

Abstract
Myocardial injury induced by diabetes has become an increasing health problem. Chrysophanol (CHR) has been widely studied as a potential treatment for many diseases due to its anti-inflammatory effects, but has not been investigated in regard to diabetes-induced myocardial injury. The present study evaluated the myocardial protective effects of CHR in C57BL/KsJ-db/db diabetic mice. C57BL/KsJ-db/db and C57BLKS/J mice were treated with vehicle, metformin (100 mg/kg/day) or CHR (50 or 100 mg/kg/day) for 28 days. An oral glucose tolerance test was performed to detect blood glucose levels. Blood lipids, triglycerides, total cholesterol, myocardial function-associated enzymes, namely creatine kinase (CK) and lactate dehydrogenase (LDH), and insulin levels were analyzed. TNF-α, interleukin (IL)-1β and IL-6 inflammatory cytokine levels in serum and myocardial tissues were determined by ELISA. Expression of silent information regulator l (SIRT1) and high mobility group box 1/NF-κB pathway-associated proteins in myocardial tissues were measured by western blot analysis and immunohistochemistry. CHR treatment at both concentrations markedly decreased blood lipid and serum insulin levels, and inhibited the myocardial enzymes CK and LDH. CHR also significantly ameliorated the cardiac pathological changes in diabetic mice. The inflammatory cytokine levels that were increased in C57BL/KsJ-db/db diabetic mice were downregulated by CHR treatment. CHR also increased SIRT1 protein expression and inhibited activation of the HMGB1/NF-κB pathway. In conclusion, the present study indicates that CHR effectively protected against diabetic myocardial injury via regulation of SIRT1 and the HMGB1/NF-κB signaling pathway.
Keywords: chrysophanol, myocardial injury, inflammation, silent information regulator l, high mobility group box 1/NF-κB
Introduction
Diabetes mellitus (DM) is a metabolic disease characterized by chronic hyperglycemia due to defects in insulin secretion and/or action (1). The global number of patients with DM was ~382 million in 2013, and is predicted to reach ~592 million by 2035 (2). DM causes abnormal metabolism of sugar, fat and protein, which can lead to serious complications such as renal failure, ketoacidosis, peripheral neuropathy, blindness and cerebral arteritis (3,4). Myocardial injury is a serious complication of DM that is attracting research interest (5,6). Myocardial injury induced by DM is primarily caused by inflammatory reactions and hepatic lipid accumulation (7). Myocardial inflammation is a key mechanism of the condition. Notably, anti-inflammatory drugs have protective effects on diabetic myocardial injury (8), and may therefore be an effective treatment.
Silent information regulator l (SIRT1) is a type of nicotinamide adenosine dinucleotide dependent deacetylase, which affects a variety of cellular processes and has an important role in tissue damage and repair (9). Recent studies have demonstrated that SIRT1 plays an important role in inflammation and autoimmunity (10,11). High mobility group box 1 (HMGB1) is a highly conserved nucleoprotein, which affects a variety of biological processes, including inflammatory diseases (12). HMGB1 in extracellular fluid also serves a role as a damage-associated molecular pattern molecule in the process of inflammation and cell migration (13). HMGB1 serves a role in the release of pro-inflammatory cytokines (14). Under inflammatory conditions, HMGB1 is passively released or actively secreted into the extracellular environment from the affected monocytes/macrophages (14), while NF-κB is activated and takes part in the inflammatory response (15). It has been identified that HMGB1 participates in various pathophysiological signaling pathways triggered by the diabetic environment; therefore, it is a promising molecular target for the treatment of DM (16).
Chrysophanol (CHR), an anthraquinone isolated from rhubarb, belongs to the anthraquinone family, which also contains emodin, aloe emodin, rhein and physcion (17,18). CHR has been demonstrated to have multiple beneficial pharmacological effects, such as anti-inflammatory (19), antitumor (20), antiviral (21) and antiproliferative effects (22). To the best of our knowledge, there has been no previous research into whether CHR has therapeutic effects on diabetic myocardial injury in db/db mice. Therefore, the aim of the present study was to evaluate the ability of CHR to attenuate diabetic myocardial injury.
Materials and methods
Animal study
A total of 48 male spontaneously diabetic mice (C57BL/KsJ-db/db mice; age, 6–8 weeks; weight, 35–40 g) and 12 male wild-type C57BLKS/J mice (age, 6–8 weeks; weight, 20–22 g) were obtained from the Model Animal Research Center of Nanjing University. Mice were maintained under standard laboratory condition, with free access to food and water and housed prior to experiments in an animal room under standard conditions (23±2°C; 60±10% humidity; 12 h light/dark cycle). All the experimental procedures were approved by and performed in accordance with Nantong Medical University.
Experimental design
Following 1 week of feeding and adaptation, the mice were divided randomly into five groups (each n=12): i) Wild, in which wild-type C57BLKS/J mice were orally administered normal saline; ii) C57BL/KsJ-db/db control group, in which C57BL/KsJ-db/db mice were orally administered normal saline; iii) metformin, in which C57BL/KsJ-db/db mice were intragastrically administered metformin (Sigma-Aldrich; Merck KGaA; 100 mg/kg/day); iv) CHR, 50 mg/kg, in which C57BL/KsJ-db/db mice were intragastrically administered CHR (50 mg/kg/day; 98% purity; National Institutes for Food and Drug Control); and v) CHR, 100 mg/kg/day, in which C57BL/KsJ-db/db mice were administered CHR by gavage at a dose of 100 mg/kg/day. All treatments were provided for 28 consecutive days. The CHR concentrations of 50 and 100 mg/kg/day were selected for use in the present study according to a preliminary experiment (data not shown). At the conclusion of the experiment, the mice were anesthetized with ketamine/xylazine (100 and 10 mg/kg respectively, 0.1 ml/25 g body weight) by intraperitoneal injection, and then ~0.8-ml blood samples were collected from the orbital plexus of the eyes. The blood samples were centrifuged at 3,000 × g for 10 min at 4°C and the serum was collected. Blood serum was collected for subsequent hematological or biochemical assays. Death of the mice was verified by the complete cessation of the heartbeat and breathing, and disappearance of reflexes. In addition, heart tissue was immediately removed, rinsed with physiological saline solution and then stored at −80°C prior to further analysis. Sections of the heart were fixed in 10% (v/v) neutral buffered formalin for hematoxylin and eosin (H&E) staining.
Oral glucose tolerance test (OGTT)
On day 29 day after the initiation of treatment, the OGTT was performed. Mice were fasted overnight and subsequently received glucose (2 g/kg) by gavage at 8:00 a.m. Blood glucose concentrations at different time points (0, 30, 60, 90 and 120 min) following glucose administration were evaluated using a glucose analyzer (SureStep®; Lifescan, Inc.).
Biochemical measurements
Creatine kinase (CK), lactate dehydrogenase (LDH), total triglyceride (TG) and total cholesterol (TC) levels in the serum were measured using commercially available standard kits (cat. nos. A032, A020-2, A110-1 and A111-1, respectively; Nanjing Jiancheng Bioengineering Institute Co., Ltd.) according to the manufacturer's instructions. The insulin concentration in serum was determined using an insulin ELISA kit (cat. no. 7544-MR; R&D Systems, Inc.).
Determination of tumor necrosis factor (TNF)-α, interleukin (IL)-1β and IL-6 levels in serum and heart tissues
Levels of cytokines in the serum and heart, namely IL-6, IL-1β and TNF-α, were analyzed using commercially available ELISA kits (cat. nos. M6000B, MLB00C and MTA00B, respectively; R&D Systems, Inc.) in accordance with the manufacturer's instructions. The optical density (OD) of each well was read at 450 nm, and the concentration of the inflammatory cytokine was quantified with reference to a standard curve.
Histological examination
Heart tissues were carefully removed and fixed in 10% (v/v) formalin at room temperature for 48 h, then embedded in paraffin wax. Samples were cut into 4-µm sections and stained with H&E (Nanjing Jiancheng Bioengineering Institute). Following dehydration with 80, 90 and 100% ethanol and n-butanol, the myocardial tissue was waxed in a 60°C wax box and then embedded in paraffin. Tissue sections (5-µm) were dried at 45°C and obtained from each paraffin block. The sections were heated at 60°C for 1 h and dewaxed with xylene. Following hydration, the sections were stained with 0.5% H&E at room temperature for 5 min, dehydrated with gradient ethanol, cleared with xylene and mounted with neutral gum. Optical microscopy (Olympus Corporation) was used to examine pathological changes of the heart tissue (magnification, ×200).
Immunohistochemistry
The expressions of HMGB1 and phosphorylated (p)-NF-κB p65 in the heart tissues were evaluated using immunohistochemistry staining. The heart tissues were embedded in paraffin and sectioned. Then, the paraffin sections were deparaffinized in xylene, rehydrated by ethanol and incubated with 3% hydrogen peroxide. Heart tissues samples were blocked at room temperature with 3% BSA (Beijing Solarbio Science & Technology Co., Ltd.) and incubated with HMGB1(1:1,00, cat. no. ab79823; Abcam) and phosphorylated (p)-NF-κB p65 (1:1,00, cat. no. ab86299; Abcam) at 4°C overnight. Samples were then washed three times with PBS, treated with horseradish peroxidase goat anti-rabbit IgG secondary antibody (1:200; cat. no. WLA037a; Wanleibio Co., Ltd.) for 20 min at 37°C and washed three times with PBS. Samples were then stained at room temperature for 2 min with 0.05% 3-3′diaminobenzidine (DAB) and a total of 10 fields were randomly selected from each sample were observed using a microscope (magnification, ×200, Olympus Corporation).
Western blot analysis
Heart tissue was homogenized, washed with PBS and lysed in radioimmunoprecipitation assay buffer (Beyotime Institute of Biotechnology). The protein concentration was determined using an Enhanced Bicinchoninic Acid Protein Assay kit (Beyotime Institute of Biotechnology). An equal amount of protein (~50 µg) was loaded per lane and separated via SDS-PAGE (Mini-Protean 3®; Bio-Rad Laboratories, Inc.) on a 10% gel. Proteins were then transferred onto a polyvinylidene difluoride membrane (EMD Millipore; Merck KGaA) and blocked with 5% skim milk at room temperature for 2 h. Membranes were incubated overnight at 4°C with primary antibodies against SIRT1 (1:1,000; cat. no. ab110304; Abcam), HMGB1 (1:1,000; cat. no. ab79823; Abcam), NF-κB p65 (1:1,000; cat. no. ab16502; Abcam), phosphorylated (p)-NF-κB p65 (1:1,000; cat. no. ab86299; Abcam), NF-κB inhibitor-α (IκBα; 1:1,000; cat. no. ab32518; Abcam), p-IκBα (1:1,000; cat. no. ab24783; Abcam) and GAPDH (1:2,000; cat. no. ab181602; Abcam). Membranes were then incubated with secondary antibody, horseradish peroxidase-labeled mouse anti-rabbit IgG (1:5,000; cat. no. 5127, Cell Signaling Technology, Inc.) or horseradish peroxidase-labeled rabbit anti-mouse IgG (1:5,000; cat. no. 58802, Cell Signaling Technology, Inc.), at room temperature for 2 h. Protein bands were visualized using an enhanced chemoluminescence staining detection kit (Bio-Rad Laboratories, Inc.) and densitometry was performed with Bandscan 5.0 software (Glycomix Ltd.). The expression of target protein was normalized to that of GAPDH.
Statistical analysis
Data are expressed as the mean ± standard deviation. Differences between multiple groups were evaluated by one-way analysis of variance with Tukey's multiple comparison test as the post hoc test using GraphPad Prism software 6.0 (GraphPad Software, Inc.). P<0.05 was considered to indicate a statistically significant difference.
Results
CHR reduces blood glucose levels in a diabetic mouse model
The C57BL/KsJ-db/db group displayed significantly higher blood glucose levels compared with the wild group (Fig. 1). Following treatment with CHR (50 and 100 mg/kg), the blood glucose levels of the C57BL/KsJ-db/db mice were significantly lower than those of the C57BL/KsJ-db/db mice treated with saline. Metformin (100 mg/kg) similarly decreased the blood glucose concentration of the C57BL/KsJ-db/db mice.
Figure 1.
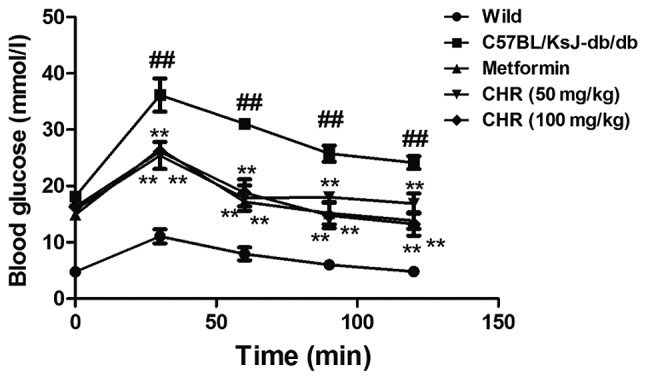
Effect of CHR on the blood glucose levels of diabetic mice determined using the oral glucose tolerance test. ##P<0.01 vs. wild group; **P<0.01 vs. C57BL/KsJ-db/db group. Wild, wild-type C57BLKS/J mice; C57BL/KsJ-db/db, spontaneously diabetic mice; CHR, chrysophanol.
CHR attenuates myocardial pathological changes in a diabetic mouse model
In the wild group, the myocardial cell membranes were intact, the myofibril structure was balanced, and adjacent myofibrils were continuous (Fig. 2). By contrast, a large number of inflammatory cells were observed in the hearts of the C57BL/KsJ-db/db group, myocardial cells were swollen or denatured, myocardial necrosis was evident, and no striations were visible (Fig. 2). CHR or metformin treatment markedly attenuated the pathological changes observed in the diabetic mouse model, especially in the high dose CHR group (Fig. 2).
Figure 2.

Effects of CHR on the heart tissue of diabetic mice. Hematoxylin and eosin staining (magnification, ×200) was performed to study inflammatory cells. Wild, wild-type C57BLKS/J mice; C57BL/KsJ-db/db, spontaneously diabetic mice; CHR, chrysophanol.
CHR reduces CK, LDH, insulin, TG and TC levels in serum
The serum TG and TC levels of C57BL/KsJ-db/db mice were significantly higher compared with those of the wild group (Fig. 3). Treatment with CHR (50 or 100 mg/kg) or metformin significantly attenuated the DM-induced increases in TG and TC levels. To explore the influence of CHR on heart function, the CK and LDH activities and serum insulin levels of the mice were also measured. In the C57BL/KsJ-db/db group, the serum insulin, CK and LDH levels were significantly increased compared with those of the wild group (Fig. 3). Treatment with CHR (50 or 100 mg/kg) or metformin significantly attenuated the DM-induced increases in insulin, CK and LDH levels (Fig. 3).
Figure 3.

Effect of CHR on TC, TG, insulin, CK and LDH in the serum of diabetic mice. ##P<0.01 vs. wild group; **P<0.01 vs. C57BL/KsJ-db/db group. Wild, wild-type C57BLKS/J mice; C57BL/KsJ-db/db, spontaneously diabetic mice; CHR, chrysophanol; TC, total cholesterol; TG total triglyceride; CK, creatine kinase; LDH, lactate dehydrogenase.
CHR reduces inflammatory cytokine levels in serum and the heart
In order to explore the influence of CHR on the inflammatory response, TNF-α, IL-6 and IL-1β levels in the serum and heart tissues of the mice were determined. The results demonstrated that inflammatory cytokine levels in the serum and heart tissues of C57BL/KsJ-db/db mice were significantly higher compared with those of the wild-type control mice (Fig. 4). However, treatment with CHR (50 or 100 mg/kg) or metformin significantly suppressed the levels of inflammatory cytokines in the serum and heart tissues of the C57BL/KsJ-db/db mice (Fig. 4).
Figure 4.

Effect of CHR on TNF-α, IL-6 and IL-1β levels in diabetic mice. Inflammatory cytokine levels in the (A) serum and (B) heart tissue. ##P<0.01 vs. wild group; **P<0.01 vs. C57BL/KsJ-db/db group. Wild, wild-type C57BLKS/J mice; C57BL/KsJ-db/db, spontaneously diabetic mice; CHR, chrysophanol; TNF-α, tumor necrosis factor-α; IL, interleukin.
CHR increases SIRT1 expression and inhibits the HMGB1/NF-κB signaling pathway in a diabetic mouse model
SIRT1 and the HMGB1/NF-κB signaling pathway participate in the regulation of inflammation (23,24). Western blot analysis was used to explore whether SIRT1 and the HMGB1/NF-κB signaling pathway participate in the effect of CHR on heart inflammation. HMGB1, p-NF-κB p65 and p-IκB were significantly upregulated while SIRT1 was downregulated in the hearts of C57BL/KsJ-db/db mice (Fig. 5). CHR (50 and 100 mg/kg) and metformin (100 mg/kg) significantly attenuated these changes (Fig. 5). Additionally, immunohistochemistry demonstrated that the expression of HMGB1 and p-NF-κB in heart tissue was higher in C57BL/KsJ-db/db mice than in their wild-type counterparts, and that treatment with 100 mg/kg CHR markedly decreased HMGB1 and p-NF-κB expression in C57BL/KsJ-db/db mice (Fig. 6).
Figure 5.

Effect of CHR on SIRT1 and the HMGB1/NF-κB signaling pathway in diabetic mice. Representative western blots and quantification of protein levels. ##P<0.01 vs. wild group; **P<0.01 vs. C57BL/KsJ-db/db group. Wild, wild-type C57BLKS/J mice; C57BL/KsJ-db/db, spontaneously diabetic mice; CHR, chrysophanol; SIRT1, silent information regulator l; HMGB1, high mobility group box 1; p-, phosphorylated; P65, NF-κB p65; IκBα, NF-κB inhibitor-α.
Figure 6.

Effects of CHR on the HMGB1/NF-κB signaling pathway in diabetic mice. Immunohistochemical staining of HMGB1 and p-NF-κB (magnification, ×200). Wild, wild-type C57BLKS/J mice; C57BL/KsJ-db/db, spontaneously diabetic mice; CHR, chrysophanol; HMGB1, high mobility group box 1; p-P65-, phosphorylated NF-κB p65.
Discussion
DM is characterized by hyperglycemia, insulin deficiency, insulin resistance and pathology in many organs, including nerves, the liver and glomeruli (25). DM accelerates atherosclerotic diseases and affects the heart, brain and lower limb arteries (26). DM and insulin resistance are important factors in DM-induced heart injury. In the present study, spontaneously diabetic mice demonstrated abnormal OGTT performance and lipid profiles, and elevated serum insulin, CK and LDH levels. The results indicated that CHR and metformin attenuated the heart injury of the diabetic mice. No significant dose-dependent effects of CHR treatment were observed when comparing low dose (50 mg/kg) and high dose (100 mg/kg) groups, which might be due to the 28-day CHR treatment duration being too short (27). Future study will involve increasing the duration of observation to 8–20 weeks to better analyze the dose-dependent effects of CHR. The blood glucose results were consistent with the observed heart histological changes, which confirmed the beneficial effect of CHR on heart injury. Therefore, the present findings suggest that CHR is an effective cardiac protection agent. In order to elucidate the protective effect of CHR on diabetic heart injury, the possible molecular mechanism was investigated. The present study determined that the TNF-α, IL-6 and IL-1β levels in the serum and heart tissue of C57BL/KsJ-db/db mice were higher than those in the wild-type mice, which suggests that inflammation was increased during DM. Treatment with CHR significantly suppressed the inflammatory cytokine levels in the serum and heart tissue of the C57BL/KsJ-db/db mice. Inflammatory cytokines have important roles in the initiation and development of heart injury (28), as they affect the infiltration of white blood cells in the liver and amplify the damage to the heart (29). IL-6 has a pathological effect on chronic inflammation, myocardial infarction and rheumatoid arthritis (30). The present study demonstrated that CHR has the ability to reduce these cytokine levels, which suggests that the anti-inflammatory effects of CHR may participate in protecting the heart.
In order to explore the anti-inflammatory molecular mechanism of CHR on heart injury, the roles of inflammation-related SIRT1 and the HMGB1/NF-κB signaling pathway were investigated. HMGB1 is a key factor during inflammation in aseptic and infection-associated reactions (31). It has common characteristics with cytokines, acts on the surface receptors of immune cells, and induces the expression of inflammatory factors and the further release of HMGB1, thereby promoting an inflammatory cascade reaction (32). When heart injury occurs, HMGB1 is passively released from damaged cells or actively secreted from activated immune cells. HMGB1 activates NF-κB and induces the expression of pro-inflammatory genes (33). It has been demonstrated that SIRT1 inhibits the transcriptional activity of NF-κB through deacetylation of the p65 subunit, thereby reducing the production and activation of inflammatory cytokines (34). Therefore, CHR might exert beneficial activities by attenuating inflammatory responses through downregulation of the HMGB1/NF-κB pathway via the upregulation of SIRT1. Decreased inflammatory responses promote the activation and recruitment of inflammatory cells, leading to the initiation of inflammation (35). In the present study, western blot analysis demonstrated that HMGB1, p-NF-κB p65 and p-IκB protein levels in the heart tissue of C57BL/KsJ-db/db mice were significantly increased compared with those in wild-type mice, and SIRT1 expression levels in the heart tissue of C57BL/KsJ-db/db mice were lower compared with those in wild-type mice. CHR intervention significantly reversed these changes, suggesting that the anti-inflammatory effect of CHR may occur via regulation of SIRT1 and inhibition of the HMGB1/NF-κB pathway.
In conclusion, the present study successfully demonstrated the protective effect of CHR against DM-induced heart damage. The possible mechanisms underlying the protective effect of CHR may be associated with the attenuation of inflammation of the heart via regulation of SIRT1 and inhibition of the HMGB1/NF-κB signaling pathway. The present findings provide preliminary evidence suggesting the potential of CHR as a therapeutic drug for DM-induced heart injury.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the present study are available from the corresponding author on reasonable request.
Authors' contributions
PX, JZ, AZ and LL contributed to conception and design of the study. HC, WT, NZ, ZY and MG contributed to acquisition, analysis and interpretation of the data and writing the manuscript. PX, JZ, MG and QW carried out the animal experiments. All authors read and approved the final manuscript.
Ethics approval and consent to participate
All the experimental procedures were approved by and perfor-med in accordance with Nantong Medical University.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
- 1.Mäkimattila S, Virkamäki A, Groop PH, Cockcroft J, Utriainen T, Fagerudd J, Yki-Järvinen H. Chronic hyperglycemia impairs endothelial function and insulin sensitivity via different mechanisms in insulin-dependent diabetes mellitus. Circulation. 1996;94:1276–1282. doi: 10.1161/01.CIR.94.6.1276. [DOI] [PubMed] [Google Scholar]
- 2.Guariguata L, Whiting DR, Hambleton I, Beagley J, Linnenkamp U, Shaw JE. Global estimates of diabetes prevalence for 2013 and projections for 2035. Diabetes Res Clin Pract. 2014;103:137–149. doi: 10.1016/j.diabres.2013.11.002. [DOI] [PubMed] [Google Scholar]
- 3.Brownlee M. The pathobiology of diabetic complications: A unifying mechanism. Diabetes. 2005;54:1615–1625. doi: 10.2337/diabetes.54.6.1615. [DOI] [PubMed] [Google Scholar]
- 4.Giacco F, Brownlee M. Oxidative stress and diabetic complications. Circ Res. 2010;107:1058–1070. doi: 10.1161/CIRCRESAHA.110.223545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Adeyemi DO, Ukwenya VO, Obuotor EM, Adewole SO. Anti-hepatotoxic activities of Hibiscus sabdariffa L. in animal model of streptozotocin diabetes-induced liver damage. BMC Complement Altern Med. 2014;14:277. doi: 10.1186/1472-6882-14-277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dias AS, Porawski M, Alonso M, Marroni N, Collado PS, González-Gallego J. Quercetin decreases oxidative stress, NF-kappaB activation, and iNOS overexpression in liver of streptozotocin-induced diabetic rats. J Nutr. 2005;135:2299–2304. doi: 10.1093/jn/135.10.2299. [DOI] [PubMed] [Google Scholar]
- 7.Matafome P, Nunes E, Louro T, Amaral C, Crisóstomo J, Rodrigues L, Moedas AR, Monteiro P, Cipriano A, Seiça R. A role for atorvastatin and insulin combination in protecting from liver injury in a model of type 2 diabetes with hyperlipidemia. Naunyn Schmiedebergs Arch Pharmacol. 2009;379:241–251. doi: 10.1007/s00210-008-0363-y. [DOI] [PubMed] [Google Scholar]
- 8.Wellen KE, Hotamisligil GS. Inflammation, stress, and diabetes. J Clin Invest. 2005;115:1111–1119. doi: 10.1172/JCI25102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Finkel T, Deng CX, Mostoslavsky R. Recent progress in the biology and physiology of sirtuins. Nature. 2009;460:587–591. doi: 10.1038/nature08197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lai T, Wen X, Wu D, Su G, Gao Y, Chen C, Wu W, Lv Y, Chen Z, Lv Q, et al. SIRT1 protects against urban particulate matter-induced airway inflammation. Int J Chron Obstruct Pulmon Dis. 2019;14:1741. doi: 10.2147/COPD.S202904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lee JH, Moon JH, Lee YJ, Park SY. SIRT1, a Class III histone deacetylase, regulates LPS-induced inflammation in human keratinocytes and mediates the anti-inflammatory effects of hinokitiol. J Invest Dermatol. 2017;137:1257–1266. doi: 10.1016/j.jid.2016.11.044. [DOI] [PubMed] [Google Scholar]
- 12.Harris HE, Andersson U, Pisetsky DS. HMGB1: A multifunctional alarmin driving autoimmune and inflammatory disease. Nat Rev Rheumatol. 2012;8:195–202. doi: 10.1038/nrrheum.2011.222. [DOI] [PubMed] [Google Scholar]
- 13.Yang H, Wang H, Chavan SS, Andersson U. High mobility group box protein 1 (HMGB1): The prototypical endogenous danger molecule. Mol Med 1(Suppl 21) 2015:S6–S12. doi: 10.2119/molmed.2015.00087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kornblit B, Munthe-Fog L, Madsen HO, Strøm J, Vindeløv L, Garred P. Association of HMGB1 polymorphisms with outcome in patients with systemic inflammatory response syndrome. Crit Care. 2008;12:R83. doi: 10.1186/cc6935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xueyang D, Zhanqiang M, Chunhua M, Kun H. Fasudil, an inhibitor of Rho-associated coiled-coil kinase, improves cognitive impairments induced by smoke exposure. Oncotarget. 2016;7:78764–78772. doi: 10.18632/oncotarget.12853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Komers R. Rho kinase inhibition in diabetic kidney disease. Br J Clin Pharmacol. 2013;76:551–559. doi: 10.1111/bcp.12196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lu CC, Yang JS, Huang AC, Hsia TC, Chou ST, Kuo CL, Lu HF, Lee TH, Wood WG, Chung JG. Chrysophanol induces necrosis through the production of ROS and alteration of ATP levels in J5 human liver cancer cells. Mol Nutr Food Res. 2010;54:967–976. doi: 10.1002/mnfr.200900265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kim SJ, Kim MC, Lee BJ, Park DH, Hong SH, Um JY. Anti-Inflammatory activity of chrysophanol through the suppression of NF-kappaB/caspase-1 activation in vitro and in vivo. Molecules. 2010;15:6436–6451. doi: 10.3390/molecules15096436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wen Q, Mei L, Ye S, Liu X, Xu Q, Miao J, Du S, Chen D, Li C, Li H. Chrysophanol demonstrates anti-inflammatory properties in LPS-primed RAW 264.7 macrophages through activating PPAR-γ. Int Immunopharmacol. 2018;56:90–97. doi: 10.1016/j.intimp.2018.01.023. [DOI] [PubMed] [Google Scholar]
- 20.Shiezadeh F, Mousavi SH, Amiri MS, Iranshahi M, Tayarani-Najaran Z, Karimi G. Cytotoxic and apoptotic potential of rheum turkestanicum janisch root extract on human cancer and normal cells. Iran J Pharm Res. 2013;12:811–819. [PMC free article] [PubMed] [Google Scholar]
- 21.Chang SJ, Huang SH, Lin YJ, Tsou YY, Lin CW. Antiviral activity of Rheum palmatum methanol extract and chrysophanol against Japanese encephalitis virus. Arch Pharm Res. 2014;37:1117–1123. doi: 10.1007/s12272-013-0325-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhi-Yun LI, Ming Z, Wei JI, et al. Protective effect of chrysophanol on hypoxic injury of rat adrenal medulla pheochromocytoma cells. Chin J Cerebrovascular Dis. 2012;9:418–427. (In Chinese) [Google Scholar]
- 23.Zhang J, Yang S, Chen F, Li H, Chen B. Ginkgetin aglycone ameliorates LPS-induced acute kidney injury by activating SIRT1 via inhibiting the NF-κB signaling pathway. Cell Biosci. 2017;7:44. doi: 10.1186/s13578-017-0173-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Qi Z, Zhang Y, Qi S, Ling L, Gui L, Yan L, Lv J, Li Q. Salidroside inhibits HMGB1 acetylation and release through upregulation of SirT1 during inflammation. Oxid Med Cell Longev. 2017;2017:9821543. doi: 10.1155/2017/9821543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brownlee M. Biochemistry and molecular cell biology of diabetic complications. Nature. 2001;414:813–820. doi: 10.1038/414813a. [DOI] [PubMed] [Google Scholar]
- 26.Gerrity RG, Natarajan R, Nadler JL, Kimsey T. Diabetes-induced accelerated atherosclerosis in swine. Diabetes. 2001;50:1654–1665. doi: 10.2337/diabetes.50.7.1654. [DOI] [PubMed] [Google Scholar]
- 27.He Q, Pu J, Yuan A, Yao T, Ying X, Zhao Y, Xu L, Tong H, He B. Liver X receptor agonist treatment attenuates cardiac dysfunction in type 2 diabetic db/db mice. Cardiovasc Diabetol. 2014;13:149. doi: 10.1186/s12933-014-0149-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bujak M, Frangogiannis NG. The role of IL-1 in the pathogenesis of heart disease. Arch Immunol Ther Exp (Warsz) 2009;57:165–176. doi: 10.1007/s00005-009-0024-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ohsuzu F. The roles of cytokines, inflammation and immunity in vascular diseases. J Atheroscler Thromb. 2004;11:313–321. doi: 10.5551/jat.11.313. [DOI] [PubMed] [Google Scholar]
- 30.Tanaka T, Narazaki M, Kishimoto T. IL-6 in inflammation, immunity, and disease. Cold Spring Harb Perspect Biol. 2014;6:a016295. doi: 10.1101/cshperspect.a016295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yang H, Tracey KJ. Targeting HMGB1 in inflammation. Biochim Biophys Acta. 2010;1799:149–156. doi: 10.1016/j.bbagrm.2009.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Andersson U, Tracey KJ. HMGB1 Is a therapeutic target for sterile inflammation and infection. Annu Rev Immunol. 2011;29:139–162. doi: 10.1146/annurev-immunol-030409-101323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lotze MT, Tracey KJ. High-mobility group box 1 protein (HMGB1): Nuclear weapon in the immune arsenal. Nat Rev Immunol. 2005;5:331–342. doi: 10.1038/nri1594. [DOI] [PubMed] [Google Scholar]
- 34.Kauppinen A, Suuronen T, Ojala J, Kaarniranta K, Salminen A. Antagonistic crosstalk between NF-kB and SIRT1 in the regulation of inflammation and metabolic disorders. Cell Signal. 2013;25:1939–1948. doi: 10.1016/j.cellsig.2013.06.007. [DOI] [PubMed] [Google Scholar]
- 35.Salem ML, Hossain MS, Nomoto K. Mediation of the immunomodulatory effect of beta-estradiol on inflammatory responses by inhibition of recruitment and activation of inflammatory cells and their gene expression of TNF-alpha and IFN-gamma. Int Arch Allergy Immunol. 2000;121:235–245. doi: 10.1159/000024323. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets used and/or analyzed during the present study are available from the corresponding author on reasonable request.