

Abstract
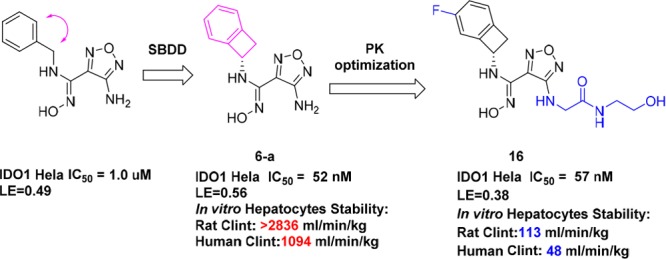
Checkpoint inhibitors have demonstrated unprecedented efficacy and are evolving to become standard of care for certain types of cancers. However, low overall response rates often hamper the broad utility and potential of these breakthrough therapies. Combination therapy strategies are currently under intensive investigation in the clinic, including the combination of PD-1/PD-L1 agents with IDO1 inhibitors. Here, we report the discovery of a class of IDO1 heme-binding inhibitors featuring a unique amino-cyclobutarene motif, which was discovered through SBDD from a known and weakly active inhibitor. Subsequent optimization efforts focused on improving metabolic stability and were greatly accelerated by utilizing a robust SNAr reaction of a facile nitro-furazan intermediate to quickly explore different polar side chains. As a culmination of these efforts, compound 16 was identified and demonstrated a favorable overall profile with superior potency and selectivity. Extensive studies confirmed the chemical stability and drug-like properties of compound 16, rendering it a potential drug candidate.
Keywords: Indoleamine-2,3-dioxygenase 1; IDO1; cyclobutarene; electrocyclic ring opening; cancer immunotherapy
The field of cancer immunotherapy, in which the power of the host immune system is leveraged against diseased tissues, has witnessed great progress in the past few years.1,2 Immune checkpoint inhibitors, such as monoclonal antibodies (mAbs) targeting programmed cell death protein 1 (PD-1), programmed death ligand 1 (PD-L1), and cytotoxic T-lymphocyte antigen 4 (CTLA-4), have demonstrated unprecedented and enduring efficacy in a variety of cancers, including subtypes typically resistant to conventional therapies.3,4 Despite these breakthroughs, the overall response rate of these novel antitumor therapies remains low, limiting their potential to benefit broad patient population.3,4 To address this issue, different combination strategies are being intensively explored, including chemotherapy and radiotherapy as well as those mechanisms capable of overcoming tumor-induced local immunosuppression.5,6
Indoleamine-2,3-dioxygenase 1 (IDO1) is a heme-containing enzyme, which catalyzes the first and rate-limiting step of tryptophan catabolism, also known as the kynurenine pathway.7−9 The initially formed metabolite N-formyl-kynurenine (NFK) undergoes further degradation and leads to the formation of several downstream metabolites, which together are called kynurenines. In the tumor microenvironment, the kynurenine pathway can be hijacked as a mechanism of immune escape. IDO1 over- or induced- expression is often associated with poor prognosis in a variety of cancer types and can lead to local depletion of tryptophan, which is essential for T cell proliferation and activity. Furthermore, kynurenines themselves are also reported to be immuno-suppressive through either activation of the AhR pathway or upregulation of regulatory T cells.10−12 Therefore, inhibition of IDO1 by a small molecule inhibitor has the potential to unleash the host immune response by blocking tumor-induced immunosuppression, and its combination with anti-PD-1 or anti-PD-L1 agents could further improve response rates and offer broader clinical efficacy.10−12 Thus, targeting IDO1 for cancer immunotherapy has attracted great attention, and a diverse class of IDO1 inhibitors has been disclosed in the past few years.10−12
In general, most of the reported inhibitors can be classified into two types depending on their mechanism of inhibition (Figure 1). The first class of inhibitors can be termed “heme-binding”, an example of which is Epacadostat.13 This class of compounds inhibits IDO1 activity through binding directly to the heme moiety present in the active site of IDO1. The second class of inhibitors can be termed “heme-displacing”, as exemplified by BMS-986205.14 This class of inhibitors functions by competing with and displacing the heme moiety, probably during the IDO1 protein synthesis and folding stage.14 Here, we describe the discovery and optimization of a class of heme-binding inhibitors guided by structural based drug design (SBDD). This class of inhibitors features a unique amino-cyclobutarene motif, which was extensively derisked for potential decomposition due to electrocyclic ring opening, confirming their suitability for further progression.
Figure 1.
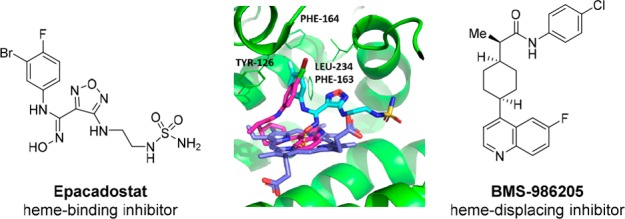
Overlay of cocrystal structures of Epacadostat (cyan) and BMS-986205 (magenta) bound to IDO1. Heme molecule depicted in blue.
We began our investigation by exploring several hit-finding approaches simultaneously, including virtual screening, high throughput screening (HTS), and SBDD drawing inspiration from literature.12 The first two approaches only resulted in identification of some weakly active hits, which consisted mainly of imidazole and its derivatives. These findings are consistent with what has been reported regarding some unique structural features around the IDO1 active binding pocket for heme-binding inhibitors. As Röhrig et al. nicely summarized in 2015,12 the majority of the known and efficient IDO1 heme-binders often contain one heme binding element (either N or O) and one aromatic ring tightly binding to a narrow lipophilic pocket (termed as A pocket). For example, Epacadostat binds the heme moiety through the oxygen atom of its hydroxyamidine moiety, while its halogenated phenyl ring situates in the A pocket and contributes significantly to its binding affinity. In light of these disappointing hits from a screening strategy, our hit finding strategy through SBDD soon evolved to be our major focus of exploration.
Furazanyl hydroxyamidine 1 was known to be a weakly active but efficient IDO1 heme binding inhibitor (Figure 2).15 Based on docking, we envisioned that benzo-fused analogs 2, in particular, 6,4-fused cyclobutarene core analogs, might be tolerated and fit even better into the narrow lipophilic A pocket. To test this hypothesis, a series of analogs with varying ring sizes were prepared, and the results are summarized in Table 1. Although 6,6-fused analog 3 was only weakly active in the IDO1 enzymatic assay, its 6,5-fused counterpart seemed more encouraging. In addition, the absolute stereochemistry was crucial and (S)-isomer 4-b was favored. Translocation of the substitution position (5) was not tolerated, which was also consistent with the modeling prediction. The 6,4-fused cyclobutarene analog further improved potency, and its more active (S)-isomer 6-a exhibited excellent potency in both enzymatic and cellular assays.
Figure 2.
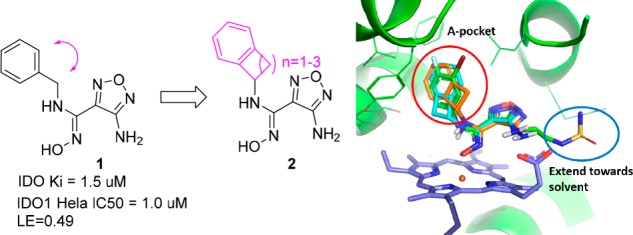
SBDD strategy and overlay of designed cyclobutarenes (cyan and orange) with Epacadostat (green).
Table 1. SAR of A-pocket Exploration.


IC50 values are the mean of at least two runs.
NA stands for not tested.
Upon identifying the 6,4-fused cyclobutarene motif as optimal, our attention shifted to introducing additional substituents around the cyclobutarene core. Modeling studies indicated that small substituents might be tolerated or even beneficial to further optimize interactions with the binding pocket.12,15 Although neither methyl nor gem-difluoro substitution on the four-membered ring (7 and 8) was allowed, fluoro substitution within the aromatic ring was well tolerated (9–11). Particularly, incorporation of a 5-F substitution (11-a) improved cellular potency 2-fold.
With the 5-F-substituted cyclobutarene moiety in hand as an optimal piece replacing the benzyl group present in compound 1, we next focused our attention toward improving metabolic stability, as furazanyl amine 11-a showed very poor in vitro stability (Table 2). Our strategy aimed at incorporating polar side chain substituents at the 3-position of the furazan ring, which had the potential of improving both metabolic stability and other ancillary physicochemical properties.16 Modeling indicated that side chains at this position likely would extend toward solvent and thus would have limited effect on binding affinity.15,17 To accelerate our SAR exploration, it was desirable to utilize the furazanyl amino group in 11-a directly as a synthetic handle for rapid exploration of various side chains. However, it was known that such furazanyl amine is highly deactivated and resistant to react under typical reductive amination or alkylation conditions, due to the strongly electron withdrawing nature of the furazanyl ring.17 Although an alternative route through a Boulton/Katrizky rearrangement18 was known and could be employed to bypass this issue, we sought to identify a more direct transformation to expedite our SAR exploration. Gratifyingly, it was soon discovered that compound 11-a could be converted to a highly versatile furanzanyl nitro intermediate 12 by following a two-step sequence19,20 (Table 2) (Need to use with caution on large scale due to the highly exothermic nature of 12 at elevated temperature!).20 The corresponding NO2 group was a superior leaving group for SNAr chemistry and was readily displaced by a variety of nucleophiles under mild conditions.21,22
Table 2. Side Chain Optimization via Facile SNAr To Improve Metabolic Stability.


IC50 values are the mean of two or more runs.
Human whole blood potency and unbound potency.
AlogP was calculated according to the method described in ref (23).
HPLC measured value.
Rat in vivo PK total and intrinsic clearance.
Rat in vivo PK mean residence time (MRT).
Unbound volume of distribution.
Human in vitro hepatocytes intrinsic clearance.
Through this modified route, a diverse set of analogs bearing different N-linked side chains were quickly prepared in parallel from 12. As expected, while these analogs (13–16) showed comparable potency in both enzymatic and cellular assays (Table 2), the varying side chains had a significant effect on the metabolic stability of the compounds. For example, parent compound 11-a suffered from a poor in vivo pharmacokinetic profile incompatible with QD dosing in human. Introduction of an AcNHCH2CH2NH– side chain (13) did not improve the in vivo pharmacokinetic profile too much, both compounds having extremely short MRTs. Switching the terminal residue in 13 from N–Ac to a more polar group (N-SO2NH2, 14) resulted in significant improvement in the rat MRT mediated by a significant reduction in the intrinsic clearance relative to minor change in the unbound volume. A similar trend was observed between analogs 15 and 16. Substituting a more polar hydroxyethyl amide (16) for the terminal primary amide in 15 further improved the MRT again through improvements in intrinsic clearance relative to minor changes in unbound volume. Compound 16 was also found to have excellent in vitro stability in human hepatocytes.
In addition to N-linked side chains, S-linked side chains were also tolerated to some extent (17–19). The S-linked analogs generally had similar overall profiles relative to the N-linked analogs. Compound 19, the S-linked analog of compound 16, had a similar intrinsic potency to compound 16, and an inferior pharmacokinetic profile (Table 2).
Having identified compound 16 as an optimal combination of potency and metabolic stability, we selected it for further profiling. Initially we were pleasantly surprised to discover that 16 displayed good cell permeability and in vivo bioavailability (Figures 3 and 4). In addition to its low calculated logP value, several other parameters including H-bond donor/acceptor counts and PSA also fall out of the typical preferred value range for oral absorption.16 A correlation analysis of calculated AlogP and rat in vivo unbound clearance (Figure 3) revealed that, within this class of compounds, metabolically more stable analogs tend to display lower AlogP values (−1.0 to +1.0) than the usual range preferred for good absorption (+1.0 to +3.0).16 This was likely due to the presence of multiple intramolecular H-bonds, which partially masks some of the polar features in the compounds.24,25 This hypothesis also aligns well with the experimentally measured HPLC logD value (Table 2) and a similar observation reported for Epacadostat.17
Figure 3.
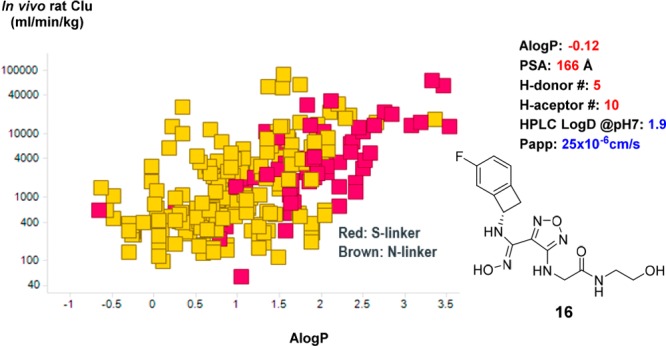
Correlation of AlogP and rat in vivo unbound clearance. AlogP was calculated according to the method described in the literature.23
Figure 4.

Overall profile of 16.
As summarized in Figure 4, compound 16 showed good IDO1 inhibition across species and superior metabolic stability across species. In addition, 16 was clean in standard off-target profiling including selectivity against a set of 108 targets in a Eurofins Panlabs panel. Compound 16 also showed a favorable profile in preclinical toxicity studies, and no significant adverse events were observed. On the basis of its good whole blood potency and superior pharmacokinetic stability, 16 was predicted to have a lower projected human dose than Epacadostat (BID), with potential for QD dosing.
We were able to obtain a cocrystal structure of compound 17 bound to IDO1 protein (Figure 5). Compound 17 adopts a similar binding mode as that of Epacadostat, binding to heme through an oxygen atom present in its hydroxyamidine motif.13 The 5-F-substituted cyclobutarene ring resides in the A pocket, and its oxadiazole ring is slightly shifted relative to Epacadostat, likely to accommodate the bulky cyclobutarene moiety. The side chains of both compounds extend toward the solvent region.
Figure 5.
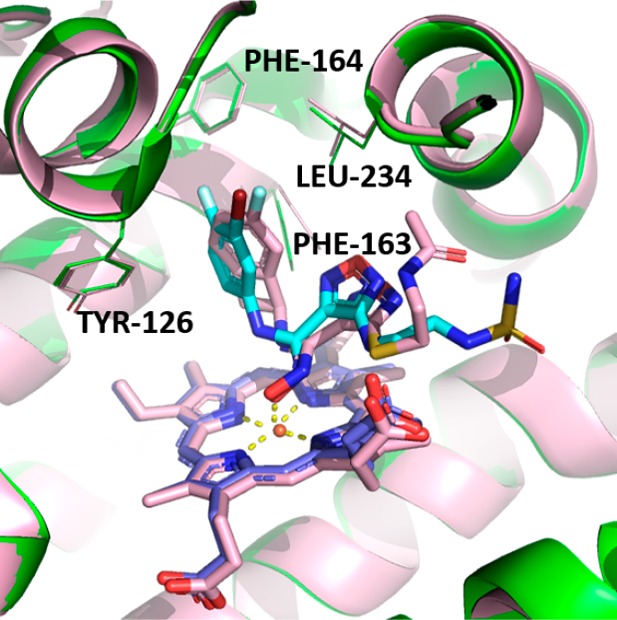
Overlay of cocrystal structures of 17 (pink) and Epacadostat (cyan) bound to IDO1.
While we were encouraged by the favorable profile of compound 16, cyclobutarene derivatives are known to undergo conrotatory electrocyclic ring opening to ortho-quinodimethanes under thermal conditions.26 The triggering temperature of such ring opening often depends on the nature of substituents on the cyclobutene ring (Figure 6).26 Electron-donating substituents such as OH or NH2 can dramatically decrease thermal stability and lead to decomposition even at ambient temperature. However, such instability can be mitigated by capping the free NH2 group with electron withdrawing groups, such as amides or carbamates.26 Hence, soon after the discovery of this class of amino-cyclobutarene analogs, we initiated studies to evaluate the chemical stability of our compounds as a key determinant of their suitability as potential drug candidates.
Figure 6.

Theoretical and experimental assessments of electrocyclic ring opening potential of amino-substituted cyclobutarenes.
The activation barriers for electrocyclic ring opening (ΔG‡) were computed by density functional theory calculations27−31 with the quasiharmonic approximation proposed by Cramer and Truhlar32 to assess whether there was correlation with experimental observation. For the free amine analog 11, the computed ΔG‡ values of 27–28 kcal/mol suggested that the analogs were prone to ring-opening at room temperature, consistent with reported experimental results.26 Protonation of 11 was predicted to increase the ring-opening activation barrier to 42.6 kcal/mol, which was well aligned with the much better stability of the HCl salt form 11 observed experimentally. Structures 20 and 21 were selected as prototypes for calculation to model the N-linked and S-linked analogs, respectively. Both were found to have estimated ΔG‡ values of ∼33 kcal/mol and thus predicted to be much more stable than the free amine analog 11.
Encouraged by these results, 16 was evaluated experimentally and indeed showed good thermal stability. After heating at 100 °C for 3 h, greater than 88% of the parent remained intact. Compound 16 was also evaluated and exhibited good stability under a variety of conditions, including acidic, basic, oxidizing, photolytic, and homolytic conditions (Table 3). These results confirmed the chemical stability and drug-like properties of this class of molecules and supported their suitability as potential drug candidates.
Table 3. Chemical Stability Assessment of 16 under Various Conditions.
| % parent (3 h)a | % parent (24 h)a | |
|---|---|---|
| Thermal, 100 °C | 88.4 | NA |
| 0.01 N HCl, 40 °C | 98.1 | NA |
| 0.1 N HCl, 40 °C | NA | 96.2 |
| 0.01 N NaOH, 40 °C | 97.4 | NA |
| 0.1 N NaOH, 40 °C | NA | 81.5 |
| H2O2, ambient | NA | 97.0 |
| AIBN, 40 °C | NA | 77.1 |
aNA stands for not tested.
Encouraged by our stability and safety results, we proceeded to evaluate 16 in the EMT6 mouse syngeneic model either alone or in combination with anti-PD-1 mAb.33 Significant therapeutic benefit was observed in the combination group when compound 16 was administered (100 mg/kg, bid) together with anti-PD-1 (5 mg/kg, twice a week) (Figure 7).
Figure 7.
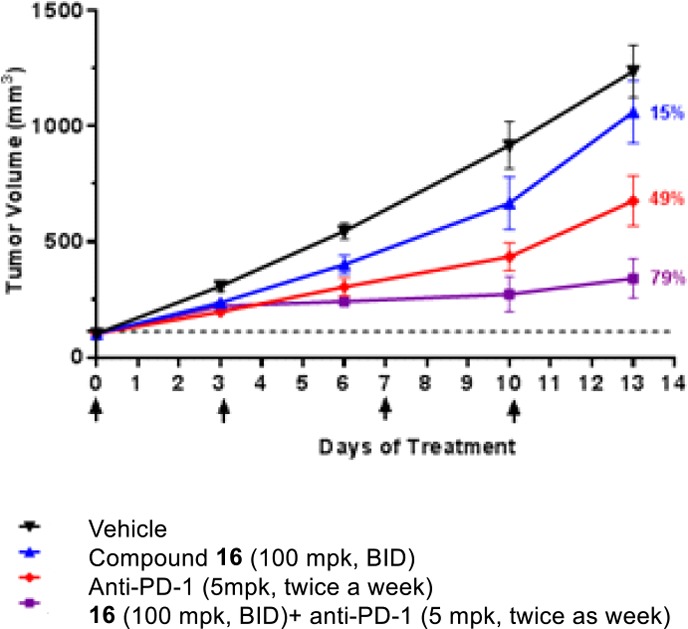
Combination treatment of compound 16 and anti-PD-1 significantly improves therapeutic benefit.
In conclusion, a class of novel IDO1 heme-binding inhibitors featuring an unprecedented amino-cyclobutarene motif in A-pocket was identified through SBDD. Further optimization of the side chain to improve metabolic stability was greatly accelerated by harnessing a nitro-furazan intermediate amenable to react with a variety of nucleophiles under mild conditions via SNAr reaction. Compound 16 was discovered and exhibited a very favorable overall profile, including excellent potency, selectivity, pharmacokinetics, and predicted human dose. Extensive theoretical and experimental studies were carried out to confirm the chemical stability of the amino-cyclobutarene motif and to reinforce its suitability as a component of potential drug candidates for further progression. Lead compound 16 demonstrated good efficacy synergy when combined with anti-PD-1 mAb in a mouse EMT6 tumor syngeneic model.
Acknowledgments
We thank Dr. Edward Sherer for assisting with VCD calculation and Drs. Matthew J. Mitcheltree, Min Lu, and Chunhui Huang for proofreading the manuscript and providing constructive suggestions. We also thank Drs. Nengxue Wang and his colleagues at Wuxi Apptec for preparation of several compounds described here.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.9b00344.
Synthetic procedures and analytical data for selected compounds, conditions for biological assays, VCD determination of the absolute stereochemistry of 16, X-ray statistics for 17, DFT calculation methods and results, and dose response curves of IC50S (PDF)
Accession Codes
The PDB code for 17 is 6pu7.
The authors declare no competing financial interest.
Supplementary Material
References
- Lizée G.; Overwijk W. W.; Radvanyi L.; Gao J.; Sharma P.; Hwu P. Harnessing the power of the immune system to target cancer. Annu. Rev. Med. 2013, 64, 71–90. 10.1146/annurev-med-112311-083918. [DOI] [PubMed] [Google Scholar]
- Yang Y. Cancer Immunotherapy: Harnessing the immune system to battle cancer. J. Clin. Invest. 2015, 125, 3335–3337. 10.1172/JCI83871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawlik A.; Machaj F.; Rosik J.; Szostak B. CTLA4 antagonists in phase I and phase II clinical trials, current status and future perspectives for cancer therapy. Expert Opin. Invest. Drugs 2019, 28, 149–159. 10.1080/13543784.2019.1559297. [DOI] [PubMed] [Google Scholar]
- Alsaab H. O.; Sau S.; Alzhrani R.; Tatiparti K.; Bhise K.; Kashaw S. K.; Iyer A. K. PD-1 and PD-L1 checkpoint signaling inhibition for cancer immunotherapy: mechanism, combinations, and clinical outcome. Front. Pharmacol. 2017, 8, 561. 10.3389/fphar.2017.00561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams J. L.; Smothers J.; Srinivasan R.; Hoos A. Big opportunities for small molecules in immuno-oncology. Nat. Rev. Drug Discovery 2015, 14, 603–622. 10.1038/nrd4596. [DOI] [PubMed] [Google Scholar]
- Huck B. R.; Kötzner L.; Urbahns K. Small molecules drive big improvements in immuno-oncology therapies. Angew. Chem., Int. Ed. 2018, 57, 4412–4428. 10.1002/anie.201707816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricciuti B.; Leonardi G. C.; Puccetti P.; Fallarino F.; Bianconi V.; Sahebkar A.; Baglivo S.; Chiari R.; Pirro M. Targeting indoleamine-2,3-dioxygenase in cancer: Scientific rationale and clinical evidence. Pharmacol. Ther. 2019, 196, 105–116. 10.1016/j.pharmthera.2018.12.004. [DOI] [PubMed] [Google Scholar]
- Brochez L.; Chevolet I.; Kruse V. The rationale of indoleamine 2,3-dioxygenase inhibition for cancer therapy. Eur. J. Cancer 2017, 76, 167–182. 10.1016/j.ejca.2017.01.011. [DOI] [PubMed] [Google Scholar]
- Prendergast G. C.; Mondal A.; Dey S.; Laury-Kleintop L. D.; Muller A. J. Inflammatory reprogramming with IDO1 inhibitors: turning immunologically unresponsive ‘cold’ tumors ‘hot’. Trends Cancer. 2018, 4, 38–58. 10.1016/j.trecan.2017.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prendergast G. C.; Malachowski W. P.; DuHadaway J. B.; Muller A. J. Discovery of IDO1 inhibitors: from bench to bedside. Cancer Res. 2017, 77, 6795–6811. 10.1158/0008-5472.CAN-17-2285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller A. J.; Manfredi M. G.; Zakharia Y.; Prendergast G. C. Inhibiting IDO pathways to treat cancer: lessons from the ECHO-301 trial and beyond. Semin. Immunopathol. 2019, 41, 41–48. 10.1007/s00281-018-0702-0. [DOI] [PubMed] [Google Scholar]
- Röhrig U. F.; Majjigapu S. R.; Vogel P.; Zoete V.; Michielin O. Challenges in the discovery of indoleamine 2,3-dioxygenase 1 (IDO1) inhibitors. J. Med. Chem. 2015, 58, 9421–9437. 10.1021/acs.jmedchem.5b00326. [DOI] [PubMed] [Google Scholar]
- Lewis-Ballester A.; Pham K. N.; Batabyal D.; Karkashon S.; Bonanno J. B.; Poulos T. L.; Yeh S. R. Structural insights into substrate and inhibitor binding sites in human indoleamine 2,3-dioxygenase 1. Nat. Commun. 2017, 8, 1693–1700. 10.1038/s41467-017-01725-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelp M. T.; Kates P. A.; Hunt J. T.; Newitt J. A.; Balog A.; Maley D.; Zhu X.; Abell L.; Allentoff A.; Borzilleri R.; Lewis H. A.; Lin Z.; Seitz S. P.; Yan C.; Groves J. T. Immune-modulating enzyme indoleamine 2,3-dioxygenase is effectively inhibited by targeting its apo-form. Proc. Natl. Acad. Sci. U. S. A. 2018, 115, 3249–3254. 10.1073/pnas.1719190115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue E. W.; Douty B.; Wayland B.; Bower M.; Liu X.; Leffet L.; Wang Q.; Bowman K. J.; Hansbury M. J.; Liu C.; Wei M.; Li Y.; Wynn R.; Burn T. C.; Koblish H. K.; Fridman J. S.; Metcalf B.; Scherle P. A.; Combs A. P. Discovery of potent competitive inhibitors of indoleamine 2,3-dioxygenase with in vivo pharmacodynamic activity and efficacy in a mouse melanoma model. J. Med. Chem. 2009, 52, 7364–7367. 10.1021/jm900518f. [DOI] [PubMed] [Google Scholar]
- Kerns E. H.; Di L.. Drug-like Properties: Concepts, Structure Design and Methods: from ADME to Toxicity Optimization; Academic Press: London, UK, 2008; Chapter 4. [Google Scholar]
- Yue E. W.; Sparks R.; Polam P.; Modi D.; Douty B.; Wayland B.; Glass B.; Takvorian A.; Glenn J.; Zhu W.; Bower M.; Liu X.; Leffet L.; Wang Q.; Bowman K. J.; Hansbury M. J.; Wei M.; Li Y.; Wynn R.; Burn T. C.; Koblish H. K.; Fridman J. S.; Emm T.; Scherle P. A.; Metcalf B.; Combs A. P. INCB24360 (Epacadostat), a highly potent and selective indoleamine-2,3-dioxygenase 1 (IDO1) inhibitor for immuno-oncology. ACS Med. Chem. Lett. 2017, 8, 486–491. 10.1021/acsmedchemlett.6b00391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frenna V.; Piccionello A. P.; Cosimelli B.; Ghelfi F.; Spinelli D. The Boulton–Katritzky reaction: a kinetic study of the effect of 5-nitrogen substituents on the rearrangement of some (Z)-phenylhydrazones of 3-benzoyl-1,2,4-oxadiazoles. Eur. J. Org. Chem. 2014, 2014, 7006–7014. 10.1002/ejoc.201402569. [DOI] [Google Scholar]
- Agosti A.; Bertolini G.; Bruno G.; Lautz C.; Glarner T.; Deichtmann W. Handling hydrogen peroxide oxidations on a Large Scale: synthesis of 5-bromo-2-nitropyridine. Org. Process Res. Dev. 2017, 21, 451–459. 10.1021/acs.oprd.6b00433. [DOI] [Google Scholar]
- Martinot T. A.; Ardolino M.; Chen L.; Lam Y.; Li C.; Maddess M. L.; Muzzio D.; Qi J.; Saurí J.; Song Z. J.; Tan L.; Vickery T.; Yin J.; Zhao R. Process Safety Considerations for the Supply of a High-Energy Oxadiazole IDO1-Selective Inhibitor. Org. Process Res. Dev. 2019, 23, 1178–1190. 10.1021/acs.oprd.9b00105. [DOI] [Google Scholar]
- Stepanov A. I.; Sannikov V. S.; Dashko D. V.; Roslyakov A. G.; Astrat’ev A. A.; Stepanova E. V. A new preparative method and some chemical properties of 4-furazan-3-carboxylic acid amidrazones. Chem. Heterocycl. Compd. 2015, 51, 350–360. 10.1007/s10593-015-1707-4. [DOI] [Google Scholar]
- Paton R. M.Product Class 7:1,2,5-Oxadiazoles. In Science of Synthesis 13: Category 2, Hetarenes and Related Ring Systems; Storr R. C., Gilchrist T. L., Eds.; Thieme: New York, 2004; Vol. 13, pp 185– 218. [Google Scholar]
- Ghose A. K.; Viswanadhan V. N.; Wendoloski J. J. Prediction of hydrophobic (lipophilic) properties of small organic molecules using fragmental methods: an analysis of AlogP and ClogP methods. J. Phys. Chem. A 1998, 102, 3762–3772. 10.1021/jp980230o. [DOI] [Google Scholar]
- a Caron G.; Kihlberg J.; Ermondi G. Intramolecular hydrogen bonding: An opportunity for improved design in medicinal chemistry. Med. Res. Rev. 2019, 39, 1707–1729. 10.1002/med.21562. [DOI] [PubMed] [Google Scholar]
- Kuhn B.; Mohr P.; Stahl M. Intramolecular Hydrogen Bonding in Medicinal Chemistry. J. Med. Chem. 2010, 53, 2601–2611. 10.1021/jm100087s. [DOI] [PubMed] [Google Scholar]
- Sadana A. K.; Saini R. K.; Billups W. E. Cyclobutarenes and related compounds. Chem. Rev. 2003, 103, 1539–1602. 10.1021/cr010022j. [DOI] [PubMed] [Google Scholar]
- All of the geometry and frequency calculations were performed using the M06-2X (ref (28)) functional with Grimme’s D3 (ref (29)) dispersion correction and the 6-31G** basis set. Single-point energies were calculated with the same functional and the def2-TZVPP (ref (30)) basis set. All of the calculations were carried out on Gaussian 09 (ref (31)).
- a Zhao Y.; Truhlar D. G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. 10.1007/s00214-007-0310-x. [DOI] [Google Scholar]
- Grimme S.; Antony J.; Ehrlich S.; Krieg H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. 10.1063/1.3382344. [DOI] [PubMed] [Google Scholar]
- Weigend F.; Ahlrichs R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. 10.1039/b508541a. [DOI] [PubMed] [Google Scholar]
- Frisch M. J.; Trucks G. W.; Schlegel H. B.; Scuseria G. E.; Robb M. A.; Cheeseman J. R.; Scalmani G.; Barone V.; Mennucci B.; Petersson G. A.; Nakatsuji H.; Caricato M.; Li X.; Hratchian H. P.; Izmaylov A. F.; Bloino J.; Zheng G.; Sonnenberg J. L.; Hada M.; Ehara M.; Toyota K.; Fukuda R.; Hasegawa J.; Ishida M.; Nakajima T.; Honda Y.; Kitao O.; Nakai H.; Vreven T.; Montgomery J. A. Jr.; Peralta J. E.; Ogliaro F.; Bearpark M.; Heyd J. J.; Brothers E.; Kudin K. N.; Staroverov V. N.; Keith T.; Kobayashi R.; Normand J.; Raghavachari K.; Rendell A.; Burant J. C.; Iyengar S. S.; Tomasi J.; Cossi M.; Rega N.; Millam J. M.; Klene M.; Knox J. E.; Cross J. B.; Bakken V.; Adamo C.; Jaramillo J.; Gomperts R.; Stratmann R. E.; Yazyev O.; Austin A. J.; Cammi R.; Pomelli C.; Ochterski J. W.; Martin R. L.; Morokuma K.; Zakrzewski V. G.; Voth G. A.; Salvador P.; Dannenberg J. J.; Dapprich S.; Daniels A. D.; Farkas Ö.; Foresman J. B.; Ortiz J. V.; Cioslowski J.; Fox D. J.. Gaussian 09, revision D.01; Gaussian, Inc.: Wallingford, CT, 2013.
- Ribeiro R. F.; Marenich A. V.; Cramer C. J.; Truhlar D. G. Use of Solution-Phase Vibrational Frequencies in Continuum Models for the Free Energy of Solvation. J. Phys. Chem. B 2011, 115, 14556–14562. 10.1021/jp205508z. [DOI] [PubMed] [Google Scholar]
- Spahn J.; Peng J.; Lorenzana E.; Kan D.; Hunsaker T.; Segal E.; Mautino M.; Brincks E.; Pirzkall A.; Kelley S.; Mahrus S.; Liu L.; Dale S.; Quiason C.; Jones E.; Liu Y.; Latham S.; Salphati L.; DeMent K.; Merchant M.; Hatzivassiliou G. Improved anti-tumor immunity and efficacy upon combination of the IDO1 inhibitor GDC-0919 with anti-PD-L1 blockade versus anti-PD-L1 alone in preclinical tumor models. J. Immunother Cancer. 2015, 3 (Suppl 2), 303. 10.1186/2051-1426-3-S2-P303. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.