructed by the “ImageAfterMath” module. The diameter range set for Nuclei identification was 4-15 pixel units. The “ExpandOrShrink” module was used to grow the nuclear segmentation area by 3 pixels to define the cellular area and the “MeasureObjectIntensity” module was used to determine the mean intensity for each cell object identified.
Light sheet fluorescence microscopy
Male embryos at 5, 7 and 11 PCW deemed devoid of morphological anomalies were dissected after overnight fixation in 4% PFA. Whole-mount and cryosection immunostaining were performed as described in Belle et al.63, with the following conditions: tissue was incubated with primary antibodies (see Supplementary Table 22 for antibody details) for 9 days at 37°C, with secondary antibody for 16 hours at 37°C using dedicated host species antibodies and reagent combination. TO-PRO-3 647 was used at 1:100 in whole embryos and 1:5000 on cryosections. Whole-mount specimens were solvent-cleared as described63, and imaged in dibenzylether with a Miltenyi Lavision Biotech ultramicroscope (Olympus MXV10 stereomicroscope and PCO Edge SCMOS CCD camera using the dedicated Imspector pro acquisition software. Four lasers (at 488, 561, 647 and 790nm wavelengths) were used to generate light sheets. IMaris (v9.2, BitPlane) was used for image conversion and processing. Photoshop (Adobe) was used to create panels. All raw files are being made available on demand through our dedicated KeenEye Technologies-hosted Platform (www.transparent-human-embryo.com, ≪request database access≫).
Statistics and reproducibility
For all analyses of fetal liver 3’ 10x data, n = 14 biologically independent samples were included. This includes Figure 1b, Figure 2a, Figure 3a-b, Figure 4b, Figure 5a-b, Figure 6a-b, Figure 6g, Extended Data 1c, Extended Data 2a, Extended Data 3a-e and h, Extended Data 4a, Extended Data 5e, Extended Data 5g-i, Extended Data 6a-e, Extended Data 7b and h, and Extended Data 8.
For all analyses of fetal liver 3’ 10x data by developmental stage, n = 4 7-8 PCW, n = 4 9-11 PCW, n = 3 12-14 PCW, and n = 3 15-17 PCW biologically independent samples were used. This includes Figure 1c, Figure 4a, Extended data 1d-e, Extended Data 5a-c and f.
For analyses including 10x sequencing data of skin, kidney, yolk sac, decidua and placenta, n = 7, 3, 3, 11 and 5 biologically independent samples were used, respectively. These analyses are shown in Figure 3b, Figure 4b, Figure 5a-b, Figure 6g, Extended Data 1a-b, Extended Data 3c-e and h, Extended Data 5g-i, and Extended Data 6c-e.
For analysis including sequencing data of cord blood and adult bone marrow, n = 8 biologically independent samples of each were used. These analyses are shown in Figure 6g and Extended Data 7h.
For all scRNA-seq data shown, all cells of a given label from indicated tissues are shown, no down-sampling or sub-setting was performed. The following cells numbers generated using scRNA-seq are displayed in each of the listed figures:
| Figure 1b-c | 113,063 fetal liver cells |
| Figure 2a | 113,063 fetal liver cells |
| Figure 3a | 104,515 fetal liver cells |
| Figure 3b | 43,507 fetal liver cells, 2,455 yolk sac cells, and 243 skin cells |
| Figure 4a | 16,919 fetal liver cells |
| Figure 4b | 16,919 fetal liver cells, 2,757 skin cells, 213 kidney cells, and 259 yolk sac cells |
| Figure 5a-b | 38,464 fetal liver cells, 6,887 yolk sac cells, and 10,008 cells from decidua and placenta |
| Figure 6a-b | 5,673 fetal liver cells |
| Figure 6g | 3,439 fetal liver HSC/MPP, 205 yolk sac progenitors, 1,082 cord blood HSCs, and 3,668 adult bone marrow HSCs |
| Ext Data 1a | 10,258 skin cells, and 17, 95 kidney cells |
| Ext Data 1b | 10,071 yolk sac cells |
| Ext Data 1c-e | 113,063 fetal liver cells |
| Ext Data 1f | 1,206 fetal liver cells from n = 2 biologically independent samples profiled using Smart-seq2 (also displayed in Extended Data 2b) |
| Ext Data 2a | 113,063 fetal liver cells |
| Ext Data 2b | 1,206 fetal liver cells from n = 2 biologically independent samples profiled using Smart-seq2; and 381 fetal liver erythroid liver erythroblastic island (EI) populations (early, mid and late erythroids, VCAM1+ EI macrophages), Kupffer cells and endothelium validated by SS2 from n = 2 biologically independent fetal liver samples |
| Ext Data 2d | 381 fetal liver erythroid liver erythroblastic island (EI) populations (early, mid and late erythroids, VCAM1+ EI macrophages), Kupffer cells and endothelium validated by SS2 from n = 2 biologically independent fetal liver samples |
| Ext Data 3a-b | 52,237 fetal liver cells |
| Ext Data 3c | 52,237 fetal liver cells, 362 skin cells, and 28 kidney cells |
| Ext Data 3d | 52,327 fetal liver cells, 362 skin cells, 28 kidney cells, and 2,793 yolk sac cells were analysed, and a maximum of 20 cells displayed of each cell type per tissue |
| Ext Data 3e | 52,237 fetal liver cells, 362 skin cells, 28 kidney cells, and 2,588 yolk sac cells |
| Ext Data 3h | 3,439 HSC/MPP, 1,342 MEMP, 11,985 Early Erythroid, 27,000 Mid Erythroid, 3,180 Late Erythroid, 3,983 Megakaryocytes, and 1,308 Mast cells from fetal liver; 55 MEMP, 51 Mid Erythroid, 137 Late Erythroid, 11 Megakaryocytes, and 108 Mast cells from skin; and 2 MEMP and 26 Megakaryocytes from kidney. |
| Ext Data 5a | 16,919 fetal liver cells |
| Ext Data 5b-c | 767 fetal liver Early lymphoid/T lymphocytes |
| Ext Data 5d | 16,666 fetal liver cells from n = 7 biologically independent samples. |
| Ext Data 5e | 7,467 fetal liver cells |
| Ext Data 5f | 32,308 fetal liver cells |
| Ext Data 5g | 16,919 fetal liver cells, 2,775 skin cells, 213 kidney cells, and 464 yolk sac cells |
| Ext Data 5h | 6,706 NK cells and 1,726 ILC precursors from fetal liver, 1,479 NK cells and 1,142 ILC precursors from skin, and 155 NK cells and 36 ILC precursors from kidney |
| Ext Data 5i | 1,726 fetal liver ILC precursors, 1,142 skin ILC precursors, and 36 kidney ILC precursors |
| Ext Data 6a | 6,606 fetal liver cells |
| Ext Data 6b | 11,653 fetal liver cells |
| Ext Data 6c | 38,646 fetal liver cells, 6,887 yolk sac cells, and 10,008 cells from decidua and placenta were analysed, and a maximum of 50 cells displayed of each cell type per tissue |
| Ext Data 6d | 43,498 fetal liver cells, 8,350 skin cells, and 1,514 kidney cells |
| Ext Data 6e | 24,841 Kupffer cells, 2,586 monocytes, 253 pDC precursors, 336 DC1, and 3,954 DC2 from fetal liver; 5,474 macrophages, 704 monocytes, 36 pDCs, 99 DC1, and 527 DC2 from skin; and 1,075 macrophages, 82 monocytes, 1 pDC, 1 DC2, and 271 DC2 from kidney |
| Ext Data 7b | 6,606 fetal liver cells and 161 of 349 scRNA-seq profiled cells (Supplementary Table 12) from n = 3 biologically independent samples, sorted as per FACS gates in Extended Data 7a |
| Ext Data 7h | 3,439 HSC/MPPs, 1,341 MEMPs, 234 Pre pro B cells, 658 Neutrophil-myeloid progenitors, 350 Monocyte precursors, 253 pDC precursors, and 330 DC precursors from fetal liver; 205 yolk sac progenitors; 1,082 cord blood HSCs; and 3,668 adult bone marrow HSCs |
| Ext Data 8 | 113,063 fetal liver cells |
Representative mini bulk RNA-seq data of 100 cells per cell state from 1 of n = 3 biologically independent fetal liver samples is shown in Extended Data 2b-c.
Cytospin images shown in Figure 2b and Extended Data 5e are representative from 1 of n = 3 biologically independent samples.
Hyperion images shown in Extended Data 2e are representative from 1 of n = 4 biologically independent 8 PCW fetal livers and 1 of n = 4 biologically independent 15 PCW fetal livers.
Immunohistochemical (IHC) staining of 8 PCW fetal skin in Extended Data 3f is representative from 1 of n = 3 biologically independent samples. IHC staining of fetal liver in Extended Data 4-b are from independent samples and both are representative from 1 of n = 3 biologically independent samples.
Light sheet fluorescence microscopy of embryo (5PCW) hand skin shown in Extended Data 3g is a representative image from 1 of n = 3 biologically independent samples.
Statistical analysis of differential gene expression was done using one-tailed Wilcoxon rank sum test with Bonferroni correction, including those shown in heatmaps and violin plots.
In Figure 6b, expression of genes in HSC/MPP 1 compared to each other cell cluster, and MEMP/neutrophil-myeloid progenitor/pre pro B cell compared to each other cell cluster was statistically significant unless specified as not significant (ns). All p < 0.001, except expression of KLF1 between HSC/MPP1 vs HSC/MPP2 (p = 0.00934), IL1RL1 between HSC/MPP 1 vs HSC/MPP 2 (p = 0.00148), GATA2 between MEMP vs HSC/MPP2 (p = 0.00802), BCL11A between HSC/MPP 1 vs HSC/MPP 7 (p = 0.00114), LYZ between HSC/MPP 1 vs HSC/MPP 5 (p = 0.00675), AZU1 between HSC/MPP 1 vs HSC/MPP 8 (p = 0.00494), CSF1R between Neutrophil-myeloid progenitors vs HSC/MPP 8 (p = 0.213). In Figure 6g, **** indicates p < 0.0001 when comparing expression of ANXA1, DUSP1, HLA-B, and HSPAIA between samples. Expression of genes in fetal liver Early lymphoid/T lymphocytes was compared across developmental stages and displayed in Extended Data 5b-c. No significant difference across stage was observed in the genes displayed in Extended 5b. The following comparisons shown in Extended Data 5c were significant: CD2 expression at 7-8 PCW vs 9-17 PCW (p < 0.0001) and vs 12-17 PCW (p < 0.0001); TRDC expression at 7-8 PCW vs 9-17 PCW (p < 0.0001) and 9-11 PCW vs 12-17 PCW (p < 0.0001); CD8A expression at 7-8 PCW vs 9-17 PCW (p = 0.00714); CD27 expression at 7-8 PCW vs 9-17 PCW (p < 0.0001) and 9-11 PCW vs 12-17 PCW (p < 0.0001); IL7R expression at 7-8 PCW vs 9-17 PCW (p < 0.0001), 9-11 PCW vs 12-17 PCW (p = 0.00168), and 12-14 PCW vs 15-17 PCW (p = 0.00216); JCHAIN expression at 7-8 PCW vs 9-17 PCW (p < 0.0001); CD3D expression at 7-8 PCW vs 9-17 PCW (p < 0.0001); KLRB1 expression at 9-11 PCW vs 12-17 PCW (p < 0.0001); TRAC expression at 7-8 PCW vs 9-17 PCW (p < 0.0001) and 9-11 PCW vs 12-17 PCW (p < 0.0001); and PRF1 expression at 9-11 PCW vs 12-17 PCW (p < 0.0001). Statistically significant gene expression shown in Extended Data 5h comparing expression in NK cells and ILC precursors across tissues was p < 0.001 where **** was shown, and p = 0.00236 for TXNIP expression in ILC precursors in liver compared to those in kidney. Expression of genes in ILC precursors in fetal liver, skin and kidney, as shown in Extended Data 5i, was compared between tissues and no significant difference was observed. Extended Data 6e shows comparisons between fetal liver, skin and kidney for macrophages, monocytes, pDCs, DC1, and DC2. The following comparisons were statistically significant (p < 0.0001) between macrophages from different tissues: CD14 expression in kidney vs liver and vs skin; CD68 expression in liver vs skin and vs kidney; CD163, RNASE1 and F13A1 expression between all tissues; and, VCAM1 expression in liver vs skin and vs kidney. The following comparisons were statistically significant between monocytes from different tissues: CD14 expression in liver vs skin (p < 0.0001); CD68 expression in liver vs skin (p < 0.0001); S100A9 expression in liver vs kidney (p < 0.0001), and skin vs kidney (p = 0.0245); FCGR3A. expression in liver vs skin (p = 0.0004); POSTN expression in liver vs skin (p < 0.0001) and vs kidney (p < 0.0001), and skin vs kidney (p = 0.0411). No significant difference was observed when comparing genes in pDCs across tissues. CLEC9A expression was statistically significantly different in liver vs skin DC1 (p < 0.0001). The following comparisons were statistically significant between DC2 from different tissues: CD1C expression in skin vs liver (p < 0.0001) and vs kidney (p < 0.0001); CLEC10A expression in kidney vs liver (p < 0.0001). and vs skin (p < 0.0001); S100B expression in skin vs liver (p < 0.0001) and vs kidney (p = 0.0162); and FCER1A and CD83 expression between each tissue (p < 0.0001).
Statistical analysis of HSC colony assays shown in Figure 6c-e, and Extended Data 7 was done by applying two-tailed Fisher’s exact tests to colony counts. Figure 6c and d, and Extended Data 7f shows 125, 217 and 124 colonies from 7-8 PCW, 12-14 PCW and 15-17 PCW fetal liver samples, respectively from n = 2 biologically independent samples per development stage. The number of colonies per sample is 93, 32, 93, 124, 84, 40, which are shown in Extended Data 7c. The number of colonies between the following stages in Figure 6c were statistically significant: Erythroid colonies in 7-8 PCW vs. 15-17 PCW (p = 0.0238), Erythroid/Megakaryocyte/Myeloid colonies in 7-8 PCW vs. 15-17 PCW (p = 0.0294), NK colonies in 7-8 PCW vs. 15-17 PCW (p = 0.0357), and Erythroid/Myeloid colonies in 7-8 PCW vs 12-14 PCW (p = 0.0188) and 15-17 PCW (p < 0.001), and 12-14 PCW vs 15-17 PCW (p = 0.0232). The number of Erythroid-containing colonies was significant between each stage shown in Figure 6d (p < 0.001). The number of colonies that differentiated along 3 lineages was significant between 7-8 PCW vs 12-14 PCW (p = 0.0041), and 7-8 PCW vs 15-17 PCW (p = 0.0027). Figure 6e shows 141, 74 and 124 colonies from 7-8 PCW, 12-14 PCW and 15-17 PCW fetal liver samples, respectively from n = 2 biologically independent samples per development stage. The number of B cell-forming colonies in Figure 6e was significant between 7-8 PCW vs 12-14 PCW (p = 0.0014) and 15-17 PCW (p = 0.0044). Extended Data 7d-e shows 163, 196, 182 colonies from n = 3 7-8 PCW, n = 2 12-14 PCW, and n = 2 15-17 PCW biologically independent fetal liver samples, respectively on the left, and 42, 74, 47, 97, 99, 59, 123 colonies by individual sample on the right. The number of Erythroid colonies compared to all other types shown in Extended Data 7d was statistically significant between 12-14 PCW and 15-17 PCW (p = 0.0307). The number of Erythroid-containing colonies was significant between 7-8 PCW vs 15-17 PCW (p = 0.0013), and 12-14 PCW vs 15-17 PCW (p = 0.0497), as shown in Extended Data 7e. Extended Data 7f shows 125, 217 and 124 colonies from 7-8 PCW, 12-14 PCW and 15-17 PCW fetal liver samples, respectively from n = 2 biologically independent samples per development stage. Extended Data 7g shows 141, 74 and 124 colonies from 7-8 PCW, 12-14 PCW and 15-17 PCW fetal liver samples, respectively from n = 2 biologically independent samples per development stage. The percentage of NK-containing colonies was statistically significant in 7-8 PCW vs 15-17PCW (p = 0.0032), and 12-4 PCW vs 15-17 PCW (p = 0.0115).
Flow cytometric analysis of cell cycle phases, as shown in Figure 6g and Extended Data 7i was performed on cells from n = 3 7-8 PCW and n = 3 12-16 PCW biologically independent fetal liver samples, and n = 2 biologically independent cord blood samples. One-way ANOVA with Tukey’s multiple comparison test was used to determine statistical significance between stages (7-8 PCW and 12-14 PCW) and samples (fetal liver and cord blood). The percent of CD34+CD38- cells in G0 was significantly higher in 12-14 PCW livers compared to 7-8 PCW livers (p = 0.0136).
Cell cycle phases determined by transcriptome analysis of fetal liver cells, fetal skin cells, and fetal kidney cells is shown in Extended Data 3h. Statistical significance of the proportion of MEMP and Megakaryocytes in each cell cycle phase was compared between fetal liver, skin and kidney using Kruskal-Wallis with Dunn’s post hoc test. Statistical significance of the proportion of Mid and Late Erythroids, and Mast cells in each cell cycle phase was compared between fetal liver and skin using two-tailed Mann-Whitney tests. The following comparisons were statistically significant: Megakaycoytes in fetal liver vs fetal kidney in G1 (p = 0.0317), G2M (p = 0.0317) and S (p = 0.0139); Megakaryocytes in fetal liver vs fetal skin in S (p = 0.0039); Mid Erythroids in fetal liver vs skin in G1 (p = 0.0031) and G2M (p < 0.0001); Late Late Erythroids in fetal liver vs fetal skin in G1 (p = 0.0021) and S (p < 0.0001); and, Mast cells in fetal liver vs fetal skin in G1 (p = 0.0248) and S (p = 0.0337).
Statistical comparison of the percent of MEMP, Mid and Late Erythroids, Megaryocytes and Mast cells expressing MKI67 in fetal liver vs NLT (skin and kidney) was performed using two-tailed Mann-Whitney tests. This is displayed in Extended Data 3h, where the following comparisons were statistically significant: Megakaryocytes in fetal liver vs fetal NLT (p = 0.007), Mid Erythroid in fetal liver vs fetal skin (p = 0.0305), and Late Erythroid in fetal liver vs fetal skin (p = 0.0368).
Statistical comparison of the percent of fetal liver HSC/MPP expressing MKI67 compared to the percent of fetal liver MEMP, Pre pro B cells, Neutrophil-myeloid progenitors, Monocyte precursors, pDC precursors and DC precursors, Yolk Sac progenitors, and HSC from cord blood and adult bone marrow expressing MKI67 was performed using Kruskal-Wallis with Dunn’s post hoc test. This is displayed in Extended Data 7h, where the percent of MKI67-expressing fetal liver HSC/MPPs was statistically significant when compared to the following populations: fetal liver MEMP (p = 0.0180), Monocyte precursors (p < 0.0001), DC precursors (p < 0.0001), cord blood HSC (p < 0.0001), and adult bone marrow HSC (p = 0.0076)
Extended Data 1g displays flow cytometric analysis of the frequency of B cells in the CD34-cells from n = 13 6-9 PCW, n = 13 9-12 PCW, n = 14 12-15 PCW and n = 9 15-19 PCW biologically independent fetal liver samples. Statistical significance across the developmental stages was compared using Kruskal-Wallis with Dunn’s multiple comparison post-test, for which the following comparisons were statistically significant: 6-9 PCW vs 12-15 PCW (p < 0.0001), 6-9 PCW vs 15-19 PCW (p = 0.0003), 9-12 PCW vs 12-15 PCW (p = 0.0157), and 9-12 PCW vs 15-19 PCW (p = 0.0287).
Statistically significant, dynamically variable genes from pseudotime were determined by Likelihood of ratio test applied in monocle (see Methods). Select genes were displayed for Erythroid, Mast cell and Megakaryocyte lineages shown in Extended Data 3b, B cell lineage shown in Extended Data 5e, and DC1, DC2 and Monocyte lineage shown in Extended Data 6b.
Investigation of potential receptor:ligand interactions between all fetal liver VCAM1+ EI macrophages (161 cells) and Early and Mid Erythroids (11,985 and 27,000 cells respectively), as shown in Extended Data 4a, was performed using CellPhoneDB (see Methods). A permutation test was applied to determine statistical significance, which is indicated by the colour of the dots. ln-normalised median expression of ITGA4 and VCAM1 in the same cells is also displayed in a violin plot (right panel).
ImageStream analysis of fetal liver cells shown in Extended Data 4c-f was performed on n = 3 biologically independent samples. Extended Data 4c-d display representative data from one sample. Extended Data 4e shows representative images of cells from 38,576 single cells and 1,945 doublets from one sample. The percent of each cell type that was observed within the doublet of singlet gate, as per Extended Data 4c, was compared using Kruskal-Wallis with Dunn’s post hoc test. The percent of doublets in VCAM1+ EI macrophages was significantly different the percent of doublets in Erythroids (p = 0.0194).
Statistical significance of TNFSF13B expression in Kupffer cells over time compared to NFKBIA in HSC/MPPs (p = 0.0245), Pre pro B cells (p = 0.0008), Pro B cells (p = 0.0004), Pre B cells (p = 0.0197) and B cells (p = 0.0343) across 4 developmental stages spanning 6-17 PCW was determined using two-tailed Pearson’s R test.
Extended Data
Extended Data Figure 1. Single cell transcriptome map of fetal liver.

a, Fetal skin and kidney haematopoietic cells visualised by UMAP. Colours indicate cell state. Inset: colours indicate tissue type. b, UMAP visualisation of yolk sac haematopoietic cells. Colours indicate cell state. Inset: colours indicate location within yolk sac. c, UMAP visualisation of 3’ liver 10x cells post batch correction, coloured by sample. d, UMAP visualisation (top) of 3’ 10x liver sample sex mixing grouped by developmental stage, and violin plots (bottom) showing ln-normalised median expression of XIST (green) and RSP4Y1 (purple), which marks female and male samples respectively. e, UMAP visualisation of fetal liver composition by developmental stage. Colours indicate cell state. f, UMAP visualisation of fetal liver cells profiled using Smart-seq2. Colours indicate cell states as shown in e. g, Frequency (mean +/- s.e.m.) of B cells in the CD34- cells detected in 6-19 PCW fetal livers by flow cytometry (* p < 0.05; *** p = 0.003; **** p < 0.001).
Extended Data Figure 2. Transcriptome validation of fetal liver cells.

a, Assessment of 48 genes from the 4,471 highly variable genes by using a Random Forest classifier to assign cell labels, where ‘true cell label’ indicates the manual annotation based on the full list of variable genes. b, Comparison of representative mini bulk RNAseq data (in coloured triangles) and liver erythroblastic island (EI) populations (early, mid and late erythroids, VCAM1+ EI macrophages), Kupffer cells and endothelium validated by SS2 (in colour) overlaid on whole liver SS2 populations (grey). c, Dot plot showing representative median scaled ln-normalised gene expression of 100 FACS-isolated liver cells based on marker gene expression in Figure 2a. Gene expression indicated by spot size and colour intensity. d, Dot plot showing median scaled ln-normalised gene expression of FACS sorted single cells from liver erythroblastic island (EI) populations (early, mid and late erythroids, VCAM1+ EI macrophages), Kupffer cells and endothelium shown as coloured dots in b based on marker gene expression in Figure 2a. Gene expression frequency (% cells within cell type expressing) indicated by spot size and expression level by colour intensity. e, Overlay pseudo-colour Hyperion representative images for 8 PCW and 15 PCW fetal liver. Far left images are shown at 5x magnification with zoom of insets on right at 20x magnification (1μm/pixel). Bile ducts are marked with an *.
Extended Data Figure 3. Fetal liver and NLT haematopoiesis.

a, PAGA analysis of fetal liver HSC/MPP, erythroid, megakaryocyte and mast cell lineages from Figure 3a. Lines symbolise connection; line thickness corresponds to the level of connectivity (thin to thick = low to high PAGA connectivity). b, Heat map showing min-max normalised expression of statistically significant (p < 0.001), dynamically variable genes from pseudotime analysis for erythroid, megakaryocyte and mast cell inferred trajectories. Transcription factors in bold, asterisk (*) mark genes not previously implicated for the respective lineages. c, FDG visualisation of fetal liver, skin and kidney HSC/MPP, MEMP, erythroid, megakaryocyte and mast cell lineages. d, Heat map showing the scaled ln-expression of selected marker genes in fetal liver, NLT and yolk sac subsets. e, PAGA connectivity scores of HSC/MPP, erythroid, megakaryocyte and mast cell lineages between fetal liver, skin, kidney (K) and yolk sac. f, Representative immunohistochemical staining of sequential sections of 8 PCW fetal skin for endothelium (CD34+) and erythroblasts (nucleated and GYPA+), nuclei stained with blue alkaline phosphatase. Zoom in of insets (right) bordered with black (top) indicate nucleated cells stained positive for GYPA within CD34+ blood vessels, and those bordered with red (bottom) indicate nucleated GYPA+ cells outside CD34+ blood vessels. Scale bar, 100μm. g, Representative light sheet fluorescence microscopy of embryo (5 PCW) hand skin. Scale bar, 5μm; TO-PRO-3/nuclei = red, GYPA = green (see also Supplementary Video 2). < indicates extravascular nucleated erythroid cells. h, Stacked barplots (right) of the mean +/- s.d. percent of fetal liver (red), skin (blue) and kidney (green) HSC/MPP, MEMP, Erythroid, Megakaryocyte and Mast cells in each stage of the cell cycle (G1 (navy), G2M (blue), and S (white) phase), and ln-normalised median expression of MKI67 transcript (right) in corresponding liver vs NLT cell types (total percent of MKI67 expressing cells stated above plots; each dot represents a single cell). * p < 0.05; ** p < 0.01; *** p < 0.005.
Extended Data Figure 4. Investigation of interactions between fetal liver macrophages and erythroid cells.

a, Representative immunohistochemical staining of fetal liver for erythroblasts and macrophages with GYPA and CD68, respectively. Scale bar, 50μm. Statistically significantly (p < 0.05) enriched receptor-ligand interactions from CellPhoneDB between VCAM1+ EI macrophages (purple) and two erythroid populations (early and mid; red) (n = 14 biologically independent samples). Asterisk (*) indicate protein complexes. Violin plots show ln-normalised median gene expression value of VCAM1 and ITGA4 in cells analysed by CellPhoneDB (marked by # in dot plot). b, Representative immunohistochemical staining of sequential sections of 8 PCW fetal liver for VCAM1+ EI macrophages (VCAM1+) and CD49d+GYPA+ cells with nuclei stained using blue alkaline phosphatase. Zoom in of insets (right) with coloured arrows indicating erythroblast (yellow) and VCAM1+ EI macrophage (purple). Scale bar, 100μm. c, Representative gating strategy used to visualise fetal liver erythroid cells, VCAM1+ EI macrophages, Kupffer cells, Mono-macs, and mast cells. d, Bright field, VCAM1 (CD106), CD34, CD45, KIT (CD117), GYPA, CD14, and HLA-DR images for each cell type within gates shown in c. e, Representative bright field images of cells found within the single cell and doublet gates. f, Barplots showing the mean +/- s.d. proportion of each cell type within the single cell gate (white) or doublet gate (grey); * p = 0.0194. g, Comparison of macrophage and erythroid gene expression in mouse macrophages (red) and EI macrophages (blue), n = 3 from Li et al.19.
Extended Data Figure 5. Lymphoid lineages in fetal liver and NLT.

a, PAGA analysis of fetal liver HSC/MPP and lymphoid cell types from Figure 1b showing changes over four developmental stages. Lines symbolise connection; line thickness corresponds to the level of connectivity (thin to thick = low to high PAGA connectivity). b, Feature plots and c, violin plots showing ln-normalised median expression of selected known NK, ILC and T cell genes over gestation for early lymphoid/T lymphocyte cluster; ** p < 0.001; *** p < 0.005; **** p < 0.0001. d, Dot plot showing median scaled ln-normalised median expression of V(D)J transcripts in fetal liver lymphoid cell types. Gene expression indicated by spot size and colour intensity. e, Heat map showing normalised expression of statistically significant, dynamically variable genes from pseudotime analysis for B cell lineage inferred trajectory (likelihood ratio test). Transcription factors are in bold. Morphology of liver Pro/Pre B cells and B cells by Giemsa stain after cytospin. f, ln-normalised expression (mean +/- s.e.m.) of TNFSF13B in Kupffer cells and NFKBIA in HSC/MPPs and cells in the B cell lineage across 4 developmental stages spanning 6-17 PCW; trend lines showing linear regression. g, PAGA connectivity scores of HSC/MPP and lymphoid cells from fetal liver, skin, kidney and yolk sac. h, ln-normalised median expression of selected known NK (left) and ILC precursor (right) marker genes and selected differentially expressed genes between liver (red), skin (blue) and kidney (green) visualised by violin plots (*** p < 0.005; **** p < 0.001). i, Violin plots showing ln-normalised median expression of selected known ILC and NK cell genes expressed in ILC precursors from fetal liver, skin, and kidney.
Extended Data Figure 6. Tissue signatures in developing myeloid cells.

a, Diffusion map of fetal liver HSC/MPP, progenitors and precursors from 1b. b, Heat map showing min-max normalised expression (p < 0.001) of dynamically variable genes from pseudotime analysis for monocyte, DC1 and DC2 inferred trajectories. Transcription factors in bold, * mark genes not previously implicated for the respective lineages. c, Heat map visualisation comparing scaled expression of the top marker genes of decidua/placenta (red), fetal liver (black) and yolk sac (purple) progenitor and myeloid populations. d, PAGA connectivity scores of HSC/MPP and myeloid cells from fetal liver, skin and kidney. e, ln-normalised median expression of 3 known marker genes & 3 differentially expressed genes in corresponding myeloid populations across fetal liver, skin and kidney visualised by violin plots (* p < 0.05; *** p < 0.005; **** p < 0.0001).
Extended Data Figure 7. HSC/MPP differentiation potential by gestation.

a, Experimental design for single cell transcriptome and culture of fetal liver cells from representative FACS gates illustrated. b, Alignment of 349 scRNA-seq profiled cells from FACS gates in a with 10x profiled HSC/MPPs and early progenitors visualised using FDG, point shape corresponds to sequencing type (triangle = SS2 plate data, circle = 10X data). c, Stacked barplot of all different types of colonies generated by single ‘HSC pool’ gate cells (gate defined in a). d, Stacked bar plot of all different types of colonies generated by single ‘HSC pool’ gate cells without MS5 stroma layer (gate defined in a) by stage (left) and in individual samples (right), * p < 0.05. e, Percentage of colonies generated by single ‘HSC pool’ cells without MS5 stroma layer containing erythroid cells (sum of Ery, Ery/Meg, Ery/Meg/My, and Ery/My colonies shown in c), ** p < 0.01. f, Percentage of colonies from single cell culture (shown in 6c) that differentiated along 3 lineages (defined as sum of Ery/NK/My and Ery/Meg/My colonies) branches (*** p < 0.005). g, Percentage of colonies containing NK cells following B/NK optimised culture of 10 cells from ‘HSC pool’ gate (* p < 0.05, ** p < 0.01). h, Percentage (Mean +/- s.e.m.) of HSC/MPP and early progenitors in fetal liver, yolk sac, cord blood and adult bone marrow expressing MKI67 (* p < 0.05, ** p < 0.01, **** p < 0.001). i, Mean +/- s.d. percentage of CD34+CD38- and CD34+CD38+cells in the indicated cell cycle phases (right) as determined by flow cytometry analysis (left, representative plot of n = 8 biologically independent samples) (G0: Ki67-DAPI-, G1: Ki67+DAPI-, S-G2-M: Ki67+DAPI+ (left)).
Extended Data Figure 8. Expression of known Primary Immunodeficiency (PID)-linked genes in fetal liver.

Dot plots showing relative expression of genes known to be associated with major PID disease categories in fetal liver cell types from Figure 1b. Early L/T L, Early lymphoid/T lymphocyte. Gene expression frequency (% cells within cell type expressing) indicated by spot size and expression level by colour intensity.
Extended Data Figure 9. FACS gating strategy for scRNA-seq analysis.

a, Gating strategy used to FACS-isolate cells for droplet-(10x) and plate-based scRNA-seq (Smart-seq2) for samples F2-F17. b, Gating strategy used to FACS-isolate cells for cytospins, scRNA-seq (Smart-seq2) and 100 cell RNA-seq. c, Flow cytometry gating strategy used to identify the colonies cultured in vitro from single cells as shown in Figure 6c. d, Flow cytometry gating strategy used to identify B and NK colonies cultured in vitro from 10 cells as shown in Figure 6e.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
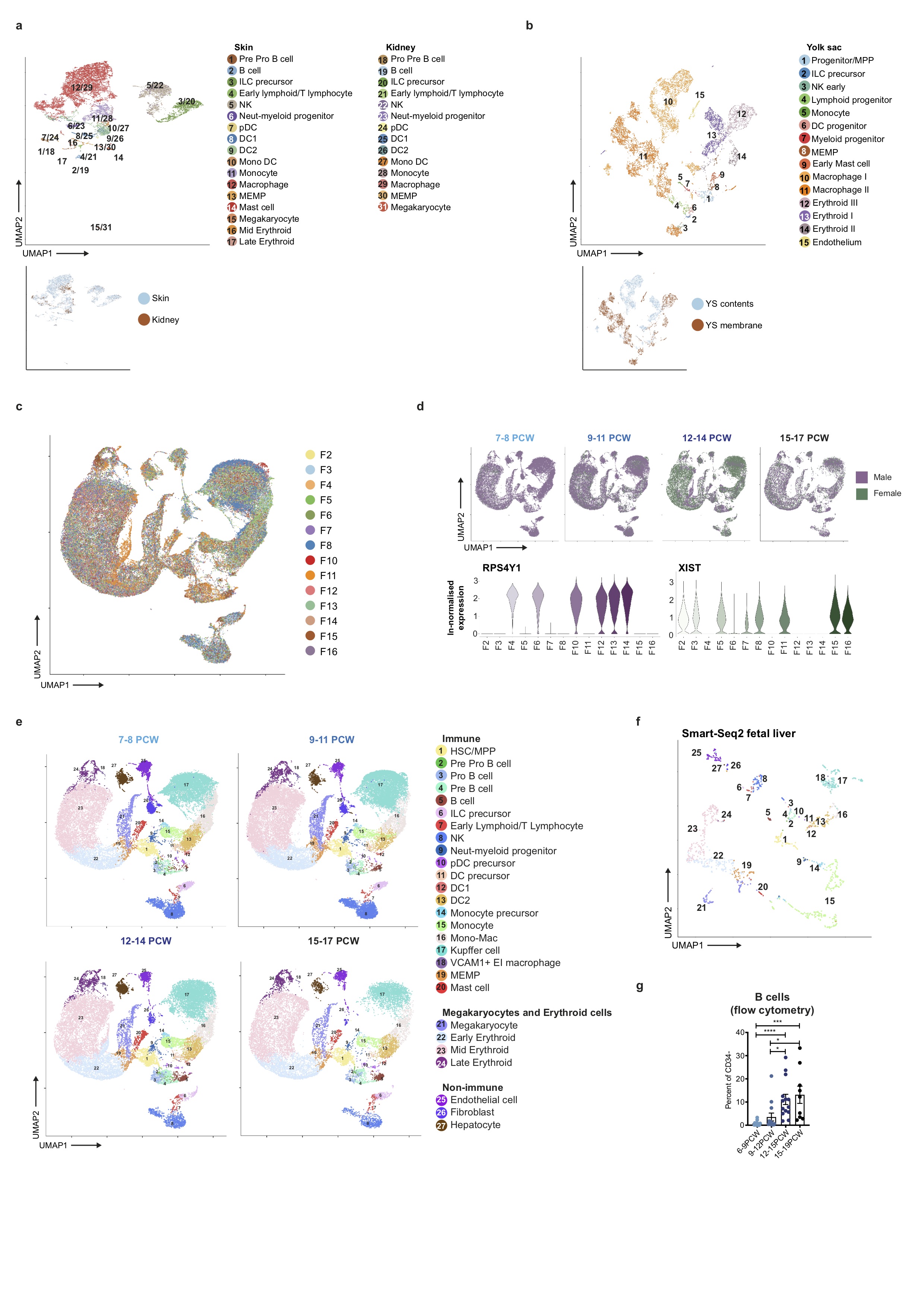{kind=link}
{kind=link}
{kind=link}
{kind=link}
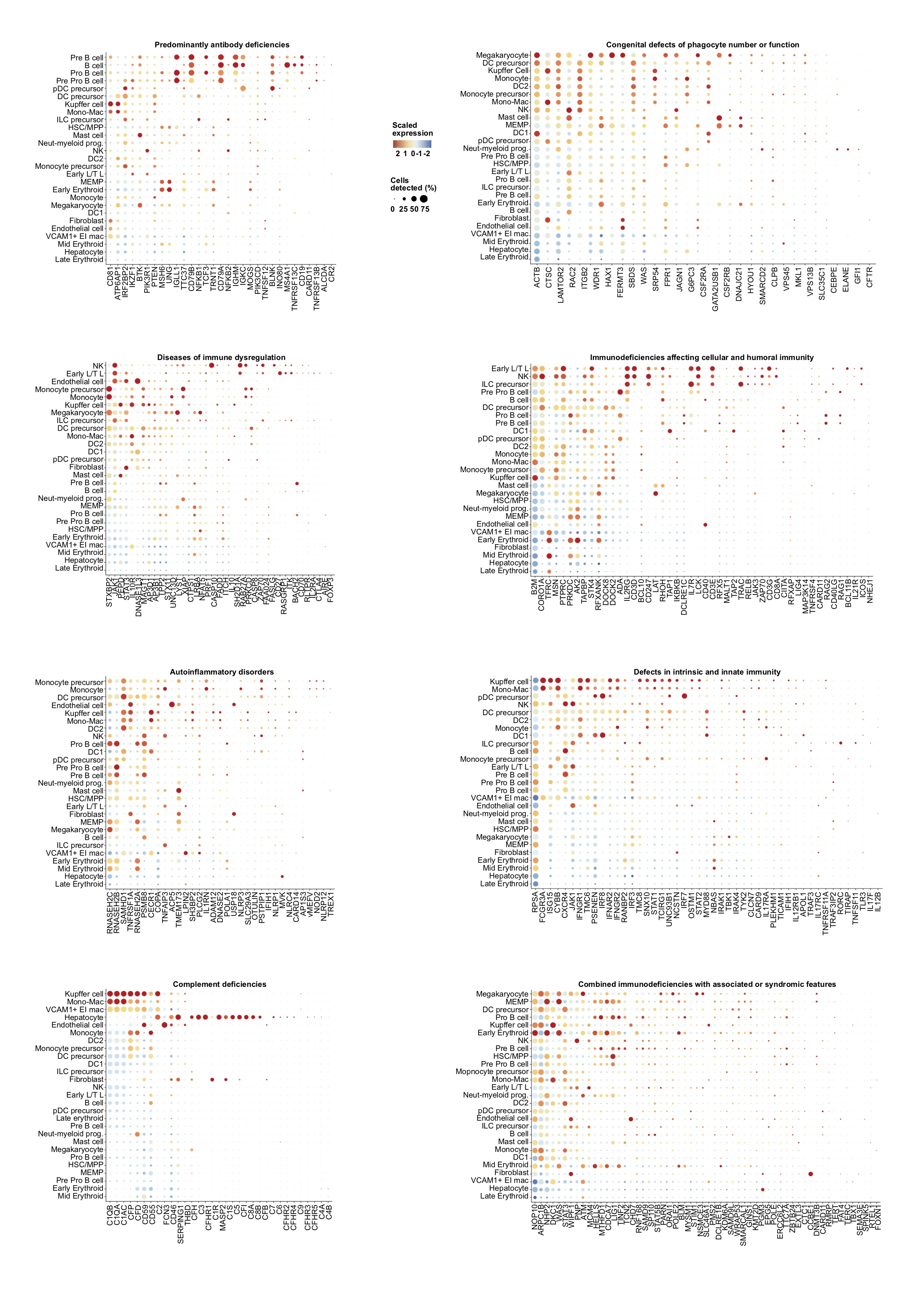{kind=link}
Acknowledgements
This publication is part of the Human Cell Atlas-www.humancellatlas.org/publications. We thank the Newcastle University Flow Cytometry Core Facility, Bioimaging Core Facility, Genomics Facility, NUIT for technical assistance, School of Computing for access to the High-Performance Computing Cluster, Newcastle Molecular Pathology Node Proximity Lab, Alison Farnworth for clinical liaison, Sophie Hambleton for primary immunodeficiency expertise, Helen Chen for immunohistochemistry assistance, and Morgane Belle and Stephane Fouquet for light sheet fluorescence microscopy assistance. The human embryonic and fetal material was provided by the Joint MRC / Wellcome (MR/R006237/1) Human Developmental Biology Resource (www.hdbr.org).
Funding
We acknowledge funding from the Wellcome Human Cell Atlas Strategic Science Support (WT211276/Z/18/Z); M.H. is funded by Wellcome (WT107931/Z/15/Z), The Lister Institute for Preventive Medicine and NIHR and Newcastle-Biomedical Research Centre; S.A.T. is funded by Wellcome (WT206194), ERC Consolidator and EU MRG-Grammar awards and; S.B. is funded by Wellcome (WT110104/Z/15/Z) and St. Baldrick’s Foundation; E.L. is funded by a Wellcome Sir Henry Dale and Royal Society Fellowships, European Haematology Association, Wellcome and MRC to the Wellcome-MRC Cambridge Stem Cell Institute and BBSRC.
Footnotes
Author contributions
M.H.; S.A.T. and E.L. conceived and directed the study. M.H.; S.A.T.; E.L. and R.A,B. designed the experiments. Samples were collected by S.L. and S.L., isolated by R.A.B. and libraries prepared by E.S.; L.M.; D.M.P.; R.V-T; J.P. and J.F. Flow cytometry and FACS experiments were performed by R.A.B.; E.F.C.; L.J.; D.M. and A.F. Imaging mass cytometry experiments were performed by M.A.; B.M.; B.I.; D.M. and A.F. Cytospins were performed by D.D.; J.F.; and in vitro culture differentiation experiments were performed by L.J.; D.M. and E.F.C. Immunohistochemistry was performed by B.I.; M.A.; F.G. and C.M.; P.C. and M.A. interpreted immunohistochemistry and developmental pathology sections. Y.G. and A.C. performed and interpreted light sheet fluorescence microscopy experiments. M.S.K.; B.L.; O.A.; M.T.; D.D.; T.L.T.; M.S.; O.R-R. and A.R. generated adult and cord blood scRNA-seq datasets. D.M.P.; K.G.; K.P.; S.W.; I.G.; M.E.; P.V.; M.Y.; Z.M. and J.B performed the computational analysis. M.H.; D.M.P.; R.A.B.; B.G.; E.L.; I.R.; A.R.; E.F.C; L.J.; A-C.V.; R.R.; E.P.; M.M.; J.P.; G.R.; K.B.M.; M.J.T.S.; A.F.; K.G.; S.W.; I.G.; S.B. and J.B. interpreted the data. M.H.; L.J.; R.A.B.; E.S.; D.M.P; B.G.; E.L.; I.R; K.R.; S.W.; I.G.; A-C.V. and A.R wrote the manuscript. All authors read and accepted the manuscript.
Competing Interests
None declared
Data and materials availability
The raw sequencing data, expression count data with cell classifications are deposited at ArrayExpress: https://www.ebi.ac.uk/arrayexpress/experiments/E-MTAB-7407/
Code availability
All scripts are available at https://github.com/haniffalab/FCA_liver
References
- 1.Jagannathan-Bogdan M, Zon LI. Hematopoiesis. Development. 2013;140:2463. doi: 10.1242/dev.083147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Parekh C, Crooks GM. Critical Differences in Hematopoiesis and Lymphoid Development Between Humans and Mice. J Clin Immunol. 2013;33:711–715. doi: 10.1007/s10875-012-9844-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ivanovs A, et al. Human haematopoietic stem cell development: from the embryo to the dish. Development. 2017;144:2323–2337. doi: 10.1242/dev.134866. [DOI] [PubMed] [Google Scholar]
- 4.Holt PG, Jones CA. The development of the immune system during pregnancy and early life. Allergy. 2001;55:688–697. doi: 10.1034/j.1398-9995.2000.00118.x. [DOI] [PubMed] [Google Scholar]
- 5.Kashem SW, Haniffa M, Kaplan DH. Antigen-Presenting Cells in the Skin. Annu Rev Immunol. 2017;35:469–499. doi: 10.1146/annurev-immunol-051116-052215. [DOI] [PubMed] [Google Scholar]
- 6.Mass E, et al. Specification of tissue-resident macrophages during organogenesis. Science. 2016 doi: 10.1126/science.aaf4238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ohls RK, et al. Neutrophil Pool Sizes and Granulocyte Colony-Stimulating Factor Production in Human Mid-Trimester Fetuses. Pediatr Res. 1995;37:806. doi: 10.1203/00006450-199506000-00022. [DOI] [PubMed] [Google Scholar]
- 8.Picard C, et al. International Union of Immunological Societies: 2017 Primary Immunodeficiency Diseases Committee Report on Inborn Errors of Immunity. J Clin Immunol. 2018;38:96–128. doi: 10.1007/s10875-017-0464-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gentek R, et al. Hemogenic Endothelial Fate Mapping Reveals Dual Developmental Origin of Mast Cells. Immunity. 2018;48:1160–1171.e5. doi: 10.1016/j.immuni.2018.04.025. [DOI] [PubMed] [Google Scholar]
- 10.Iskander D, et al. Elucidation of the EP defect in Diamond-Blackfan anemia by characterization and prospective isolation of human EPs. Blood. 2015;125:2553. doi: 10.1182/blood-2014-10-608042. [DOI] [PubMed] [Google Scholar]
- 11.MacParland SA, et al. Single cell RNA sequencing of human liver reveals distinct intrahepatic macrophage populations. Nat Commun. 2018;9:4383. doi: 10.1038/s41467-018-06318-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.An X, et al. Global transcriptome analyses of human and murine terminal erythroid differentiation. Blood. 2014;123:3466. doi: 10.1182/blood-2014-01-548305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gautier E-F, et al. Comprehensive Proteomic Analysis of Human Erythropoiesis. Cell Rep. 2016;16:1470–1484. doi: 10.1016/j.celrep.2016.06.085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dedhia P, Kambayashi T, Pear WS. Notch2 paves the way to mast cells by Hes1 and Gata3. Proc Natl Acad Sci. 2008;105:7629. doi: 10.1073/pnas.0803439105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Okada Y, et al. Homeodomain proteins MEIS1 and PBXs regulate the lineage-specific transcription of the platelet factor 4 gene. Blood. 2003;101:4748. doi: 10.1182/blood-2002-02-0380. [DOI] [PubMed] [Google Scholar]
- 16.Vento-Tormo R, et al. Single-cell reconstruction of the early maternal–fetal interface in humans. Nature. 2018;563:347–353. doi: 10.1038/s41586-018-0698-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Klei TRL, Meinderts SM, van den Berg TK, van Bruggen R. From the Cradle to the Grave: The Role of Macrophages in Erythropoiesis and Erythrophagocytosis. Front Immunol. 2017;8:73. doi: 10.3389/fimmu.2017.00073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kessel KU, et al. Emergence of CD43-Expressing Hematopoietic Progenitors from Human Induced Pluripotent Stem Cells. Transfus Med Hemotherapy. 2017;44:143–150. doi: 10.1159/000477357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li W, et al. Identification and transcriptome analysis of erythroblastic island macrophages. Blood. 2019 doi: 10.1182/blood.2019000430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ebert BL, et al. An Erythroid Differentiation Signature Predicts Response to Lenalidomide in Myelodysplastic Syndrome. PLOS Med. 2008;5:e35. doi: 10.1371/journal.pmed.0050035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gale RP. Development of the immune system in human fetal liver. In: Touraine J-L, Gale RP, Kochupillai V, editors. Fetal liver transplantation. Springer; Netherlands: 1987. pp. 45–56. [DOI] [Google Scholar]
- 22.Phillips JH, et al. Ontogeny of human natural killer (NK) cells: fetal NK cells mediate cytolytic function and express cytoplasmic CD3 epsilon,delta proteins. J Exp Med. 1992;175:1055. doi: 10.1084/jem.175.4.1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Forkel M, et al. Composition and functionality of the intrahepatic innate lymphoid cell-compartment in human nonfibrotic and fibrotic livers. Eur J Immunol. 2017;47:1280–1294. doi: 10.1002/eji.201646890. [DOI] [PubMed] [Google Scholar]
- 24.Haynes BF, Heinly CS, Early human T. cell development: analysis of the human thymus at the time of initial entry of hematopoietic stem cells into the fetal thymic microenvironment. J Exp Med. 1995;181:1445–1458. doi: 10.1084/jem.181.4.1445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sánchez MJ, Spits H, Lanier LL, Phillips JH. Human natural killer cell committed thymocytes and their relation to the T cell lineage. J Exp Med. 1993;178:1857. doi: 10.1084/jem.178.6.1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Darrasse-Jèze G, Marodon G, Salomon BL, Catala M, Klatzmann D. Ontogeny of CD4+CD25+ regulatory/suppressor T cells in human fetuses. Blood. 2005;105:4715. doi: 10.1182/blood-2004-10-4051. [DOI] [PubMed] [Google Scholar]
- 27.Wucherpfennig KW, et al. Structural requirements for binding of an immunodominant myelin basic protein peptide to DR2 isotypes and for its recognition by human T cell clones. J Exp Med. 1994;179:279. doi: 10.1084/jem.179.1.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Spits H, et al. Innate lymphoid cells — a proposal for uniform nomenclature. Nat Rev Immunol. 2013;13:145. doi: 10.1038/nri3365. [DOI] [PubMed] [Google Scholar]
- 29.Chen L, et al. CD56 Expression Marks Human Group 2 Innate Lymphoid Cell Divergence from a Shared NK Cell and Group 3 Innate Lymphoid Cell Developmental Pathway. Immunity. 2018;49:464–476.e4. doi: 10.1016/j.immuni.2018.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Roy A, et al. Perturbation of fetal liver hematopoietic stem and progenitor cell development by trisomy 21. Proc Natl Acad Sci. 2012;109:17579. doi: 10.1073/pnas.1211405109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Almaden JV, et al. B-cell survival and development controlled by the coordination of NF-κB family members RelB and cRel. Blood. 2016;127:1276. doi: 10.1182/blood-2014-10-606988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stremmel C, et al. Yolk sac macrophage progenitors traffic to the embryo during defined stages of development. Nat Commun. 2018;9:75. doi: 10.1038/s41467-017-02492-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ginhoux F, Jung S. Monocytes and macrophages: developmental pathways and tissue homeostasis. Nat Rev Immunol. 2014;14:392. doi: 10.1038/nri3671. [DOI] [PubMed] [Google Scholar]
- 34.Murphy TL, et al. Transcriptional Control of Dendritic Cell Development. Annu Rev Immunol. 2016;34:93–119. doi: 10.1146/annurev-immunol-032713-120204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tusi BK, et al. Population snapshots predict early haematopoietic and erythroid hierarchies. Nature. 2018;555:54. doi: 10.1038/nature25741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rodrigues PF, et al. Distinct progenitor lineages contribute to the heterogeneity of plasmacytoid dendritic cells. Nat Immunol. 2018;19:711–722. doi: 10.1038/s41590-018-0136-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Seu KG, et al. Unraveling Macrophage Heterogeneity in Erythroblastic Islands. Front Immunol. 2017;8:1140. doi: 10.3389/fimmu.2017.01140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McGovern N, et al. Human Dermal CD14+ Cells Are a Transient Population of Monocyte-Derived Macrophages. Immunity. 2014;41:465–477. doi: 10.1016/j.immuni.2014.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sun J-B, et al. Deficiency in Calcium-Binding Protein S100A4 Impairs the Adjuvant Action of Cholera Toxin. Front Immunol. 2017;8:1119. doi: 10.3389/fimmu.2017.01119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Janelsins BM, Lu M, Datta SK. Altered inactivation of commensal LPS due to acyloxyacyl hydrolase deficiency in colonic dendritic cells impairs mucosal Th17 immunity. Proc Natl Acad Sci. 2014;111:373. doi: 10.1073/pnas.1311987111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McGovern N, et al. Human fetal dendritic cells promote prenatal T-cell immune suppression through arginase-2. Nature. 2017;546:662. doi: 10.1038/nature22795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Grün D, et al. De Novo Prediction of Stem Cell Identity using Single-Cell Transcriptome Data. Cell Stem Cell. 2016;19:266–277. doi: 10.1016/j.stem.2016.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Velten L, et al. Human haematopoietic stem cell lineage commitment is a continuous process. Nat Cell Biol. 2017;19:271. doi: 10.1038/ncb3493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Belluschi S, et al. Myelo-lymphoid lineage restriction occurs in the human haematopoietic stem cell compartment before lymphoid-primed multipotent progenitors. Nat Commun. 2018;9:4100. doi: 10.1038/s41467-018-06442-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cabezas-Wallscheid N, et al. Identification of Regulatory Networks in HSCs and Their Immediate Progeny via Integrated Proteome, Transcriptome, and DNA Methylome Analysis. Cell Stem Cell. 2014;15:507–522. doi: 10.1016/j.stem.2014.07.005. [DOI] [PubMed] [Google Scholar]
- 46.Pietras EM, et al. Functionally Distinct Subsets of Lineage-Biased Multipotent Progenitors Control Blood Production in Normal and Regenerative Conditions. Cell Stem Cell. 2015;17:35–46. doi: 10.1016/j.stem.2015.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Doulatov S, et al. Induction of Multipotential Hematopoietic Progenitors from Human Pluripotent Stem Cells via Respecification of Lineage-Restricted Precursors. Cell Stem Cell. 2013;13:459–470. doi: 10.1016/j.stem.2013.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Morrison SJ, Hemmati HD, Wandycz AM, Weissman IL. The purification and characterization of fetal liver hematopoietic stem cells. Proc Natl Acad Sci. 1995;92:10302. doi: 10.1073/pnas.92.22.10302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bowie MB, et al. Hematopoietic stem cells proliferate until after birth and show a reversible phase-specific engraftment defect. J Clin Invest. 2006;116:2808–2816. doi: 10.1172/JCI28310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Copley MR, et al. The Lin28b–let-7–Hmga2 axis determines the higher self-renewal potential of fetal haematopoietic stem cells. Nat Cell Biol. 2013;15:916. doi: 10.1038/ncb2783. [DOI] [PubMed] [Google Scholar]
- 51.Gerrelli D, Lisgo S, Copp AJ, Lindsay S. Enabling research with human embryonic and fetal tissue resources. Development. 2015;142:3073. doi: 10.1242/dev.122820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bullen P, Wilson D. The Carnegie staging of human embryos: a practical guide. Mol Genet Early Hum Dev. 1997:27–35. [Google Scholar]
- 53.Hern WM. Correlation of fetal age and measurements between 10 and 26 weeks of gestation. Obstet Gynecol. 1984;63 [PubMed] [Google Scholar]
- 54.Roy A, et al. High resolution IgH repertoire analysis reveals fetal liver as the likely origin of life-long, innate B lymphopoiesis in humans. Clin Immunol. 2017;183:8–16. doi: 10.1016/j.clim.2017.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Villani A-C, et al. Single-cell RNA-seq reveals new types of human blood dendritic cells, monocytes, and progenitors. Science. 2017;356 doi: 10.1126/science.aah4573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pijuan-Sala B, et al. A single-cell molecular map of mouse gastrulation and early organogenesis. Nature. 2019;566:490–495. doi: 10.1038/s41586-019-0933-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wolock SL, Lopez R, Klein AM. Scrublet: Computational Identification of Cell Doublets in Single-Cell Transcriptomic Data. Cell Syst. 2019;8:281–291.e9. doi: 10.1016/j.cels.2018.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Korsunsky I, et al. Fast, sensitive, and accurate integration of single cell data with Harmony. bioRxiv. 2018 doi: 10.1101/461954. 461954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Subramanian A, et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci. 2005;102:15545. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jia L-H, et al. AnimalTFDB 3.0: a comprehensive resource for annotation and prediction of animal transcription factors. Nucleic Acids Res. 2018;47:D33–D38. doi: 10.1093/nar/gky822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Efremova M, Vento-Tormo M, Teichmann SA, Vento-Tormo R. CellPhoneDB v2.0: Inferring cell-cell communication from combined expression of multi-subunit receptor-ligand complexes. bioRxiv. 2019 doi: 10.1101/680926. 680926. [DOI] [PubMed] [Google Scholar]
- 62.The 1000 Genomes Project Consortium et al. A global reference for human genetic variation. Nature. 2015;526:68. doi: 10.1038/nature15393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Belle M, et al. Tridimensional Visualization and Analysis of Early Human Development. Cell. 2017;169:161–173.e12. doi: 10.1016/j.cell.2017.03.008. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.