

Abstract
Background
Non-small cell lung cancer (NSCLC) is one of the causes of carcinomas mortality worldwide. Ecliptasaponin A (ES), a natural product extracted from the plant known as Eclipta prostrata, has been reported as an anti-cancer drug against various cancer cell lines. However, the exact mechanisms of ES have not yet been fully characterized.
Methods
Numerous studies have been done to support that ES has a powerful inhibiting effect on the growth of cancers via the activation of apoptosis and autophagy. To explore the underlying mechanisms of anti-cancer and investigate the relationships of the apoptosis and autophagy, we used apoptosis signal-regulating kinase 1 (ASK1) inhibitor (GS-4997), c-Jun N-terminal kinase (JNK) inhibitor (SP600125), and autophagy inhibitor [chloroquine (CQ) and 3-methyladenine (3-MA)].
Results
ES could potently suppress cell viability and induces apoptotic cell death of human lung cancer cells H460 and H1975. ES activated apoptosis via ASK1/JNK pathway, GS-4997 and SP600125 can attenuated these effects. Furthermore, ES could triggered autophagy in lung cancer cell lines, and the autophagy inhibitor 3-MA and CQ reversed ES-induced apoptosis in H460 and H1975 cells. Furthermore, SP600125 can inhibit autophagy.
Conclusions
This study showed that ES induces apoptosis in human lung cancer cells by triggering enhanced autophagy and ASK1/JNK pathway, which may thus be a promising agent against lung cancer.
Keywords: Lung cancer, Ecliptasaponin A (ES), apoptosis signal-regulating kinase 1 (ASK1), c-Jun N-terminal kinase (JNK), autophagy, apoptosis
Introduction
Lung cancer is one of the main causes of global cancer mortality and cancer-related death (1,2). Even though there has been development in treatment over the past 20 years, the five-year survival rate has levelled at <20% (1). Consequently, preferable therapeutic regimens and strategies with high efficiency for this deadly disease to eradicate non-small cell lung cancer (NSCLC) are urgent and vital.
Natural products from pure plant extracts used in the treatment of cancers have long and rich experience, and are currently getting increasing attention from researchers around the world (3-5). Nevertheless, though Ecliptasaponin A (ES) has a clear structure (Figure 1A), the exact role and mechanism of it on carcinoma of lung remains unclear.
Figure 1.

ES inhibits cell viability in human lung carcinoma cells. (A) Chemical structure of ES; (B) cell viability was evaluated by MTT assay, H460 and H1975 cells were treated with ES for 24 and 48 h; (C) cells were treated with ES to form colonies. All data are expressed as mean ± SD. *, P <0.05; **, P<0.01; ***, P<0.001 compared to the control group. ES, Ecliptasaponin A.
Apoptosis is often triggered by oncotherapy agents and is a key player in clinical treatment of malignant tumors, which are regulated by a number of genes and signaling pathways related to apoptosis (6-8).
Autophagy is an evolutionarily conserved cellular self-digesting process with the characteristic of autophagosome formation (9), has recently been suggested as a “double edge sword” in the development and progress of tumor (10), may be either cytoprotective (11,12) or cytotoxic during therapy (13,14). In other words, given the appropriate circumstances, overabundant autophagy may induce cell death, or apoptosis (15,16), while other evidences have suggested that autophagy can protect tumor cells from apoptosis (17,18).
Many pathological processes are influenced by autophagy and apoptosis, which, in turn, mutually modify one another (19). Accumulating studies have demonstrated that numerous antitumor agents can induce apoptosis while trigger autophagy (20).
Apoptosis signal-regulating kinase 1 (ASK1) is a member of the mitogen-activated protein kinase (MAPK) family. MAPKs induce cells apoptosis, which leads to downstream signaling of MAPKs, c-Jun N-terminal kinases (JNKs) and p38 MAPKs (21). ASK1 is involved in many different stress responses, such as apoptosis (22-24), and ASK1 has been reported to be involved in many diseases, such as cancer (25-27).
JNK is part of the MAPK family, which is involved in modulating cell survival, proliferation, and apoptosis (28,29). And, in many cases of tumorigenesis and development, JNK has been demonstrated to regulate the apoptotic response of cells to pro-inflammatory cytokines and environmental stresses (30).
In this study, we aim to confirm the effect of ES on lung cancer cells and to elucidate its underlying molecular mechanisms.
Methods
Reagents and antibodies
ES was dissolved in Dimethylsulfoxide (DMSO). 3-(4,5-dimethylthiazol-yl)-2,5-diphenyltetrazolium bromide (MTT), DMSO, hoechst33342, 3-methyladenine (3-MA), chloroquine (CQ) and paraformaldehyde were purchased from Abcam (Cambridge, MA, USA). GS-4997, SP600125 and Z-VAD-FMK were acquired from Selleck Chemicals (Houston, TX, USA). Fetal Bovine serum and RPMI 1640 medium were acquired from Gibco Life Technologies (Grand Island, NY, USA). Primary antibodies (i.e. cleaved caspase-3, LC3B, Beclin-1, phosphor-JNK) were purchased from Cell Signaling Technology, Inc. (Beverly, MA, USA).
Cell culture
H460 and H1975 cells were acquired from the Cell Bank of the Chinese Academy of Science (Shanghai, China), and were subsequently cultured in RPMI 1640 medium with 10% FBS and in a 5% CO2 incubator at 37 °C.
Cell viability assay
H460 and H1975 were seeded at 104 cells/well into 96—well plates and treated with growing concentrations of ES for 24 and 48 h. At the end of incubation, the medium was removed, and replaced by 0.5 mg/mL MTT, which was followed by a 4-h incubation. The crystal formazan was dissolved in 150 µL DMSO and reading of the absorbance was completed at 490 nm using a microplate reader.
Clone formation assay
We seeded cells at 1,000–2,000 cells per well into six-well plates. The medium was changed and refreshed in 2-day interims. Finally, the we fixed the colonies with paraformaldehyde and stained them using Giemsa staining.
Annexin V-FITC/propidium iodide (PI) staining assay
Apoptosis was identified through an Annexin V-FITC/PI assay kit (BD Biosciences, San Diego, CA, USA). We acquired about 10,000 cells for each sample. Annexin V-FITC and PI fluorescence were detected and analyzed by using a flow cytometer.
Western blot assay
Cells were collected and lysed with RIPA buffer after being treated for 24 h under the prescribed conditions. Total proteins 20–40 µg were loaded into each well and separated by 12% separation gel. And then was transferred to PVDF membrane at 100 V for 120 min. For 1 hour, we blocked the membranes in 5% non-fat milk at room temperature. They were subsequently incubated with specific primary antibodies at room temperature for 2 hours or overnight at 4 °C. After washing with TBST, the HRP-conjugated secondary antibodies for 1 hour at room temperature. A chemiluminescence detection system visualized and analyzed the bolts (Bio-Rad, Hercules, CA, USA).
Xenograft nude mouse model in vivo
Male BALB-c nude mice (4-week-old) obtained from the Animal Center of the Chinese Academy of Science (Shanghai, China) were the subjects in this study. Subcutaneously, H460 cells (2×106 cells/each) with a 100 µL suspension were injected into the right flanks of the mice. As the tumors grew to roughly 50–100 mm3, the mice were randomly placed into the three following groups: control, ES (25 mg/kg), ES (50 mg/kg) were all intraperitoneally injected. At 3-day intervals, the body weights and tumor sizes of each mouse were monitored, with tumor volume being determined via a formula: length × width 2/2. On day 21, the mice were sacrificed, and we the tumors were frozen in liquid nitrogen or formalin-fixed for further analysis.
Statistical analysis
All experiments were completed in triplicate with data being expressed as mean ± SD. We applied an analysis of variance (ANOVA), and values of *P <0.05, **P<0.01, and ***P<0.001 constituted statistical significance.
Results
ES induces lung carcinoma cell death
We use MTT assay to evaluate the cell viability which were exposed to increasing concentrations of ES for 24 and 48 h. As shown in Figure 1B, H460 and H1975 were significantly induced cell death by ES in both dose-dependent and time-dependent ways.
To confirm that the cell viability of H460 and H1975 were inhibited by the treatment of ES, we observed that the ability of the cells to form colonies was prevented by ES in a concentration-dependent method, which supported an anti-cancer effect of ES (Figure 1C).
To explore the manner of cell death, flow cytometry was carried out to label H460 and H1975 cells that had been subjected to apoptosis, with ES being discovered as inducing apoptosis in H460 and H1975 cells in a dose-dependent manner (Figure 2A).
Figure 2.

ES induces cell apoptosis in human lung carcinoma cells. (A) Apoptosis was analyzed by FACS using an Annexin V-FITC/PI cell apoptosis kit; (B) stained with Hoechst33342 solution and captured by fluorescence microscopy. White arrows indicate condensed nuclei; (C) expression levels of indicated proteins were detected by western blot assay; (D) pretreated with Z-VAD-FMK (50 µmol/L) for 1 h, apoptosis was analyzed by FACS using an Annexin V-FITC/PI cell apoptosis kit. All data are expressed as mean ± SD. *, P <0.05; **, P<0.01; ***, P<0.001 compared to the control group. ES, Ecliptasaponin A; FACS, fluorescence-activated cell sorting; PI, propidium iodide.
To sum up, these findings indicated that ES promoted H460 and H1975 cell death through triggering apoptosis.
Figure 2B (under a fluorescent microscope with 30 µM concentrations after staining with Hoechst 33342) shows H460 and H1975 exhibiting a post-treatment decreased cell count, chromatin condensation, and cell size (31), which all point to the occurrence of apoptosis.
The early stages of apoptosis are characterized by the activation of caspase (32). Cleavage of caspase-3,8,9 was detected by western blot assay in order to determine the involvement of caspase activation in ES-induced apoptosis. Figure 2C shows that, compared to the control group, ES exposure groups significantly elevated the caspase-3,8,9 activities with a dose-dependent increase. To verify these findings, we pretreated H460 and H1975 cells with the pan-caspase inhibitor, Z-VAD-FMK, which functioned as a caspase-activity blocker. Flow cytometry findings give credence to the ability of Z-VAD-FMK to reverse ES-induced cell death (Figure 2D).
Therefore, ES could induce apoptosis in human lung carcinoma H460 and H1975 cells through the caspase-dependent apoptotic pathway.
ES stimulates autophagy in human lung carcinoma cells
Initially, shown in Figure 3A, the increased formation of autophagosomes with the characteristic of expressing the mRFP-GFP-LC3 was visualized stably in the presence of 30µM ES. Then, a hallmark of autophagy (33), LC3B I/II’s protein expression level was identified by the western blot assay. Exposed to the indicated concentrations of ES for 24 hours (Figure 3B), other autophagy-related proteins, such as Beclin-1 and P62/SQSTM1 also saw increased expression. From the findings above, in can be concluded that ES can indeed activate autophagy in human lung carcinoma cells.
Figure 3.

ES induces autophagy, which contributes to cell death. (A) Images of mRFP-GFP-LC3 adenovirus infection monitored the autophagy flux in H460 and H1975 cells under fluorescence microscope (200× magnification); (B) the expression levels of indicated proteins were analyzed by western blot assay; (C) pretreated with 3-MA (5 mmol/L) or CQ (20 µmol/L) for 1 h, apoptosis was analyzed by FACS using an Annexin V-FITC/PI cell apoptosis kit; (D) cells were pretreated with 3-MA (5 mmol/L) or CQ (20 µmol/L) for 1 h. Expression levels of indicated proteins were detected by western blot assay. All data are expressed as mean ± SD. *, P <0.05; **, P<0.01; ***, P<0.001 compared to the control group. ES, Ecliptasaponin A; 3-MA, 3-methyladenine; CQ, chloroquine; FACS, fluorescence-activated cell sorting; PI, propidium iodide.
Apoptosis and autophagy are two approaches that seal cell fate (34); therefore, the link between autophagy and ES-induced apoptosis was further explored. As shown in Figure 3C,D, we cotreated H460 and H1975 cells with ES (30 µM) and autophagy inhibitors, and showed that ES and 3-MA in combination or CQ notably reduced the proportion of apoptotic cells in comparison to those treated with ES by itself. 3-MA blocks autophagy at the early stage, while CQ blocks autophagy at the late stage, however, combination inhibitor led to the inactivation of caspases, as shown as cleaved caspase-3.
Collectively, these data further confirmed autophagy’s pro-apoptotic involvement in human lung carcinoma cells treated with ES.
ASK1/JNK pathway play an important role in apoptosis with ES treated human lung carcinoma cells
MAPK-related pathways were involved in cancer cell survival and cell death and to further clarify the relationship of ES and MAPK pathways in lung cancer, Western blot was used to determine the levels of MAPK-related pathway proteins and their phosphorylated forms. The phosphorylation levels of ASK1, P38, JNK and AKT (S473 and T308) were showed a marked increase while the phosphorylation levels of ERK were declined in response to ES treatment (Figure 4).
Figure 4.

ES induces MAPK pathways in human lung carcinoma cells. Western blot assay for MAPK pathways in ES-treated cells. ES, Ecliptasaponin A; MAPK, mitogen-activated protein kinase.
To verify the roles of p-ASK1 and p-JNK in the apoptotic cells induced by ES, we combined ES with GS-4997 (specific inhibitor for ASK1) or SP600125 (specific inhibitor for JNK) in the treatment. As shown in Figure 5A,B, combination with GS4997 or SP600125 significantly downregulated the number of apoptotic cells, and in Figure 5C,D, the expression levels of cleaved caspase-3 decreased in co-treatment than that of ES treatment alone.
Figure 5.

Reversed the activation of ASK1-JNK pathway can attenuate ES-induced apoptosis and JNK plays a vital role in autophagy induced by ES. (A,B) Pretreated with GS-4997 (GS; 1 µmol/L) and SP600125 (SP; 20 µmol/L) for 1 h, apoptosis was analyzed by FACS using an Annexin V-FITC/PI cell apoptosis kit; (C,D) pretreated with GS-4997 (GS; 1 µmol/L) and SP600125 (SP; 20 µmol/L) for 1 h, expression levels of indicated proteins were detected by western blot assay. All data are expressed as mean ± SD. **, P<0.01; ***, P<0.001 compared to the control group. ASK1, Apoptosis signal-regulating kinase 1; JNK, c-Jun N-terminal kinase; ES, Ecliptasaponin A; FACS, fluorescence-activated cell sorting; PI, propidium iodide.
Furthermore, SP600125 can inhibit autophagy (Figure 5D). Being in combination treatment using ES and SP600125, the expression of LC3B-II was significantly lower than treatment with ES alone. Collectively, these data confirmed that the activation of ASK1/JNK pathways contributed to promoting ES-induced apoptosis in human lung carcinoma cells and activating autophagy may be mediated by JNK pathway.
ES exhibits effective anti-cancer effect in vivo
To verify the part that ES has in regulating lung cancer cell growth in vivo, a xenograft tumor model that injected H460 cells subcutaneously into the right flanks of nude mice was made. In Figure 6A,B, the tumor size was in a visual reduction in the groups of treatment with ES compared to the control group. Consistently, tumor weight and volume of ES-treated at doses of 25 and 50 mg/kg were lower than the control group (Figure 6C,D). However, the body weight was not significantly altered among the three groups (Figure 6E), which indicated no significant biological toxicity of ES in vivo at certain concentration. These data imply that ES can be considered as an anti-cancer drug for lung carcinoma with few side effects.
Figure 6.

ES suppress the growth of tumor xenografts in vivo. (A,B) Image of human lung cancer xenograft from the control group and ES-treated groups; (C) tumor weight of the three groups; (D,E) tumor volume and body weight were measured every three days. All data are expressed as mean ± SD. **, P<0.01; ***, P<0.001 compared to the control group. ES, Ecliptasaponin A.
Discussion
ES is a plant-derived natural product and has been associated with potent anti-cancer effects. In the present study, we first reported how ES inhibits growth in lung cancer cells and elucidated the potential mechanisms involved in its anti-tumor effect. ES inhibits the viability of H460 and H1975 cells in both a time-dependent and dose-dependent manner, and ES regulated the apoptosis, autophagy through MAPKs signaling pathway.
Apoptosis is a form of cell death, the induction of which is presumed to factor prominently in most anti-tumor therapy mechanisms. The execution of apoptosis was caspases, which is from a family of proteases (35). Apoptosis is realized either via the caspase-8-induced death receptor pathway or the caspase-9-activated mitochondrial apoptosis pathway. Subsequently, apoptosis mediated by Bcl-2 family members is induced by caspase-3 or by cleaving its substrates, poly ADP-ribose polymerase (PARP) (36,37). As we all know, the activation of Caspase-3 is led by the activation of Caspase-8 or Caspase-9 and is a crucial link in the process of apoptosis (38,39).
Flow cytometry and western blots results suggested that ES induced apoptosis in H460 and H1975 cells, which increased amount of cleaved caspase-3,8,9. These data suggested that ES induced apoptosis of H460 and H1975 cells through caspase activation.
Autophagy is a conserved pathway characterized by compromised or and deleterious cellular components being taken into autophagosomes, and ending up in the lysosome (40).
Our results found that the expression level of autophagy-related factors, such as LC3, Beclin1 and P62, three prominent autophagy markers involved in autophagy, all increased in a dose-dependent manner. Specifically, the conversion of LC3-I to LC3-II has positive correlations to the number of autophagosomes and the autophagic activity of cells (33,41). Beclin1 functions to form a trimer and induce the aggregation of proteins related to autophagy in the nucleation phase (42). P62 is a marker of autophagic degradation which is consumed during the elongation phase. It is able to hasten the transfer of ubiquitinated substrates to autophagosomes (43). At the most, P62 expression level is negative correlated with the degree of autophagy induction (44).
Compared to treatment with ES alone, the apoptosis was decreased in response to the 3-MA or CQ treatment with ES. These results suggested that although apoptosis and autophagy were induced by ES in lung cancer cells, a pro-apoptotic effect might still be exercised by autophagy.
It is well known that the MAPK pathways figure prominently in intrinsic and extrinsic apoptosis in human lung cancer (45). We supposed that JNK was activated by ASK1, using the S-4997 and SP600125 regulators to verify the dependence of ES treatment effect on the function of ASK1/JNK in lung cancers. In accordance with these findings, JNK is a key play in lung cancer, figuring both in apoptosis and autophagy. As we know, the activation of JNK can stimulate its downstream, such as c-Jun transcription factor, which can result Beclin-1 rising and promoting autophagy further (46).
Here, in lung cancer-bearing nude mice, treatment with ES was shown to cause a clear suppression of tumor growth. Furthermore, treatment with ES at doses of 25 and 50 mg/kg had marked inhibitory effects on the tumor growth of lung cancer in vivo.
Conclusions
To sum up, the present research is the first to illuminate the molecular mechanisms of ES suppressing the growth of lung cancer cells. Briefly, ES induce apoptosis partly through autophagy and the ASK1/JNK signaling pathway (Figure 7). Moreover, ES exert a powerful anti-cancer effect in vivo. These findings demonstrated that ES is a novel and promising clinical molecular targeted therapeutic drug of human lung cancer.
Figure 7.
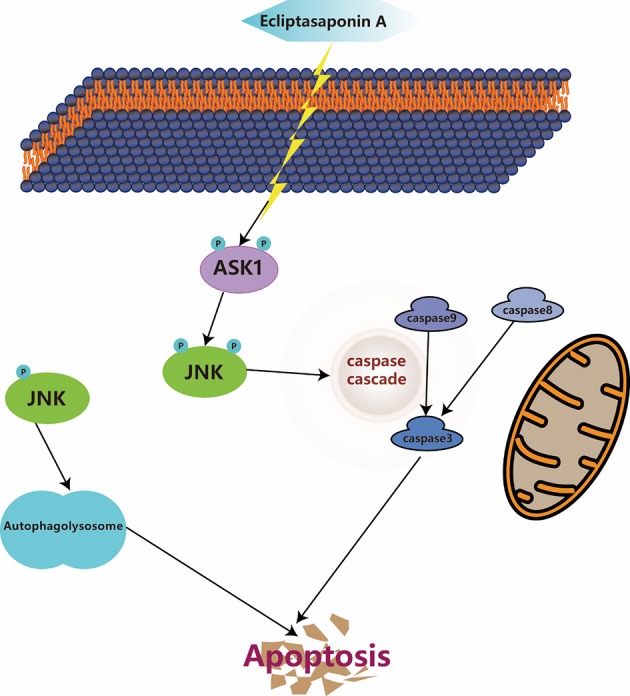
Schematic overview of the hypothesized molecular mechanism underlying of ES effects on lung cancer cells. ES, Ecliptasaponin A.
Acknowledgments
Funding: This study was funded by Key research project of traditional Chinese medicine science and technology plan in Zhejiang Province (2015ZZ007) and Key discipline of traditional Chinese medicine (integrated traditional Chinese and western medicine) in Zhejiang Province (2017YFC0113500). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Ethical Statement: The authors are accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. This study has been approved by the ethics committee of the First Affiliated Hospital of Zhejiang University in China (No. 20191084) and written informed consent was obtained from eligible guardians.
Footnotes
Conflicts of Interest: The authors have no conflicts of interest to declare.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin 2019;69:7-34. 10.3322/caac.21551 [DOI] [PubMed] [Google Scholar]
- 2.Miller KD, Nogueira L, Mariotto AB, et al. Cancer treatment and survivorship statistics, 2019. CA Cancer J Clin 2019;69:363-85. 10.3322/caac.21565 [DOI] [PubMed] [Google Scholar]
- 3.Mignani S, Rodrigues J, Tomas H, et al. Dendrimers in combination with natural products and analogues as anti-cancer agents. Chem Soc Rev 2018;47:514-32. 10.1039/C7CS00550D [DOI] [PubMed] [Google Scholar]
- 4.Fontana F, Raimondi M, Di Domizio A, et al. Unraveling the molecular mechanisms and the potential chemopreventive/therapeutic properties of natural compounds in melanoma. Semin Cancer Biol 2019. [Epub ahead of print]. 10.1016/j.semcancer.2019.06.011 [DOI] [PubMed] [Google Scholar]
- 5.Wattanathamsan O, Hayakawa Y, Pongrakhananon V. Molecular mechanisms of natural compounds in cell death induction and sensitization to chemotherapeutic drugs in lung cancer. Phytother Res 2019. [Epub ahead of print]. 10.1002/ptr.6422 [DOI] [PubMed] [Google Scholar]
- 6.Cao K, Tait SWG. Apoptosis and Cancer: Force Awakens, Phantom Menace, or Both? Int Rev Cell Mol Biol 2018;337:135-52. 10.1016/bs.ircmb.2017.12.003 [DOI] [PubMed] [Google Scholar]
- 7.Burke PJ. Mitochondria, Bioenergetics and Apoptosis in Cancer. Trends Cancer 2017;3:857-70. 10.1016/j.trecan.2017.10.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Croce CM, Reed JC. Finally, An Apoptosis-Targeting Therapeutic for Cancer. Cancer Res 2016;76:5914-20. 10.1158/0008-5472.CAN-16-1248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yang Z, Klionsky DJ. Mammalian autophagy: core molecular machinery and signaling regulation. Curr Opin Cell Biol 2010;22:124-31. 10.1016/j.ceb.2009.11.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.White E, Mehnert JM, Chan CS. Autophagy, Metabolism, and Cancer. Clin Cancer Res 2015;21:5037-46. 10.1158/1078-0432.CCR-15-0490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li J, Hou N, Faried A, et al. Inhibition of autophagy augments 5-fluorouracil chemotherapy in human colon cancer in vitro and in vivo model. Eur J Cancer 2010;46:1900-9. 10.1016/j.ejca.2010.02.021 [DOI] [PubMed] [Google Scholar]
- 12.Katayama M, Kawaguchi T, Berger MS, et al. DNA damaging agent-induced autophagy produces a cytoprotective adenosine triphosphate surge in malignant glioma cells. Cell Death Differ 2007;14:548-58. 10.1038/sj.cdd.4402030 [DOI] [PubMed] [Google Scholar]
- 13.Xiong HY, Guo XL, Bu XX, et al. Autophagic cell death induced by 5-FU in Bax or PUMA deficient human colon cancer cell. Cancer Lett 2010;288:68-74. 10.1016/j.canlet.2009.06.039 [DOI] [PubMed] [Google Scholar]
- 14.Guo WJ, Zhang YM, Zhang L, et al. Novel monofunctional platinum (II) complex mono-Pt induces apoptosis-independent autophagic cell death in human ovarian carcinoma cells, distinct from cisplatin. Autophagy 2013;9:996-1008. 10.4161/auto.24407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yuan J, Kroemer G. Alternative cell death mechanisms in development and beyond. Genes Dev 2010;24:2592-602. 10.1101/gad.1984410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Denton D, Nicolson S, Kumar S. Cell death by autophagy: facts and apparent artefacts. Cell Death Differ 2012;19:87-95. 10.1038/cdd.2011.146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kaza N, Kohli L, Roth KA. Autophagy in brain tumors: a new target for therapeutic intervention. Brain Pathol. 2012;22:89-98. 10.1111/j.1750-3639.2011.00544.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mora R, Regnier-Vigouroux A. Autophagy-driven cell fate decision maker: activated microglia induce specific death of glioma cells by a blockade of basal autophagic flux and secondary apoptosis/necrosis. Autophagy 2009;5:419-21. 10.4161/auto.5.3.7881 [DOI] [PubMed] [Google Scholar]
- 19.Maiuri MC, Zalckvar E, Kimchi A, et al. Self-eating and self-killing: crosstalk between autophagy and apoptosis. Nat Rev Mol Cell Biol 2007;8:741-52. 10.1038/nrm2239 [DOI] [PubMed] [Google Scholar]
- 20.Sun WL, Chen J, Wang YP, et al. Autophagy protects breast cancer cells from epirubicin-induced apoptosis and facilitates epirubicin-resistance development. Autophagy 2011;7:1035-44. 10.4161/auto.7.9.16521 [DOI] [PubMed] [Google Scholar]
- 21.Ichijo H, Nishida E, Irie K, et al. Induction of apoptosis by ASK1, a mammalian MAPKKK that activates SAPK/JNK and p38 signaling pathways. Science 1997;275:90-4. 10.1126/science.275.5296.90 [DOI] [PubMed] [Google Scholar]
- 22.Kanamoto T, Mota M, Takeda K, et al. Role of apoptosis signal-regulating kinase in regulation of the c-Jun N-terminal kinase pathway and apoptosis in sympathetic neurons. Mol Cell Biol 2000;20:196-204. 10.1128/MCB.20.1.196-204.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tobiume K, Matsuzawa A, Takahashi T, et al. ASK1 is required for sustained activations of JNK/p38 MAP kinases and apoptosis. EMBO Rep 2001;2:222-8. 10.1093/embo-reports/kve046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Noguchi T, Ishii K, Fukutomi H, et al. Requirement of reactive oxygen species-dependent activation of ASK1-p38 MAPK pathway for extracellular ATP-induced apoptosis in macrophage. J Biol Chem 2008;283:7657-65. 10.1074/jbc.M708402200 [DOI] [PubMed] [Google Scholar]
- 25.Iriyama T, Takeda K, Nakamura H, et al. ASK1 and ASK2 differentially regulate the counteracting roles of apoptosis and inflammation in tumorigenesis. EMBO J 2009;28:843-53. 10.1038/emboj.2009.32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hayakawa Y, Hirata Y, Nakagawa H, et al. Apoptosis signal-regulating kinase 1 and cyclin D1 compose a positive feedback loop contributing to tumor growth in gastric cancer. Proc Natl Acad Sci U S A 2011;108:780-5. 10.1073/pnas.1011418108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hayakawa Y, Hirata Y, Nakagawa H, et al. Apoptosis signal-regulating kinase 1 regulates colitis and colitis-associated tumorigenesis by the innate immune responses. Gastroenterology 2010;138:1055-67.e1. 10.1053/j.gastro.2009.11.015 [DOI] [PubMed] [Google Scholar]
- 28.Davis RJ. Signal transduction by the JNK group of MAP kinases. Cell 2000;103:239-52. 10.1016/S0092-8674(00)00116-1 [DOI] [PubMed] [Google Scholar]
- 29.Yuan D, Huang S, Berger E, et al. Kupffer Cell-Derived Tnf Triggers Cholangiocellular Tumorigenesis through JNK due to Chronic Mitochondrial Dysfunction and ROS. Cancer Cell 2017;31:771-789.e6. 10.1016/j.ccell.2017.05.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Davies C, Tournier C. Exploring the function of the JNK (c-Jun N-terminal kinase) signalling pathway in physiological and pathological processes to design novel therapeutic strategies. Biochem Soc Trans 2012;40:85-89. 10.1042/BST20110641 [DOI] [PubMed] [Google Scholar]
- 31.Wong RS. Apoptosis in cancer: from pathogenesis to treatment. J Exp Clin Cancer Res 2011;30:87. 10.1186/1756-9966-30-87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hu W, Kavanagh JJ. Anticancer therapy targeting the apoptotic pathway. Lancet Oncol 2003;4:721-9. 10.1016/S1470-2045(03)01277-4 [DOI] [PubMed] [Google Scholar]
- 33.Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagy research. Cell 2010;140:313-26. 10.1016/j.cell.2010.01.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Debnath J, Baehrecke EH, Kroemer G. Does autophagy contribute to cell death? Autophagy 2005;1:66-74. 10.4161/auto.1.2.1738 [DOI] [PubMed] [Google Scholar]
- 35.Budihardjo I, Oliver H, Lutter M, et al. Biochemical pathways of caspase activation during apoptosis. Annu Rev Cell Dev Biol 1999;15:269-90. 10.1146/annurev.cellbio.15.1.269 [DOI] [PubMed] [Google Scholar]
- 36.Green DR. Apoptotic pathways: paper wraps stone blunts scissors. Cell 2000;102:1-4. 10.1016/S0092-8674(00)00003-9 [DOI] [PubMed] [Google Scholar]
- 37.Pop C, Salvesen GS. Human caspases: activation, specificity, and regulation. J Biol Chem 2009;284:21777-81. 10.1074/jbc.R800084200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shi Y. Mechanisms of caspase activation and inhibition during apoptosis. Mol Cell 2002;9:459-70. 10.1016/S1097-2765(02)00482-3 [DOI] [PubMed] [Google Scholar]
- 39.Shalini S, Dorstyn L, Dawar S, et al. Old, new and emerging functions of caspases. Cell Death Differ 2015;22:526-39. 10.1038/cdd.2014.216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mahoney E, Lucas DM, Gupta SV, et al. ER stress and autophagy: new discoveries in the mechanism of action and drug resistance of the cyclin-dependent kinase inhibitor flavopiridol. Blood 2012;120:1262-73. 10.1182/blood-2011-12-400184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Alirezaei M, Flynn CT, Wood MR, et al. Coxsackievirus can exploit LC3 in both autophagy-dependent and -independent manners in vivo. Autophagy 2015;11:1389-407. 10.1080/15548627.2015.1063769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kang R, Zeh HJ, Lotze MT, et al. The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ 2011;18:571-80. 10.1038/cdd.2010.191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sahani MH, Itakura E, Mizushima N. Expression of the autophagy substrate SQSTM1/p62 is restored during prolonged starvation depending on transcriptional upregulation and autophagy-derived amino acids. Autophagy 2014;10:431-41. 10.4161/auto.27344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pankiv S, Clausen TH, Lamark T, et al. P62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem 2007;282:24131-45. 10.1074/jbc.M702824200 [DOI] [PubMed] [Google Scholar]
- 45.Ahmad S, Khan MY, Rafi Z, et al. Oxidation, glycation and glycoxidation-The vicious cycle and lung cancer. Semin Cancer Biol 2018; 49:29-36. 10.1016/j.semcancer.2017.10.005 [DOI] [PubMed] [Google Scholar]
- 46.Lorin S, Pierron G, Ryan KM, et al. Evidence for the interplay between JNK and p53-DRAM signalling pathways in the regulation of autophagy. Autophagy 2010;6:153-4. 10.4161/auto.6.1.10537 [DOI] [PubMed] [Google Scholar]