

Abstract

A medicinal chemistry effort focused on identifying a structurally diverse candidate for phosphoinositide 3-kinase delta (PI3Kδ) led to the discovery of clinical candidate INCB050465 (20, parsaclisib). The unique structure of 20 contains a pyrazolopyrimidine hinge-binder in place of a purine motif that is present in other PI3Kδ inhibitors, such as idelalisib (1), duvelisib (2), and INCB040093 (3, dezapelisib). Parsaclisib (20) is a potent and highly selective inhibitor of PI3Kδ with drug-like ADME properties that exhibited an excellent in vivo profile as demonstrated through pharmacokinetic studies in rats, dogs, and monkeys and through pharmacodynamic and efficacy studies in a mouse Pfeiffer xenograft model.
Keywords: Parsaclisib, INCB050465, PI3Kδ, phosphoinositide 3-kinase delta, dezapelisib, INCB040093
Phosphoinositide 3-kinase delta (PI3Kδ) is a lipid kinase that phosphorylates the D-3 position of the inositol ring of phosphoinositides.1 By acting as a downstream mediator of signaling from receptor tyrosine kinases, including the B-cell receptor, Toll-like receptors, and cytokine receptors,2 PI3Kδ plays a primary role in PI3K-AKT-mTOR pathway activation. When dysregulated, PI3Kδ can lead to the development of hematologic malignancies including acute myeloid leukemia3 and chronic lymphocytic leukemia.4 PI3Kδ is also central in the regulation of immune cell function (e.g., B cells, T cells, neutrophils, and mast cells),2,5 and PI3Kδ activation therefore may promote B-cell malignancies via both direct tumor and immunomodulatory mechanisms. Importantly, by the same virtue, PI3Kδ is also garnering interest as a therapeutic target in inflammatory respiratory diseases (e.g., chronic obstructive pulmonary disorder, asthma), autoimmune diseases (e.g., systemic lupus erythematosus, rheumatoid arthritis, type 1 diabetes), and activated PI3Kδ syndrome.2,6,7 As such, there are currently intense efforts toward identifying inhibitors of PI3Kδ to treat hematologic malignancies as well as other cancers and immunologic diseases.
The first PI3Kδ-selective inhibitor, idelalisib (1), was approved for the treatment of hematologic malignancies (Figure 1).8,9 More recently, duvelisib (2), an inhibitor of both PI3Kδ and PI3Kγ, was also approved for the same indications.10 The efforts at Incyte to find a selective inhibitor of PI3Kδ led initially to INCB04009311 (3, dezapelisib). Dezapelisib (3) was advanced into clinical trials for the treatment of patients with previously treated B-cell malignancies.12 Even though these compounds demonstrate proof-of-concept that inhibition of PI3Ks are effective in treating cancer, the clinical trials of 1, 2, and 3 all revealed similar liver toxicity that has limited their full potential and there remains a need to develop alternative inhibitors with better safety profiles.13−15
Figure 1.

Structures of idelalisib (1), duvelisib (2), INCB040093 (3, dezapelisib), and INCB050465 (20, parsaclisib).
Dezapelisib (3) is a potent inhibitor of PI3Kδ (IC50 = 3.4 nM) with very good selectivity over the other isoforms of PI3K and over 200 other kinases,11 and it displayed cellular activity in SU-DHL-6 viability (IC50 = 18 nM) and Pfeiffer proliferation (IC50 = 14 nM) assays (Table 1). To estimate the clinical plasma concentration (total drug) needed to achieve half maximal inhibitory concentration (IC50), the IC50 value from the Pfeiffer cell assay was corrected for the fraction unbound in media to approximate IC50, and then corrected for the fraction unbound in human plasma. The calculated protein binding adjusted (PBadj) IC50 in the Pfeiffer assay further supported 3 was indeed a potent inhibitor of PI3Kδ.
Table 1. Selected SAR of 3–17.
| Cmpd | PI3Kδ SPA IC50 (nM) | SU-DHL-6 Viability IC50 (nM) | Pfeiffer Proliferation IC50 (nM) | Pfeiffer PBadj IC50 (nM) | % fu in human plasma |
|---|---|---|---|---|---|
| 3 | 3.4 | 18 | 14 | 66 | 17 |
| 4a | 430 | 1800 | 490 | 3200 | 10 |
| 5 | 8.4 | 140 | 50 | 1000 | 1.4 |
| 6 | 13 | 73 | 26 | 270 | 5.9 |
| 7a | 99 | 1300 | 660 | ND | 0.64 |
| 8a | 5.3 | 60 | 8.7 | ND | ND |
| 9 | 2.8 | 100 | 120 | 950 | 2.9 |
| 10 | 3.6 | 170 | 130 | 3100 | 1.3 |
| 11 | 1.7 | 14 | 9.5 | 40 | 12 |
| 12 | 0.99 | 3.1 | 3.4 | 17 | 14 |
| 13 | 1.8 | 21 | 17 | 61 | 22 |
| 14a | 120 | 840 | 610 | 4500 | 9.2 |
| 15 | 0.93 | 7.4 | 8.6 | 61 | 8.9 |
| 16a | 11 | 44 | 11 | 53 | 19 |
| 17 | 1.9 | 2.4 | 1.5 | 3.6 | 42 |
Racemates. fu = fraction unbound; ND = not determined.
The pharmacokinetic (PK) studies of 3 in the cynomolgus monkey (cyno) demonstrated that 3 exhibited good oral exposure (AUC of 4.9 μM.h dosed at 10 mg/kg), but also had a short half-life (1.3 h). Improvements in potency, PK, and toxicity were the goals of a second-generation inhibitor of PI3Kδ. As a result of the encouraging properties outlined above, 3 provided a good starting point to explore structurally diverse scaffolds. These studies culminated with the design of INCB050465 (20, parsaclisib), a highly potent and selective PI3Kδ inhibitor that is currently in clinical trial studies for diffuse large B-cell lymphoma (DLBCL),16 follicular lymphoma,17 marginal zone lymphoma,18 and mantle cell lymphoma.19 Described herein are the obstacles that were encountered and the solutions that were found which eventually led to 20.
One of the first ideas examined was to create a new core by breaking the thiazole ring of 3 and forming a new 6,6-bicyclic heterocycle (Figure 2) while maintaining the purine hinge-binder that was needed for PI3Kδ potency. The nitrogen in the quinoline ring (4) was designed to replace binding pocket interactions from the carbonyl oxygen of 3. The synthesis of this purine series provided analogs that were either racemic or scalemic. For all single isomer compounds, the stereogenic center was assigned to be the same stereochemistry as C-1′ of 3 (the opposite enantiomer was known to be inactive). The stereochemistry of 3 was assigned S based on the X-ray crystal structure data of an intermediate (unpublished data). Quinoline 4 was significantly less active compared with 3 (Table 1). It was hypothesized that potency could be restored by introducing small lipophilic groups at the 5-position of the quinoline ring to mimic the removed thiazole ring. In particular, chloro-substitution (5) dramatically improved the potency of 4 (IC50 = 8.4 nM vs 430 nM). Compound 5 was similarly potent to 3 but the low free fraction in human plasma of 5 translated into a PBadj IC50 in the Pfeiffer assay that was 15-fold less potent than 3. Introducing more polarity to the quinoline ring with the addition of one nitrogen at the 2-position of quinoline 5 gave cinnoline 6. Compound 6 displayed better human plasma free fraction, higher Caco2 permeability (Supporting Information, Table S15), and similar potency compared with 5. While 6 still exhibited a higher PBadj IC50 in the Pfeiffer assay compared with 3, this was offset with improved rat PK that revealed an increase in oral exposure (AUC of 13 μM.h (6) vs AUC of 4.4 μM.h (3) for the same dose of 5 mg/kg). Even though the potency of 6 was weaker compared with 3, the improvements in PK with 6 warranted further advancement. Unfortunately, 6 exhibited toxicity when dosed in dogs and its progression was halted.
Figure 2.

Design of quinoline and cinnoline inhibitors 4–6.
Unsuccessful attempts to find alternative bicyclic cores with better potency or properties as the cinnoline led to the design of inhibitors that featured a monocyclic core ring instead of a bicyclic ring. Opening the pyridazine ring of cinnoline 6 (Figure 3) and installing a cyano group at the 3-position of the resultant phenyl ring provided 7. While 7 was less potent than 6 toward PI3Kδ in binding and cellular assays, structure–activity relationship (SAR) studies revealed that replacement of the cyano group in 7 with a heteroaryl group could impact potency significantly. The methylpyrazole group of 8 afforded improved potency in binding and cellular assays compared with 7. Although 8 was very potent, two aromatic groups adjacent to each other on the phenyl ring were believed to be less than ideal for achieving the physical properties needed for a drug candidate. SAR studies at the 2-position of the phenyl ring revealed that the solvent exposed 3-fluorophenyl group could be significantly simplified to the methoxy ether 9 without loss of potency in the enzyme binding assay.
Figure 3.

Monocyclic purine inhibitors 7–11.
Truncating the phenyl group to methoxy at the 2-position enabled an expanded examination of SAR at the 3-position. Substitution of the pyrazole ring for the sulfonyl phenyl analog 10 afforded similar potency with acceptable Caco2 permeability and human intrinsic clearance (hIntCl) values (Supporting Information, Table S15). Unfortunately, 10 had low free fraction in human plasma which translated into a very high PBadj IC50 in the Pfeiffer assay (IC50 = 3100 nM). The more polar pyridyl analog 11 provided a 9-fold improvement in human plasma free fraction compared with 10. The oral exposure of 11 in rat PK was not as good as 6 (AUC of 13 μM.h (6) vs AUC of 3.0 μM.h (11) for the same dose of 5 mg/kg) which could have been caused by the low Caco2 permeability (0.5 × 10–6 cm/s) of 11. While many analogs were prepared in this series, finding the right balance of properties remained a challenge. Using the monocyclic phenyl core, alternative hinge-binders were considered as a potential solution.
The purine hinge-binder was replaced with 4,6-diaminopyrimidine-5-carbonitrile (Figure 4), which proved to be an effective substitute. Compound 12 offered a slight enhancement in potency compared with 11 but also a large improvement in Caco2 permeability (7.9 vs 0.5 × 10–6 cm/s). Taken together, these properties delivered excellent oral exposure in rat PK (AUC of 10 μM.h dosed at 4 mg/kg) for 12. Unfortunately, 12 had elevated levels of inhibition in the HEK293 and hERG counter screens (Supporting Information, Table S14). The strategy to reduce HEK293 and hERG inhibition with polar amide groups resulted in the examination of a variety of pyridyl carboxamides. The dimethyl amide found in 13 provided the best combination of potency and physical properties with decreased HEK293 and hERG levels. Amide 13 exhibited good oral exposure in rat PK (AUC of 7.7 μM.h dosed at 4 mg/kg) and was advanced into cyno PK where it surprisingly showed a very short half-life (0.2 h) and very poor oral exposure (AUC of 0.2 μM.h dosed at 2 mg/kg). A number of other compounds that contained the aminocyanopyrimidine hinge-binder also exhibited poor cyno PK. Metabolic studies revealed that the aminocyanopyrimidine hinge-binder was susceptible to oxidation in cynos by aldehyde oxidase (AO) at the 2′-position. The aminocyanopyrimidine was abandoned and the focus shifted back to hinge-binder optimization.
Figure 4.

Evolution of purine inhibitor 11 to aminocyanopyrimidine inhibitors 12 and 13 to pyrazolopyrimidine inhibitors 14 and 15.
It was reasoned that blocking the 2′-position of the pyrimidine ring could prevent AO oxidation. Unfortunately, a variety of functional groups that were examined resulted in either loss of potency or compounds that did not have optimal ADME properties (data not shown). Several other monocyclic and bicyclic hinge-binders were examined and the most promising was the pyrazolopyrimidine of 14 which was essentially a cyclized version of the aminocyanopyrimidine (Figure 4). The potency of 14 could be substantially improved with the addition of a methyl group at the 3′-position of the pyrazolopyrimidine (15) which resulted in a >100-fold increase in PI3Kδ inhibition. The overall profile of 15 appeared encouraging and SAR studies at the 3-position of the central phenyl ring continued with aryl groups. However, a parallel effort to identify saturated rings at the 3-position had initiated and, in general, provided compounds with better physical properties.
A number of saturated rings were synthesized, and selected examples are shown in Figure 5. Excitingly, piperidine 16 was only slightly less potent than pyridyl amide 15 with a similar intrinsic clearance, but 16 had a higher free fraction in human plasma. Azetidines typically had a better profile than piperidines and extensive SAR studies led to azetidine 17. Compound 17 was very potent in the biochemical and cellular assays and it had a very high free fraction in human plasma that translated into a PBadj IC50 in the Pfeiffer assay that was 18-fold more potent than 3. Compound 17 was studied in rat and cyno PK where it exhibited good oral exposure in rats (AUC of 6.8 μM.h dosed at 5 mg/kg) but only moderate oral exposure in cyno (AUC of 4.1 μM.h dosed at 1 mg/kg). Initial toxicology studies indicated that 17 was tolerated in dogs, but unfortunately, the 28-day investigational new drug (IND)-enabling toxicology study revealed testicular lesions which halted further development.
Figure 5.

Monocyclic pyrazolopyrimidine inhibitors 16–18 with 3-substituted saturated cyclic rings.
One challenge observed with saturated heterocycles containing a basic nitrogen was their propensity to inhibit hERG. This could be mitigated with nearby polar groups such as the secondary alcohol found in 17. Another approach to mitigate hERG inhibition was to remove the basic nitrogen altogether, which led to the design of the five-membered lactam ring of 18. As the result of reduced hERG inhibition and similar potency to 17, lactam 18 was deemed to be a good lead for further investigation and several analogs (18–25) were synthesized (Supporting Information, Schemes S13–S16). Both diastereoisomers of the lactam ring provided compounds that were potent, although the lactam stereogenic center was usually not assigned. The more potent of the lactam diastereoisomers are presented in Table 2 (18–21). The corresponding less potent diastereoisomers (22–25) can be found in the Supporting Information.
Table 2. SAR of Lactam Pyrazolopyrimidines 18–21.
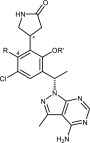
| Cmpda | * | R | R′ | PI3Kδ SPA IC50 (nM) | SU-DHL-6 Viability IC50 (nM) | Pfeiffer Proliferation IC50 (nM) | Pfeiffer PBadj IC50 (nM) | % fu in human plasma | IntCL (L/h/kg) | Caco2 Pm (×10–6 cm/s) |
|---|---|---|---|---|---|---|---|---|---|---|
| 18 | Me | Me | 2.4 | 13 | 51 | 480 | 10 | 0.7 | 2.8 | |
| 19 | Me | Et | 1.4 | 2.1 | 4.8 | 47 | 6.5 | 0.7 | 1.9 | |
| 20 | R | F | Et | <1.0 | 1.6 | 2.5 | 4.2 | 5.8 | 0.7 | 1.0 |
| 21 | CN | Et | <1.0 | 0.47 | 1.7 | 4.1 | 23 | 0.7 | 0.3 |
Single isomers. fu = fraction unbound.
SAR studies of the lactam series focused around the central phenyl ring. Converting the methoxy of 18 to ethoxy (19) resulted in a slight increase in potency. Replacement of the methyl at the 4-position of 19 with fluoro (20) resulted in another small boost in potency. The most potent analog was cyano 21 which was about 3-fold more potent in the cellular assays than 19. The intrinsic clearance values of these lactams were all very similar. The cyano analog (21) had the highest free fraction (23%); however, it also had the lowest Caco2 permeability (0.3 × 10–6 cm/s). The fluoro analog (20), which had the same PBadj IC50 in the Pfeiffer assay as cyano 21, but with slightly better Caco2 permeability (1 × 10–6 cm/s), offered the best balance of properties. Compound 20 was selected for further profiling which revealed that it is potent in the PI3Kδ filter binding assay and is very selective over the other PI3K isoforms (Table 3). In addition, 20 exhibits selectivity over a panel of 197 kinases20 and is very potent in the RAMOS cellular and human whole blood basophil assays. Compound 20 did not inhibit hERG or the major isoforms of human cytochrome P450, and it has very good solubility at pH 7.4 (see Supporting Information, Table S13, for additional data).
Table 3. Summary of In Vitro Profile of 20.
| assay | result | N |
|---|---|---|
| PI3Kδ SPA IC50 (nM) | <1.0 | 7 |
| PI3Kδ FB IC50 (nM) | 1.1 ± 0.50 | 11 |
| PI3Kα FB IC50 (nM) | >20,000 | 4 |
| PI3Kβ FB IC50 (nM) | >20,000 | 4 |
| PI3Kγ FB IC50 (nM) | >10,000 | 5 |
| SU-DHL-6 IC50 (nM) | 1.6 ± 0.90 | 22 |
| Pfeiffer IC50 (nM) | 2.5 ± 0.65 | 8 |
| RAMOS IC50 (nM) | 1.1 ± 0.85 | 11 |
| WB IC50 (nM) | 2.0 ± 1.2 | 5 |
| hERG patch IC50 (nM) | 75,000 | 1 |
| Solubility at pH 7.4 (μg/mL) | 210 ± 72 | 3 |
The modeling of 20 with PI3Kδ (Figure 6), which was based on the X-ray cocrystal structure (PDB ID 6G6W) of a different inhibitor with PI3Kδ,21 revealed a similar induced fit exhibited by other PI3Kδ-selective inhibitors.22 The pyrazolopyrimidine in 20 binds the hinge region of PI3Kδ forming hydrogen bonds with the NH of Val828 and CO of Glu826. The phenyl ring is positioned in the induced specificity pocket formed by the side chains of Trp760 and Met752, which adopts this alternate conformation to accommodate the phenyl ring. The chloro and fluoro groups on the phenyl ring of 20 effectively mimic the second ring commonly found in the bicyclic core structure of PI3Kδ inhibitors (e.g., 3). The carbonyl of the lactam also forms hydrogen-bonding interactions with both Thr750 and Lys708.
Figure 6.
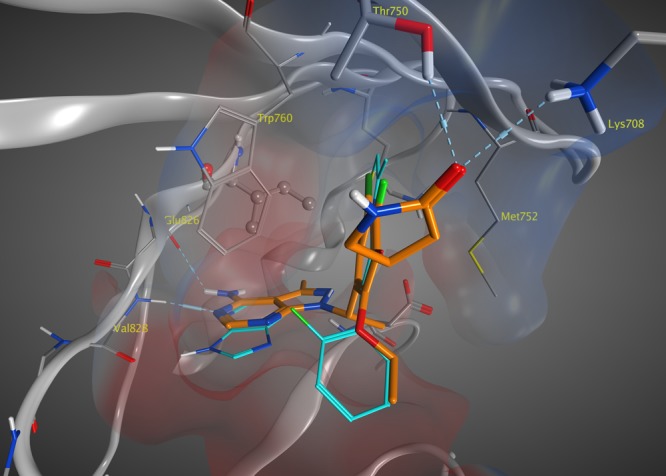
Proposed binding model of 20 (orange) with PI3Kδ overlaid with 3 (cyan).
The PK of 20 have been studied in rats, dogs, and monkeys (Supporting Information, Table S20). Following intravenous (IV) administration, 20 displayed low systemic clearance, representing 26%, 2%, and 5% of the hepatic blood flow in rats, dogs, and monkeys, respectively. The steady-state volume of distribution was moderate in rats (1.54 L/kg) and monkeys (0.72 L/kg), but low in dogs (0.32 L/kg). The terminal elimination half-life values were favorable after IV dosing, ranging from 4.0 h (rat) to 7.3 h (monkey) with similar values obtained after oral dosing. Following oral administration, 20 was rapidly absorbed with Tmax (time taken to reach the maximum concentration) values of 0.3 h (rat) to 2.5 h (monkey). Oral exposure was high and the bioavailability of 20 was complete in dogs (100%), and high in monkeys (79%) and rats (74%).
To determine the effects of 20 in vivo, the Pfeiffer DLBCL subcutaneous mouse xenograft model was used. Pfeiffer cells are highly sensitive to PI3Kδ inhibition in vitro with IC50 values for 20 in proliferation and signaling assays less than 5 nM.20 Compound 20 dosed orally twice daily significantly inhibited Pfeiffer xenograft tumor growth at 1 mg/kg (Figure 7A; p < 0.05)). This correlated with profound inhibition of the phosphorylation of AKT at Ser473, a marker of PI3Kδ activity, observed in mice (Figure 7B). Descriptions of additional pharmacologic, pharmacokinetic, and pharmacodynamic analyses in multiple models are described elsewhere.20
Figure 7.
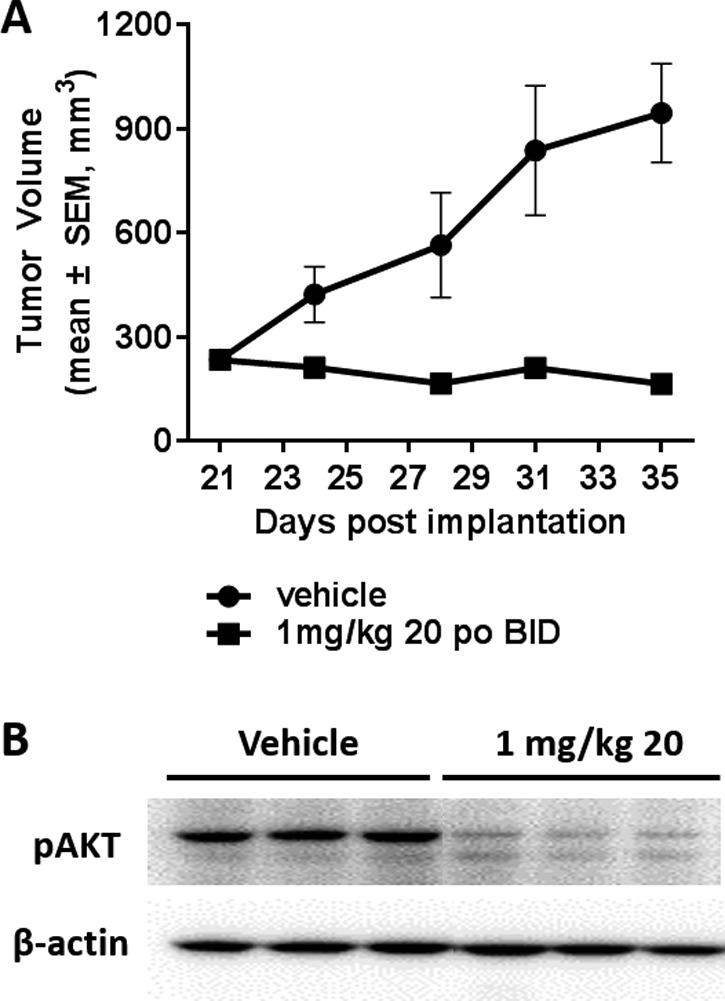
(A) Compound 20 suppresses tumor growth in the Pfeiffer mouse xenograft model. Compound 20 was administered orally twice daily for 14 days. The percentage of tumor growth inhibition (TGI) was measured on the final day of study. (B) Analysis of mice from the Pfeiffer xenograft model revealed inhibition of the phosphorylation of AKT at Ser473.
Preclinical 28-day IND-enabling toxicology studies in rats and dogs demonstrated no liver toxicities and suggested 20 was suitable for advancement into clinical trials. Thus far, 20 has shown reduced liver-related toxicities in clinical trials,23 which supports the hypothesis that the hepatotoxicity displayed by 1, 2, and 3 could be related to the purine group that is common to all three first-generation compounds.20
In summary, a potent and selective inhibitor of PI3Kδ was discovered. The bicyclic thiazolopyrimidinone core of 3 could be truncated to a single phenyl ring. Replacement of the purine hinge-binder of 3 with pyrazolopyrimidine provided compounds that were slightly more potent with improved cellular permeability. Further optimization revealed the five-membered lactam ring was a good replacement of aryl rings at the 3-position of the core phenyl ring that led to the discovery of 20. The excellent pharmacokinetics of 20 in various species, its efficacy in inhibiting tumor growth in a Pfeiffer xenograft model, and its lack of preclinical toxicity allowed it to be advanced into clinical development. Parsaclisib (20) is currently in Phase 2 clinical trials for the treatment of various cancers.16−19
Acknowledgments
This manuscript is dedicated to the memory of our friend and colleague Dr. Yun-Long Li. Yun-Long was a passionate and creative medicinal chemist who initiated the PI3Kδ program and led the team that identified INCB040093. The discovery of Parsaclisib could not have been possible without the pivotal insights from Yun-Long. We thank Laurine Galya, Mei Li, James Doughty, and Karl Blom for their analytical assistance. We thank Ravi Jalluri for creating the molecular modeling graphics. We thank Jordan Fridman for discussions related to pharmacology. We thank Daniel Levy, Neil Lajkiewicz, and Matthew McCammant for proofreading this manuscript. Medical writing assistance was provided by Simon J. Slater, PhD, CMPP, of Envision Pharma Group (Philadelphia, PA), funded by Incyte Corporation.
Glossary
Abbreviations
- ADME
absorption, distribution, metabolism, and excretion
- AKT
protein kinase B
- AUC
area under the plasma drug concentration–time curve
- Cmax
maximum drug concentration
- F
fraction of an administered dose of unchanged drug that reaches the systemic circulation
- FB
filter binding
- hERG
human ether-a-go-go-related gene
- IV
intravenous
- mTOR
mammalian target of rapamycin
- SPA
scintillation proximity assay
- t1/2
time taken for half the initial dose of medicine administered to be eliminated from the body
- Vdss
volume of distribution at steady state
- WB
whole blood
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.9b00334.
Biological and ADME assays; General experimental procedures and analytical characterization methods; Synthetic schemes; Experimental procedures and analytical data for compounds 4–25; X-ray crystal structure of 20; HPLC purity analysis; In vitro profile and physiochemical properties of 20; In vitro assays and ADME of Compounds 3–25; Pharmacokinetic parameters (PDF)
Author Contributions
Design/conception of study or data acquisition/analysis and/or interpretation: all authors. Drafting manuscript or providing critical review of content: all authors. Approval of final draft for publication: all authors. Accountable for all aspects of work: all authors. Yun-Long Li is deceased (April 20, 2016) and Brian Wayland is deceased (May 27, 2018).
Incyte Corporation, Wilmington, DE, USA.
The authors declare no competing financial interest.
Supplementary Material
References
- Vanhaesebroeck B.; Leever S. J.; Ahmadi K.; Timms J.; Katso R.; Driscoll P. C.; Woscholski R.; Parker P. J.; Waterfield M. D. Synthesis and Function of 3-Phosphorylated Inositol Lipids. Annu. Rev. Biochem. 2001, 70, 535–602. 10.1146/annurev.biochem.70.1.535. [DOI] [PubMed] [Google Scholar]
- Puri K. D.; Gold M. R. Selective Inhibitors of Phosphoinositide 3-Kinase Delta: Modulators of B-Cell Function with Potential for Treating Autoimmune Inflammatory Diseases and B-Cell Malignancies. Front. Immunol. 2012, 3, 1–16. 10.3389/fimmu.2012.00256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billottet C.; Grandage V. L.; Gale R. E.; Quattropani A.; Rommel C.; Vanhaesebroeck B.; Khwaja A. A Selective Inhibitor of the p110d isoform of PI 3-Kinase Inhibits AML Cell Proliferation and Survival and Increases the cytotoxic effects of VP16. Oncogene 2006, 25, 6648–6659. 10.1038/sj.onc.1209670. [DOI] [PubMed] [Google Scholar]
- Fruman D. A.; Rommel C. PI3Kδ Inhibitors in Cancer: Rationale and Serendipity Merge in the Clinic. Cancer Discovery 2011, 1, 562–572. 10.1158/2159-8290.CD-11-0249. [DOI] [PubMed] [Google Scholar]
- Fruman D. A.; Chiu H.; Hopkins B. D.; Bagrodia S.; Cantley L. C.; Abraham R. T. The PI3K Pathway in Human Disease. Cell 2017, 170, 605–635. 10.1016/j.cell.2017.07.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michalovich D.; Nejentsev S. Activated PI3 Kinase Delta Syndrome: From Genetics to Therapy. Front. Immunol. 2018, 9, 1–6. 10.3389/fimmu.2018.00369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stokes C. A.; Condliffe A. M. Phosphoinositide 3-Kinase δ (PI3Kδ) in Respiratory Disease. Biochem. Soc. Trans. 2018, 46, 361–369. 10.1042/BST20170467. [DOI] [PubMed] [Google Scholar]
- Markham A. Idelalisib: First Global Approval. Drugs 2014, 74, 1701–1707. 10.1007/s40265-014-0285-6. [DOI] [PubMed] [Google Scholar]
- Yang Q.; Modi P.; Newcomb T.; Queva C.; Gandhi V. Idelalisib: First-in-Class Delta Inhibitor for the Treatment of Chronic Lymphocytic Leukemia, Small Lymphocytic Leukemia, and Follicular Lymphoma. Clin. Cancer Res. 2015, 21, 1537–1542. 10.1158/1078-0432.CCR-14-2034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blair H. Duvelisib: First Global Approval. Drugs 2018, 78, 1847–1853. 10.1007/s40265-018-1013-4. [DOI] [PubMed] [Google Scholar]
- Shin N.; Li Y.-L.; Mei S.; Wang K. H.; Hall L.; Katiyar K.; Wang Q.; Yang G.; Rumberger B.; Leffet L.; He X.; Rupar M.; Bowman K.; Favata M.; Li J.; Liu M.; Li Y.; Covington M.; Koblish H.; Soloviev M.; Shuey D.; Burn T.; Diamond S.; Fridman J.; Combs A.; Yao W.; Yeleswaram S.; Hollis G.; Vaddi K.; Huber R.; Newton R.; Scherle P. INCB040093 is a Novel PI3Kδ Inhibitor for the Treatment of B Cell Lymphoid Malignancies. J. Pharmacol. Exp. Ther. 2018, 364, 120–130. 10.1124/jpet.117.244947. [DOI] [PubMed] [Google Scholar]
- Incyte . Study of INCB040093 in Subjects with Previously Treated B-Cell Malignancies. Available from https://clinicaltrials.gov/ct2/show/NCT01905813. NLM identifier: NCT01905813. Accessed October 16, 2019.
- Coutré S. E.; Barrientos J. C.; Brown J. R.; de Vos S.; Furman R. R.; Keating M. J.; Li D.; O’Brien S. M.; Pagel J. M.; Poleski M. H.; Sharman J. P.; Yao N.-S.; Zelenetz A. D. Management of Adverse Events Associated with Idelalisib Treatment: Expert Panel Opinion. Leuk. Lymphoma 2015, 56, 2779–2786. 10.3109/10428194.2015.1022770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flinn I. W.; Patel M.; Oki Y.; Horwitz S.; Foss F. F.; Allen K.; Douglas M.; Stern H.; Sweeney J.; Kharidia J.; Kelly P.; Kelly V. M.; Kahl B. Duvelisib, an Oral Dual PI3K-δ, γ Inhibitor, Shows Clinical Activity in Indolent Non-Hodgkin Lymphoma in a Phase 1 Study. Am. J. Hematol. 2018, 93, 1311–1317. 10.1002/ajh.25228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips T. J.; Forero-Torres A.; Sher T.; Diefenbach C. S.; Johnston P.; Talpaz M.; Pulini J.; Zhou L.; Scherle P.; Chen X.; Barr P. M. Phase 1 Study of the PI3Kd Inhibitor INCB040093 ± JAK1 Inhibitor Itacitinib in Relapsed/Refractory B-Cell Lymphoma. Blood 2018, 132, 293–306. 10.1182/blood-2017-10-812701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Incyte . A Phase 2 Safety and Efficacy Study of INCB050465 in Subjects With Relapsed or Refractory Diffuse Large B-Cell Lymphoma (CITADEL-202). Available from https://clinicaltrials.gov/ct2/show/NCT02998476. NLM identifier: NCT02998476. Accessed October 16, 2019.
- Incyte . An Open-Label Study of INCB050465 in Relapsed or Refractory Follicular Lymphoma (CITADEL-203). Available from https://clinicaltrials.gov/ct2/show/NCT03126019. NLM identifier: NCT03126019. Accessed October 16, 2019.
- Incyte . A Study of INCB050465 in Subjects With Relapsed or Refractory Marginal Zone Lymphoma (CITADEL-204). Available from https://clinicaltrials.gov/ct2/show/NCT03144674. NLM identifier: NCT03144674. Accessed October 16, 2019.
- Incyte . A Study of INCB050465 in Relapsed or Refractory Mantle Cell Lymphoma Previously Treated With or Without a BTK Inhibitor (CITADEL-205). Available from https://clinicaltrials.gov/ct2/show/NCT03235544. NLM identifier: NCT03235544. Accessed October 16, 2019.
- Shin N.; Stubbs M.; Koblish H.; Yue E. W.; Soloviev M.; Douty B.; Wang K. H.; Wang Q.; Gao M.; Feldman P.; Yang G.; Hall L.; Hansbury M.; O’Connor S.; Leffet L.; Collins R.; Katiyar K.; He X.; Waeltz P.; Collier P.; Lu J.; Li Y.-L.; Li Y.; Liu P. C. C.; Burn T.; Covington M.; Diamond S.; Shuey D.; Roberts A.; Yeleswaram S.; Hollis G.; Metcalf B.; Yao W.; Huber R.; Combs A.; Newton R.; Scherle P.. Parsaclisib (INCB050465) is a Next-Generation PI3Kδ Inhibitor with Reduced Hepatotoxicity and Potent Antitumor and Immunomodulatory Activities in Models of B-Cell Malignancy. Manuscript submitted. [DOI] [PubMed] [Google Scholar]
- Erra M.; Taltavull J.; Bernal F. J.; Caturla J. F.; Carrascal M.; Pagès L.; Mir M.; Espinosa S.; Gràcia J.; Domínguez M.; Sabaté M.; Paris S.; Maldonado M.; Hernández B.; Bravo M.; Calama E.; Miralpeix M.; Lehner M. D.; Calbet M. Discovery of a Novel Inhaled PI3Kδ Inhibitor for the Treatment of Respiratory Diseases. J. Med. Chem. 2018, 61, 9551–9567. 10.1021/acs.jmedchem.8b00873. [DOI] [PubMed] [Google Scholar]
- Berndt A.; Miller S.; Williams O.; Le D. D.; Houseman B. T.; Pacold J. I.; Gorrec F.; Hon W.-C.; Ren P.; Liu Y.; Rommel C.; Gaillard P.; Ruckle T.; Schwarz M. K.; Shokat K. M.; Shaw J. P.; Williams R. L. The p110δ Structure: Mechanisms for Selectivity and Potency of New PI(3)K Inhibitors. Nat. Chem. Biol. 2010, 6, 117–124. 10.1038/nchembio.293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forero-Torres A.; Ramchandren R.; Yacoub A.; Wertheim M. S.; Edenfield W. J.; Caimi P.; Gutierrez M.; Akard L.; Escobar C.; Call J.; Persky D.; Iyer S.; DeMarini D. J.; Zhou L.; Chen X.; Dawkins F.; Phillips T. J. Blood 2019, 133, 1742–1752. 10.1182/blood-2018-08-867499. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.