

Abstract
Small molecules that inhibit the metabolic enzyme NAMPT have emerged as potential therapeutics in oncology. As part of our effort in this area, we took a scaffold morphing approach and identified 3-pyridyl azetidine ureas as a potent NAMPT inhibiting motif. We explored the SAR of this series, including 5 and 6 amino pyridines, using a convergent synthetic strategy. This lead optimization campaign yielded multiple compounds with excellent in vitro potency and good ADME properties that culminated in compound 27.
Keywords: NAMPT, azetidine, structure based drug design, scaffold morphing, amino pyridine
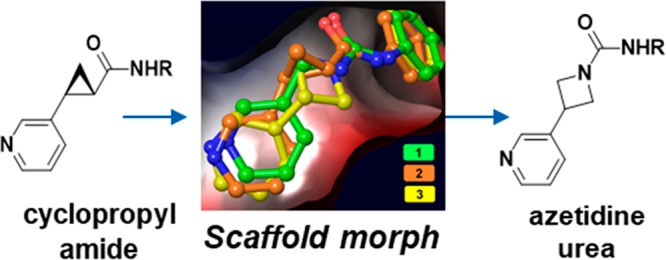
The production of nicotinamide adenine dinucleotide (NAD+) is a critical biochemical process in mammalian cells. NAD+ is a substrate for enzymes such as poly(ADP-ribose) polymerases (PARPs) and sirtuins and is a critical component of cellular metabolism.1 Given the importance of this biochemical, mammalian cells have evolved multiple methods to produce NAD+,2 but the predominant pathway in mammals involves the conversion of nicotinamide (NAM) and phosphoribosyl pyrophosphate (PRPP) into nicotinamide mononucleotide (NMN) by the enzyme nicotinamide phosphoribosyltransferase (NAMPT) followed by a second reaction catalyzed by NMN adenylyltransferase (NMNAT, Figure 1A).3,4 The NAMPT catalyzed reaction is the overall rate limiting step in this process, and it therefore controls the availability of NAD+. Cancer cells are highly active metabolically, and as a consequence they consume NAD+ at a higher rate than noncancerous cells.5 In this vein, small molecule inhibition of NAMPT in cancerous cells has been shown to deplete cellular NAD+ and lead to cell death, and together these lines of evidence point to NAMPT as a promising target to develop novel therapies for oncology.6−8 As shown in Figure 1B, we previously reported the elaboration of urea 1 to the 3-pyridyl (S,S) cyclopropyl carboxamide 2.9 The 3-pyridyl (S,S) carboxamide was a promising motif that potently inhibits NAMPT and led to a compound that demonstrated in vivo efficacy in a mouse xenograft model. Furthermore, the cellular potency of these molecules enabled their translation into payloads for antibody-drug conjugates (ADC).10 We were curious, though, if we could identify an alternative warhead that could provide even greater potency for use as a cytotoxic payload in antibody–drug conjugates because of the known risk for on-target toxicity with this mechanism.11,12 With this goal in mind, we reconsidered urea 1 in the context of the cellular potency exhibited by the cyclopropane analogue 2. We reasoned that if small rings are tolerated in this portion of the molecule, then we may be able to enclose the left-hand nitrogen of the urea into a ring. As shown in Figure 1B and the crystal structure in Figure 2, this concept led to azetidine urea 3, and this compound was found to be very potent in CellTiterGlo (CTG) assays in A2780 and COR-L23 cells. With our scaffold morphing hypothesis validated, we wanted to further explore this SAR of these ring containing ureas.13
Figure 1.

(A) The rate limiting step in the synthesis of NAD+ is the NAMPT catalyzed synthesis of NMN from NAM and PRPP. (B) Our initial identification of the azetidine ureas came from a scaffold morph of the previously established cyclopropyl carboxamide NAMPT inhibitors. (C) Synthesis of the five and six membered ring ureas demonstrates the superiority of the azetidine urea for NAMPT inhibition.
Figure 2.
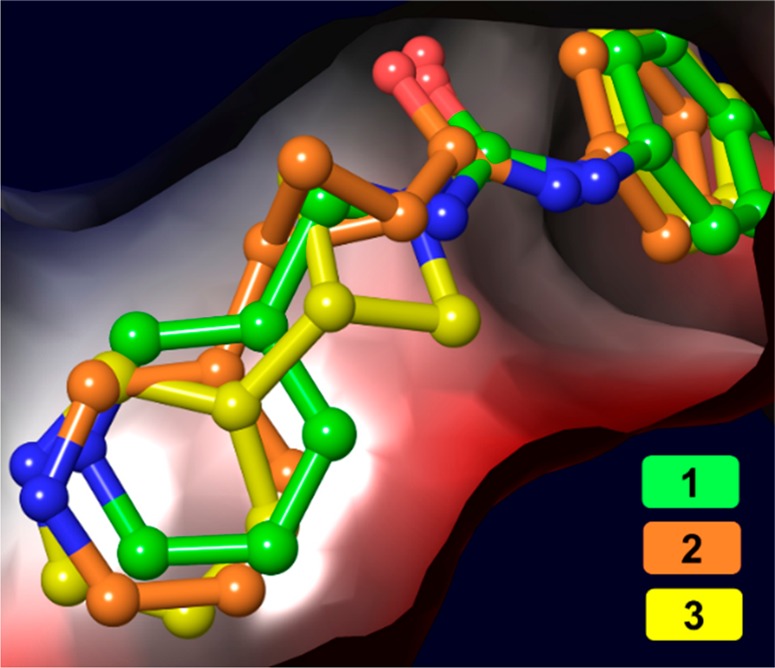
Overlay of urea 1 (PDB: 6ATB), cyclopropane 2 (PDB: 6AZJ), and urea 3 (PDB: 6PEB) in the NAM binding pocket of NAMPT. As shown above, there is a large degree of spatial overlap for the three pyridine rings.
Before commencing an optimization campaign for this scaffold, we designed experiments to confirm that urea 3 is functioning directly through NAMPT inhibition. First, we added the biochemical product of NAMPT, NMN, to our cell based assays and found that this completely rescues A2780 and COR-L23 cells from cell death induced by urea 3. Next, we demonstrated that 3 directly inhibits NAMPT activity in a biochemical inhibition assay (Figure 1B, IC50: 2.7 nM). Furthermore, we obtained a cocrystal structure of urea 3 bound to NAMPT, as shown in Figure 2, and found that the pyridine of 3 resides in the NAM binding pocket, consistent with 3 acting as a NAMPT substrate-based inhibitor.14 Interestingly, when urea 1, cyclopropane 2, and urea 3 are overlaid in the NAMPT active site (Figure 2), all three pyridine nitrogens are very closely aligned, further evidence that the trajectory of the nitrogen is critical for the substrate-based mechanism of these compounds,14 and collectively, these data strongly suggest that NAMPT is the cellular efficacy target of urea 3. Confident that analogs of 3 would also directly inhibit NAMPT, we chose to drive our compound optimization program through cell based assays given that our ultimate goal was a small molecule with in vivo efficacy.
As summarized in Figure 1C, we next generated the isoindol-1-one azetidine urea 4 and found that removing one of the pthalimide oxygens improves the activity of the compound approximately 10-fold in A2780 cells. We then determined if the four-membered nitrogen ring was the ideal ring size for cellular activity. To this end, we prepared both enantiomers of the pyrrolidine ring, compounds 5 and 6, as well as the achiral piperidine urea 7. In our primary assays, we found that all three compounds were several orders of magnitude less potent against both A2780 and COR-L23 cells. With this clarifying result in hand, we decided to focus on the azetidine urea and further elaborate the SAR of this series.
To further enable additional investigations into the structure activity relationship (SAR) of this series, we first prepared the key 3-pyridyl azetidine building blocks 16,1517, and 18 shown in Scheme 1. The synthesis begins with either 3-bromo pyridine 8 or the bis-halogenated pyridines 9 and 10. A Negishi coupling with 3-iodo N-Boc azetidine provides the essential carbon–carbon bond for these compounds and yields the Boc protected intermediates 11, 12, and 13. To then install an amine at either the 5 or 6 position of the ring (see below for further details), we employed benzophenonimine as a nitrogen source using Buchwald–Hartwig conditions followed by deprotection to generate anilines 14 and 15. Finally, the Boc group was removed with trifluoroacetic acid to provide the azetidine building blocks 16, 17, and 18.
Scheme 1.

Reagents and conditions: (a) Zn, I2, Pd2(dba)3, N-Boc-3-iodoazetidine, tri(o-tolyl)phosphine, DMF, 50 °C, 90 min (Compounds 8 and 9); (b) Zn, I2, Pd2(dba)3, N-Boc-3-iodoazetidine, tri-2-furylphosphine; (c) benzophenonimine, sodium tert-butoxide, Pd2(dba)3, toluene, 100 °C, 3 h, then 50% hydroxylamine in water, MeOH, 25 °C, 18 h; (d) TFA, DCM, 25 °C, 3 h.
For our optimization campaign, we were interested in the (S)-phenylethyl amide motif that we had previously identified9 to determine if it was also optimal for the azetidine ureas. In this vein, we prepared the collection of (S)-phenylethyl amide building blocks 21a–f shown in Scheme 2. The synthesis of these compounds began with commercially available (S)-1-(4-nitrophenyl)ethan-1-amine 19 that was functionalized either through a reductive amination/condensation sequence for intermediate 20a or through an amide bond formation for intermediates 20b–f. Next, the aryl nitro group was reduced with zinc to set the stage for the final urea bond formation. Anilines 21a–f and pyridyl azetidines 16–18 were then combined with phenyl chloroformate in this convergent route to produce pyridines 22a–f, 6-amino pyridines 23a–f, and 5-amino pyridines 24a–f.
Scheme 2.

Reagents and conditions: (a) methyl 2-formylbenzoate, DIPEA, sodium triacetoxyborohydride, DCM, 25 °C, 14 h (for 20a); (b) HATU, or COMU, DIPEA, DMF, 25 °C, 2 h; (c) Zn, NH4Cl, EtOH/H2O, 50 °C, 1 h; (d) amine, DIPEA, phenyl chloroformate, DCE 0 °C, 30 min then azetidine, 40 °C, 16 h.
Upon completion, the compounds were tested in our primary assays, and the results are shown in Table 1. In addition to NAMPT activity, we obtained physiochemical (solubility, permeability) and ADME data (mouse microsomal clearance, CYP3A4 inhibition). We found that cellular activity was broadly tolerant of structural modifications to the (S)-phenylethylamide moiety. Initially, we were interested if the (S)-phenylethylamine led to enhanced activity relative to the unsubstituted benzyl amine. Comparing the matched benzylamine isoindol-1one 4 (Figure 1B) and the (S)-phenylethyl isoindol-1-one (22a, Table 1), we found that the methyl group in this series afforded similar cellular potency unlike the clear improvement observed in our previous cyclopropyl carboxamide series.6 Even though a potency enhancement was not observed, phenyl containing amides were still sufficiently active in the A2780 and COR-L23 assays as demonstrated by 22a and 3-fluoro phenyl amide 22b, but they were both moderately potent in the CYP3A4 assay (3.04 and 3.11 μM, respectively). A heteroaryl amide such as pyrimidine 22c eliminated CYP3A4 inhibition, but this substitution also led to an approximately 10-fold decrease in potency and a significant reduction in passive permeability as measure by the MDCK assay.
Table 1. Cellular Activity and in Vitro ADME Assays for Compounds 22a–22f.
| Compound | A2780 IC50 (nM) | COR-L23 IC50 (nM) | MLMa (μL min–1 mg–1) | CYP3A4 (midazolim, μM) | HTb solubility pH 6.8 (mM) | MDCK MDR1 LEc A to B (×10–6 cm/s) |
|---|---|---|---|---|---|---|
| 22a | 1.5 | 8 | 123.3 | 3.04 | 0.130 | 16.26 |
| 22b | 2.3 | 10 | 60.9 | 3.11 | 0.011 | 12.5 |
| 22c | 36 | 130 | 28.9 | >25 | 0.930 | 0.66 |
| 22d | 17 | 58 | 58 | >25 | 0.380 | 0.89 |
| 22e | 5.7 | 38 | 40.8 | >25 | >1.0 | 0.65 |
| 22f | 25 | 120 | 31.4 | >25 | 0.700 | 1.99 |
| 23a | 5.9 | 19 | <25 | >25 | 0.220 | 5.89 |
| 23b | 7.6 | 27 | <25 | >25 | 0.032 | 4.58 |
| 23c | 68 | 160 | <25 | >25 | 0.30 | 0.55 |
| 23d | 46 | 110 | <25 | >25 | 0.310 | 0.51 |
| 23e | 11 | 41 | <25 | >25 | 0.760 | 0.83 |
| 23f | 58 | 120 | 22 | >25 | 0.760 | 0.68 |
| 24a | 45 | 130 | 157.8 | 4.69 | 0.035 | 3.04 |
| 24b | 240 | 630 | 27.4 | >25 | 0.017 | 1.05 |
| 24c | 340 | 1380 | 83.4 | >25 | 0.300 | 0.58 |
| 24d | 500 | 970 | 60.2 | >25 | 0.440 | 0.70 |
| 24e | 34 | 200 | 47.4 | >25 | 0.820 | 0.66 |
| 24f | 770 | 1580 | 56.9 | >25 | 0.230 | 0.71 |
MLM: mouse liver microsomes.
High throughput.
Madin–Darby canine kidney multidrug resistance low expressing.
We next investigated the activity of compounds with aliphatic amides, as exemplified by pyran 22d, N-methyl proline 22e, and ethyl 22f. The aliphatic groups were less active in the cell based assays than their aromatic counterparts, but this was compensated by improvement in other dimensions. For example, all three of these compounds showed no measurable activity in the CYP3A4 inhibition assay. In addition, these compounds had a high aqueous solubility, especially when compared to 3-fluorophenyl analogue 22b. In contrast, however, the passive permeability of the aliphatic amides significantly decreased relative to the aromatic amides (Table 1). Combined, the reduced permeability and cellular activity of the aliphatic amides prevented their further progression.
Intrigued by the promising cellular activity of the aromatic amides, we explored other avenues to improve NAMPT activity while reducing CYP3A4 inhibition. Since 6-aminopyridine had been shown to reduce CYP inhibition in the NAMPT inhibitor literature,16 we wanted to investigate the role of this motif in the cellular activity of our scaffold. We also wanted to investigate 5-amino pyridines because, to our knowledge, this substructure had not been utilized in a NAMPT inhibitor. The results of both of these SAR investigations are summarized in Table 1.
For the 6-amino pyridines, 23a–f, these compounds generally suffer a moderate potency loss compared to the des-amino analogues. Examining the most potent matched pair as an example, the des-amino 22a has cellular IC50’s of 1.5 and 8 nM against A2780 and COR-L23, respectively, while the analogous values for 6-amino 23a are 5.9 and 19 nM. However, this loss in potency is compensated by a significant decrease in mouse microsomal clearance (22a: 123 μL/min mg, 23a: < 25 μL/min mg) and CYP3A4 inhibition (22a: 3.04 μM, 23a: >25 μM). In contrast to the improvements in these ADME properties, adding the amino group to the scaffold led to a significant reduction in passive permeability (compare the permeability of 22a and 23a for example). Therefore, despite the attractive improvement in clearance and CYP inhibition, the reduction in cellular activity and permeability relative to the des-amino analogues led to their deprioritization.
Next, the cellular activities of the 5-amino compounds 24a–f were interrogated. This substitution led to a significant reduction in cellular activity relative to the des-amino or 6-amino analogues. For example the A2780 IC50 of 3-fluoro phenyl 24b is 240 nM, which is a 100-fold drop in potency relative to des-amino 22b. Similar declines in potency were observed across the 5-amino analogues, and they were deprioritized from further investigations.
Since we were unable to improve the potency of this scaffold by modifying the pyridine ring, we returned to the potential of the amide to improve cellular activity. The potency of the fused isoindol-1-one stood out to us, and we were curious if changing the position of the fused aryl ring would impact activity. Furthermore, we were inspired by the use of heterocycles as amide replacements in the angiotensin II receptor blocker literature,17 and we hypothesized that replacing the amide could improve the passive permeability of the series. Finally, we wanted to revisit compounds that lacked the (S)-phenylethylamine since this modification did not offer any advantage in terms of cellular potency when comparing matched pairs 4 (A2780 IC50: 0.6 nM) and 22a (A2780 IC50: 1.5 nM). To this end, we prepared the compounds shown in Table 2 using a similar route as depicted in Scheme 2 (see Supporting Information for further details of the syntheses).
Table 2. SAR of Amide Replacements.

Altering the orientation of the fused aryl ring had the desired effect, and compounds 25–28 are among the more potent analogs that we synthesized in this series of NAMPT inhibitors but do not significantly exceed the potency of our previously identified cyclopropylcarboxamides.9 Furthermore, we found that replacing the amide with a heterocycle as in compounds 26–28 led to high levels of passive permeability, validating our hypothesis. Given the single digit nanomolar potency of benzoimidazole 27, we next wanted to determine the suitability of this compound for in vivo studies. Time course oral pharmacokinetics in nude mice was obtained for 27 up to 30 mg/kg, and this data is plotted in Figure 3A as the fraction unbound (27 is 96.2% bound to mouse plasma protein). Comparing this exposure data to the COR-L23 IC50 (3.9 nM), we found that we would need to dose at least 5 mg/kg twice daily in order to maintain exposure above this level in mice. In advance of an in vivo efficacy experiment, we first tested the tolerability of nude mice to twice daily repeat oral dosing of 27. As shown in Figure 3B, at all doses tested, there was a significant amount of body weight loss in the mice relative to vehicle. This is in contrast to our previous experience,9 and one potential explanation lies in the differences in PK of the two compounds. At a 10 mg/kg oral dose 27 has both a higher Cmax (29,112 nmol/L) and AUC0–7 h (67,413 nmol·hr/L) compared to cyclopropane SI-9 (Cmax = 12,691 nmol/L; AUC0–7 h = 19,187 nmol·hr/L, see Supporting Information for more details).9 It is therefore possible that mutiple dosing of 27 led to toxic levels of NAMPT inhibition yielding the observed intolerability in mice. These data are consistent with the known risk of on-target toxicity with this mechanism11,12 and are likely due to direct target inhibition rather than an event specific to the azetidine urea motif. Based on the data in Figure 3A, a twice daily dose of 0.3 or 1 mg/kg could potentially provide the required target coverage, but we did not have an opportunity to test the tolerability of these doses. Given the weight loss data for 27, we decided to stop further exploration of this compound in vivo.
Figure 3.

(A) Time course of calculated unbound plasma concentrations of 27 at oral doses of 30, 5, 1, and 0.3 mg/kg. 27 is 96.2% bound to mouse plasma protein. (B) Percent weight loss induced by oral dosing of 27 to nude mice. 27 was dosed twice per day.
In summary, a scaffold morph from a cyclopropyl carboxamide led to the identification of 3-azetidine urea NAMPT inhibitors. Through biochemical experiments we were able to show that these compounds were acting directly on NAMPT function and initiated a lead optimization campaign based on this substructure. A convergent synthetic route was developed, including the highly versatile bifunctional pyridine building blocks 17 and 18, to explore this SAR. We found that a broad range of substituents was tolerated on the eastern portion of the molecule and that the unsubstituted pyridine provided the best blend of cellular potency and ADME properties. These efforts culminated in the highly potent NAMPT inhibitor 27, though we were unable to achieve our stated aim of improving upon the potency of the cyclopropane series. This compound was tested in a repeat dose tolerability study in nude mice in preparation for an in vivo efficacy experiment, but we found that it was not tolerated at 5 mg/kg twice daily. Based on these data that highlight the potential for on-target NAMPT mediated intolerability, we decided not to pursue further evaluation of this compound.
Acknowledgments
We thank the in vitro ADME group for generating the data in Tables 1 and 2, Scott Busby for the biochemical inhibition data in Figure 1, Giuliano Berellini for contributing to Figure 3A, and Dallas Bednarczyk for data analysis.
Glossary
Abbreviations
- NAD
nicotinamide adenine dinucleotide
- PARP
poly (ADP-ribose) polymerase
- NAM
nicotinamide
- PRPP
phosphoribosyl pyrophosphate
- NMN
nicotinamide mononucleotide
- NAMPT
nicotinamide phosphoribosyltransferase
- NMNAT
nicotinamide mononucleotide adenylyltransferase
- ADC
antibody–drug conjugate
- ADME
absorption, distribution, metabolism, and excretion
- CTG
Cell Titer Glo
- SAR
structure–activity relationship
- MLM
mouse liver microsome
- CYP
cytochrome P450
- HT
high throughput
- MDCK
Madin–Darby canine kidney
- MDR1
multidrug resistance
- LE
low expressing
- PK
pharmacokinetics
- MTD
maximum tolerated dose.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.9b00325.
Author Present Address
§ (D.B.) Casma Therapeutics, 400 Technology Square, Suite 201, Cambridge, MA 02139.
This research was funded by Novartis Inc.
The authors declare the following competing financial interest(s): The authors were all employees of the Novartis Institutes for Biomedical Research during the time when this work was completed.
Supplementary Material
References
- Belenky P.; Bogan K. L.; Brenner C. NAD+ metabolism in health and disease. Trends Biochem. Sci. 2007, 32, 12–19. 10.1016/j.tibs.2006.11.006. [DOI] [PubMed] [Google Scholar]
- Bogan K. L.; Brenner C. Nicotinic acid, nicotinamide, and nicotinamide riboside: a molecular evolution of NAD+ precursor vitamins in human nutrition. Annu. Rev. Nutr. 2008, 28, 115–130. 10.1146/annurev.nutr.28.061807.155443. [DOI] [PubMed] [Google Scholar]
- Revollo J. R.; Grimm A. A.; Imai S. The NAD biosynthesis pathway mediated by nicotinamide phosphoribosyltransferase regulates Sir2 activity in mammalian cells. J. Biol. Chem. 2004, 279, 50754–63. 10.1074/jbc.M408388200. [DOI] [PubMed] [Google Scholar]
- Rongvaux A.; Galli M.; Denanglaire S.; Van Gool F.; Drèze P. L.; Szpirer C.; Bureau F.; Andris F.; Leo O. Nicotinamide phosphoribosyl transferase/pre-B cell colony-enhancing factor/visfatin is required for lymphocyte development and cellular resistance to genotoxic stress. J. Immunol. 2008, 181, 4685–4695. 10.4049/jimmunol.181.7.4685. [DOI] [PubMed] [Google Scholar]
- Mei S. C.; Brenner C. NAD as a Genotype-Specific Drug Target. Chem. Biol. 2013, 20, 1307–1308. 10.1016/j.chembiol.2013.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sampath D.; Zabka T. S.; Misner D. L.; O’Brien T.; Dragovich P. S. Inhibition of nicotinamide phosphoribosyltransferase (NAMPT) as a therapeutic strategy in cancer. Pharmacol. Ther. 2015, 151, 16–31. 10.1016/j.pharmthera.2015.02.004. [DOI] [PubMed] [Google Scholar]
- Galli U.; Travelli C.; Massarotti A.; Fakhfouri G.; Rahimian R.; Tron G. C.; Genazzani A. A. Medicinal Chemistry of nicotinamide phosphoribosyltransferase (NAMPT) inhibitors. J. Med. Chem. 2013, 56, 6279–6296. 10.1021/jm4001049. [DOI] [PubMed] [Google Scholar]
- Montecucco F.; Cea M.; Bauer I.; Soncini D.; Caffa I.; Lasigliè D.; Nahimana A.; Uccelli A.; Bruzzone S.; Nencioni A. Nicotinamide phosphoribosyltransferase (NAMPT) inhibitors as therapeutics: rationales, controversies, clinical experience. Curr. Drug Targets 2013, 14, 637–643. 10.2174/1389450111314060003. [DOI] [PubMed] [Google Scholar]
- Palacios D. S.; Meredith E.; Kawanami T.; Adams C.; Chen X.; Darsigny V.; Geno E.; Palermo M.; Baird D.; Boynton G.; Busby S. A.; George E. L.; Guy C.; Hewett J.; Tierny L.; Thigale S.; Weihofen W.; Wang L.; White N.; Yin M.; Argikar U. A. Structure based design of nicotinamide phosphoribosyltransferase (NAMPT) inhibitors from a phenotypic screen. Bioorg. Med. Chem. Lett. 2018, 28, 365–370. 10.1016/j.bmcl.2017.12.037. [DOI] [PubMed] [Google Scholar]
- Karpov A. S.; Abrams T.; Clark S.; Raikar A.; D’Alessio J. A.; Dillon M. P.; Gesner T. G.; Jones D.; Lacaud M.; Mallet W.; Martyniuk P.; Meredith E.; Mohseni M.; Nieto-Oberhauber C. M.; Palacios D. S.; Perruccio F.; Piizzi G.; Zurini M.; Bialucha C. U. Nicotinamide Phosphoribosyltransferase Inhibitor as a Novel Payload for Antibody-Drug Conjugates. ACS Med. Chem. Lett. 2018, 9, 838–842. 10.1021/acsmedchemlett.8b00254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zabka T. S.; Singh J.; Dhawan P.; Liederer B. M.; Oeh J.; Kauss M. A.; Xiano Y.; Zak M.; Lin T.; McCray B.; La N.; Nguyen T.; Beyer J.; Farman C.; Uppal H.; Dragovich P. S.; O’Brien T.; Sampath D.; Misner D. L. Retinal toxicity, in vivo and in vitro, associated with inhibition of nicotinamide phosphoribosyltransferase. Toxicol. Sci. 2015, 144, 163–172. 10.1093/toxsci/kfu268. [DOI] [PubMed] [Google Scholar]
- Tarrant J. M.; Dhawan P.; Singh J.; Zabka T. S.; Clarke E.; DosSantos G.; Dragovich P. S.; Sampath D.; Lin T.; McCray B.; La N.; Nguyen T.; Kauss A.; Dambach D.; Misner D. L.; Diaz D.; Uppal H. Preclinical models of nicotinamide phosphoribosyltransferase inhibitor-mediated hematotoxicity and mitigation by co-treatment with nicotinic acid Toxicol. Toxicol. Mech. Methods 2015, 25, 201–211. 10.3109/15376516.2015.1014080. [DOI] [PubMed] [Google Scholar]
- Azetidine urea based NAMPT inhibitors have also been described in the patent literature: Curtin A. L.; Longenecker K.; Hansen T. M.; Clark R. F.; Sorenson B.; Heyman H. R.; Ji Z.. NAMPT Inhibitors. U.S. Patent 0336168, November 13, 2014.
- Oh A.; Ho Y.-C.; Zak M.; Liu Y.; Chen X.; Yuen P.-W.; Zheng X.; Liu Y.; Dragovich P. S.; Wang W. Structural and Biochemical Analyses of the Catalysis and Potency Impact of Inhibitor Phosphoribosylation by Human Nicotinamide Phosphoribosyltransferase. ChemBioChem 2014, 8, 1121–1130. 10.1002/cbic.201402023. [DOI] [PubMed] [Google Scholar]
- Claffey M. M.; Helal C. J.; Verhoest P. R.. Amino-heterocyclic compounds. U.S. Patent 01990771, July 29, 2010.
- Gunzner-Toste J.; Zhao G.; Bauer P.; Baumeister T.; Buckmelter A. J.; Caligiuri M.; Clodfelter K. H.; Fu B.; Han B.; Ho Y.-C.; Kley N.; Liang X.; Liederer B. M.; Lin J.; Mukadam S.; O’Brien T.; Oh A.; Reynolds D. J.; Sharma G.; Skelton N.; Smith C. C.; Sodhi J.; Wang W.; Wang Z.; Xiao Y.; Yuen P.-w.; Zak M.; Zhang L.; Zheng X.; Bair K. W.; Dragovich P. S. Discovery of potent and efficacious urea-containing nicotinamide phosphoribosyltransferase (NAMPT) inhibitors with reduced CYP2C9 inhibition properties. Bioorg. Med. Chem. Lett. 2013, 23, 3531–3538. 10.1016/j.bmcl.2013.04.040. [DOI] [PubMed] [Google Scholar]
- Wexler R. R.; Greenlee W. J.; Irvin J. D.; Goldberg M. R.; Prendergast K.; Smith R. D.; Timmermans P.B.M.W.M. Nonpeptide Angiotensin II Receptor Antagonists: The Next Generation in Antihypertensive Therapy. J. Med. Chem. 1996, 39, 625–656. 10.1021/jm9504722. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.