

Abstract
Allosteric kinase inhibitors represent a promising new therapeutic strategy for targeting kinases harboring oncogenic driver mutations in cancers. Here, we report the discovery, optimization, and structural characterization of allosteric mutant-selective EGFR inhibitors comprising a 5,10-dihydro-11H-dibenzo[b,e][1,4]diazepin-11-one scaffold. Our structure-based medicinal chemistry effort yielded an inhibitor (3) of the EGFR(L858R/T790M) and EGFR(L858R/T790M/C797S) mutants with an IC50 of ∼10 nM and high selectivity, as assessed by kinome profiling. Further efforts to develop allosteric dibenzodiazepinone inhibitors may serve as the basis for new therapeutic options for targeting drug-resistant EGFR mutations.
Keywords: EGFR, kinase inhibitor, allosteric inhibitor, dibenzodiazepinone, mutant-selective, non-small cell lung cancer
Activating mutations of the epidermal growth factor receptor (EGFR), e.g., L858R and in-frame exon 19 deletions, give rise to non-small cell lung cancer and confer sensitivity to EGFR-targeted tyrosine kinase inhibitors (TKIs) such as gefitinib and erlotinib.1,2 Acquired resistance to these TKIs occurs predominantly by the acquisition of a T790M “gatekeeper” mutation, which enhances ATP binding to the EGFR kinase.3,4 Selective inhibition of T790M-positive tumors is accomplished with third-generation TKIs (e.g., WZ4002, osimertinib) that irreversibly form a covalent bond with C797 at the edge of the ATP binding site.5,6 Tumors acquire resistance to these inhibitors, in ∼20–25% of cases, by the further acquisition of a C797S mutation rendering these inhibitors ineffective by preventing the formation of the potency-conferring covalent bond.7,8 Therefore, continued development of inhibitors is required to address mutations that confer resistance to first- and third-generation EGFR-targeting TKIs.
Accordingly, we sought to discover small-molecule inhibitors of activating EGFR mutations that act through an alternative, allosteric binding mode.9 To that end, we recently reported a mutant-selective EGFR allosteric inhibitor (EAI001Figure 1) that binds a pocket adjacent to the ATP-binding site, affording exquisite selectivity for the mutant kinase compared to WT EGFR.10 Initial optimization of this hit produced EAI045 (Figure 1), which exhibited 1000-fold selectivity for L858R/T790M EGFR compared to WT. However, EAI045 showed minimal cellular activity owing to this compound’s inability to bind the allosteric pocket of the active state, where the αC-helix is positioned inward, on the receiver subunit of the active EGFR asymmetric kinase dimer.11EAI045 was rendered effective at regressing L858R/T790M/C797S tumors in vivo upon cotreatment with the EGFR monoclonal antibody cetuximab, which disrupts the formation of active EGFR dimers.10 Further efforts produced a more potent analog (JBJ-04-125-02, Figure 1), which incorporates a phenylpiperazine on the C6 position of the EAI045 isoindolinone moiety and is capable of acting as a single-agent inhibitor in EGFR L858R/T790M/C797S cells and genetically engineered mouse models.12 Furthermore, dual targeting of EGFR with JBJ-04-125-02 and osimertinib was found to be more effective in vitro and in vivo than either single agent alone.12
Figure 1.

Chemical structures of EAI001, EAI045, JBJ-04-125-02, and 5,10-dihydro-11H-dibenzo[b,e][1,4]diazepin-11-one allosteric EGFR inhibitors described in this work.
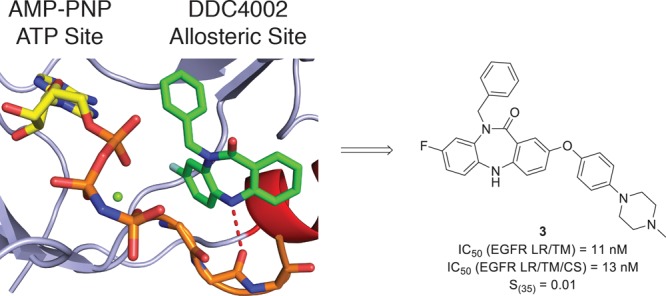
In this study, we sought to evaluate a second series of allosteric mutant-selective EGFR inhibitors. As previously described, EAI001 was discovered in a screen for mutant-selective inhibitors of EGFR(L858R/T790M).10,13 The same screen also yielded EAI002 comprising a 5,10-dihydro-11H-dibenzo[b,e][1,4]diazepin-11-one scaffold (Figure 1), which selectively inhibited L858R/T790M with a biochemical IC50 of 52 nM compared to >1000 nM for WT. Subtle optimization of this hit compound via a fluorine shift afforded DDC4002 (Figure 1), which exhibited mutant-selective nanomolar biochemical IC50 values against L858R/T790M and L858R/T790M/C797S compared to WT (Table 1). Intriguingly, DDC4002 resembles previously reported ATP-competitive, selective checkpoint kinase 1 (Chk1) inhibitors;14 however, selective benzylation of the core amide precludes ATP site binding by abrogating critical hydrogen bonding contacts to the hinge likely favoring redirection to the allosteric site.
Table 1. Biochemical Activities and Antiproliferative Activities of a Panel of EGFR Allosteric Inhibitors.
| EGFR
biochemical activity IC50 (nM)a |
antiproliferative
activity Ba/F3 + Cetuximab IC50 (μM)b |
|||||||
|---|---|---|---|---|---|---|---|---|
| Compound ID | WT | L858R | L858R/T790M | L858R/T790M/C797S | WT | L858R | L858R/T790M | L858R/T790M/C797S |
| EAI045 | >1000 | 8.8 ± 0.9 | 2.0 ± 0.5 | 4.7 ± 0.3 | >10 | 0.84 ± 0.7 | 0.47 ± 0.2 | 0.25 ± 0.2 |
| DDC4002 | >1000 | 690 ± 120 | 39 ± 4 | 59 ± 8 | 9.7 ± 0.5 | >10 | 1.5 ± 0.4 | 1.2 ± 0.3 |
| 1 | >1000 | 150 ± 23 | 31 ± 2 | 19 ± 3 | 4.1 ± 1 | 3.7 ± 0.1 | 0.77 ± 0.1 | 0.93 ± 0.2 |
| 2 | >1000 | 130 ± 12 | 12 ± 0.9 | 23 ± 4 | 4.0 ± 1 | 3.8 ± 0.5 | 0.35 ± 0.07 | 0.35 ± 0.2 |
| 3 | >1000 | 154 ± 15 | 11 ± 2 | 13 ± 0.8 | 3.2 ± 0.8 | 2.7 ± 1 | 0.36 ± 0.2 | 0.20 ± 0.08 |
IC50 values were measured from a single experiment in triplicate. ATP concentration was 100 μM. Errors are reported as ± standard error.
IC50 values were measured from a single experiment with three replicates. Errors are reported as ± standard deviations.
To define the inhibitor binding mode, we determined crystal structures of EGFR(T790M/V948R) in complex with EAI002 and DDC4002 (Figures 2 and S1–S4). Expectantly, both inhibitors are bound to the kinase domain in an allosteric pocket adjacent to the ATP-binding site (Figure 2), as observed previously for EAI001(10) and JBJ-04-125-02(12). The 7-membered diazepinone ring of DDC4002 is puckered inward toward the αC-helix (Figure 2B) with the 8-fluorobenzene ring bound within the hydrophobic back pocket and the unsubstituted benzene ring along the αC-helix positioned out toward the solvent. The benzyl substituent extends toward the kinase N-lobe, bound in between AMP-PNP and side chains of K745, L788, and the T790M gatekeeper mutation. While the inhibitor mostly forms hydrophobic interactions, the diazepinone N–H forms an H-bond with the backbone carbonyl of F856 in the DFG motif (red dotted line Figure 2B). Additionally, the crystal structure of EAI002 contains four EGFR chains in the asymmetric unit all with EAI002 bound in the allosteric site, but with only one AMP-PNP and three AMP bound in the ATP site, presumably from AMP-PNP hydrolysis. The binding of either AMP-PNP or AMP does not impact the EAI002 binding mode.
Figure 2.

Structure and binding mode of a dibenzodiazepinone EGFR allosteric inhibitor. (A) Overall view of the structure of EGFR(T790M/V948R) bound to DDC4002 and AMP-PNP (PDB 6P1D). The V948R mutation enables the kinase domain to crystallize in the inactive state. DDC4002 is shown in CPK spheres with green carbon atoms. (B) Detailed view of DDC4002 bound to the allosteric pocket with AMP-PNP. P-loop and A-loop segments are hidden for clarity. (C) View of DDC4002 (green) and EAI045 (PDB 6P1L, white) from the overlay of crystal structures.
Additionally, we determined a crystal structure of EGFR(T790M/V948R) in complex with the phenylglycine EAI045 (Figures S1B and S4). Similar to EAI001(10) and JBJ-04-125-02,12EAI045 binds exclusively in the R configuration. Structural alignment of the kinase domains reveals that, despite distinct chemical structures, the binding mode of the dibenzodiazepinone inhibitors has significant overlap with that of the phenylglycines (Figures 2C and S3). Specifically, both scaffolds exhibit H-bonding to the backbone carbonyl of F856 as well as fluorobenzene moieties positioned toward the hydrophobic cleft at the back of the pocket. These conserved interactions confirm that these apparently unrelated scaffolds are anchored to the allosteric site through conserved interactions. Additionally, a recent EAI045 crystal structure bound to T790M/C797S/V948R in the absence of AMP-PNP shows limited variance in EAI045 binding mode, indicating that allosteric inhibitor binding to EGFR is structurally agnostic to the presence of ATP binding.15
To swiftly access the selectively substituted fused [6–7–6] tricyclic core, we streamlined our original route (Route A, Scheme 1) to a versatile, concise 2-step synthesis involving a tandem copper(I)-catalyzed intramolecular Ullmann condensation (Route B, Scheme 1).16 Based on the binding mode of DDC4002 (Figure 2B), we hypothesized that functionalization at the C2 position would be capable of enhancing the biochemical potency of these inhibitors, in a similar manner to that observed for EAI045 and JBJ-04-125-02.12 Following the latter example, coupling of a 4-(piperazinyl)phenyl substituent to C2 (1, Scheme 1) productively enhanced the potency of this scaffold. Structure–activity relationships revealed that engineering flexibility at this position via an Ullmann biaryl ether linkage (2 and 3, Scheme 1) modestly improved the effectiveness of these inhibitors, with 3 exhibiting biochemical potencies similar to EAI045 against L858R/T790M and L858R/T790M/C797S.
Scheme 1. Synthetic Routes (A,B) for Synthesis of 5,10-Dihydro-11H-dibenzo[b,e][1,4]diazepin-11-ones DDC4002 and Compounds 1–3.

Reagents and conditions: [i] SOCl2, DMF, Δ; [ii] 5-fluoro-2-iodoaniline, Et3N, CH2Cl2, 0 °C to RT, 81%, two steps; [iii] BnBr, NaH, THF, 0 to 40 °C; [iv] Fe, NH4Cl, THF/MeOH/H2O, 50 °C, 72%, two steps; [v] CuI, K2CO3, DMSO, 135 °C, 64%; [vi] benzylamine, EDC·HCl, HOBt, DIEA, DMF, 87%; [vii] 4-fluoro-2-iodoaniline, CuI, K2CO3, DMSO, 80 to 135 °C, 44%; [viii] BBr3, CH2Cl2, −20 °C to RT, 85%; [ix] tert-butyl 4-(4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl)piperazine-1-carboxylate, PdCl2(dppf), XPhos, 2 N Na2CO3, 1,4-dioxane, 100 °C; [x] TFA, CH2Cl2, 45%, two steps; [xi] 1-(4-iodophenyl)-4-methylpiperazine (3) or tert-butyl 4-(4-iodophenyl)piperazine-1-carboxylate (13), CuI, l-proline, K2CO3, DMSO, 80 °C, (3 36%); [xii] TFA, CH2Cl2, 30%, two steps.
To establish that these compounds inhibit EGFR through an allosteric mechanism, biochemical IC50 values were measured at varying ATP concentrations spanning 1 to 1000 μM (Table S1). Indeed, compounds 2 and 3 showed no significant variance with [ATP] consistent with allosteric inhibition. This further supports that the dibenzodiazepinones are effective inhibitors of mutant EGFR, operating in an allosteric mechanism as characterized crystallographically (Figure 2).
We additionally determined the impact of EGFR inhibition on cellular proliferation in transformed murine Ba/F3 cells. All compounds were found to be ineffective at limiting Ba/F3 proliferation due to EGFR dimer-induced resistance as we have observed previously (Table S2).10 Cotreatment with cetuximab (1 μg/mL) establishes this to be the case and leads to productive inhibition of L858R/T790M. Consistent with their biochemical potencies, dibenzodiazepinones with biaryl ether moieties (2 and 3) exhibit the best effect in combination with cetuximab with IC50 of ∼0.2–0.4 μM against L858R/T790M and L858R/T790M/C797S cells. Compounds 1–3 were not amenable to crystallographic characterization, but we expect that the phenylpiperazine group extends along the αC-helix to enhance the potency of 1–3 relative to DDC4002 in a manner analogous to that observed for JBJ-04-125-02.12 While these dibenzodiazepinone-based inhibitors are effective mutant-selective inhibitors of EGFR in a cellular context, they are still critically reliant on cotreatment with cetuximab.
We next sought to assess the selectivity of the best performing inhibitor (3) against a panel of 468 kinases via KINOMEscan profiling (DiscoverX). At a concentration of 10 μM, 3 displayed excellent selectivity across the human kinome with S-Score(35) = 0.01 (Figure S5, Table S4). While the KINOMEscan shows binding of 3 to EGFR WT and mutants, the results from activity and cellular assays indicate more reliable and robust selectivity for the oncogenic mutant targets (Table 1). Except for the expected WT EGFR and EGFR mutants, only two additional targets, SLK and KIT(V559D/V654A), were identified. We confirmed these hits to be false positives of 3 with SLK and KIT(V559D/V654A) (Kd > 10 μM; KINOMEscan KdELECT, DiscoverX) and confirmed no impact on SLK enzymatic activity (IC50 > 10 μM; Invitrogen, LanthaScreen). Although it is a valuable survey tool for assessing kinase selectivity, in our experience, KINOMEScan profiling does not correlate with enzymatic and cellular potencies as we have recently shown in the case of JBJ-04-125-02.12
In conclusion, we have discovered and optimized an allosteric mutant-selective EGFR inhibitor based on the dibenzodiazepinone scaffold. As the presently established mutant-selective allosteric EGFR inhibitors consist of a phenylglycine scaffold,10,12 the compounds described here demonstrate that diverse chemical scaffolds are capable of acting as mutant-selective EGFR inhibitors while preserving essential structural elements that anchor the inhibitors to the allosteric pocket. Therefore, this discovery expands the opportunity to discover additional chemical series as allosteric EGFR inhibitors. Our structure-based medicinal chemistry effort yielded an inhibitor (3) of the EGFR(L858R/T790M) and EGFR(L858R/T790M/C797S) mutants with an IC50 of ∼10 nM and high selectivity, as assessed by kinome profiling. Cotreatment with cetuximab resulted in antiproliferative activity in EGFR mutant Ba/F3 cells. Together with the previously reported dibenzodiazepine-based inhibitors of PAK1, which bind a closely related but distinct allosteric pocket, these compounds potentially indicate a broader application of benzodiazepine compounds as allosteric kinase inhibitors.17 Additionally, dibenzodiazepinone compounds represent new additions to the growing list of allosteric inhibitors for kinase targets, as previously explored for MEK,18,19 BCR-ABL1,20 and others.21,22 We plan to further optimize physicochemical and pharmacokinetic properties to produce more effective mutant-selective allosteric EGFR inhibitors, which is the subject of ongoing efforts.
Acknowledgments
We acknowledge Dr. Yong Jia (Novartis) for conducting high-throughput screening. We also thank Drs. Kunhua Li and Tyler S. Beyett for insightful discussions.
Glossary
ABBREVIATIONS
- EGFR
epidermal growth factor receptor
- EAI
EGFR allosteric inhibitor
- TKI
tyrosine kinase inhibitor
- EDC
1-ethyl-3-(3-dimethylaminopropyl)carbodiimide
- HOBt
hydroxybenzotriazole
- DIEA
N,N-diisopropylethylamine
- Dppf
1,1′-Bis(diphenylphosphino)ferrocene
- XPhos
2-dicyclohexylphosphino-2′,4′,6′-triisopropylbiphenyl
- AMP-PNP
adenylyl-imidodiphosphate
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.9b00381.
Chemistry, biological assay, and X-ray crystallography data (PDF)
Author Contributions
○ These authors contributed equally. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
This work was supported by the National Institutes of Health grants R01CA201049 (to M.J.E., N.S.G., and P.A.J.), R35CA242461 (to M.J.E.), R01CA116020 (to M.J.E.), P01CA154303 (to M.J.E., N.S.G., and P.A.J.), R01CA135257 (to P.A.J.), and R35 CA220497 (to P.A.J.). This work is based upon research conducted at the Northeastern Collaborative Access Team beamlines, which are funded by the National Institute of General Medical Sciences from the National Institutes of Health (P30 GM124165). The Pilatus 6M detector on the 24-ID-C beamline is funded by a NIH-ORIP HEI grant (S10 RR029205). This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02–06CH11357.
The authors declare the following competing financial interest(s): P.A.J. reports receiving commercial research grants from AstraZeneca, Boehringer Ingelheim, Astellas Pharmaceuticals, PUMA, Eli Lilly, and Takeda Oncology, has ownership interest (including stock, patents, etc.) in Gatekeeper Pharmaceuticals and LOXO Oncology, is a consultant or advisory board member for AstraZeneca, Boehringer Ingelheim, Daiichi Sankyo, Roche/Genentech, Pfizer, Merrimack Pharmaceuticals, Chugai Pharmaceuticals, Araxes Pharmaceuticals, Mirati, Ignyta, and LOXO Oncology, and has received other remuneration from Labcorp. M.J.E. reports receiving a commercial research grant from Novartis Institutes for Biomedical Research, reports receiving other commercial research support from Takeda, and has been a consultant for Novartis Institutes for Biomedical Research. N.S.G. reports receiving a commercial research grant from Takeda and is a consultant or advisory board member for C4, Petra, Syros, Soltego, and B2S Bio. No potential conflicts of interest were disclosed by the other authors.
Supplementary Material
References
- Paez J. G.; Jänne P. A.; Lee J. C.; Tracy S.; Greulich H.; Gabriel S.; Herman P.; Kaye F. J.; Lindeman N.; Boggon T. J.; Naoki K.; Sasaki H.; Fujii Y.; Eck M. J.; Sellers W. R.; Johnson B. E.; Meyerson M. EGFR Mutations in Lung Cancer: Correlation with Clinical Response to Gefitinib Therapy. Science 2004, 304 (5676), 1497–1500. 10.1126/science.1099314. [DOI] [PubMed] [Google Scholar]
- Lynch T. J.; Bell D. W.; Sordella R.; Gurubhagavatula S.; Okimoto R. A.; Brannigan B. W.; Harris P. L.; Haserlat S. M.; Supko J. G.; Haluska F. G.; Louis D. N.; Christiani D. C.; Settleman J.; Haber D. A. Activating Mutations in the Epidermal Growth Factor Receptor Underlying Responsiveness of Non–Small-Cell Lung Cancer to Gefitinib. N. Engl. J. Med. 2004, 350 (21), 2129–2139. 10.1056/NEJMoa040938. [DOI] [PubMed] [Google Scholar]
- Yun C.-H.; Mengwasser K. E.; Toms A. V.; Woo M. S.; Greulich H.; Wong K.-K.; Meyerson M.; Eck M. J. The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proc. Natl. Acad. Sci. U. S. A. 2008, 105 (6), 2070–2075. 10.1073/pnas.0709662105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi S.; Boggon T. J.; Dayaram T.; Jänne P. A.; Kocher O.; Meyerson M.; Johnson B. E.; Eck M. J.; Tenen D. G.; Halmos B. EGFR mutation and resistance of non–small-cell lung cancer to gefitinib. N. Engl. J. Med. 2005, 352 (8), 786–792. 10.1056/NEJMoa044238. [DOI] [PubMed] [Google Scholar]
- Zhou W.; Ercan D.; Chen L.; Yun C.-H.; Li D.; Capelletti M.; Cortot A. B.; Chirieac L.; Iacob R. E.; Padera R. Novel mutant-selective EGFR kinase inhibitors against EGFR T790M. Nature 2009, 462 (7276), 1070. 10.1038/nature08622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cross D. A.; Ashton S. E.; Ghiorghiu S.; Eberlein C.; Nebhan C. A.; Spitzler P. J.; Orme J. P.; Finlay M. R. V.; Ward R. A.; Mellor M. J.; et al. AZD9291, an irreversible EGFR TKI, overcomes T790M-mediated resistance to EGFR inhibitors in lung cancer. Cancer Discovery 2014, 4 (9), 1046–1061. 10.1158/2159-8290.CD-14-0337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thress K. S.; Paweletz C. P.; Felip E.; Cho B. C.; Stetson D.; Dougherty B.; Lai Z.; Markovets A.; Vivancos A.; Kuang Y.; et al. Acquired EGFR C797S mutation mediates resistance to AZD9291 in non–small cell lung cancer harboring EGFR T790M. Nat. Med. 2015, 21 (6), 560. 10.1038/nm.3854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niederst M. J.; Hu H.; Mulvey H. E.; Lockerman E. L.; Garcia A. R.; Piotrowska Z.; Sequist L. V.; Engelman J. A. The allelic context of the C797S mutation acquired upon treatment with third-generation EGFR inhibitors impacts sensitivity to subsequent treatment strategies. Clin. Cancer Res. 2015, 21 (17), 3924–3933. 10.1158/1078-0432.CCR-15-0560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu P.; Clausen M. H.; Nielsen T. E. Allosteric small-molecule kinase inhibitors. Pharmacol. Ther. 2015, 156, 59–68. 10.1016/j.pharmthera.2015.10.002. [DOI] [PubMed] [Google Scholar]
- Jia Y.; Yun C.-H.; Park E.; Ercan D.; Manuia M.; Juarez J.; Xu C.; Rhee K.; Chen T.; Zhang H.; Palakurthi S.; Jang J.; Lelais G.; DiDonato M.; Bursulaya B.; Michellys P.-Y.; Epple R.; Marsilje T. H.; McNeill M.; Lu W.; Harris J.; Bender S.; Wong K.-K.; Jänne P. A.; Eck M. J. Overcoming EGFR(T790M) and EGFR(C797S) resistance with mutant-selective allosteric inhibitors. Nature 2016, 534, 129. 10.1038/nature17960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X.; Gureasko J.; Shen K.; Cole P. A.; Kuriyan J. An allosteric mechanism for activation of the kinase domain of epidermal growth factor receptor. Cell 2006, 125 (6), 1137–1149. 10.1016/j.cell.2006.05.013. [DOI] [PubMed] [Google Scholar]
- To C.; Jang J.; Chen T.; Park E.; Mushajiang M.; De Clercq D. J. H.; Xu M.; Wang S.; Cameron M. D.; Heppner D. E.; Shin B. H.; Gero T. W.; Yang A.; Dahlberg S. E.; Wong K.-K.; Eck M. J.; Gray N. S.; Jänne P. A. Single and dual targeting of mutant EGFR with an allosteric inhibitor. Cancer Discovery 2019, 9 (7), 926–943. 10.1158/2159-8290.CD-18-0903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia Y.; Juarez J.; Li J.; Manuia M.; Niederst M. J.; Tompkins C.; Timple N.; Vaillancourt M.-T.; Pferdekamper A. C.; Lockerman E. L.; et al. EGF816 exerts anticancer effects in non–small cell lung cancer by irreversibly and selectively targeting primary and acquired activating mutations in the EGF receptor. Cancer Res. 2016, 76 (6), 1591–1602. 10.1158/0008-5472.CAN-15-2581. [DOI] [PubMed] [Google Scholar]
- Wang L.; Sullivan G. M.; Hexamer L. A.; Hasvold L. A.; Thalji R.; Przytulinska M.; Tao Z.-F.; Li G.; Chen Z.; Xiao Z.; Gu W.-Z.; Xue J.; Bui M.-H.; Merta P.; Kovar P.; Bouska J. J.; Zhang H.; Park C.; Stewart K. D.; Sham H. L.; Sowin T. J.; Rosenberg S. H.; Lin N.-H. Design, Synthesis, and Biological Activity of 5,10-Dihydro-dibenzo[b,e][1,4]diazepin-11-one-Based Potent and Selective Chk-1 Inhibitors. J. Med. Chem. 2007, 50 (17), 4162–4176. 10.1021/jm070105d. [DOI] [PubMed] [Google Scholar]
- Zhao P.; Yao M.-Y.; Zhu S.-J.; Chen J.-Y.; Yun C.-H. Crystal structure of EGFR T790M/C797S/V948R in complex with EAI045. Biochem. Biophys. Res. Commun. 2018, 502 (3), 332–337. 10.1016/j.bbrc.2018.05.154. [DOI] [PubMed] [Google Scholar]
- Gawande S. D.; Kavala V.; Zanwar M. R.; Kuo C. W.; Huang W. C.; Kuo T. S.; Huang H. N.; He C. H.; Yao C. F. Synthesis of Dibenzodiazepinones via Tandem Copper (I)-Catalyzed C-N Bond Formation. Adv. Synth. Catal. 2014, 356 (11–12), 2599–2608. 10.1002/adsc.201301020. [DOI] [Google Scholar]
- Karpov A. S.; Amiri P.; Bellamacina C.; Bellance M.-H.; Breitenstein W.; Daniel D.; Denay R.; Fabbro D.; Fernandez C.; Galuba I.; et al. Optimization of a dibenzodiazepine hit to a potent and selective allosteric PAK1 inhibitor. ACS Med. Chem. Lett. 2015, 6 (7), 776–781. 10.1021/acsmedchemlett.5b00102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y.; Adjei A. A. The clinical development of MEK inhibitors. Nat. Rev. Clin. Oncol. 2014, 11 (7), 385. 10.1038/nrclinonc.2014.83. [DOI] [PubMed] [Google Scholar]
- Zhao Z.; Xie L.; Bourne P. E. Insights into the binding mode of MEK type-III inhibitors. A step towards discovering and designing allosteric kinase inhibitors across the human kinome. PLoS One 2017, 12 (6), e0179936 10.1371/journal.pone.0179936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoepfer J.; Jahnke W.; Berellini G.; Buonamici S.; Cotesta S.; Cowan-Jacob S. W.; Dodd S.; Drueckes P.; Fabbro D.; Gabriel T.; Groell J.-M.; Grotzfeld R. M.; Hassan A. Q.; Henry C.; Iyer V.; Jones D.; Lombardo F.; Loo A.; Manley P. W.; Pellé X.; Rummel G.; Salem B.; Warmuth M.; Wylie A. A.; Zoller T.; Marzinzik A. L.; Furet P. Discovery of Asciminib (ABL001), an Allosteric Inhibitor of the Tyrosine Kinase Activity of BCR-ABL1. J. Med. Chem. 2018, 61 (18), 8120–8135. 10.1021/acs.jmedchem.8b01040. [DOI] [PubMed] [Google Scholar]
- Weisner J.; Gontla R.; van der Westhuizen L.; Oeck S.; Ketzer J.; Janning P.; Richters A.; Mühlenberg T.; Fang Z.; Taher A.; et al. Covalent-allosteric kinase inhibitors. Angew. Chem., Int. Ed. 2015, 54 (35), 10313–10316. 10.1002/anie.201502142. [DOI] [PubMed] [Google Scholar]
- Mobitz H.; Jahnke W.; Cowan-Jacob S. Expanding the opportunities for modulating kinase targets with allosteric approaches. Curr. Top. Med. Chem. 2016, 17 (1), 59–70. 10.2174/1568026616666160719165314. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.