

Abstract
Genome-wide single nucleotide polymorphism (SNP) arrays can be used to explore homozygosity segments, where two haplotypes inherited from the parents are identical. In this study, we identified a total of 27,358 runs of homozygosity (ROH) with an average of 153 ROH events per animal in Chinese local cattle. The sizes of ROH events varied considerably ranging from 0.5 to 66 Mb, with an average length of 1.22 Mb. The highest average proportion of the genome covered by ROH (~11.54% of the cattle genome) was found in Nanda cattle (NDC) from South China, whereas the lowest average proportion (~3.1%) was observed in Yanhuang cattle (YHC). The average estimated FROH ranged from 0.03 in YHC to 0.12 in NDC. For each of three ROH classes with different sizes (Small 0.5–1 Mb, Medium 1–5 Mb and Large >5 Mb), the numbers and total lengths of ROH per individual showed considerable differences across breeds. Moreover, we obtained 993 to 3603 ROH hotspots (which were defined where ROH frequency at a SNP within each breed exceeded the 1% threshold) among eight cattle breeds. Our results also revealed several candidate genes embedded with ROH hotspots which may be related to environmental conditions and local adaptation. In conclusion, we generated baselines for homozygosity patterns in diverse Chinese cattle breeds. Our results suggested that selection has, at least partially, played a role with other factors in shaping the genomic patterns of ROH in Chinese local cattle and might provide valuable insights for understanding the genetic basis of economic and adaptive traits.
Subject terms: Genomics, Animal breeding
Introduction
Runs of homozygosity (ROH) are defined by contiguous homozygous segments where the two haplotypes transmitted from the parents are identical1. The occurrence of ROH within a population can be influenced by both reduced effective population size in the past and recent inbreeding. In addition, potential effect from genetic drift may also contribute to the formation of ROH. The availability of high-density SNP arrays has promoted the recent studies of regions of autozygosity. Initially, ROH analysis was utilized as a strategy to map recessive diseases genes in human2,3. Genome-wide analysis of homozygosity and their variation across individuals can offer important insights into genetic diversity and demographic history4. Abundance of ROH and their directional dominance on complex traits have been demonstrated in diverse human population5. Previous studies have investigated both genomic and geographic distribution of ROH and their contribution to population history. In human, recent studies suggested the total number and length of ROH per individual display considerable variation in worldwide human populations6, and the genomic distribution of ROH followed an obvious South to North gradient which was in consistent with European population history7.
ROH has also been widely utilized in farm animals to characterize population structure and demography history. Also, analyses of ROH can help to disclose the genetic relationships among individuals, measure the genome inbreeding level, and identify the region associated with economically important trait8–20. In cattle, Purfield et al. assessed the level of ROH in a wide range of daily and beef cattle breeds11, and their findings suggested different distribution patterns of ROH length may imply variations in breed origins and recent management among them. Extensive studies have been performed for evaluation of inbreeding and their negative effects on production and reproductive ability based on ROH in commercial daily cattle15,16,18. Also, several studies have been conducted to estimate inbreeding depression and investigate climatic adaptation in local cattle14,21.
Previous studies suggested the occurrence of ROH is not randomly distributed across the genomes, and ROH hotspots appear to be shared among individuals, which is probably caused by selection pressure22. The autozygosity across genome can help to improve the understanding of selection process and identify potential candidate genes associated with important traits in cattle23–29. The formation of ROH of various sizes (long vs. short) may reflect different evolutionary events on recent vs. ancient inbreeding6,12, with long ROH indicate more recent inbreeding, while short ROH suggest more ancient inbreeding. The investigation of these ROH segments can further improve our understanding of selection histories for special traits among diverse populations23,24.
Most of Chinese local cattle have been isolated in local feed environments, thus the potential homozygosity regions could be generated among them, which may also be influenced by adaptive selection and other factors in specific environment30–32. Despite several studies have conducted on analysis of population structures and admixture in Chinese local cattle31,33, no comprehensive study has been reported to explore the ROH pattern using high density SNP arrays. The aims of the present study were to (i) investigate genome-wide autozygosity patterns and genomic inbreeding level in Chinese local cattle; (ii) characterize profiles of ROH with different sizes and their gene contents with ROH hotspots; (iii) evaluate potential diverse selection based on the landscape of ROH across genome.
Results
Genomic ROH distribution
We identified a total of 27,358 ROH with an average of 153 per individual. The ROH size varied considerably from 0.5 to 66 Mb, with an average size of 1.22 Mb across all autosomes (Supplementary File 1: Table S1). The descriptive statistics of ROH and nonredundant ROH region per breed were presented in Table 1. Based on geographical locations, 8 breeds can be divided into 4 groups including North group (YHC and MGC), Northwest group (CDM), Southwest group (PWC, LSC and ZTC) and South group (WSC and NDC). We found cattle breeds from South and Southwest groups had the higher average ROH numbers, while cattle from North group had lower average numbers. The number of ROH per animal varied from 87 in CDM to 351 in NDC cattle (Table 1). Also, we observed the highest proportion of the genome covered by ROH in NDC (average ROHlength per individual was 323 Mb, corresponding to ~11.54% of cattle genome) from South China, whereas the lowest was found in YHC (average ROHlength per individual was 87 Mb, corresponding to ~3.1%). As expected, the proportion of the genome coverage by ROH for each breed showed high correlation with their average observed heterozygosity (Ho) with r = 0.71. In addition, we found the numbers and sizes of ROH events per individual varied across populations. NDC had larger average numbers and sizes of ROH events than other breeds (Fig. 1).
Table 1.
The descriptive statistics of ROH and ROH region for eight Chinese local cattle breeds.
| Sample Size | Total Numbera | Average Number per ind | Total length (Mb)b | Average length Per ind (Mb) | ROH-region length (Mb)c | ROH-region numberd | Genome Coveragee | |
|---|---|---|---|---|---|---|---|---|
| YHC | 21 | 1889 | 90 | 1821 | 87 | 1135 | 759 | 0.41 |
| MGC | 21 | 2156 | 103 | 4859 | 231 | 1906 | 423 | 0.68 |
| CDM | 25 | 2167 | 87 | 4900 | 196 | 2078 | 329 | 0.74 |
| PWC | 23 | 2601 | 113 | 3389 | 147 | 1675 | 721 | 0.60 |
| LSC | 22 | 2873 | 131 | 2397 | 109 | 1297 | 1011 | 0.46 |
| ZTC | 23 | 2861 | 124 | 3496 | 152 | 1800 | 650 | 0.64 |
| WSC | 21 | 4741 | 226 | 4935 | 235 | 1957 | 755 | 0.70 |
| NDC | 23 | 8070 | 351 | 7431 | 323 | 2129 | 770 | 0.76 |
Note:
aThe total number of ROH events for each breed.
bThe total length of ROH events across individuals for each breed.
cThe length of ROH region for each breed.
dThe number of nonredundant ROH region for each breed.
eThe proportion of ROH coverage of genome for each breed.
Figure 1.

Individual patterns of ROH. The distributions of ROH statistics per individual for eight Chinese local cattle breeds, including YHC (n = 21), MGC (n = 21), CDM (n = 25), PWC (n = 23), LSC (n = 22), ZTC (n = 23), WSC (n = 21), and NDC (n = 23). Groups are divided based on geographic locations in China (North, Northwest, South and Southwest). (A) The length of ROH events per individual. (B) The number of ROH events per individual.
ROH region and inbreeding coefficients
ROH regions were determined by merging ROH identified across all individuals using previously published protocols implemented in BEDTools34. Totally, we identified 86 nonredundant ROH regions in Chinese local cattle, covering 2.47 Gb of the cattle genome (Supplementary File 2: Table S2). We observed the total length of merged ROH regions ranged from 1.13 Gb (~40.53% of genome) in YHC to 2.12 Gb (~76.02% of genome) in NDC (Table 1). Next, we estimated inbreeding coefficients within each population using three methods including FROH, FHOM and FGRM. The individual FROH values varied from 0.01 to 0.34, while the highest average FROH (0.12) was estimated in NDC. The FGRM values were lower when compared to FROH, which ranged from −0.07 to 0.36, whereas the coefficient for FHOM ranged from −0.15 to 0.33. The highest correlation (r = 0.85, P < 2.2 × 10−16) was observed between FROH vs. FHOM, and the significant correlations were found for FROH vs. FGRM (r = 0.78, P < 2.2 × 10−16) and FHOM vs. FGRM (r = 0.81, P < 2.2 × 10−16).
Genomic patterns of homozygosity
To investigate the ROH pattern, we divided them into three size classes: A. Small (500 kb to 1 Mb), B. Medium (1 Mb to 5 Mb), and C. Large (>5 Mb), as described in the previous study12. The ROH distributions by total length and number for each group were shown in Fig. 2. The proportion of the genome in ROH of different lengths (thus inferred to be autozygous) varied clearly among breeds.
Figure 2.
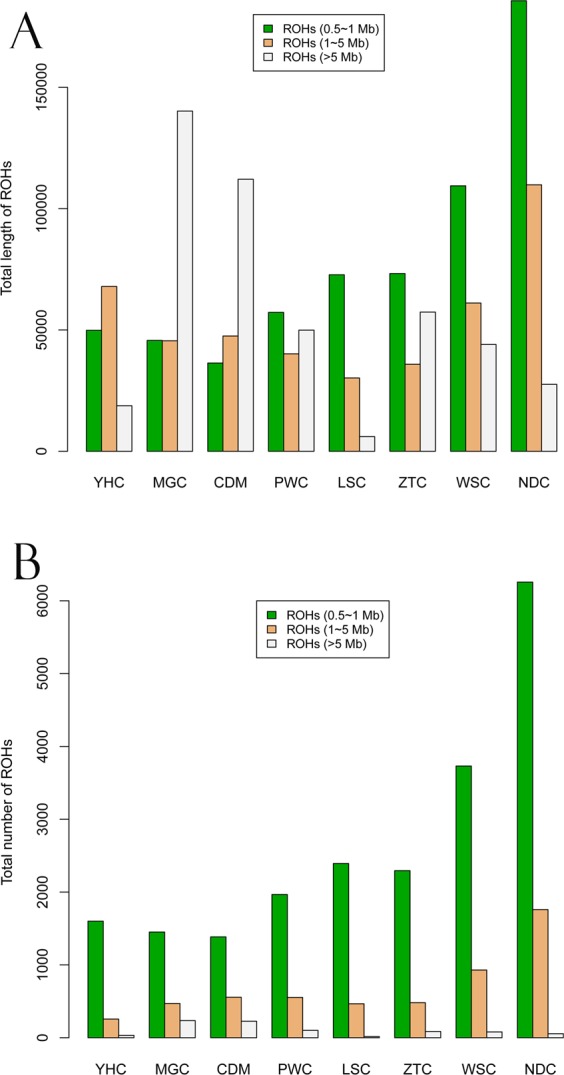
Total length and number of ROH in Chinese local cattle. (A) The total length of ROH belonging to three size classes including Small (0.5 to 1 Mb), Medium (1 to 5 Mb) and Large (>5 Mb) size classes for each of the different populations. (B) The total number of ROH belonging to three size classes.
Our result showed two breeds (MGC and CDM) had the largest total lengths of Large ROH (>5 Mb, Fig. 2A) as compared to the other breeds, while the LSC had the smallest total length. Moreover, Small and Medium ROH were found to be predominant in the South group breeds (NDC and WSC) by their total lengths. Our results also revealed that Small ROH predominated across Southwest groups of Chinese local cattle (Fig. 2B). The total numbers of Small and Median ROH showed increasing trends for cattle from North to South groups.
As previously reported, the likelihood of ROH occurrence at a particular chromosomal position generally depends on the length of ROH12. We found that the distributions of ROH occurrence along the autosomes vary among eight breeds. The high proportion of ROH mostly appeared in the middle of the chromosomes with the low recombination rates, and much less towards the telomeric regions (Fig. 3). Additionally, NDC and WSC have high proportion of ROH across chromosomes when compared to other breeds.
Figure 3.
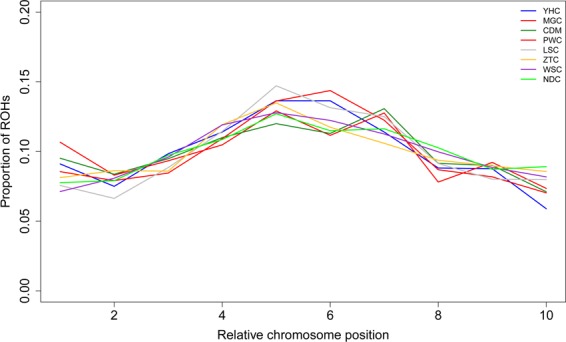
ROH distribution over relative chromosomal position in Chinese local cattle, and the distributions of ROH bins are averaged over all chromosomes.
ROH profiles across populations
To study the demographic history, we firstly plotted the numbers of ROH per individual against the total length of them (Fig. 4A). We observed ROH profiles (length vs. number) displayed different patterns among eight breeds. Out of 8 cattle breeds, YHC showed a small number of ROH and short total length in ROH with most points locating in the left corner of the plot (Fig. 4A). MGC, CDM, ZTC, and PWC had most points in the higher left corner of the plot, indicating a small number of ROH, but a large sum of total ROH length. WSC and NDC were extreme cases with large numbers of Small and Medium ROH and large total ROH lengths. Also, we observed the different patterns of ROH when ROH were classified into three class by sizes, including Large (Fig. 4B), Medium (Fig. 4C) and Small classes (Fig. 4D). Our results suggested Medium and Large ROH showed large differences among individuals.
Figure 4.

Evaluation of number of ROH and ROH total length for eight cattle populations. The number of ROH found for each individual genome (x-axis) is plotted against ROH total length (i.e. the length of Mb covered by ROH in each genome, y-axis). We analyzed cattle from YHC, MGC, CDM, LSC, PWC, ZTC, WSC and NDC. (A) All ROH; (B) size class Large (>5 Mb); (C) size class Medium (1 to 5 Mb) and (D) size class Small (0.5 to 1 Mb).
To assess the genomic distribution patterns of total ROH length, we also conducted pairwise comparisons among the total lengths of three classes of Small, Medium and Large ROH in individual cattle genomes (Fig. 5). We found the total lengths of Small and Medium were highly correlated (r = 0.72, P < 2.2 × 10−16), while none of them is strongly correlated with the total length of Large (r = 0.36 with P < 5.36 × 10−7 and r = −0.19 with p < 0.0106, respectively).
Figure 5.

Comparison of total lengths of ROH per individual across size classes, including Small, Medium and Large. Pairwise correlations and their significance levels between the total lengths of Small, Medium, and Large ROH in individual genomes were estimated using Pearson correlations. (A) Large versus Medium (r = 0.72). (B) Large versus Small (r = 0.36). (C) Medium versus Small (r = −0.19).
ROH involved with selection signatures
To investigate effect of selection in diverse cattle, we explored the occurrences of ROH across genome. The frequency of a SNP locus (%) in those ROH was assessed for each breed, and then plotted against the position of the SNP along autosomes (Fig. 6). The threshold for each breed were 0.29, 0.48, 0.4,0.30,0.32,0.30, 0.57, 0.74 for YHC, MGC, CDM, PWC, LSC, ZTC, WSC and NDC cattle. Our results suggested that the occurrences of ROH varied clearly within the genome among diverse cattle. We observed 3603, 1542, 1055, 2195, 3216, 1839, 1444 and 993 hotspots at top 1% in YHC, MGC, CDM, PWC, LSC, ZTC, WSC and NDC cattle, respectively. Within the genomic regions with a high level of autozygosity under potential selection, we identified 382 unique genes across eight breeds. Among them, twelve genes including CTNNA1, LRRTM2, SIL1, SNHG4, MATR3, PAIP2, MZB1, SPATA24, DNAJC18, ECSCR, TMEM173, UBE2D2 were shared across eight breeds. The DAVID annotations for these top annotated genes for each of eight breeds were presented in Supplementary File 3: Table S3. Most genes identified in North and Norwest group, including MGC and CDM, were enriched in lysozyme activity, cytolysis, defense response to Gram-negative bacterium and cell wall macromolecule catabolic process.
Figure 6.

Manhattan plot of the distribution of runs of homozygosity (ROH) hotspots in the Chinese local cattle genome. The x-axis represents the SNP genomic coordinate, and the y-axis shows the frequency (%) of overlapping ROH shared among individuals. (A) YHC; (B) MGC; (C) CDM; (D) PWC; (E) LSC; (F) ZTC; (G) WSC; and (H) NDC.
Among 382 unique genes, a total of 137 were identified in two and more populations. In YHC, two regions were detected on BTA7 and BTA21, including two genes (ETF1 and HSPA9) at 51.5 Mb on BTA7 and three genes (TMEM266, ETFA and ISL2) at 32 Mb on BTA21. In MGC, we detected top three regions on BTA7, BTA11 and BTA23: the top region on BTA7 overlapped with six genes CTNNA1, LRRTM2, SIL1, SNHG4, MATR3 and PAIP2, while no known gene was found within the regions on BTA11 and BTA23. Also, we observed the top three regions on BTA5, BTA13 and BTA14 in CDM. Among them, the top region on BTA5 contained genes YEATS4 and LYZ1. We found 17 genes were included in the region ranging from 54.48 to 54.85 Mb on BTA13, while no known gene was found within the region on BTA14. In the LSC cattle, we observed one significant region on each of BTA5, BTA7 and BTA16, which overlapped with many genes and spanned 0.6 Mb, 7 Mb and 0.1 Mb, respectively. While in the ZTC, only one region surpassed the 50% occurrence of ROH which ranged from 51.6 to 52.7 Mb, containing the important gene like CTNNA1, ISL2, DNAJC18, and SPATA24. Finally, no significant region (>50% occurrence) was detected for the PWC in Southwest groups. For two breeds in South group, we observed that multiple regions on BTA2, BTA5, BTA7, BTA11, BTA16, BTA14 and BTA22 were shared by WSC and NDC within the regions of above 50% occurrences.
In addition, a total of 247 genes were identified in unique breed based on the occurrence of ROH, of those, 56, 9, 15, 69, 28, 38, 24, 8 genes were uniquely identified in YHC, MGC, CDM, LSC, PWC, ZTC, WSC and NDC, respectively.
Discussion
Cattle represent important genetic resources that contribute to local economy. Many studies have been explored the ROH pattern and its associated inbreeding depression related to traits at the genomic level in multiple cattle population8. However, the ROH patterns and their distributions in Chinese local cattle remain largely unexplored. To our knowledge, this study is the first attempt to investigate the occurrence and distribution of ROH in Chinese local cattle using high-density SNP arrays. The high density SNP arrays are more sensitive for the determination of small segments, as previously reported35, while the BovineSNP50K array may underestimate the numbers of segments of 1 to 4 Mb in length.
We estimated inbreeding coefficients within each population using three methods including FROH, FHOM and FGRM. In general, the correlations among FROH, FHOM and FGRM varied dramatically across studies. Our study revealed the relative high correlation (0.78~0.85) among them, and high correlations between FROH and the two other estimates of genomic inbreeding (FGRM and FHOM) can be considered as an accurate estimator of the IBD genomic proportion17. On the other hand, several other studies have found a low to moderate FGRM-FROH correlation for dairy breeds36,37, while moderate to high correlations were reported in Holstein cattle and Jersey populations15,38. We suspected that these different results could be related to ROH type, sample populations and SNP array platforms.
In this study, we found clear differences in terms of total number and length of ROH in diverse Chinese local cattle, and the relationship between them varied considerably among individuals. This was consistent with previous studies with large-scale datasets in cattle, their findings suggested that ROH are often differentially distributed across breeds, and the patterns of ROH (number and length) may imply differences in breed origins and recent management11. The British Isles breeds including Hereford, Guernsey, Angus and Jersey cattle displayed the highest sum of ROH per animal compared other breeds, while African breeds displayed high variability in total number of ROH among breeds11.
A recent study revealed that the average total length of ROH ranged from 106 Mb in Piedmontese to 371 Mb in Brown cattle36, and their study suggested that dairy breeds have longer and larger size ROH than beef and dual-purpose cattle, and inbreeding is more recent in these dairy breeds. In our study, we found some breeds like WSC and NDC in South China displayed larger ROH size, which may reflect the effects of current inbreeding. We suspected that both breeds may be selected in isolated conditions with relative small effective population sizes. This finding also implied that continuing isolation and a reduced population size might play an essential role in the formation of ROH.
The distribution of ROH numbers and lengths can illustrate the genetic diversity of studied population. We also observed diverse pattern between the total number and length of ROH in Chinese local cattle, which were agree with the previous goat and sheep studies20,28. A large proportion of individuals with large number and length were identified in NDC and WSC (Fig. 4A). In contrast, for the Small ROHs in each individual, the total length of Small ROH in NDC and WSC mostly ranged from 100 to 250 Mb and none of them was less than 100 Mb (Fig. 4D). This finding reflected that both ancient and recent inbreeding had a critical impact on cattle genomes. This ROH pattern might be caused by the breeding isolation of these populations. Additionally, the NDC and WSC have relatively small populations and might have experienced bottlenecks as indicated by high fraction of the genome in ROH. On the other hand, YHC showed very low amounts of ROH (Fig. 4A). This is consistent with recent admixture in the individuals from YHC. Otherwise, we observed medium level ROH in terms of both total length and number of ROH in CDM and PWC. This probably reflects an absence of admixture in those breeds. The comparison for distribution of ROH numbers and lengths may be useful to demonstrate the genetic diversity among cattle breeds.
A recent study assessed the pattern of homozygosity and they found the average coverage of genome with autozygosity was 175 Mb in Gyr (Bos indicus) cattle17, while our study found the average value of ROH coverage per animal were 323, 235 and 231 Mb in NDC, WSC and MGC, respectively. We also observed the cumulative ROH length was dominated by an abundance of Small to Medium ROH in WSC as well as NDC, which implied either of them is an isolated breed based on a source population of substantial size. Abundance of ROH due to genetic isolation has also been reported in European cattle populations13 and Japanese wild boar12. The accumulation of large ROH in individuals of MGC and CDM could be caused by potential selection pressure for special environmental conditions of these populations, which has also been reported in Spanish local goat breeds20. Three ROH class (Large, Small and Medium) may be influenced via different processes, including recent inbreeding and recombination on different evolutionary scales6. Clustering ROH into different size classes make it possible to detect and interpret genomic difference among breeds. In addition, previous study reported that long ROH are mostly enriched in regions with deleterious mutations, and inbreeding can elevate the occurrence of rare recessive diseases that represent homozygotes for deleterious mutations39. Therefore, control the increase of inbreeding can effectively promote the implement conservation programs and maintain genetic diversity in local cattle breeds.
Also, pairwise comparisons between the total lengths of Small, Medium, and Large ROH in individual cattle genomes suggested the total lengths of Small and Medium ROH are highly correlated (r = 0.72, P < 2.2 × 10−16). Neither class Small or Medium is strongly correlated with the total length of Large ROH (r = 0.36 with P < 5.36 × 10−7 and r = −0.19 with P < 0.0106, respectively). Our study revealed that most of ROH belongs to the Short to Medium ROH categories. Compared with breeds from Southwest and South, our results suggested that two breeds (MGC and CDM) have been high inbred based on high coverage of Large ROH (Fig. 2A), probably due to a relatively small population size and close mating for each of two breeds. Because long homozygosity segments across chromosome can be interrupted by recombination events, Large ROH are usually generated by recent inbreeding, while Small/Medium ROH are indicative of more ancient relatedness. For instance, a previous study highlighted ROH preferentially occur in regions of decreased recombination activity12. And another study reported the existence of recombination hotspots across the cattle genome can influence ROH11. Our finding was consistent with these results, and the identified regions with ROH hotspots frequently coincided with those with lower recombination rates. Moreover, we also found the studied populations display different proportions of ROH across the chromosomes (Fig. 3).
The occurrence of ROH hotspots in genomic regions that harbor candidate genes may be involved in selection pressure in response to environmental condition. Previous study has investigated homozygosity in Polish Red cattle, which are characterized by high fertility and easy calving, good immunity and health, as well as high milk quality29. Their finding revealed four genes regulating signaling lymphocytic activation including SLAMF1, CD48, Ly9 and SLAMF3. In our study, we found several genes within ROH hotspots, which have been previously reported under selection in cattle40–45. Among them, we identified Heat Shock Protein Family A (Hsp70) Member 9 (HSPA9) in seven Chinese local breed, this gene is immune-related genes, and also shows differentially expressed during bovine muscle development46. Previous studies suggested that HSPA9 abundance in muscle had positive correlation with beef tenderness47,48. This regulation of HSPA9 on meat tenderness is likely due to its anti-apoptotic effect, which prevents the formation of protein aggregates49. Moreover, we observed several genes like CTNNA1, SIL1, PAIP2 in eight breed, and ZBTB46 and GMEB2 in CDM located within/near the ROH hotspots, which are also involved in selection in goat and sheep50–52.
To assesses the breed-specific differences in ROH occurrence related to selection, we investigated the genes within or near the ROH islands. In our study, we identified one gene named EEF1A2 in CDM, which was reported to be related to fatty acid and potentially regulate meat tenderness53–55. Several genes SIRT6, CREB3L3 and B3GNT5 were specifically detected in YHC cattle, which are reported related to energy meat quality, homeostasis and glycan synthesis56–59. Two genes TCF12 and TXNDC9 uniquely detected in LSC which were related to muscle lipid composition and marbling in beef cattle60,61. Moreover, we identified breed-specific genes (BMP2, FGF9 and CD14) related to growth traits and immune process in PWC, WSC and ZTC, respectively62–64. Using the ROH approach, we detected many genes which show potential effects for important traits as reported in many previous studies. Our finding further supported that pattern of ROH can be utilized to explore genomic regions and genes under specific selection, and the shared ROH may potentially contain alleles associated with important traits. Despite many genes were identified in our ROH analysis, functional validation of them is still warranted in the future.
It was noted that we compared our settings to some other studies which used PLINK under its default settings11,17,21, and found out our settings generally agreed with the most of published studies using the Bovine HD SNP array. For example, Purfield et al. tested a minimum run length of 58 SNPs was needed to produce <5% randomly generated ROH and the maximum gap between two consecutive homozygous SNPs in a run was set at a 100 kb11. We found their options used was like ours, which slides a window of 50 SNPs, in one SNP intervals, across the genome estimating homozygosity. Although a previous simulation study has tested out ROH calling programs and their settings and reported the best program (PLINK) and its optimal settings65, the ROH calling methods and their settings are still challenging11,35, thus these results including ours should be interpreted by cautions. Also, to avoid bias and overestimation for ROH detection using PLINK, more options should be considered carefully according to the genome size and the number of SNPs genotyped.
In summary, our study revealed the occurrence of autozygosity varied largely across breeds in Chinese local cattle as different recent and ancient events. We further speculate that selection has partially contributed to landscape of ROH and their functions in the Chinese local cattle.
Materials and Methods
Ethics statement
All animal experiments were approved by the Chinese Academy of Agricultural Sciences (CAAS, Beijing, China). All animal procedures were performed in strict accordance with the guidelines proposed by the China Council on Animal Care and the Ministry of Agriculture of People’s Republic of China.
Genotyped samples
The genome-wide SNP data of 179 samples from eight populations including Yanhuang (YHC), Menggu (MGC), Caidamu(CDM), Liangshan (LSC), Pingwu(PWC), Zhaotong (ZTC), Wenshan (WSC) and Nandan (NDC) were genotyped by Illumina BovineHD SNPs array, which was retrieved from our previous study32. The full names, associated abbreviation for each breeds and additional information on the locations of the sampling areas was presented in Supplementary File 4: Table S4. To minimize genetic relationships among samples, we removed closely related individuals if the PI-HAT value was greater than 0.25. Only autosomal SNPs passed following filters were used for subsequently analyses, and all selected samples displayed a genotype calling rate of more than 99%. Individuals with more than 5% of missing SNPs were removed from further analyses. Moreover, the SNPs were filtered with MAF < 0.05 (a minor allele frequency higher than 0.95) and geno <0.1 (only SNPs with a 90% genotyping rate or higher).
ROH estimation
ROH were detected across autosomes for each individual using PLINK v1.0766. The ROH were defined by a minimum of 0.5 Mb in length to avoid short and common ROH that occur throughout the genome due to LD. The PLINK software detect homozygous segments by scanning along genotype for each individual. The following six criteria were considered to define a ROH: (i) a sliding window of 50 SNPs across the genome; (ii) the proportion of homozygous overlapping windows was 0.05; (iii) the minimum number of consecutive SNPs included in a ROH was 100; (vi) a required minimum density was set at 50 kb/SNP; (v) the minimum length of a ROH was set to 500 kb, the maximum gap between consecutive homozygous SNPs was 100 kb; and (vi) a maximum of two SNPs with missing genotypes and up to one heterozygous genotype were allowed in a ROH.
ROH classification and inbreeding coefficient
ROH were divided into three classes based on length: 0.5–1 Mb, 1–5 Mb and >5 Mb and were identified as Small, Medium and Large, respectively. Three metrics were used to estimate levels of inbreeding: (1) FHOM was assessed based on the proportion of homozygotes using PLINK v1.07 software. (2) FROH was calculated as described by McQuillan et al. (an individual’s summed ROH length was normalized by the length of the autosomal genome covered by SNPs)67. (3) FGRM was estimated based on the variance of additive genotypes using GCTA 1.19.2 method according to Yang et al.68. Correlations of inbreeding coefficient for three methods were estimated using cor.test function in R v3.2.4.
Statistical analysis of the genomic ROH across breeds
The correlations between the average proportion of the genome coverage by ROH and average observed heterozygosity (Ho) for each breed were estimated by cor.test functions in R v3.4.2. The ROH pattern based on the total number of ROH per individual and the total length of those ROH were measured for three classes. For analysis of distribution of ROH across genome, each chromosome was divided into 20 equal size segments, and their relative distribution over these chromosomal segments were calculated across breeds. All plots in current study were generated with the R programing (v.3.2.4).
Genomic regions within ROH
To investigate genomic regions that were associated with occurrences of ROH within each breed, the fraction of SNPs in ROH was estimated based on the frequency of a SNP in them across individuals. ‘ROH hotspot’ was identified as a region of adjacent SNPs with an ROH frequency per SNP above the threshold of 1%. Genomic coordinates for all identified ROH regions were used to annotate genes using the UMD3.1 genome assembly. The function of these genes and GO terms were further assessed using DAVID Functional Annotation Bioinformatics Microarray Analysis platform69,70.
Supplementary information
Supplementary File1_Table S1, Supplementary File2_Table S2, Supplementary File3_Table S3, Supplementary File4_Table S4
Acknowledgements
This study was supported by the Basic R&D Fund for the Central Level Scientific Research Institute (2016ywf-yb-6), National Beef Cattle Industrial Technology System (CARS-37), the Agricultural Science and Technology Innovation Program in Chinese Academy of Agricultural Sciences (ASTIP-IAS-TS-16 and ASTIP-IAS03). L.Y.X was supported by the Elite Youth Program in Chinese Academy of Agricultural Sciences. The project was also partly supported by Beijing City Board of Education Foundation (PXM2016_014207_000012) for the data analysis and interpretation of the study.
Author contributions
Conceived and designed the experiments: L.Y.X., J.Y.L. and G.E.L. Analyzed the data: G.Y.Z., L.Y., H.J.G., B.Z. and L.Y.X. Contributed reagents/materials/analysis tools: L.P.Z., Y.C. and X.G. Wrote the paper: L.Y.X., G.E.L. and J.Y.L. All authors read and approved the final manuscript.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Lingyang Xu and Guoyao Zhao.
Contributor Information
Lingyang Xu, Email: xulingyang@163.com.
Junya Li, Email: lijunya@caas.cn.
Supplementary information
is available for this paper at 10.1038/s41598-019-53274-3.
References
- 1.Ceballos Francisco C., Joshi Peter K., Clark David W., Ramsay Michèle, Wilson James F. Runs of homozygosity: windows into population history and trait architecture. Nature Reviews Genetics. 2018;19(4):220–234. doi: 10.1038/nrg.2017.109. [DOI] [PubMed] [Google Scholar]
- 2.Lander ES, Botstein D. Homozygosity mapping: a way to map human recessive traits with the DNA of inbred children. Science. 1987;236:1567–1570. doi: 10.1126/science.2884728. [DOI] [PubMed] [Google Scholar]
- 3.Rudan I, Campbell H, Carothers AD, Hastie ND, Wright AF. Contribution of consanguinuity to polygenic and multifactorial diseases. Nature genetics. 2006;38:1224–1225. doi: 10.1038/ng1106-1224. [DOI] [PubMed] [Google Scholar]
- 4.Kirin M, et al. Genomic runs of homozygosity record population history and consanguinity. PloS one. 2010;5:e13996. doi: 10.1371/journal.pone.0013996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Joshi PK, et al. Directional dominance on stature and cognition in diverse human populations. Nature. 2015;523:459–462. doi: 10.1038/nature14618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pemberton TJ, et al. Genomic patterns of homozygosity in worldwide human populations. American journal of human genetics. 2012;91:275–292. doi: 10.1016/j.ajhg.2012.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nothnagel M, Lu TT, Kayser M, Krawczak M. Genomic and geographic distribution of SNP-defined runs of homozygosity in Europeans. Human molecular genetics. 2010;19:2927–2935. doi: 10.1093/hmg/ddq198. [DOI] [PubMed] [Google Scholar]
- 8.Peripolli E, et al. Runs of homozygosity: current knowledge and applications in livestock. Animal genetics. 2017;48:255–271. doi: 10.1111/age.12526. [DOI] [PubMed] [Google Scholar]
- 9.Mastrangelo S, et al. Runs of homozygosity reveal genome-wide autozygosity in Italian sheep breeds. Animal genetics. 2018;49:71–81. doi: 10.1111/age.12634. [DOI] [PubMed] [Google Scholar]
- 10.MacLeod IM, Larkin DM, Lewin HA, Hayes BJ, Goddard ME. Inferring demography from runs of homozygosity in whole-genome sequence, with correction for sequence errors. Molecular biology and evolution. 2013;30:2209–2223. doi: 10.1093/molbev/mst125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Purfield DC, Berry DP, McParland S, Bradley DG. Runs of homozygosity and population history in cattle. BMC genetics. 2012;13:70. doi: 10.1186/1471-2156-13-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bosse M, et al. Regions of homozygosity in the porcine genome: consequence of demography and the recombination landscape. PLoS genetics. 2012;8:e1003100. doi: 10.1371/journal.pgen.1003100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Upadhyay MR, et al. Genetic origin, admixture and population history of aurochs (Bos primigenius) and primitive European cattle. Heredity. 2017;119:469. doi: 10.1038/hdy.2017.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Reverter A, et al. Genomic inbreeding depression for climatic adaptation of tropical beef cattle. Journal of animal science. 2017;95:3809–3821. doi: 10.2527/jas2017.1643. [DOI] [PubMed] [Google Scholar]
- 15.Bjelland DW, Weigel KA, Vukasinovic N, Nkrumah JD. Evaluation of inbreeding depression in Holstein cattle using whole-genome SNP markers and alternative measures of genomic inbreeding. Journal of dairy science. 2013;96:4697–4706. doi: 10.3168/jds.2012-6435. [DOI] [PubMed] [Google Scholar]
- 16.Martikainen K, Tyriseva AM, Matilainen K, Poso J, Uimari P. Estimation of inbreeding depression on female fertility in the Finnish Ayrshire population. Journal of animal breeding and genetics = Zeitschrift fur Tierzuchtung und Zuchtungsbiologie. 2017;134:383–392. doi: 10.1111/jbg.12285. [DOI] [PubMed] [Google Scholar]
- 17.Peripolli E, et al. Assessment of runs of homozygosity islands and estimates of genomic inbreeding in Gyr (Bos indicus) dairy cattle. BMC genomics. 2018;19:34. doi: 10.1186/s12864-017-4365-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Signer-Hasler H, et al. Population structure and genomic inbreeding in nine Swiss dairy cattle populations. Genetics, selection, evolution: GSE. 2017;49:83. doi: 10.1186/s12711-017-0358-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gomez-Raya L, Rodriguez C, Barragan C, Silio L. Genomic inbreeding coefficients based on the distribution of the length of runs of homozygosity in a closed line of Iberian pigs. Genetics, selection, evolution: GSE. 2015;47:81. doi: 10.1186/s12711-015-0153-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Manunza A, et al. A genome-wide perspective about the diversity and demographic history of seven Spanish goat breeds. Genetics, selection, evolution: GSE. 2016;48:52. doi: 10.1186/s12711-016-0229-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Upadhyay MR, et al. Genetic origin, admixture and population history of aurochs (Bos primigenius) and primitive European cattle. Heredity. 2017;118:169–176. doi: 10.1038/hdy.2016.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang Q, Guldbrandtsen B, Bosse M, Lund MS, Sahana G. Runs of homozygosity and distribution of functional variants in the cattle genome. BMC genomics. 2015;16:542. doi: 10.1186/s12864-015-1715-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Forutan M, et al. Inbreeding and runs of homozygosity before and after genomic selection in North American Holstein cattle. BMC genomics. 2018;19:98. doi: 10.1186/s12864-018-4453-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim ES, Sonstegard TS, Rothschild MF. Recent artificial selection in U.S. Jersey cattle impacts autozygosity levels of specific genomic regions. BMC genomics. 2015;16:302. doi: 10.1186/s12864-015-1500-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kim ES, et al. Effect of artificial selection on runs of homozygosity in u.s. Holstein cattle. PloS one. 2013;8:e80813. doi: 10.1371/journal.pone.0080813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ferencakovic M, et al. Estimates of autozygosity derived from runs of homozygosity: empirical evidence from selected cattle populations. Journal of animal breeding and genetics = Zeitschrift fur Tierzuchtung und Zuchtungsbiologie. 2013;130:286–293. doi: 10.1111/jbg.12012. [DOI] [PubMed] [Google Scholar]
- 27.Metzger J, et al. Runs of homozygosity reveal signatures of positive selection for reproduction traits in breed and non-breed horses. BMC genomics. 2015;16:764. doi: 10.1186/s12864-015-1977-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mastrangelo S, et al. Genome-wide scan for runs of homozygosity identifies potential candidate genes associated with local adaptation in Valle del Belice sheep. Genetics, selection, evolution: GSE. 2017;49:84. doi: 10.1186/s12711-017-0360-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Szmatoła T, et al. Characteristics of runs of homozygosity in selected cattle breeds maintained in Poland. Livestock Science. 2016;188:72–80. doi: 10.1016/j.livsci.2016.04.006. [DOI] [Google Scholar]
- 30.Mei Chugang, Wang Hongcheng, Liao Qijun, Wang Lizhong, Cheng Gong, Wang Hongbao, Zhao Chunping, Zhao Shancen, Song Jiuzhou, Guang Xuanmin, Liu George E, Li Anning, Wu Xueli, Wang Chongzhi, Fang Xiaodong, Zhao Xin, Smith Stephen B, Yang Wucai, Tian Wanqiang, Gui Linsheng, Zhang Yingying, Hill Rodney A, Jiang Zhongliang, Xin Yaping, Jia Cunling, Sun Xiuzhu, Wang Shuhui, Yang Huanming, Wang Jian, Zhu Wenjuan, Zan Linsen. Genetic Architecture and Selection of Chinese Cattle Revealed by Whole Genome Resequencing. Molecular Biology and Evolution. 2017;35(3):688–699. doi: 10.1093/molbev/msx322. [DOI] [PubMed] [Google Scholar]
- 31.Gao Y, et al. Species composition and environmental adaptation of indigenous Chinese cattle. Scientific reports. 2017;7:16196. doi: 10.1038/s41598-017-16438-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yang L, et al. Genome-wide analysis reveals differential selection involved with copy number variation in diverse Chinese Cattle. Scientific reports. 2017;7:14299. doi: 10.1038/s41598-017-14768-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang W, et al. Genome-wide assessment of genetic diversity and population structure insights into admixture and introgression in Chinese indigenous cattle. BMC genetics. 2018;19:114. doi: 10.1186/s12863-018-0705-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ferencakovic M, Solkner J, Curik I. Estimating autozygosity from high-throughput information: effects of SNP density and genotyping errors. Genetics, selection, evolution: GSE. 2013;45:42. doi: 10.1186/1297-9686-45-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Marras G, et al. Analysis of runs of homozygosity and their relationship with inbreeding in five cattle breeds farmed in Italy. Animal genetics. 2015;46:110–121. doi: 10.1111/age.12259. [DOI] [PubMed] [Google Scholar]
- 37.Mastrangelo S, et al. Genomic inbreeding estimation in small populations: evaluation of runs of homozygosity in three local dairy cattle breeds. Animal: an international journal of animal bioscience. 2016;10:746–754. doi: 10.1017/S1751731115002943. [DOI] [PubMed] [Google Scholar]
- 38.Pryce JE, Haile-Mariam M, Goddard ME, Hayes BJ. Identification of genomic regions associated with inbreeding depression in Holstein and Jersey dairy cattle. Genetics, selection, evolution: GSE. 2014;46:71. doi: 10.1186/s12711-014-0071-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Szpiech ZA, et al. Long runs of homozygosity are enriched for deleterious variation. American journal of human genetics. 2013;93:90–102. doi: 10.1016/j.ajhg.2013.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Porto-Neto LR, et al. Genomic divergence of zebu and taurine cattle identified through high-density SNP genotyping. BMC genomics. 2013;14:876. doi: 10.1186/1471-2164-14-876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Randhawa IA, Khatkar MS, Thomson PC, Raadsma HW. A Meta-Assembly of Selection Signatures in Cattle. PloS one. 2016;11:e0153013. doi: 10.1371/journal.pone.0153013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lee T, et al. Genetic variants and signatures of selective sweep of Hanwoo population (Korean native cattle) BMB reports. 2013;46:346–351. doi: 10.5483/BMBRep.2013.46.7.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Xu L, et al. Genomic signatures reveal new evidences for selection of important traits in domestic cattle. Molecular biology and evolution. 2015;32:711–725. doi: 10.1093/molbev/msu333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Taye M, et al. Exploring evidence of positive selection signatures in cattle breeds selected for different traits. Mammalian genome: official journal of the International Mammalian Genome Society. 2017;28:528–541. doi: 10.1007/s00335-017-9715-6. [DOI] [PubMed] [Google Scholar]
- 45.Bahbahani H, et al. Signatures of positive selection in East African Shorthorn Zebu: A genome-wide single nucleotide polymorphism analysis. Scientific reports. 2015;5:11729. doi: 10.1038/srep11729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.He H, Chen S, Liang W, Liu X. Genome-wide proteomics analysis on longissimus muscles in Qinchuan beef cattle. Animal genetics. 2017;48:131–140. doi: 10.1111/age.12508. [DOI] [PubMed] [Google Scholar]
- 47.Picard B, et al. Inverse relationships between biomarkers and beef tenderness according to contractile and metabolic properties of the muscle. Journal of agricultural and food chemistry. 2014;62:9808–9818. doi: 10.1021/jf501528s. [DOI] [PubMed] [Google Scholar]
- 48.Guillemin N, Bonnet M, Jurie C, Picard B. Functional analysis of beef tenderness. Journal of proteomics. 2011;75:352–365. doi: 10.1016/j.jprot.2011.07.026. [DOI] [PubMed] [Google Scholar]
- 49.Rodrigues RT, et al. Differences in Beef Quality between Angus (Bos taurus taurus) and Nellore (Bos taurus indicus) Cattle through a Proteomic and Phosphoproteomic Approach. PloS one. 2017;12:e0170294. doi: 10.1371/journal.pone.0170294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lai FN, et al. Whole-genome scanning for the litter size trait associated genes and SNPs under selection in dairy goat (Capra hircus) Scientific reports. 2016;6:38096. doi: 10.1038/srep38096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Guan D, et al. Scanning of selection signature provides a glimpse into important economic traits in goats (Capra hircus) Scientific reports. 2016;6:36372. doi: 10.1038/srep36372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Liu Z, et al. Genome-wide analysis reveals signatures of selection for important traits in domestic sheep from different ecoregions. BMC genomics. 2016;17:863. doi: 10.1186/s12864-016-3212-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Perez R, Canon J, Dunner S. Genes associated with long-chain omega-3 fatty acids in bovine skeletal muscle. Journal of applied genetics. 2010;51:479–487. doi: 10.1007/BF03208877. [DOI] [PubMed] [Google Scholar]
- 54.Hiller B, Angulo J, Olivera M, Nuernberg G, Nuernberg K. How selected tissues of lactating holstein cows respond to dietary polyunsaturated fatty acid supplementation. Lipids. 2013;48:357–367. doi: 10.1007/s11745-012-3737-3. [DOI] [PubMed] [Google Scholar]
- 55.Hiller B, Herdmann A, Nuernberg K. Dietary n-3 fatty acids significantly suppress lipogenesis in bovine muscle and adipose tissue: a functional genomics approach. Lipids. 2011;46:557–567. doi: 10.1007/s11745-011-3571-z. [DOI] [PubMed] [Google Scholar]
- 56.Rincon G, Farber EA, Farber CR, Nkrumah JD, Medrano JF. Polymorphisms in the STAT6 gene and their association with carcass traits in feedlot cattle. Animal genetics. 2009;40:878–882. doi: 10.1111/j.1365-2052.2009.01934.x. [DOI] [PubMed] [Google Scholar]
- 57.Gui LS, et al. Genetic variants in the SIRT6 transcriptional regulatory region affect gene activity and carcass quality traits in indigenous Chinese beef cattle (Bos taurus) BMC genomics. 2018;19:785. doi: 10.1186/s12864-018-5149-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Poulsen NA, Robinson RC, Barile D, Larsen LB, Buitenhuis B. A genome-wide association study reveals specific transferases as candidate loci for bovine milk oligosaccharides synthesis. BMC genomics. 2019;20:404. doi: 10.1186/s12864-019-5786-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ha NT, et al. Liver transcriptome analysis reveals important factors involved in the metabolic adaptation of the transition cow. Journal of dairy science. 2017;100:9311–9323. doi: 10.3168/jds.2016-12454. [DOI] [PubMed] [Google Scholar]
- 60.Dunner S, et al. Genes involved in muscle lipid composition in 15 European Bos taurus breeds. Animal genetics. 2013;44:493–501. doi: 10.1111/age.12044. [DOI] [PubMed] [Google Scholar]
- 61.Lee SH, et al. Use of a bovine genome array to identify new biological pathways for beef marbling in Hanwoo (Korean Cattle) BMC genomics. 2010;11:623. doi: 10.1186/1471-2164-11-623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Seabury CM, et al. Genome-wide association study for feed efficiency and growth traits in U.S. beef cattle. BMC genomics. 2017;18:386. doi: 10.1186/s12864-017-3754-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Totty ML, Morrell BC, Spicer LJ. Fibroblast growth factor 9 (FGF9) regulation of cyclin D1 and cyclin-dependent kinase-4 in ovarian granulosa and theca cells of cattle. Molecular and cellular endocrinology. 2017;440:25–33. doi: 10.1016/j.mce.2016.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Beecher C, et al. Polymorphisms in bovine immune genes and their associations with somatic cell count and milk production in dairy cattle. BMC genetics. 2010;11:99. doi: 10.1186/1471-2156-11-99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Howrigan DP, Simonson MA, Keller MC. Detecting autozygosity through runs of homozygosity: a comparison of three autozygosity detection algorithms. BMC genomics. 2011;12:460. doi: 10.1186/1471-2164-12-460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Purcell S, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. American journal of human genetics. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.McQuillan R, et al. Runs of homozygosity in European populations. American journal of human genetics. 2008;83:359–372. doi: 10.1016/j.ajhg.2008.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yang J, Lee SH, Goddard ME, Visscher PM. GCTA: a tool for genome-wide complex trait analysis. American journal of human genetics. 2011;88:76–82. doi: 10.1016/j.ajhg.2010.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nature protocols. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 70.Huang da W, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic acids research. 2009;37:1–13. doi: 10.1093/nar/gkn923. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary File1_Table S1, Supplementary File2_Table S2, Supplementary File3_Table S3, Supplementary File4_Table S4