

Abstract
Interleukin-1β is a potent proinflammatory cytokine that plays a key role in the pathogenesis of the brain aging and diverse range of neurological diseases including Alzheimer’s disease, Parkinson’s disease, stroke and persistent pain. Activated microglia are the main cellular source of interleukin-1β in the brain. Cathepsin B is associated with the production and secretion of interleukin-1β through pyrin domain-containing protein 3 inflammasome-independent processing of procaspase-3 in the phagolysosomes. The leakage of cathepsin B from the endosomal-lysosomal system during aging is associated with the proteolytic degradation of mitochondrial transcription factor A, which can stabilize mitochondrial DNA. Therefore, microglial cathepsin B could function as a major driver for inflammatory brain diseases and brain aging. Orally active and blood-brain barrier-permeable specific inhibitors for cathepsin B can be potentially effective new pharmaceutical interventions against inflammatory brain diseases and brain aging.
Keywords: brain aging, caspase-1, cathepsin B, inflammatory brain diseases, interleukin-1β, microglia, mitochondrial transcription factor A, neuroinflammation; nuclear factor-κβ, oxidative stress
Introduction
A group of proteases in the endosomal-lysosomal system has been designated as cathepsin, which is derived from the Greek kathepsein (to digest). There are 15 human cathepsins, including 11 cysteine proteases (cathepsins B, C, F, H, K, L, O, S, V, X and W), two serine proteases (cathepsins A and G) and two aspartic proteases (cathepsins E and D). There is increasing evidence that cathepsins produced by microglia, the resident mononuclear phagocyte population in the brain, play important roles in neuroinflammation.
Neuroinflammation constitutes a fundamental process involved in the progression of several neurodegenerative disorders including Alzheimer’s disease, Parkinson’s disease and multiple sclerosis, while its role is still a matter of debate. Activated microglia that play important roles in neuroinflammation has the dual roles in terms of neurodegeneration and neuroprotection (Cappellano et al., 2013). The dual activity of activated microglia appears to be linked to change from an inflammatory to an anti-inflammatory phenotype. During neuroinflammation, activated microglia also increasingly express and secrete several different cathepsins, which support various inflammatory and immune functions of activated microglia (Nakanishi, 2003; Lowry and Klegeris, 2018).
Among several different cathepsins, cathepsin B (EC 3.4.22.1), a typical cysteine lysosomal protease, upregulated in primed or activated microglia is especially associated with neuroinflammatory response and oxidative stress in inflammatory brain diseases and brain aging. It is known that the activity of cathepsin B at a neutral pH outside the lysosome is limited. However, there is evidence that cathepsin B is active at cytosolic pH. We have also detected the enzymatic activity of cathepsin B using z-Arg-Arg cresyl violet, a fluorogenic substrate of cathepsin B, in the cytosol of cathepsin B-overexpressing microglial cells after treatment with a lysosomotropic agent (Ni et al., 2019).
I herein review current understanding of the roles of microglial cathepsin B in inflammatory brain diseases and brain aging through both intracellular and extracellular proteolytic mechanisms.
After a systematic search of the Medline databases, relevant articles on the roles of microglial cathepsin B in inflammatory brain diseases and brain aging from 1989 to 2019 were reviewed using following conditions: cathepsin B (MeSH Terms) OR microglia (MeSH Terms). The results were further screened by title and abstract to select the articles described the catalytic properties and biological functions of cathepsin B or the roles of microglial cathepsin B.
Microglial Cathepsin B-Mediated Phagolysosomal Pathway of Procaspase-1 Activation
A potential role of cathepsin B has been proposed in the pyrin domain-containing protein 3 (NLRP3) inflammasome-independent production of interleulin (IL)-1β in microglia (Terada et al., 2010; Sun et al., 2012). Cathepsin B may directly contribute to proteolytic maturation of procaspase-1. In addition, cathepsin B is able to cleave procaspase-11 in a cell-free system at a neutral pH, but only cleaves procaspase-1 at an acidic pH (Sun et al., 2012). Further cleavage is considered to be necessary for the full maturation of procaspase-1 after its proteolytic cleavage by cathepsin B. The fragments generated by cathepsin B cleavage are still larger than the mature caspase-1, suggesting that cathepsin B is involved in the procaspase-1 activation through its direct activation of procaspase-11, which in turn activates procaspase-1 (Terada et al., 2010). Genetic deletion or pharmacological inhibition of cathepsin B prevents the activation of precursor forms of IL-1β and IL-18 in microglial cell cultures following treatment with chromogranin A (CGA), a glycophosphoprotein secreted by neurons, through inhibition of proteolytic maturation of procaspase-1 (Terada et al., 2010). CGA can produce IL-1β in microglia without leakage of cathepsin B or activation of NOD-, LRR- and pyrin domain-containing protein 3 (NLRP3)-inflammasome (Sun et al., 2012).
It is considered that cathepsin B-containing enlarged lysosomes are phagolysosomes formed by the fusion of scavenger receptors class A-mediated phagosomes and primary lysosomes in activated microglia (Sun et al., 2012). Therefore, procaspase-1 and the inactive forms of IL-1β and IL-18 in the cytoplasm may be trapped in phagosomes, which are fused with cathepsin B-containing primary lysosomes to form the mature phagolysosomes. Following activation of procaspase-1 by cathepsin B, mature caspase-1 processes pro-IL-1β in the phagolysosomes. Finally, mature IL-1β is rapidly secreted from microglia (Figure 1A). It is also noted that cathepsin B is increased exclusively in microglia during aging (Nakanishi, 2003). Therefore, a pharmacological inhibition of cathepsin B could be a potent strategy for slowing inflammatory brain diseases and brain aging through inhibition of the procaspase-1 activation in microglia, and resultant reduction of neuroinflammation.
Figure 1.
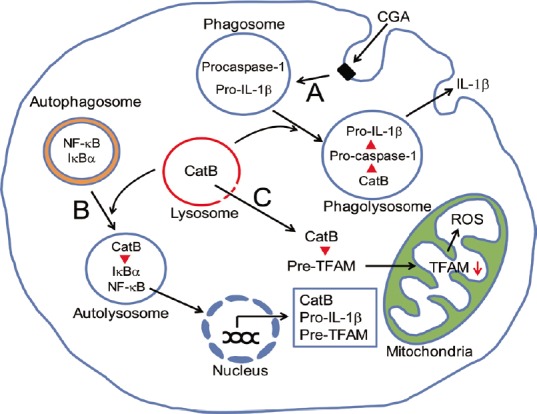
Schematic illustration representing critical roles of microglial cathepsin B (CatB) in chronic inflammatory and oxidative responses.
(A) CatB-dependent phagosomal pathway for proteolytic processing and secretion of interleulin (IL)-1β through proteolytic activation of procaspase-1. (B) CatB-dependent autophagic pathway for chronic nuclear factor-κB (NF-κB) activation through proteolytic degradation of NF-κB inhibitor α (IκBα). (C) CatB leakage-dependent pathway for increased mitochondria-derived ROS generation through proteolytic degradation of pre-TFAM in the cytosol. CGA: Chromogranin A; ROS: reactive oxygen species; TFAM: mitochondrial transcription factor A.
Microglial Cathepsin B and Inflammatory Pain
Genetic deletion or pharmacological inhibition of cathepsin B was found to abrogate CGA-induced activation of procaspase-1 and subsequent processing of pro-IL-1β and pro-IL-18 to their mature forms in microglia. Furthermore, the existence of multiple pathways that can activate proforms of IL-1β and IL-18 in microglia was further demonstrated, probably indicating an existence of backup mechanism to execute the production of these cytokines. Cathepsin B but not NLRP3 is required for the CGA-induced processing of both pro-IL-1β and pro-IL-18, whereas NLRP3 inflammasome but not cathepsin B is required for ATP-induced their processing. These observations in cultured microglia prompted us to further investigate the role of cathepsin B in chronic pain generation, because both IL-1β and IL-18 play important roles in the initiation of inflammatory and pain hypersensitivity.
We used a peripheral inflammatory pain mouse model. Following intraplantar injection of complete Freund’s adjuvant (CFA), both hyperalgesia (an augmented pain response to noxious stimulation) and allodynia (a pain produced by normally non-painful stimulation) develop in the injected paw. As expected, genetic deletion or pharmacological inhibition of cathepsin B significantly inhibited both CFA-induced mechanical allodynia and thermal hyperalgesia without affecting peripheral inflammation. In contrast, cathepsin B deficiency had no significant effect on spinal nerve injury-induced mechanical allodynia. In the spinal microglia of cathepsin B-deficient mice, mature IL-1β was not expressed following treatment with CFA (Sun et al., 2012). Microglial activation and inflammatory immune responses in the spinal cord are involved in CFA-induced mechanical allodynia and thermal hyperalgesia, because minocycline, a microglial activation inhibitor, completely inhibited the development of CFA-induced mechanical allodynia and thermal hyperalgesia (Sun et al., 2012).
Therefore, peripheral CFA-treatment may trigger cathepsin B-dependent caspase-1 activation pathways for the pro-IL-1β processing in spinal microglia, leading to pain hypersensitivity. On the other hand, spinal nerve-injury activates cathepsin B-independent pathways for the pro-IL-1β processing in spinal microglia (Sun et al., 2012). A matrix metallopeptidase 9-dependent mechanism has been proposed as an alternative pathway for the pro-IL-1β processing in the spinal cord following nerve injury. Following the peripheral inflammation, IL-1β is secreted from activated spinal microglia in a cathepsin B-dependent manner. Furthermore, it has been indicated that IL-1β induces the cyclooxygenase 2 expression in spinal neurons to synthesize prostaglandin E2, which activates protein kinase A. Protein kinase A then inhibits glycine receptors in spinal neurons through phosphorylation of the α3 subunit, leading to disinhibition of spinal neurons. Furthermore, IL-1β increases neuronal excitability through the inhibition of Ca2+-induced K+ channels activated by Ca2+ influx through both N-methyl-D-aspartate receptors and voltage-gated Ca2+ channels in acutely dissociated mouse hippocampal neurons.
Microglial Cathepsin B-Mediated Chronic Nuclear Factor-κB Activation
There is a functional proteolytic relay through cathepsins E and B in activated microglia for activation of nuclear factor-κB (NF-κB), a critical transcription factor in the regulation of the innate immune inflammatory response (Ni et al., 2015). Cathepsin E, a lysosomal aspartic protease, increases the expression of cathepsin B in microglia after hypoxic-ischemic brain damage of neonatal mice through the proteolytic liberation of tumor necrosis factor-related apoptosis-inducing ligand (TRAIL), which in turn induces the NF-κB activation in a proteasome-dependent manner. TRAIL significantly increased cathepsin B in a proteasome-dependent manner. The proteolytic degradation of NF-κB inhibitor α (IκBα), an endogenous negative regulator of NF-κB, promotes nuclear translocations of NF-κB. The ubiquitin-proteasome system is generally involved in the signal-induced IκBα degradation. However, there is accumulating evidence demonstrating that the autophagy machinery is also involved in the IκBα degradation and subsequent NF-κB nuclear translocation following treatment with tumor necrosis factor-α. In microglia, cathepsin B-mediated autophagy machinery promotes the IκBα degradation and subsequent NF-κB nuclear translocation following hypoxic-ischemic brain injury of neonatal mice (Ni et al., 2015). These observations indicate that NF-κB can be maintained in a persistently activated state through the cathepsin B-dependent autophagic degradation of IκBα in microglia (Figure 1B).
Therefore, the cathepsin E/TRAIL system is responsible for increase in cathepsin B expression through proteasome-dependent NF-κB activation during the early phase of hypoxic-ischemic brain damage. After the early NF-κB activation, autophagy may cause a delayed but long-lasting NF-κB activation. It may be concluded that NF-κB can enhance the expression of cathepsin B and beclin 1, a central regulator of autophagy, to promote autophagy during the late phase of hypoxic-ischemic brain damage of neonatal mice, because both cathepsin B and beclin 1 genes possess the NF-κB binding site. Therefore, a proteolytic relay through modulator actions of cathepsins B and E could work as a phenotypic switch in microglia along the M1–M2 phenotypic continuum through the dynamics of NF-κB activity.
Microglial Cathepsin B and Oxidative Responses: Degradation of Pre-mitochondrial Transcription Factor A
A recent report of Elmore et al. (2018) has shown that replacement of aged microglia by pharmacological inhibition of the colony-stimulating factor 1 receptor reverses the cognitive decline in aged mice. The most profound finding in this report is that acute microglial elimination and repopulation in aged mice increased gene expressions that are associated with actin cytoskeleton remodeling and synaptogenesis. According to Liebig’s law of the minimum, well known as a principle developed in agricultural science, one deficient factor can be sufficient to impair the healthy growth of algae. When applied to the brain aging, dysfunction of microglia can be sufficient to impair healthy brain function during aging.
Recently, we have also provided evidence that genetic or pharmacological inhibition of cathepsin B may prevent the age-related deficits in cognition through rejuvenation of aged microglia (Ni et al., 2019). A leakage of cathepsin B into the cytosol may trigger these responses, as there are some reports showing the increased lysosomal membrane permeability and the resultant leakage of lysosomal enzymes in aged rat brain (Nakanishi and Wu, 2009). Furthermore, the immunoreactivity for cathepsin B was markedly increased in the hippocampal microglia of mice during aging (Wu et al., 2017). Our previous studies showed that subunit c of the mitochondrial ATP synthase complex most intensively accumulated in activated microglia of cathepsin D-deficient mice (Nakanishi and Wu, 2009). Subunit c is considered to accumulate only in the lysosomes of long-lived cells. Therefore, microglia must be long-lived cells with very slow turnover. In fact, a recent report using in vivo single cell imaging has revealed the long-live nature of resident microglia that can survive the entire mouse life span (Füger et al., 2017). Thus, the fragility of the endosomal-lysosomal system of especially long-lived microglia is markedly increased during aging.
The lysosomal leakage of cathepsin B plays a critical role in a fall in the mitochondrial membrane potential, leading to mitochondria-derived reactive oxygen species (ROS) generation in culture microglia following treatment with L-leucyl-L-leucine methyl ester (LLOMe), a lysosomotropic agent. After leakage, cathepsin B processes the full-length Bid, a pro-apoptosis BH3-only member of the Bcl-2 family, to truncated Bid (t-Bid), which is essential for permeabilization of the outer mitochondria membrane (Stoka et al., 2016). However, t-Bid was not detected in the soluble extract from cultured microglia after treatment with LLOMe, indicating that the lysosomal leakage of cathepsin B and the resultant generation of ROS were not associated with apoptotic process of microglia (Ni et al., 2019). Therefore, cathepsin B may cleave cytosolic substrates other than Bid, resulting in an increase in oxidative stress and inflammatory responses.
Mitochondrial transcription factor A (TFAM) is synthesized as a full-length precursor protein (pre-TFAM) in the cytosol and then transported into the mitochondrial matrix through interaction with the protein import machinery. Once inside the matrix, presequence is cleaved by the mitochondrial processing peptidase and then mature TFAM binds with mitochondrial DNA (mtDNA). Besides the maintenance of mtDNA number as a transcription factor, TFAM can stabilize mtDNA through the formation of a nucleoid structure within the mitochondria (Nakanishi and Wu, 2009). To stabilize a ratio of TFAM to mtDNA, Lon, the major protease in the mitochondrial matrix, selectively degrades excess TFAM. Moreover, the overexpression of human TFAM in mice significantly inhibits age-dependent accumulation of mtDNA damage in microglia, resulting in a decrease in the ROS generation and the subsequent activation of NF-κB-mediated inflammatory responses and thereby leading to the improvement of age-dependent motor and memory decline (Nakanishi and Wu, 2009). Therefore, pre-TFAM synthesized in the cytosol is a potential target substrate for cathepsin B leaked in the cytosol of microglia during aging. The mean amount of pre-TFAM in cathepsin B-overexpressing cultured microglia after treatment with LLOMe was significantly lower than that in non-treated cells. Furthermore, cathepsin B can degrade human recombinant TFAM even under the neutral pH (Ni et al., 2019). Given these results, it is likely to speculate that cathepsin B leaked in the cytosol of microglia during aging induces the mitochondrial disruption through degradation of TFAM. Subnormal level of TFAM in the mitochondrial matrix may increase mitochondria-derived ROS generation, leading to the increased oxidative damage of mtDNA and the subsequent decline of the complex I activity. Declined complex I activity further increases mitochondria-derived ROS generation and inflammatory responses (Figure 1C).
The role of cathepsin B in the cognitive function remains controversial. Consistent with our findings, pharmacological or genetic inhibition of cathepsin B decreases β-amyloid levels and improves the memory function in mouse models of Alzheimer’s disease (Kindy et al., 2012). On the other hand, the beneficial effects of cathepsin B on cognition have been also reported. The overexpression of cathepsin B in hippocampal neurons using adeno-associated virus serotype 2/1 ameliorates Alzheimer’s disease-like pathologies, including β-amyloidosis and impairments of learning and memory in mice (Embury et al., 2017). Furthermore, Moon et al. (2016) have reported that cathepsin B enhances hippocampal neurogenesis and spatial memory in mice following exercise. Although they showed a significant increase of cathepsin B in tissue derived from whole brain after intravenous injection, the precise mechanism underlying the permeability of peripheral cathepsin B across the blood-brain barrier remains unclear.
Other Roles of Microglial Cathepsin B: Cell Death and β-Amyloid Clearance
Microglia are known to release a number of soluble molecules that can influence neuronal signaling and survival. Kingham and Pocock (2001) found that the culture medium of microglia induced neuronal death following stimulation with CGA. They have demonstrated that cathepsin B plays a central role in neuronal death, because neutralizing anti-cathepsin B antibodies significantly inhibited neuronal death induced by the CGA-treated microglial culture medium. Furthermore, activated BV2 microglial cells released toxic factors that caused significant neuronal death following stimulation with freshly sonicated Aβ42 (Gan et al., 2004). To identify the toxic molecules secreted from Aβ42-stimulated microglia, a large scale expression profiling analysis was conducted using filter-based cDNA arrays made from BV2 cDNA libraries enriched for Aβ42-activated microglial genes. Cathepsin B was finally identified to be one of the 554 genes transcriptionally induced by freshly sonicated Aβ42. Moreover, either siRNA-mediated gene silencing or pharmacological inhibition of cathepsin B completely abolished the neurotoxicity induced by Aβ42-activated BV2 microglial cells. These observations suggest that cathepsin B is responsible for neuronal death induced by Aβ-activated inflammatory responses (Gan et al., 2004). However, additional experiments are necessary to identify the mechanism underlying secretion of cathepsin B and its extracellular substrates.
On the other hand, Wendt et al. (2009) analyzed the role of cathepsin B in neurotoxicity of conditioned medium from lipopolysaccharide-activated microglia using both membrane-permeable and membrane-impermeable cathepsin B inhibitors. They have demonstrated that intracellular cathepsin B triggers the release of neurotoxic factors from lipopolysaccharide-activated microglia, because neurotoxicity of conditioned medium from lipopolysaccharide-activated microglia is inhibited by a membrane-permeable inhibitor of cathepsin B, but not by membrane-impermeable one. In fact, cathepsin B is necessary for the trafficking of tumor necrosis factor-α-containing vesicles to the plasma membrane of macrophages (Ha et al., 2008). Cathepsin B can also induce apoptosis in mouse hepatocytes after leakage into the cytosol to process full-length Bid to t-Bid, which is essential for permeabilization of the outer mitochondria membrane to secrete cytochrome c (Stoka et al., 2016). The release of cathepsins to the cytosol is known as lysosomal membrane permeabilization.
Cathepsin B has specific roles in Aβ degradation and clearance primarily through C-terminal truncation (Wang et al., 2012). Furthermore, cathepsin B ablation significantly increased the plaque deposition and Aβ42 levels in the hippocampus. On the other hand, lentivirus-mediated overexpression of cathepsin B in the hippocampus significantly reduced Aβ deposition in aged human amyloid precursor protein transgenic mice. Acidic pH favors rapid sequential conversion of Aβ42 into Aβ40 and then finally to the less-amyloidogenic Aβ38 (Mueller-Steiner et al., 2006). Reaction rates are slower at neutral pH but not completely diminished. These observations strongly suggest that cathepsin B is secreted from microglia accumulated around the senile plaques, and is involved in the degradation of Aβ42 and reduction of established plaques. Cathepsin B-dependent degradation of Aβ by microglia also contributes to protect neuronal homeostasis, because the persistent deposition of Aβ favors a detrimental inflammation (Cappellano et al., 2013; Comi and Tondo, 2017). Therefore, it is likely to consider that phagocytic clearance of Aβ by microglia may play a significant role in promoting the resolution of chronic neuroinflammation in Alzheimer’s disease.
Conclusion
We have proposed “microglia-aging” hypothesis demonstrating microglia as major drivers for brain aging (Nakanishi and Wu, 2009; Wu and Nakanishi, 2015; Wu et al., 2016). In the aging brain, there is an impairment of electron transfer in some mitochondrial complex, shifting the intracellular redox balance towards a more oxidized state. Microglia with highly branched fine processes are now being considered as active players in the normal healthy brain. This energy consuming behavior may responsible for the accumulation of damaged ROS-hypergenerating mitochondria in microglia. Increased intracellular ROS further activate the redox-sensitive NF-κB to provoke excessive inflammation. Importantly, microglial cathepsin B plays critical roles in both chronic inflammatory response and mitochondria-derived ROS generation in inflammatory brain diseases and brain aging. The growing understanding of the proteolytic system of cathepsin B in microglia could contribute to development of orally active and blood-brain barrier-permeable specific inhibitors for cathepsin B as therapeutic interventions against inflammatory brain diseases and brain aging.
Additional file: Open peer review reports 1 (74.8KB, pdf) and 2 (73.5KB, pdf) .
Footnotes
Conflicts of interest: The author declares no conflicts of interest.
Financial support: This project was founded by JSPS KAKENHI, No. 24390416, JP15H05015, 15K15684 and JP16H01304 (all to HN).
Copyright license agreement: The Copyright License Agreement has been signed by the author before publication.
Plagiarism check: Checked twice by iThenticate.
Peer review: Externally peer reviewed.
Open peer reviewers: Cristoforo Comi, University of Piemonte Orientale, Italy; Maria E. Figueiredo-Pereira, Hunter College of City University of New York, USA.
Funding: This project was founded by JSPS KAKENHI, No. 24390416, JP15H05015, 15K15684 and JP16H01304 (all to HN).
P-Reviewers: Comi C, Figueiredo-Pereira ME; C-Editors: Zhao M, Yu J; T-Editor: Jia Y
References
- 1.Cappellano G, Carecchio M, Fleetwood T, Magistrelli L, Cantello R, Dianzani U, Comi C. Immunity and inflammation in neurodegenerative diseases. Am J Neurodegener Dis. 2013;2:89–107. [PMC free article] [PubMed] [Google Scholar]
- 2.Comi C, Tondo G. Insights into the protective role of immunity in neurodegenerative disease. Neural Regen Res. 2017;12:64–65. doi: 10.4103/1673-5374.198980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Elmore MRP, Hohsfield LA, Kramar EA, Soreq L, Lee RJ, Pham ST, Najafi AR, Spangenberg EE, Wood MA, West BL, Green KN. Replacement of microglia in the aged brain reverses cognitive, synaptic, and neuronal deficits in mice. Aging Cell. 2018;17:e12832. doi: 10.1111/acel.12832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Embury CM, Dyavarshetty B, Lu Y, Wiederin JL, Ciborowski P, Gendelman HE, Kiyota T. Cathepsin B improves β-amyloidosis and learning and memory in models of Alzheimer’s disease. J Neuroimmune Pharmacol. 2017;12:340–352. doi: 10.1007/s11481-016-9721-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Füger P, Hefendehl JK, Veeraraghavalu K, Wendeln AC, Schlosser C, Obermüller U, Wegenast-Braun BM, Neher JJ, Martus P, Kohsaka S, Thunemann M, Feil R, Sisodia SS, Skodras A, Jucker M. Microglia turnover with aging and in an Alzheimer’s model via long-term in vivo single-cell imaging. Nat Neurosci. 2017;20:1371–1376. doi: 10.1038/nn.4631. [DOI] [PubMed] [Google Scholar]
- 6.Gan L, Ye S, Chu A, Anton K, Yi S, Vincent VA, von Schack D, Chin D, Murray J, Lohr S, Patthy L, Gonzalez-Zulueta M, Nikolich K, Urfer R. Identification of cathepsin B as a mediator of neuronal death induced by Aβ-activated microglial cells using a functional genomics approach. J Biol Chem. 2004;279:5565–5572. doi: 10.1074/jbc.M306183200. [DOI] [PubMed] [Google Scholar]
- 7.Ha SD, Martins A, Khazaie K, Han J, Chan BM, Kim SO. Cathepsin B is involved in the trafficking of TNF-α-containing vesicles to the plasma membrane in macrophages. J Immunol. 2008;181:690–697. doi: 10.4049/jimmunol.181.1.690. [DOI] [PubMed] [Google Scholar]
- 8.Kindy MS, Yu J, Zhu H, El-Amouri SS, Hook V, Hook GR. Deletion of the cathepsin B gene improves memory deficits in a transgenic Alzheimer’s disease mouse model expressing AβPP containing the wild-type β-secretase site sequence. J Alzheimers Dis. 2012;29:827–840. doi: 10.3233/JAD-2012-111604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kingham PJ, Pocock JM. Microglial secreted cathepsin B induces neuronal apoptosis. J Neurochem. 2001;76:1475–1484. doi: 10.1046/j.1471-4159.2001.00146.x. [DOI] [PubMed] [Google Scholar]
- 10.Lowry JR, Klegeris A. Emerging roles of microglial cathepsins in neurodegenerative disease. Brain Res Bull. 2018;139:144–156. doi: 10.1016/j.brainresbull.2018.02.014. [DOI] [PubMed] [Google Scholar]
- 11.Moon HY, Becke A, Berron D, Becker B, Sah N, Benoni G, Janke E, Lubejko ST, Greig NH, Mattison JA, Duzel E, van Praag H. Running-induced systemic cathepsin B secretion is associated with memory function. Cell Metab. 2016;24:332–340. doi: 10.1016/j.cmet.2016.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mueller-Steiner S, Zhou Y, Arai H, Roberson ED, Sun B, Chen J, Wang X, Yu G, Esposito L, Mucke L, Gan L. Antiamyloidogenic and neuroprotective functions of cathepsin B: implications for Alzheimer’s disease. Neuron. 2006;51:703–714. doi: 10.1016/j.neuron.2006.07.027. [DOI] [PubMed] [Google Scholar]
- 13.Nakanishi H. Neuronal and microglial cathepsins in aging and age-related diseases. Ageing Res Rev. 2003;2:367–381. doi: 10.1016/s1568-1637(03)00027-8. [DOI] [PubMed] [Google Scholar]
- 14.Nakanishi H, Wu Z. Microglia-aging: roles of microglial lysosome- and mitochondria-derived reactive oxygen species in brain aging. Behav Brain Res. 2009;201:1–7. doi: 10.1016/j.bbr.2009.02.001. [DOI] [PubMed] [Google Scholar]
- 15.Ni J, Wu Z, Peterts C, Yamamoto K, Qing H, Nakanishi H. The critical role of proteolytic relay through cathepsins B and E in the phenotypic change of microglia/macrophage. J Neurosci. 2015;35:12488–12501. doi: 10.1523/JNEUROSCI.1599-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ni J, Wu Z, Stoka V, Meng J, Hayashi Y, Peters C, Qing H, Turk V, Nakanishi H. Increased expression and altered subcellular distribution of cathepsin B in microglia induce cognitive impairment through oxidative stress and inflammatory response in mice. Aging Cell. 2019;18:e12856. doi: 10.1111/acel.12856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stoka V, Turk V, Turk B. Lysosomal cathepsins and their regulation in aging and neurodegeneration. Ageing Res Rev. 2016;32:22–37. doi: 10.1016/j.arr.2016.04.010. [DOI] [PubMed] [Google Scholar]
- 18.Sun L, Wu Z, Hayashi Y, Peters C, Tsuda M, Inoue K, Nakanishi H. Microglial cathepsin B contributes to the initiation of peripheral inflammation-induced chronic pain. J Neurosci. 2012;32:11330–11342. doi: 10.1523/JNEUROSCI.0677-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Terada K, Yamada J, Hayashi Y, Wu Z, Uchiyama Y, Peters C, Nakanishi H. Involvement of cathepsin B in the processing and secretion of interleukin-1β in chromogranin A-stimulated microglia. Glia. 2010;58:114–124. doi: 10.1002/glia.20906. [DOI] [PubMed] [Google Scholar]
- 20.Wang C, Sun B, Zhou Y, Grubb A, Gan L. Cathepsin B degrades amyloid-β in mice expressing wild-type human amyloid precursor protein. J Biol Chem. 2012;287:39834–39841. doi: 10.1074/jbc.M112.371641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wendt W, Schulten R, Stichel CC, Lubbert H. Intra- versus extracellular effects of microglia-derived cysteine proteases in a conditioned medium transfer model. J Neurochem. 2009;110:1931–1941. doi: 10.1111/j.1471-4159.2009.06283.x. [DOI] [PubMed] [Google Scholar]
- 22.Wu Z, Nakanishi H. Lessons from microglia aging for the link between inflammatory bone disorders and Alzheimer’s disease. J Immunol Res. 2015;2015:471342. doi: 10.1155/2015/471342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wu Z, Yu J, Zhu A, Nakanishi H. Nutrients, microglia aging, and brain aging. Oxid Med Cell Longev. 2016;2016:7498528. doi: 10.1155/2016/7498528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wu Z, Ni J, Liu Y, Teeling JL, Takayama F, Collcutt A, Ibbett P, Nakanishi H. Cathepsin B plays a critical role in inducing Alzheimer’s disease-like phenotypes following chronic systemic exposure to lipopolysaccharide from Porphyromonas gingivalis in mice. Brain Behav Immun. 2017;65:350–361. doi: 10.1016/j.bbi.2017.06.002. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.