

Abstract

Cystic fibrosis (CF) is a genetic disorder that affects multiple tissues and organs. CF is caused by mutations in the CFTR gene, resulting in insufficient or impaired cystic fibrosis transmembrane conductance regulator (CFTR) protein. The deletion of phenylalanine at position 508 of the protein (F508del-CFTR) is the most common mutation observed in CF patients. The most effective treatments of these patients employ two CFTR modulator classes, correctors and potentiators. CFTR correctors increase protein levels at the cell surface; CFTR potentiators enable the functional opening of CFTR channels at the cell surface. Triple-combination therapies utilize two distinct corrector molecules (C1 and C2) to further improve the overall efficacy. We identified the need to develop a C2 corrector series that had the potential to be used in conjunction with our existing C1 corrector series and provide robust clinical efficacy for CF patients. The identification of a pyrrolidine series of CFTR C2 correctors and the structure–activity relationship of this series is described. This work resulted in the discovery and selection of (2S,3R,4S,5S)-3-(tert-butyl)-4-((2-methoxy-5-(trifluoromethyl)pyridin-3-yl)methoxy)-1-((S)-tetrahydro-2H-pyran-2-carbonyl)-5-(o-tolyl)pyrrolidine-2-carboxylic acid (ABBV/GLPG-3221), which was advanced to clinical trials.
Keywords: Cystic fibrosis, ABBV/GLPG-3221, cystic fibrosis transmembrane conductance regulator protein (CFTR)
Cystic fibrosis (CF) is a genetic disorder that affects multiple tissues and organs. CF impacts more than 70 000 people worldwide.1 It is caused by mutations in the CFTR gene, resulting in insufficient or impaired cystic fibrosis transmembrane conductance regulator (CFTR) protein.2 CFTR is an anion channel that is capable of transporting both Cl– and HCO3– and is expressed in many epithelial cells. It is also responsible for regulating the balance of water and salt in multiple tissues. The impairment of the normal CFTR channel function in the lungs results in the buildup of a thick mucus that leads to bacterial colonization, resulting in the loss of lung function in CF patients. The loss of this lung function is the leading cause of death in these CF patients. These patients are also affected by potentially life-threatening pancreatic insufficiency and gastrointestinal issues.
Over 2000 single nucleotide polymorphisms (SNPs) have been identified in the CFTR protein of CF patients, 336 of which are disease-causing.3 The complications that result from these mutations fall into six classes affecting the stability, function, trafficking, or production of CFTR.4 The deletion of phenylalanine at position 508 of the protein (F508del-CFTR) is the most common mutation observed in CF patients. Approximately 85% of CF patients have at least one copy of the F508del-CFTR allele, and the homozygous F508del-CFTR mutation is observed in ∼50% of CF cases. This deletion prevents the nascent protein from folding correctly and translocating to the plasma membrane for normal anion transport.5,6 As a result, far fewer F508del-CFTR channels are expressed on the cell membrane compared with wild-type CFTR (WT-CFTR)-expressing cells.7 Furthermore, F508del-CFTR channels are less effective at channel gating compared with WT-CFTR.8 The most effective treatments of these patients employ two CFTR modulator classes.9 CFTR correctors increase protein levels at the cell surface;10 CFTR potentiators enable the functional opening of CFTR channels at the cell surface. Dual-combination therapies such as Orkambi and Symdeko have been approved,11−14 restoring lung function to a modest degree in F508del homozygous patients. Triple combinations utilizing two distinct corrector molecules and a potentiator molecule have demonstrated transformational clinical benefit in patients with a F508del-CFTR mutation on at least one allele.15−18 A New Drug Application has recently been filed to the FDA for one such triple combination.19,14 However, how universal this will be across the multitude of genotypes remains to be assessed.
Herein we report the identification of a pyrrolidine series of CFTR C2 correctors and the structure–activity relationship (SAR) of this series, which leads to the identification of ABBV-3221 (also known as GLPG-3221). This CFTR C2 corrector has been advanced to clinical trials.
We have previously reported a novel series of CFTR C1 corrector compounds, including ABBV-2222 (Figure 1),20 which are currently in clinical trials in CF patients. These C1 correctors restore the early folding process of the protein to increase the number of CFTR channels expressed on the cell membrane. C2 correctors work by any complementary mechanism to further increase CFTR at the cell surface.15 In an effort to develop a CF triple therapy, we initiated efforts to develop a CFTR C2 corrector.
Figure 1.

Structures of previously reported CFTR C1 corrector compounds.
Two cell-based assays were employed to determine the activity of CFTR modulators. CFTR correctors are a class of modulators that correct the misfolding mutant CFTR protein, increasing the CFTR channel expression on the cell surface. The CFTR expression was quantified using immortalized bronchial epithelial cells (CFBE41o)21 transduced with F508del-CFTR fused with horseradish peroxidase (HRP). See the Supporting Information for experimental details. Test corrector compounds were incubated with or without a cocorrector, 2 μM of compound 1, overnight, followed by the measurement of the HRP activity to detect matured F508del-CFTR levels.22 This CSE-HRP (cell surface expression–HRP) assay was used for primary high-throughput screens (HTSs) and as a primary assay for driving the medicinal chemistry efforts.
The objective of this research is to increase the membrane expression and improve the stability and function of the corrected F508del-CFTR channels. We measured the function of the channels using a HBE-TECC (human bronchial epithelial–transepithelial current clamp) assay. Primary human bronchial epithelial (HBE) cells from F508del CF patients were used, and an electrophysiology (E-Phys) measurement of the CFTR chloride ion current across the apical membrane was taken. See the Supporting Information for experimental details. In brief, the assay uses a transepithelial current clamp (TECC)23 instrument to measure the function of the F508del-CFTR channel after the overnight incubation of the test corrector compounds in the presence of a potentiator24 and cocorrector and activated with forskolin. The assay uses negative and positive controls (0.15 μM of ABBV-2222) to measure the correction of the mutated CFTR channel.25
An HTS was conducted, with the objective of identifying compounds that could serve as hits for the development of a CFTR C2 corrector series using the CSE-HRP assay. The desired compound profile would improve the cell surface expression with and without cocorrector 1 present. Compound 2, a pentasubstituted pyrrolidine, was identified as a potential hit. It had an average EC50 of 2.72 μM, with a maximum response of 186% relative to compound 1 alone (Table 1). In the presence of 1, compound 2 had an average EC50 of 2.11 μM, with a maximum response of 681% relative to compound 1 (Table 1). To confirm the potency of this hit, the CSE-HRP assay was repeated four times with and without compound 1 as a C1 corrector. This resulted in an EC50 range of 2.3 to 3.2 μM alone and an EC50 range of 1.7 to 2.7 μM in the presence of compound 1.
Table 1. CSE-HRP SAR for the Initial Series Hits.

CSE-HPR assay was run using compound 1 (3 μM) as the positive control. All values are the geometric means of at least two determinations.
This format of the CSE-HPR assay was run with compound 1 (3 μM) as a C1 corrector and as the positive control. All values are the geometric means of at least two determinations.
The exact stereochemistry of 2 was not specified at the time of registration in 1982, and the compound was clearly a mixture of diastereomers. One of the diastereomers of 2 was synthesized and isolated as a racemic mixture, compound 3. It had an average EC50 of 2.66 μM (151%) and an average EC50 of 1.97 μM (527%) in the presence of compound 1 (Table 1). This key hit compound was used as a positive control in the CSE-HRP assay and was tested over 300 times with and without cocorrector 1. In an effort to improve the properties of compound 3, by increasing the fraction of sp3-hybridized carbons (Fsp3), the phenyl group of the N-benzoyl moiety was replaced with a cyclohexane, resulting in compound 4. This compound had comparable potency, with greatly improved efficacy in the CSE assay. The ketone moiety of compounds 2–4 was flagged for replacement. Moving from the ketone in 4 to the benzyl ether in 5 increased the Fsp3 and removed any issues of reactivity associated with the acetophenone group.26 Compound 5 maintained reasonable potency. The individual enantiomers of 5 were isolated by chiral preparative supercritical fluid chromatograpy (SFC) to afford enantiomers 6 and 7. The majority of the potency and the efficacy of 5 clearly resides in the (2S,3R,4S,5S)-isomer, 7 (Table 1). Running the CSE-HRP assay in the presence of C1 corrector, 1, made the EC50 slightly more potent while greatly improving the efficacy. On the basis of the data presented in Table 1, we decided to focus our efforts on benzyl ether analogs with the (2S,3R,4S,5S) stereochemistry, analogs to compound 7.
Compound 7 was thus identified as submicromolar in the CSE-HRP assay and was then advanced to testing in human bronchial epithelial (HBE) cells and evaluated in the TECC assay. It showed good potency (173 nM) and efficacy (590%) relative to the positive control, ABBV-2222 (0.15 μM) in the presence of potentiator GLPG1837 (0.75 μM) (Table 2). Initially, we chose to evaluate the SAR of the benzyl ether portion of this series (Table 2). The cLogP of compound 7 is 6.01, and we were interested in introducing a heteroatom into the aromatic ring of 7, such as that found in pyridine 8. The cLogP of compound 8 was reduced to 5.27, and the compound had improved potency in the HBE-TECC assay (150 nM, 650%) (Table 2). Replacing the trifluoromethyl group of compound 8 with a cyclopentane or a tert-butyl group led to compounds that were both less potent and had higher cLogP values (5.95, 9; 6.12, 10).
Table 2. HBE-TECC SAR for Ether Analogs 7–10.
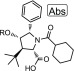

HBE-TECC assay was run using ABBV-2222 (0.15 μM) and GLPG1837 (0.75 μM) as the positive controls. All values are the geometric means of at least two determinations.
The further interrogation of compound 8 indicated that it is a CYP3A4 inducer in an induction assay compared with the positive control rifampin (Table 3). In this assay (Supporting Information), the compound is tested at 10 or 1 μM in triplicate, and the response is compared with that of 10 μM rifampin, a CYP3A4 inducer. Compounds with a response of <20% of rifampin at 10 μM are considered to have a low induction risk. Compound 8 had a response of 74% at 10 μM.
Table 3. HBE-TECC and CYP3A4 Induction SAR for N1 Analogs 8 and 11–13.
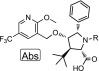

HBE-TECC assay was using ABBV-2222 (0.15 μM) and GLPG1837 (0.75 μM) as the positive controls. All values are the geometric means of at least two determinations.
All values are the means of at least three determinations.
Analogs of compound 8 were synthesized that differed in the substitution off of the pyrrolidine nitrogen. Switching from the cyclohexanecarboxamide of 8, to the isopropyl carbamate found in 11 led to a compound that was slightly more potent but less efficacious and, very importantly, not a CYP3A4 inducer (Table 3). The diastereomeric pyran compounds 12 and 13 were prepared as direct analogs of compound 8. The (S)-pyran analog 12 was more potent and much more efficacious than the (R)-pyran analog 13. Compound 12 was advanced to the CYP3A4 induction assay, where it did not induce CYP3A4 (Table 3). Compared with cyclohexane 8, pyran 12 was somewhat less potent (213 vs 150 nM), but more efficacious (720 vs 650%).
Because of the improvement in CYP3A4 induction, we focused on optimizing compounds with the (S)-pyran substitution. These analogs varied the aromatic group off the C5 position of the pyrrolidine ring (Table 4). The introduction of large alkyl groups at the ortho position of the aromatic ring (compounds 14, 15, 17, and 18) led to a dramatic improvement in potency (35–66 vs 213 nM, compound 12) while maintaining reasonable efficacy. All of these compounds were, however, potent CYP3A4 inducers whether they were tested at 1 or 10 μM. Chloro substitution at the ortho position (16) afforded a compound that did not show induction at 1 μM but did not have the desired potency (Table 4). Finally, the substitution of a methyl group at the ortho position resulted in a compound with good balance of characteristics (Compound 19, Table 4). Whereas compound 19 was somewhat less potent than compounds with a larger alkyl substitution, it still had an EC50 = 105 nM and, importantly, was not found to have a high CYP3A4 induction risk (Table 4).
Table 4. HBE-TECC and CYP3A4 Induction SAR for Aromatic Analogs 10 and 12–17.


HBE-TECC assay using ABBV-2222 (0.15 μM) and GLPG1837 (0.75 μM) as the positive controls. All values are the geometric means of at least two determinations.
All values are the means of at least three determinations.
The detailed dose response HBE-TECC data for compound 19 are shown in Figure 2. The 100% response is set by the combination of ABBV-2222 (0.15 μM) and GLPG1837 (0.75 μM). The addition of compound 19 to the combination of ABBV-2222 and GLPG1837 increased the CFTR current with EC50 = 105 nM and a maximum response of 585% compared with the double combination alone.
Figure 2.
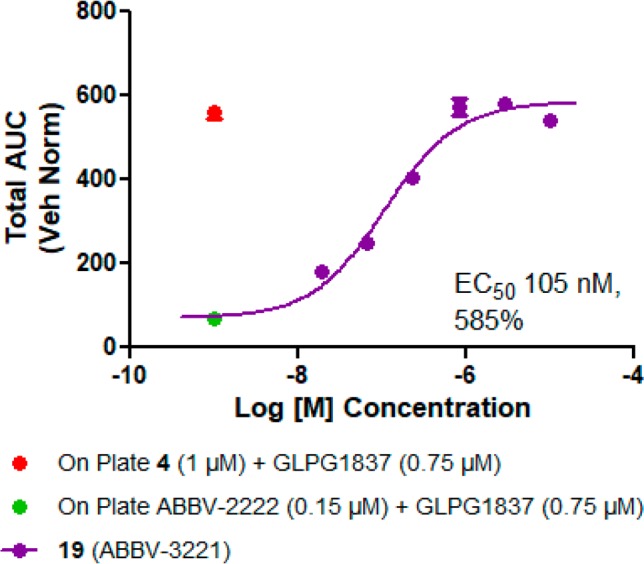
Dose–response graph for 19 in triple combination format. The data are a combination from two separate experiments run on different days with cells from one CF donor with homozygous F508delCFTR mutation, with n = 3 replicates at each concentration.
Compound 19 was selected for advancement into additional characterization. An optimized discovery scale synthesis is illustrated in Scheme 1. Commercially available glycine ethyl ester hydrochloride (20) was condensed with 2-methylbenzaldehyde to afford imine 21. An enantioselective [3 + 2] cycloaddition was an ideal method to forge the stereogenic pyrrolidine core (22) from imine 21 and (E)-3,3-dimethyl-1-nitrobut-1-ene.27 This was achieved using a Cu-catalyzed enantioselective cycloaddition catalyzed by a Cu(I)/(2-(bis(3,5-bis(trifluoromethyl)phenyl)phosphino)-3-((S)-4-isopropyl-4,5-dihydrooxazol-2-yl)cyclopenta-2,4-dien-1-yl)(cyclopenta-2,4-dien-1-yl)iron complex. Reacting pyrrolidine 22 with allyl chloroformate gave the Alloc-protected intermediate 23. A Nef reaction converted the nitro moiety of compound 23 into the ketone of 24. Sodium borohydride in ethanol gave the selective reduction of the ketone to alcohol 25. The alkylation of alcohol 25 with 3-(bromomethyl)-2-methoxy-5-(trifluoromethyl)pyridine using potassium tert-butoxide as the base in dimethylformamide (DMF) generated ether 26. The Alloc group of 26 was removed with catalytic Pd to afford the NH pyrrolidine 27 that was readily acylated with the acid chloride generated in situ from tetrahydropyran-2-carboxylic acid to yield ester 28. The ester was saponified to the acid using lithium hydroxide in MeOH/THF to afford compound 19, ABBV-3221. Optimizations required for a multikilogram scale process will be reported.28
Scheme 1. Discovery Synthesis of ABBV/GLPG-3221 (19).

Reagents and conditions: (a) MgSO4, TEA, 2-methylbenzaldehyde, DCM; (b) (2-(bis(3,5-bis(trifluoromethyl)phenyl)phosphino)-3-((S)-4-isopropyl-4,5-dihydrooxazol-2-yl)cyclopenta-2,4-dien-1-yl)(cyclopenta-2,4-dien-1-yl)iron, Cu(OTf)2, KOtBu, (E)-3,3-dimethyl-1-nitrobut-1-ene, THF; (c) allyl chloroformate, toluene; (d) PDC, 6 N HCl, Zn dust; (e) NaBH4, EtOH; (f) KOtBu, 3-(bromomethyl)-2-methoxy-5-(trifluoromethyl)pyridine, DMF; (g) 1,3-dimethlbarbituric acid, Pd(PPh3)4, EtOAc/DCM; (h) tetrahydropyran-2-carboxylic acid, oxalyl chloride, DMF, DCM; and (i) LiOH, MeOH/THF.
Compound 19 was also found to have a good pharmacokinetic (PK) profile in both rats and dogs, which is summarized in Table 5.
Table 5. Pharmacokinetic Properties of 19 in Rat and Doga.
Parameters were determined using a 1 mg/kg dose.
Clearance (L/h/kg).
Half life (h).
On the basis of the data presented here, along with additional preclinical safety evaluations, compound 19 (ABBV-3221) was identified as a novel, potent, and efficacious corrector of CFTR with a reasonable safety and PK profile. Compound 19 provides substantial CFTR functional activity improvement over a dual combination of GLPG-1837 and ABBV-2222 when used as a triple combination in in vitro studies; therefore, it was selected to advance into clinical trials to evaluate its potential for treating cystic fibrosis.
Acknowledgments
We acknowledge Clinton Yeung for the preparation of intermediate compounds.
Glossary
Abbreviations
- Alloc
allyloxycarbonyl
- CF
cystic fibrosis
- CFTR
cystic fibrosis transmembrane conductance regulator protein
- CSE-HRP
cell surface expression–horseradish peroxidase
- CYP3A4
cytochrome P450 3A4
- DCM
dichloromethane
- DDI
drug–drug interaction
- DMF
dimethylformamide
- DMSO
dimethyl sulfoxide
- E-Phys
electrophysiology
- Emax
maximum percent activity
- ER
endoplasmic reticulum
- FEV1
forced expiratory volume in 1 s
- Fsp3
sp3-hybridized carbons
- HBE
human bronchial epithelial
- HBE-TECC
human bronchial epithelial–transepithelial current clamp
- HRP
horseradish peroxidase
- HTS
high-throughput screen
- IEQ
equivalent CFTR current
- PDC
potassium dichromate
- PK
pharmacokinetic
- SAR
structure–activity relationship
- SNPs
single nucleotide polymorphisms
- SFC
supercritical fluid chromatography
- TEA
triethylamine
- TECC
transepithelial current clamp
- THF
tetrahydrofuran
- WT-CFTR
wild-type CFTR
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.9b00377.
Synthetic procedures and compound characterization data for ether compounds 5–19 and intermediates as well as assay procedures (PDF)
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The design, study conduct, and financial support for this research were provided by AbbVie and Galapagos.
The authors declare the following competing financial interest(s): All authors are current AbbVie employees.
Supplementary Material
References
- Cutting G. R. Cystic fibrosis genetics: from molecular understanding to clinical application. Nat. Rev. Genet. 2015, 16, 45–56. 10.1038/nrg3849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerem B.-S.; Rommens J. M.; Buchanan J. A.; Markiewicz D.; Cox T. K.; Chakravarti A.; Buchwald M.; Tsui L.-C. Identification of the cystic fibrosis gene: genetic analysis. Science 1989, 245, 1073–1080. 10.1126/science.2570460. [DOI] [PubMed] [Google Scholar]
- Welsh M. J.; Smith A. E. Molecular mechanisms of CFTR chloride channel dysfunction in cystic fibrosis. Cell 1993, 73, 1251–1254. 10.1016/0092-8674(93)90353-R. [DOI] [PubMed] [Google Scholar]
- Zielenski J.; Tsui L. C. Cystic fibrosis: genotypic and phenotypic variations. Annu. Rev. Genet. 1995, 29, 777–807. 10.1146/annurev.ge.29.120195.004021. [DOI] [PubMed] [Google Scholar]
- Marinko J. T.; Huang H.; Penn W. D.; Capra J. A.; Schlebach J. P.; Sanders C. R. Folding and misfolding of human membrane proteins in health and disease: from single molecules to cellular proteostasis. Chem. Rev. 2019, 119, 5537–5606. 10.1021/acs.chemrev.8b00532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukacs G. L.; Verkman A. S. CFTR: folding, misfolding and correting the ΔF508 conformational defect. Trends Mol. Med. 2012, 18, 81–91. 10.1016/j.molmed.2011.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasyk E. A.; Foskett J. K. Mutant (ΔF508) cystic fibrosis transmembrane conductance regulator Cl– channel is functional when retained in endoplasmic reticulum of mammalian cells. J. Biol. Chem. 1995, 270, 12347–12350. 10.1074/jbc.270.21.12347. [DOI] [PubMed] [Google Scholar]
- Veit G.; Avramescu R. G.; Chiang A. N.; Houck S. A.; Cai Z.; Peters K. W.; Hong J. S.; Pollard H. B.; Guggino W. B.; Balch W. E.; Skach W. R.; Cutting G. R.; Frizzell R. A.; Sheppard D. N.; Cyr D. M.; Sorscher E. J.; Brodsky J. L.; Lukacs G. L. From CFTR biology toward combinatorial pharmacotherapy: expanded classification of cystic fibrosis mutations. Mol. Biol. Cell 2016, 27, 424–433. 10.1091/mbc.e14-04-0935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowe S. M.; Verkman A. S. Cystic fibrosis transmembrane regulator correctors and potentiators. Cold Spring Harbor Perspect. Med. 2013, 3, a009761. 10.1101/cshperspect.a009761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spano V.; Montalbano A.; Carbone A.; Scudieri P.; Galietta L. J. V.; Barraja P. An overview on chemical structures as ΔF508-CFTR correctors. Eur. J. Med. Chem. 2019, 180, 430–448. 10.1016/j.ejmech.2019.07.037. [DOI] [PubMed] [Google Scholar]
- Wainwright C. E.; Elborn J. S.; Ramsey B. W.; Marigowda G.; Huang X.; Cipolli M.; Colombo C.; Davies J. C.; De Boeck K.; Flume P. A.; Konstan M. W.; McColley S. A.; McCoy K.; McKone E. F.; Munck A.; Ratjen F.; Rowe S. M.; Waltz D.; Boyle M. P. Lumacaftor-Ivacaftor in patients with cystic fibrosis homozygous for Phe508del CFTR. N. Engl. J. Med. 2015, 373, 220–231. 10.1056/NEJMoa1409547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Goor F.; Hadida S.; Grootenhuis P. D.; Burton B.; Stack J. H.; Straley K. S.; Decker C. J.; Miller M.; McCartney J.; Olson E. R.; Wine J. J.; Frizzell R. A.; Ashlock M.; Negulescu P. A. Correction of the F508del-CFTR protein processing defect in vitro by the investigational drug VX-809. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 18843–18848. 10.1073/pnas.1105787108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Goor F.; Hadida S.; Grootenhuis P. D.; Burton B.; Cao D.; Neuberger T.; Turnbull A.; Singh A.; Joubran J.; Hazlewood A.; Zhou J.; McCartney J.; Arumugam V.; Decker C.; Yang J.; Young C.; Olson E. R.; Wine J. J.; Frizzell R. A.; Ashlock M.; Negulescu P. Rescue of CF airway epithelial cell function in vitro by a CFTR potentiator, VX-770. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 18825–18830. 10.1073/pnas.0904709106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donaldson S. H.; Pilewski J. M.; Griese M.; Cooke J.; Viswanathan L.; Tullis E.; Davies J. C.; Lekstrom-Himes J. A.; Wang L. T. Tezacaftor/Ivacaftor in subjects with cystic fibrosis and F508del/F508del-CFTR or F508del/G551D-CFTR. Am. J. Respir. Crit. Care Med. 2018, 197, 214–224. 10.1164/rccm.201704-0717OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H.; Pesce E.; Sheppard D. N.; Singh A. K.; Pedemonte N. Therapeutic approaches to CFTR dysfunction: From discovery to drug development. J. Cystic Fibrosis 2018, 17, S14–S21. 10.1016/j.jcf.2017.08.013. [DOI] [PubMed] [Google Scholar]
- Holguin F. H. Triple CFTR modulator therapy for cystic fibrosis. N. Engl. J. Med. 2018, 379, 1671–1672. 10.1056/NEJMe1811996. [DOI] [PubMed] [Google Scholar]
- Keating D.; Marigowda G.; Burr L.; Daines C.; Mall M. A.; McKone E. F.; Ramsey B. W.; Rowe S. M.; Sass L. A.; Tullis E.; McKee C. M.; Moskowitz S. M.; Robertson S.; Savage J.; Simard C.; Van Goor F.; Waltz D.; Xuan F.; Young T.; Taylor-Cousar J. L. VX-445—Tezacaftor—Ivacaftor in patients with cystic fibrosis and one or two Phe508del Alleles. N. Engl. J. Med. 2018, 379, 1612–1620. 10.1056/NEJMoa1807120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies J. C.; Moskowitz S. M.; Brown C.; Horsley A.; Mall M. A.; McKone E. F.; Plant B. J.; Prais D.; Ramsey B. W.; Taylor-Cousar J. L.; Tullis E.; Uluer A.; McKee C. M.; Robertson S.; Shilling R. A.; Simard C.; Van Goor F.; Waltz D.; Xuan F.; Young T.; Rowe S. M. VX-659—Texacaftor—Ivacaftor in patients with cystic fibrosis and one or two Phe508del Alleles. N. Engl. J. Med. 2018, 379, 1599–1611. 10.1056/NEJMoa1807119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vertex Selects Triple Combination Regimen of Vx-445, Tezacaftor and Ivacaftor to Submit for Global Regulatory Approvals in Cystic Fibrosis. May 30, 2019.Https://Investors.Vrtx.Com/News-Releases/News-Release-Details/Vertex-Selects-Triple-Combination-Regimen-Vx-445-Tezacaftor-And (accessed October 24, 2019).
- Wang X.; Liu B.; Searle X.; Yeung C.; Bogdan A.; Greszler S.; Singh A.; Fan Y.; Swensen A. M.; Vortherms T.; Balut C.; Jia Y.; Desino K.; Gao W.; Yong H.; Tse C.; Kym P. Discovery of 4-[(2R,4R)-4-({[1-(2,2-difluoro-1,3-benzodioxol-5-yl)cyclopropyl]carbonyl}amino)-7-(difluoromethoxy)-3,4-dihydro-2H-chromen-2-yl]benzoic acid (ABBV/GLPG-2222), a potent cystic fibrosis transmembrane conductance regulator (CFTR) corrector for the treatment of cystic fibrosis. J. Med. Chem. 2018, 61, 1436–1449. 10.1021/acs.jmedchem.7b01339. [DOI] [PubMed] [Google Scholar]
- Ehrhardt C.; Collnot E. M.; Baldes C.; Becker U.; Laue M.; Kim K. J.; Lehr C. M. Towards an in vitro model of cystic fibrosis small airway epithelium: Characterisation of the human bronchial epithelial cell line CFBE41o. Cell Tissue Res. 2006, 323, 405–415. 10.1007/s00441-005-0062-7. [DOI] [PubMed] [Google Scholar]
- Veit G.; Avramescu R. G.; Perdomo D.; Phuan P. W.; Bagdany M.; Apaja P. M.; Borot F.; Szollosi D.; Wu Y. S.; Finkbeiner W. E.; Hegedus T.; Verkman A. S.; Lukacs G. L. Some gating potentiators, including VX-770, diminish deltaF508-CFTR functional expression. Sci. Transl. Med. 2014, 6, 246ra97. 10.1126/scitranslmed.3008889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vu C. B.; Bridges R. J.; Pena-Rasgado C.; Lacerda A. E.; Bordwell C.; Sewell A.; Nichols A. J.; Chandran S.; Lonkar P.; Picarella D.; Ting A.; Wensley A.; Yeager M.; Liu F. Fatty acid cysteamine conjugates as novel and potent autophagy activators that enhance the correction of misfolded F508del-cystic fibrosis transmembrane conductance regulator (CFTR). J. Med. Chem. 2017, 60, 458–473. 10.1021/acs.jmedchem.6b01539. [DOI] [PubMed] [Google Scholar]
- Van der Plas S. E.; Kelgtermans H.; De Munck T.; Martina S. L. X.; Dropsit S.; Quinton E.; De Blieck A.; Joannesse C.; Tomaskovic L.; Jans M.; Christophe T.; van der Aar E.; Borgonovi M.; Nelles L.; Gees M.; Stouten p.; Van Der Schueren J.; Mammoliti O.; Conrath K.; Andrews M. Discovery of N-(3-carbamoyl-5,5,7,7-tetramethyl-5,7-dihydro-4H-thieno[2,3-c]pyran-2-yl)-lH-pyrazole-5-carboxamide (GLPG1837), a novel potentiator which can open class III mutant cystic fibrosis transmembrane conductance regulator (CFTR) channels to a high extent. J. Med. Chem. 2018, 61, 1425–1435. 10.1021/acs.jmedchem.7b01288. [DOI] [PubMed] [Google Scholar]
- Van Goor F.; Hadida S.; Grootenhuis P. D.; Burton B.; Cao D.; Neuberger T.; Turnbull A.; Singh A.; Joubran J.; Hazlewood A.; Zhou J.; McCartney J.; Arumugam V.; Decker C.; Yang J.; Young C.; Olson E. R.; Wine J. J.; Frizzell R. A.; Ashlock M.; Negulescu P. Rescue of CF airway epithelial cell function in vitro by a CFTR potentiator, VX-770. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 18825–18830. 10.1073/pnas.0904709106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altenbach R. J.; Bogdan A.; Desroy N.; Greszler S. N.; Koenig J. R.; Kym P. R.; Liu B.; Scanio M. J.; Searle X. B.; Wang X.; Yeung M. C.; Zhao G.. Substituted Pyrrolidines and Method of Use. U.S. Patent Appl. 20180099932, April 12, 2018.
- Hashimoto T.; Maruoka K. Recent advances of catalytic asymmetric 1,3-dipolar cycloadditions. Chem. Rev. 2015, 115, 5366–5412. 10.1021/cr5007182. [DOI] [PubMed] [Google Scholar]
- Hartung J.; Greszler S. N.; Klix R. C.; Kallemeyn J. M. Development of an Enantioselective [3 + 2] cycloaddition to synthesize the pyrrolidine core of ABBV-3221 on multi-kilogram scale. Org. Process Res. Dev. 2019, 10.1021/acs.oprd.9b00292. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.