

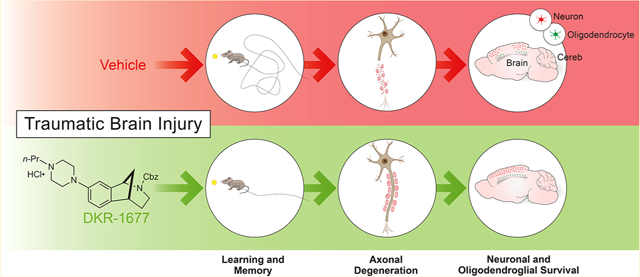
Abstract
Compounds targeting the sigma 2 receptor, which we recently cloned and showed to be identical with transmembrane protein 97 (σ2R/TMEM97), are broadly applicable therapeutic agents currently in clinical trials for imaging in breast cancer and for treatment of Alzheimer’s disease and schizophrenia. These promising applications coupled with our previous observation that the σ2R/TMEM97 modulator SAS-0132 has neuroprotective attributes and improves cognition in wild-type mice suggests that modulating σ2R/TMEM97 may also have therapeutic benefits in other neurodegenerative conditions such as traumatic brain injury (TBI). Herein, we report that DKR-1677, a novel derivative of SAS-0132 with increased affinity and selectivity for σ2R/Tmem97 (Ki = 5.1 nM), is neuroprotective after blast-induced and controlled cortical impact (CCI) TBI in mice. Specifically, we discovered that treatment with DKR-1677 decreases axonal degeneration after blast-induced TBI and enhances survival of cortical neurons and oligodendrocytes after CCI injury. Furthermore, treatment with DKR-1677 preserves cognition in the Morris water maze after blast TBI. Our results support an increasingly broad role for σ2R/Tmem97 modulation in neuroprotection and suggest a new approach for treating patients suffering from TBI.
Keywords: Neuroprotection, traumatic brain injury, blast injury, controlled cortical impact injury, σ2R/TMEM97
Graphical Abstract
INTRODUCTION
The sigma 2 receptor (σ2R), which we recently cloned and identified as the endoplasmic reticulum-resident transmembrane protein 97 (TMEM97),1 is expressed in the central nervous system and in a number of peripheral tissues.2 σ2R/TMEM97 plays a major role in regulating cholesterol trafficking in cellular proliferation and homeostasis3,4 and has long been implicated in cancer.5,6 More recently, σ2R/TMEM97 has also been implicated in neurological disorders and diseases, including Alzheimer’s disease,7–10 schizophrenia,2 alcohol use disorder,11 pain,12 and Niemann-Pick Type C1 (NPC1) disease.4 Indeed, σ2R/TMEM97 modulators are currently in clinical trials for imaging in breast cancer13 and for treating Alzheimer’s disease (AD)14 and schizophrenia.15 Through our ongoing work on design and synthesis of small molecules that bind selectively to σ2R/TMEM97,16–18 we recently discovered a number of novel compounds that exhibit promising attributes in several neurological disorders. For example, SAS-0132 (Figure 1) is efficacious in a transgenic animal model of AD and shows neuroprotective properties in nematodes.7 We also recently discovered that σ2R/TMEM97 may be a promising new target for treating pain11 and alcohol use disorder.12 For example, we showed that UKH-1114 relieves mechanical hypersensitivity in an animal model of neuropathic pain11 and that JVW-1034 reduces alcohol consumption in alcohol-dependent rodents.12 The additional finding that SAS-0132 is neuroprotective and improves cognitive performance in wild-type mice7 prompted us to embark on the syntheses of novel derivatives of SAS-0132 that might be of therapeutic utility in other neuropsychiatric indications related to impaired cognition. Here, we report the evaluation of our lead candidate from this effort, DKR-1677, in two mouse models of traumatic brain injury (TBI): blast-mediated injury and controlled cortical impact (CCI) injury. Both of these preclinical models are established as relevant to human conditions of TBI for which there are currently no established disease-modifying therapeutics.
Figure 1.

Structures of racemic SAS-0132, JVW-1034, UKH-1114, and DKR-1677 and their binding affinities for σ1R and σ2R/Tmem97, which were determined by the Psychoactive Drug Screening Program (PDSP) at Chapel Hill, North Carolina. Average Ki values are shown for each ligand at σ1R and σ2R/Tmem97. Selected pharmacokinetic parameters (intraperitoneal administration) are given for DKR-1677.
RESULTS
Pharmacologic and Pharmacokinetic Characterization of DKR-1677.
Inspired by the anti-inflammatory and procognitive properties of SAS-0132 (Figure 1) in an animal model of Alzheimer’s disease,7 we wondered whether similar compounds might be effective in animal models of TBI. Accordingly, we prepared a number of analogues of SAS-0132 using methods previously developed in our laboratories.16–18 Of the modulators of σ2R/TMEM97 we tested, DKR-1677, which has higher affinity and greater selectivity for σ2R/Tmem97 (rat PC12 cells) than many other compounds, (Figure 1, Table 1) emerged as a promising candidate.18 Notably, DKR-1677 is 45-fold more selective for σ2R/Tmem97 than σ1R, and with the exception of histamine-1 (20-fold) and the alpha-2A adrenergic (68-fold) receptors, it generally exhibits greater than 100-fold selectivity against a panel of more than 50 proteins relevant to the central nervous system (CNS) (Table 1). DKR-1677 also has many properties that align with known CNS drugs,19 making it an ideal tool compound for use in animal experiments to assess a novel approach to treat TBI. In particular, it has a low efflux ratio and rapidly crosses the blood–brain barrier, achieving good brain exposure and brain/plasma ratios (Figure 1). One drawback that might mitigate its future development is its modest half-life (t1/2 = 1.2 h). We also examined DKR-1677 for cytotoxic effects in cultured HEK293T cells under normal and stressed (serum withdrawal) conditions. We observed no difference in cytotoxicity-induced cell death following 24–48 h of 0.125 μM DKR-1677 treatment as compared to vehicle controls in either condition (data not shown).
Table 1.
DKR-1677 Binding Profile at Non-Sigma Receptor Sitesa
| target | Ki (nM) | target | Ki (nM) |
|---|---|---|---|
| 5HT1A | 1187 | Beta2 | >10 000 |
| 5HT1B | >10 000 | Beta3 | >10 000 |
| 5HT1D | 1913 | BZP rat brain | 4767 |
| 5HT1e | 7741 | D1 | >10 000 |
| 5HT2A | 1274 | D2 | >3253 |
| 5HT2B | 868 | D3 | 4426 |
| 5HT2C | 2138 | D4 | >10 000 |
| 5HT3 | >10 000 | D5 | 4888 |
| 5HT5a | 3238 | DAT | 2080 |
| 5HT6 | 2510 | DOR | >10 000 |
| 5HT7 | 766 | GabaA | >10 000 |
| A2B2 | b | H1 | 100 |
| A2B4 | b | H2 | 1766 |
| A3B2 | b | H3 | 644 |
| A3B4 | b | H4 | >10 000 |
| A4B2 | b | hERG | 695 |
| A4B2c | b | KOR | 8515 |
| A4B4 | b | M1 | 4062 |
| A7 | b | M2 | 4326 |
| A7c | b | M3 | 1114 |
| Alpha1a | 2667 | M4 | 1634 |
| Alpha1b | >10 000 | M5 | 1395 |
| Alpha1d | 1905 | MOR | 8798 |
| Alpha2a | 340 | NET | 3845 |
| Alpha2b | 1799 | NMDA | 10 000 |
| Alpha2c | 539 | PBR | >10 000 |
| Beta1 | 6282 | SERT | 8630 |
Receptor binding assays were performed by the Psychoactive Drug Screening Program (PDSP)at Chapel Hill, North Carolina. The assay protocol book can be accessed free of charge at: https://pdspdb.unc.edu/pdspWeb/content/PDSP%20Protocols%20II%202013-03-28.pdf. Values represented are single Ki determination.
<50% inhibition of radioligand binding at 10 μM.
Sourced from rodent brain.
DKR-1677 Protects Mice from Cognitive Impairment after Blast-Mediated TBI.
To assess whether DKR-1677 might have therapeutic benefits in TBI, we first turned to the blast injury model of TBI in which a blast wave is propagated through the mouse’s head. As our initial measure of efficacy, we evaluated hippocampal-dependent learning and memory in the Morris water maze task after TBI as a function of daily intraperitoneal (IP) administration of escalating doses of DKR-1677 (0.3, 1, 3, or 10 mg/kg/day) or vehicle, with treatment initiated 30 min after injury and continuing throughout behavioral testing until the day of sacrifice. Consistent with our previous observations that blast TBI does not affect learning in a similar hippocampal-dependent behavioral paradigm, the Barnes maze,20 mice in all TBI groups learned the task equally well compared with the sham-injury vehicle-treated group over a 5-day training period that was initiated 7 days post injury (dpi) (Figure 2a). However, a profound deficit was seen in the probe test of memory at 12 days post injury (dpi), as TBI animals treated with vehicle crossed the previous platform location only about 20% of the time as compared to the sham-injury group receiving vehicle (Figure 2b). Administration of daily 0.3 mg/kg DKR-1677 to blast TBI mice modestly increased the number of platform crossings without a statistically significant difference from the TBI group receiving vehicle (Figure 2b). Higher doses of DKR-1677, however, did achieve a protective effect with statistical significance relative to TBI vehicle animals (Figure 2b). Specifically, IP administration of 1 and 3 mg/kg/day of DKR-1677 to TBI mice elicited a number of platform crossings equivalent to the blast-mediated TBI vehicle group and statistically greater than the TBI vehicle group. The 10 mg/kg/day DKR-1677 group also showed an apparent trend of increased crossings over the sham-injury vehicle group as well (Figure 2b). This effect was recapitulated with a dose–response effect when the less stringent, but more traditional, measure of probe test memory was employed: % time in quadrant area where the platform had been located. Here, all four treatment groups showed statistically significant improvement in memory over the blast-mediated TBI group (Figure 2c). It is important to note that swim speed did not vary in any of the groups, which indicates that general activity levels of the animals did not affect their performance in this task (sham-injury vehicle = 2.7 ± 0.4 m/second (m/s), TBI-veh = 2.4 ± 0.9 m/s, TBI-DKR-1677 0.3 mg = 2.2 ± 0.4 m/s, 1 mg = 3.0 ± 0.5 m/s, 3 mg = 3.1 ± 0.5 m/s and 10 mg = 3.2 ± 0.6 m/s).
Figure 2.

Treatment with DKR-1677 protects mice from memory deficit after blast-mediated TBI (b-TBI). Morris water maze was used to assess hippocampal-dependent learning and memory after blast-mediated TBI. (a) Latency to find the hidden platform was used as a learning measure, and all groups showed equal ability to learn the task. (b) Platform crossing and (c) % of time spent in the target quadrant were used to measure memory in the probe test. Sham-veh (n = 16), b-TBI-veh (n = 11), b-TBI-DKR-1677 0.3 mg (n = 19), 1 mg (n = 19), 3 mg (n = 19), and 10 mg (n = 16). Values are presented as mean ± SEM. Significance was determined by one-way ANOVA with multiple comparisons. *p < 0.05, **p < 0.01, **** p < 0.0001.
DKR-1677 Protects Mice from Neurodegeneration after Blast-Mediated TBI.
We previously showed that impaired performance in hippocampal-dependent learning and memory tasks after blast-mediated TBI is correlated with axonal degeneration.20,21 To evaluate axonal degeneration in the three main groups (sham-injury vehicle, TBI vehicle, and TBI + the highest effective dose of DKR-1677), we applied silver staining to identify degenerating axons in the brain tissue of injured and sham-injury animals. Animals were sacrificed at 2 weeks following injury, after completion of behavioral testing. As shown in Figure 3, prominent axonal degeneration was visualized in the hippocampus and cortex of TBI vehicle animals relative to sham-injury vehicle animals. This degeneration was blocked in animals treated with 10 mg/kg/day of DKR-1677 for the entire postinjury interval until sacrifice (Figure 3). Representative pictures are shown in Figure 3a, and quantification of the signal with optical densitometry (Figure 3b,c) confirmed the statistical significance of this effect. Thus, the protective efficacy of DKR-1677 for learning and memory in a hippocampal-dependent task correlated anatomically with protection of animals from neurodegeneration after TBI.
Figure 3.

Treatment with DKR-1677 (10 mg/kg/day) protects mice from neurodegeneration after Blast-Mediated TBI (b-TBI). Silver staining was used to determine neurodegeneration. (a) Representative pictures of the silver stained hippocampus (HPC) and cortex (CTX) of the sham-veh, b-TBI-veh, and b-TBI-DKR-1677 (10 mg/kg/day) groups. Silver staining quantification was performed measuring the % area covered by the staining (b and c). Scale bar = 100 μm in larger box and 10 μM in smaller box, as shown. Higher magnified inset is of the white-outlined box in the lower magnification image. Values are presented as mean ± SEM. Significance was determined by one-way ANOVA with multiple comparisons (n = 5/group). **p < 0.01, *** p < 0.001.
DKR-1677 Protects Mice from Neuronal Cell Loss after CCI-Mediated TBI.
Controlled cortical impact (CCI) injury is a severe brain injury that leads to acute neuron and oligodendrocyte cell death in the cortex and underlying white matter tracts. To assess whether DKR-1677 was also neuroprotective in a CCI injury model of TBI, we examined whether treatment with DKR-1677, initiated at 1 h postinjury, resulted in improved neuron and oligodendrocyte survival after CCI injury. Accordingly, DKR-1677 (3 mg/kg/day, IP) was administered once per day for 7 days followed by blinded, nonbiased stereological counts of cortical neurons in the injury penumbra as well as oligodendrocyte counts in the cortex, corpus callosum, and external capsule of mice expressing green fluorescent protein (GFP) under the proteolipid (PLP) oligodendrocyte promoter (ie. PLP-GFP mice). We observed a significant reduction in cortical neurons identified by anti-NeuN immunoreactivity (Figure 4a) as well as a trend toward reduced numbers of cortical GFP-positive oligodendrocytes in vehicle treated CCI injured mice (Figure 4b). However, no significant differences were observed between sham and CCI injury mice treated with 3 mg/kg/day DKR-1677 (Figure 4a and b). We observed a similar effect with oligodendrocytes residing in all three white matter regions quantified, in which the external capsule residing below the injury epicenter showed a significant reduction in vehicle-treated mice (Figure 4c). Representative low magnification confocal images show differences in tissue sparing between vehicle- (Figure 4g) and DKR-1677- treated (Figure 4j) CCI-injured mice as compared to vehicle-treated sham controls (Figure 4d), where DKR-1677 administration led to a potential increase in tissue sparing. Higher magnification images of the penumbra and corpus callosum reveal differences in neuron (Figures 4e, h, and k) and oligodendrocyte (Figures 4f, i, and l) cell density mediated by DKR-1677 treatment. We also observed greater numbers of surviving oligodendrocytes in DKR-1677 treated mice, which were not significantly different from either sham or vehicle-treated CCI injury groups (Figure 4c). Together, our findings suggest DKR-1677 elicits pro-survival effects in both neurons and oligodendrocytes after CCI injury.
Figure 4.

Administration of DKR-1677 (3 mg/kg/day) reduces neuronal and oligodendrocyte cell loss at 7 days following CCI injury. Cell quantification was performed using blinded, unbiased stereology for (a) neurons in the cortical penumbra, (b) oligodendrocytes in the cortical penumbra, and (c) underlying white matter tract. Values are presented as mean ± SEM. Significance was determined by one-way ANOVA with multiple comparisons (n = 6–9/group). *p < 0.05, ***p < 0.001. Representative confocal images of vehicle-treated sham (d–f), vehicle-treated CCI injury (g–i), and DKR-1677-treated CCI injury (j–l) showing low-magnification images (d, g, and j) and high-magnification images (e, f, h, i, k, and l) of the yellow-boxed area where anti-NeuN (red) and PLP-GFP (green) fluorescence was used for quantification. Dashed line represents divisions in quantification between the corpus callosum and external capsules in panel g. CC is corpus callosum; EC is external capsule; epi-EC is external capsule under injury epicenter; IEC is external capsule lateral to injury epicenter; mPEN is medial penumbra; lPEN is lateral penumbra; Hipp is Hippocampus. Scale bar = 500 μM in d, g, and j and 20 μM in e, f, h, i, k, and l).
DISCUSSION
TBI represents a leading cause of death and disability worldwide, with an estimated 1.7 million new cases reported every year in the United States.22 Patients who survive TBI frequently suffer chronically progressive physical and neurologic impairment, including pathologic and cognitive changes resembling Alzheimer’s disease as well as post-traumatic stress disorder, mood disorders, and suicide.23 Tragically, there are currently no FDA-approved treatments for TBI that protect the brain from neurodegeneration.24 To address this critical unmet need, we examined efficacy of a novel σ2R/TMEM97 modulator in animal models of TBI.
As described previously, we discovered a variety of structurally distinct σ2R/TMEM97 modulators that are efficacious in various animal models of neuropsychiatric disorders, including AD, neuropathic pain, and alcohol use disorder.7,11,12 Based upon this body of work from our group and others,8,9 we surmised that DKR-1677, which is closely related to SAS-0132, would be an attractive candidate to test in animal models of TBI. In particular, we queried whether the protective effects associated with σ2R/TMEM97 modulation that we had previously reported in transgenic AD models of neurodegeneration would be recapitulated in acute injury in wild-type mice.
A major challenge in studying TBI and discovering new avenues for treating patients is the wide heterogeneity of injury. For example, primary injury in TBI can occur with any combination of insults, including direct impact, penetration, pressure wave exposure, and acceleration/deceleration forces. Each of these can induce a wide variation of consequences such as diffuse axonal injury and cerebral contusion, edema, and hematoma. Therefore, to establish the translational potential of any putative pharmacologic treatment for TBI, efficacy must be shown in an assortment of experimental models. With this in mind, we evaluated the efficacy of DKR-1677 in two different models of TBI: blast and CCI injury.
The major primary event in blast injury is diffuse and widespread axonal degeneration, leading to neuropsychiatric dysfunction and nerve cell death, which can be modeled in the laboratory with the overpressure chamber.20,21,25–30 Here, we observed dose-dependent protective efficacy for wild-type mice after blast injury in the memory probe test of the Morris water maze. When daily treatment was initiated 30 min after injury, doses of 1, 3, and 10 mg/kg of DKR-1677 all resulted in preserved memory in this task in the most stringent measure of platform crossings. In terms of the less stringent, but more traditional, measure of percentage of time spent in the target quadrant where the platform was previously located prior to the probe test, all tested doses (0.3, 1, 3, and 10 mg/kg) of DKR-1677 were efficacious. This effect was seen in the absence of any effect of the test compound on motor behavior, as measured by swim speed.
We also examined neurodegeneration in the 10 mg/kg dose of DKR-1677 to determine whether efficacy in protecting behavior correlated with protection from neurodegeneration through measuring axonal degeneration via silver staining. Indeed, vehicle-treated animals show substantially greater silver staining in the hippocampus and cortex than sham-injured vehicle-treated animals, and the extent of silver staining in blast-injured animals treated with DKR-1677 was significantly less than the vehicle-treated blast injury group and indistinguishable from sham injury vehicle-treated mice. Hence, DKR-1677 protects against axonal degeneration after blast injury.
In contrast to a pressure wave traveling through the brain, CCI injury provides a model of concussive impact to the brain through application of a device that rapidly accelerates a rod to impact the exposed brain surface. This system produces localized cell death and other histological and neurobehavioral alterations comparable to those observed clinically.31 After severe brain injury, cell death results from a number of distinct mechanisms, including alterations in cellular homeostasis that can lead to necrosis and apoptosis. Implementing a CCI injury force of 4 m/s, we observed significant loss of neurons and oligodendrocytes in the cortex and/or underlying white matter tracts, which was partially mitigated when animals were treated daily with 3 mg/kg/day DKR-1677.
The cellular mechanism whereby DKR-1677 regulates pro-survival effects after blast and CCI TBI is presently unknown. However, given the affinity of DKR-1677 for σ2R/TMEM97 relative to the σ1R (45-fold) and other targets in the CNS (Table 1), we posit that these beneficial effects are mediated primarily by modulation of σ2R/TMEM97. However, neuroprotective effects arising from binding to other receptors cannot be excluded. The role of σ2R/TMEM97 in cholesterol trafficking and metabolism provides some initial insights to potential mechanisms.3,4,32 Specifically, TMEM97 was identified as a functional regulator of cholesterol homeostasis that is localized to endo/lysosomal compartments where it binds the cholesterol transporter-regulating protein Niemann-Pick1 (NPC1).3 More recent studies have now shown that TMEM97 may contribute to regulating NPC1 availability in cells, which in turn alters intracellular cholesterol trafficking and storage.4 As cholesterol is the most abundant CNS sterol,33 its requirement to modulate membrane fluidity and permeability is critical to cell survival and function. Unfortunately, there is a significant gap in our understanding of the cellular and subcellular regulation of cholesterol after TBI. The protective effects of DKR-1677 on both axon stability and survival would support the possibility that this compound functions to stabilize sterol homeostasis in the plasma membrane and/or other intracellular compartments such as the lysosome or mitochondria. Additional studies are needed to further examine the effects of DKR-1677 on cholesterol metabolism after TBI. It is known that the majority of neuronal and oligodendrocyte cell death occurs within the first week following TBI.34–36 Accordingly, we administered DKR-1677 once daily over this period of time; however, the relatively short half-life (t1/2) of 1.2 h may have limited our ability to adequately block cell death. Future studies are required to determine a therapeutic profile for compound efficacy. It will also be important to determine whether DKR-1677 might help improve cognition in other forms of acute TBI or in conditions of chronic TBI, such as chronic traumatic encephalopathy (CTE).
In conclusion, there is critical need for a therapeutic agent that mitigates the pathological processes that ensue following TBI, which could thereby lead to improved functional outcomes for patients. Collectively, our studies suggest that σ2R/TMEM97 is involved in regulating pathological processes associated with TBI, suggesting that pharmacological modulation of this receptor may represent a new treatment paradigm. We are developing improved σ2R/TMEM97 ligands to further explore their therapeutic potential in TBI and other neurological conditions.
METHODS
Animals.
For blast injury experiments, all animal procedures were performed in accordance with the protocol approved by the University of Iowa Institutional Animal Care and Use Committee. Male, seven-week-old, C57BL/6J (Stock No: 000664) mice were obtained from Jackson Laboratories. Animals were grouped house with water and food provided ad libitum in a 12 h light/dark cycle and temperature-controlled conditions. For CCI injury experiments, male PLP-EGFP transgenic mice (a gift from Dr. Wendy Macklin)37 were bred on a C57Bl/6J background. All animal procedures were performed using male PLP-EGFP mice between the age of 2–4 months. Animals were kept under normal 12 h light/dark cycle conditions, and all procedures were approved by the University of Miami Animal Care and Use Committee (IACUC). Following surgical procedures, animals were housed singly without environmental enrichment.
Blast-Mediated TBI.
Eight-week old male C57/Bl6J mice were anesthetized with ketamine (100 mg/kg) and xylazine (10 mg/kg) via intraperitoneal (IP) injection and settled in an enclosed blast chamber (183 cm long and 66 cm wide). The blast chamber contained a pressurized compartment with a 15 cm opening covered with a mylar membrane and an unpressurized side with a restraint area positioned 18 cm away from the membrane. The head of the anesthetized mouse was freely moving, and the body was protected with a foam tube and positioned inside a metal shield in the restraint area. The pressurized compartment of the chamber was filled with air until the mylar membrane ruptured at 20 psi, which generated a blast wave that impacted the left side of the mouse head. The mouse’s body was shielded from the blast wave by a metal tube. The intensity of the blast wave was 149.8 ± 2.09 kPa, and the duration of total pressure (blast wave + wind gust) was ~10–15 ms. The sham-injury group was anesthetized under the same conditions but not subjected to blast injury.
CCI-Mediated TBI.
Mice were anesthetized with ketamine (100 mg/kg) and xylazine (10 mg/kg) via intraperitoneal (IP) injection and placed on a heating pad to maintain body temperature. The animal’s head was shaved and then placed on a stereotaxic frame where an incision was made through the skin, exposing the skull. For CCI injured mice, a ~5 mm diameter craniectomy was made over the right parietal cortex (bregma: −2.0 mm; lateral −2.5 mm), leaving the dura intact. Mice were then subjected to a moderate CCI injury with a piston velocity of 4.0 m/s and depth of 0.55 mm using an eCCI-6.0 device (Custom Design & Fabrication, Virginia Commonwealth, VA, United States), which contains a pressurized stainless steel piston to deliver the impact. Sham controls underwent an identical surgical procedure with the absence of the craniotomy and injury. The incision was closed using 4–0 REDISILK black braided silk nonabsorbable sutures (Ethicon, Inc., Piscataway, NJ, United States), and mice were placed in a clean, single housed cage on a heating pad. For hydration and analgesia, animals were administered 1 mL of a 100:1, lactated ringer: 0.3 mg/mL buprenorphine, solution subcutaneously 1 h postinjury (hpi) as well as 2 and 3 days postinjury (dpi).
DKR-1677 Compound Formulation.
DKR-1677 was dissolved in DMSO (10 mg/mL) and diluted to the desired concentration with a final formulation of 10% DMSO (Fischer Scientific), 10% Kolliphor (Sigma-Aldrich), and 80% D5W (5% dextrose in water, pH 7.2). The compound was administered to mice by IP injection.
HEK293T Cell Survival Assay.
HEK293T cells were grown and passaged in Falcon polystyrene plates in 10% fetal bovine serum (FBS) and Dulbecco’s modified Eagle’s medium (DMEM) (0.4% penicillin/streptomycin). Transformed HEK293T cells were seeded in 24 well plates at a density of 80 000 cells per well. Following 24 or 48 h, the media was either replaced with fresh 10% FBS DMEM or replaced with DMEM without serum (serum withdrawal) for an additional 24 h in the presence of 0.125 μM DKR1677 or vehicle control. Cell survival was determined by Trypan Blue uptake followed by automated cell counting (Bio-Rad TC20 Automated Cell Counter).
Morris Water Maze.
Morris water maze testing was conducted in a 128 cm diameter tank filled with 19 cm of water (room temperature) and mixed with white nontoxic paint to reduce platform visibility. Four different cues (shapes and color), equally spaced, were placed inside the tank for orientation of a submerged 9 cm diameter platform. Each animal was subjected to 4 days of training, consisting of four trials per day. During the probe test on day 5, the platform was removed and each animal was tested for 1 min. Any-Maze video tracking software (Stoelting Co.) was used to measure latency to find the hidden platform through the training and platform crossing, percentage of time spent in the target quadrant and average speed in the probe day.
Silver Staining of Axon Degeneration.
Mice were anesthetized and euthanized by transcardial perfusion with 1× phosphate-buffered saline (PBS: 137 mM NaCl, 2.7 mM KCl, 8 mM Na2HPO4, 2 mM KH2PO4) followed by 4% paraformaldehyde in PBS at pH 7.4. Brains were collected and postfixed in the same fixative solution used for perfusion overnight at 4 °C. Brains were then immersed in 30% sucrose in PBS for 72 h at 4 °C for cryoprotection. Brains were cut coronally (40 μM sections), processed and stained with FD NeuroSilver Kit (FD NeuroTechnologies) by FD NeuroTechnologies Inc. (Columbia, MD, United States). Brightfield images were acquired using Zeiss Axiolmager.M2 microscope, keeping light intensity and exposition time constant. The % of area covered by silver staining (black staining) was quantified using the National Institutes of Health (NIH) ImageJ software (Bethesda, MD, United States) with the plugin of the color deconvolution method as previously described.38
Neuron and Oligodendrocyte Counts and Anti-NeuN Immunohistochemistry.
At 7 dpi, mice were anesthetized with a ketamine/xylazine cocktail and transcardially perfused with 0.01 M PBS (pH 7.4) followed by 4% paraformaldehyde (PFA, pH 7.4). Brains were harvested and postfixed overnight in 4% PFA at 4 °C. The following day, brains were transferred to 30% sucrose in PBS. Brains were then placed in molds, mounted in Tissue-Tek Cryo-OCT (Fisher Scientific, United States), and serially cryo-sectioned at 30 μm thickness. Neurons were detected by immunoreactivity with anti-NeuN antibodies (1:300 Cell Signaling cat. no. 24307) and oligodendrocytes by enhanced green fluorescent protein somal fluorescence. Cell quantification was blinded and performed using Micro Bright-Field Stereo Investigator software using the Optical Fractionator Probe (3 sections per animal, 25 sections spaced apart) (MBF Bioscience, Williston, VT, United States). The medial cortex, lateral cortex, corpus callosum, external capsule below injury epicenter, and lateral external capsule were contoured under 4× magnification. The counting frame for all contours was 75:75 μm with an SRS Grid Layout of 175:175 μm. Once contoured, the cell counts were performed at 63× in immersion oil. Counts with a Gundersen Coefficient of error value of <0.1 were deemed reliable and included in the study.
Statistical Analysis.
Values are presented as mean ± SEM. One-way ANOVA followed by Bonferroni’s multiple comparison test was performed using GraphPad Prism version 7.00 for Mac.
ACKNOWLEDGMENTS
We thank Yan Shi at the Miami Project to Cure Paralysis Imaging Core for assistance acquiring representative confocal images as well as X. P. Huang of the Psychoactive Drug Screening Program (PDSP), which is directed by Bryan L. Roth at the University of North Carolina at Chapel Hill (US National Institute of Mental Health Contract HHSN-271-2013-00017-C) for receptor binding assays for DKR-1677. S.F.M. is also grateful to Dr. Richard J. Pederson (Materia, Inc.) for catalyst support and to CEM Corporation for microwave instrumentation support.
Funding
This work was supported by funds to A.A.P. from the Brockman Medical Research Foundation, from an anonymous donor to the Mary Alice Smith Fund for Neuropsychiatry Research, from the Titan Neurology Research Fund, and from the Department of Veterans Affairs Merit Review 1IO1BX002444. The contents of this manuscript do not represent the views of the U.S. Department of Veterans Affairs or the U.S. Government. This work was also supported by the Robert A. Welch Foundation (F-0652) to S.F.M., the Dell Medical School’s Texas Health Catalyst program to S.F.M. and J.J.S., NIH/NINDS NS098740 to D.J.L., and the Miami Project to Cure Paralysis to D.J.L.
Footnotes
The authors declare the following competing financial interest(s): S.F.M., J.J.S., and T.R.H. report being coinventors on pending patent applications related to work described in this article. S.F.M. and J.J.S. also report being cofounders of NuvoNuro, Inc.
REFERENCES
- (1).Alon A, Schmidt HR, Wood MD, Sahn JJ, Martin SF, and Kruse AC (2017) Identification of the gene that codes for the σ2 receptor. Proc. Natl. Acad. Sci. U. S. A 114, 7160–7165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Guo L, and Zhen X (2015) Sigma-2 receptor ligands: neurobiological effects. Curr. Med. Chem 22, 989–1003. [DOI] [PubMed] [Google Scholar]
- (3).Bartz F, Kern L, Erz D, Zhu M, Gilbert D, Meinhof T, Wirkner U, Erfle H, Muckenthaler M, Pepperkok R, and Runz H (2009) Identification of cholesterol-regulating genes by targeted RNAi screening. Cell Metab. 10, 63–75. [DOI] [PubMed] [Google Scholar]
- (4).Ebrahimi-Fakhari D, Wahlster L, Bartz F, WerenbeckUeding J, Praggastis M, Zhang J, Joggerst-Thomalla B, Theiss S, Grimm D, Ory DS, and Runz H (2016) Reduction of TMEM97 increases NPC1 protein levels and restores cholesterol trafficking in Niemann-pick type C1 disease cells. Hum. Mol. Genet 25, 3588–3599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Mach RH, Zeng C, and Hawkins WG (2013) The sigma2 receptor: a novel protein for the imaging and treatment of cancer. J. Med. Chem 56, 7137–7160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Huang YS, Lu HL, Zhang LJ, and Wu Z (2014) Sigma-2 receptor ligands and their perspectives in cancer diagnosis and therapy. Med. Res. Rev 34, 532–566. [DOI] [PubMed] [Google Scholar]
- (7).Yi B, Sahn JJ, Ardestani PM, Evans AK, Scott LL, Chan JZ, Iyer S, Crisp A, Zuniga G, Pierce JT, Martin SF, and Shamloo M (2017) Small molecule modulator of sigma 2 receptor is neuroprotective and reduces cognitive deficits and neuroinflammation in experimental models of Alzheimer’s disease. J. Neurochem 140, 561–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Izzo NJ, Staniszewski A, To L, Fa M, Teich AF, Saeed F, Wostein H, Walko T 3rd, Vaswani A, Wardius M, Syed Z, Ravenscroft J, Mozzoni K, Silky C, Rehak C, Yurko R, Finn P, Look G, Rishton G, Safferstein H, Miller M, Johanson C, Stopa E, Windisch M, Hutter-Paier B, Shamloo M, Arancio O, LeVine H 3rd, and Catalano SM (2014) Alzheimer’s therapeutics targeting amyloid beta 1–42 oligomers I: Abeta 42 oligomer binding to specific neuronal receptors is displaced by drug candidates that improve cognitive deficits. PLoS One 9, No. e111898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Izzo NJ, Xu J, Zeng C, Kirk MJ, Mozzoni K, Silky C, Rehak C, Yurko R, Look G, Rishton G, Safferstein H, Cruchaga C, Goate A, Cahill MA, Arancio O, Mach RH, Craven R, Head E, LeVine H 3rd, Spires-Jones TL, and Catalano SM (2014) Alzheimer’s therapeutics targeting amyloid beta 1–42 oligomers II: Sigma-2/PGRMC1 receptors mediate Abeta 42 oligomer binding and synaptotoxicity. PLoS One 9, No. e111899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Martin SF, Sahn JJ, Scott L, and Pierce-Shimomura JT (2015) Compounds and methods for treating cancer, neurological disorders, ethanol withdrawal, anxiety, depression, and neuropathic pain. January 22, 2015, WO2015009742 A2.
- (11).Sahn JJ, Mejia GL, Ray PR, Martin SF, and Price TJ (2017) Sigma 2 receptor/Tmem97 agonists produce long lasting antineuropathic pain effects in mice. ACS Chem. Neurosci 8, 1801–1811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Scott LL, Sahn JJ, Ferragud A, Yen RC, Satarasinghe PN, Wood MD, Hodges TR, Shi T, Prakash BA, Friese KM, Shen A, Pierce JT, Sabino V, and Martin SF (2018) Novel small molecule modulator of σ2R/Tmem97 reduces alcohol withdrawal-induced behaviors. Neuropsychopharmacology 43, 1867–1875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Abramson Cancer Center of the University of Pennsylvania (2014) Imaging of in vivo sigma-2 receptor expression with 18F ISO-1 positron emission tomography (PET/CT) in primary breast cancer. Clinical trial NCT02762110. https://clinicaltrials.gov/ct2/show/NCT02762110?term=NCT02762110&rank=1 (accessed November 2, 2016).
- (14).Cognition Therapeutics (2016) Clinical trial of CT1812 in mild to moderate Alzheimer’s Disease. Clinical trial NCT02907567. https://clinicaltrials.gov/ct2/show/NCT02907567?term=NCT02907567&rank=1 (accessed November 2, 2016).
- (15).Minerva Neurosciences, Inc. (2014) Study to evaluate the efficacy, safety, and tolerability of MIN-101 in patients with negative symptoms of schizophrenia. Clinical trial 2014-004878-42. Available at https://www.clinicaltrialsregister.eu/ctr-search/trial/2014-00487842/LV (accessed November 2, 2016).
- (16).Sahn JJ, and Martin SF (2012) Expedient synthesis of norbenzomorphan library via multicomponent assembly process coupled with ring-closing reactions. ACS Comb. Sci 14, 496–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Sahn JJ, Hodges TR, Chan JZ, and Martin SF (2016) Norbenzomorphan framework as a novel scaffold for generating sigma 2 receptor/PGRMC1 subtype-selective ligands. ChemMedChem 11, 556–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Sahn JJ, Hodges TR, Chan JZ, and Martin SF (2017) Norbenzomorphan scaffold: Chemical tool for modulating sigma receptor-subtype selectivity. ACS Med. Chem. Lett 8, 455–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Wager TT, Chandrasekaran RY, Hou X, Troutman MD, Verhoest PR, Villalobos A, and Will Y (2010) Defining desirable central nervous system drug space through the alignment of molecular properties, in vitro ADME, and safety attributes. ACS Chem. Neurosci 1, 420–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Yin TC, Britt JK, De Jesús-Cortés H, Lu Y, Genova RM, Khan MZ, Voorhees JR, Shao J, Katzman AC, Huntington PJ, Wassink C, McDaniel L, Newell EA, Dutca LM, Naidoo J, Cui H, Bassuk AG, Harper MM, McKnight SL, Ready JM, and Pieper AA (2014) P7C3 neuroprotective chemicals block axonal degeneration and preserve function after traumatic brain injury. Cell Rep. 8, 1731–1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Yin TC, Voorhees JR, Genova RM, Davis KC, Madison AM, Britt JK, Cintrón-Pérez CJ, McDaniel L, Harper MM, and Pieper AA (2016) Acute axonal degeneration drives development of cognitive, motor, and visual deficits after blast-mediated traumatic brain injury in mice. eNeuro 3, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Faul M, and Coronado V (2015) Epidemiology of traumatic brain injury. Handb. Clin. Neurol 127, 3–13. [DOI] [PubMed] [Google Scholar]
- (23).Madsen T, Erlangsen A, Orlovska S, Mofaddy R, Nordentoft M, and Benros ME (2018) Association between traumatic brain injury and risk of suicide. JAMA 320, 580–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Roozenbeek B, Lingsma HF, and Maas AI (2012) New considerations in the design of clinical trials for traumatic brain injury. Clin. Invest. (London, U. K.) 2, 153–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Dutca LM, Stasheff SF, Hedberg-Buenz A, Rudd DS, Batra N, Blodi FR, Yorek MS, Yin T, Shankar M, Herlein JA, Naidoo J, Morlock L, Williams N, Kardon RH, Anderson MG, Pieper AA, and Harper MM (2014) Invest. Ophthalmol. Vis. Sci 55, 8330–8341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Hoge CW, McGurk D, Thomas JL, Cox AL, Engel CC, and Castro CA (2008) Mild traumatic brain injury in U.S. soldiers returning from Iraq. N. Engl. J. Med 358, 453–463. [DOI] [PubMed] [Google Scholar]
- (27).Wolf SJ, Bebarta VS, Bonnett CJ, Pons PT, and Cantrill SV (2009) Blast Injuries. Lancet 374, 405–415. [DOI] [PubMed] [Google Scholar]
- (28).Goldstein LE, Fisher AM, Tagge CA, Zhang XL, Velisek L, Sullivan JA, Upreti C, Kracht JM, Ericsson M, Wojnarowicz MW, Goletiani CJ, Maglakelidze GM, Casey N, Moncaster JA, Minaeva O, Moir RD, Nowinski CJ, Stern RA, Cantu RC, Geiling J, Blusztajn JK, Wolozin BL, Ikezu T, Stein TD, Budson AE, Kowall NW, Chargin D, Sharon A, Saman S, Hall GF, Moss WC, Cleveland RO, Tanzi RE, Stanton PK, and McKee AC (2012) Chronic traumatic encephalopathy in blast-exposed military veterans and a blast neurotrauma mouse model. Sci. Transl. Med 16, 134ra60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Magnuson J, Leonessa F, and Ling GS (2012) Neuropathology of explosive blast traumatic brain injury. Curr. Neurol. Neurosci. Rep 12, 570–579. [DOI] [PubMed] [Google Scholar]
- (30).Nakagawa A, Manley GT, Gean AD, Ohtani K, Armonda R, Tsukamoto A, Yamamoto H, Takayama K, and Tominaga T (2011) Mechanisms of primary blast-induced traumatic brain injury: insights from shock-wave research. J. Neurotrauma 28, 1101–1119. [DOI] [PubMed] [Google Scholar]
- (31).Osier N, and Dixon CE (2017) Mini review of controlled cortical impact: a well-suited device for concussion research. Brain Sci. 7, No. 88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Sanchez-Pulido L, and Ponting CP (2014) TM6F2 and MAC30, new enzyme homologs in sterol metabolism and common metabolic disease. Front. Genet 5, 439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Petrov AM, Kasimov MR, and Zefirov AL (2016) Brain cholesterol metabolism and its defects: linkage to neurodegenerative diseases and synaptic dysfunction. Acta Naturae 8, 58–73. [PMC free article] [PubMed] [Google Scholar]
- (34).Raghupathi R (2004) Cell death mechanisms following traumatic brain injury. Brain Pathol. 14, 215–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Sullivan GM, Mierzwa AJ, Kijpaisalratana N, Tang H, Wang Y, Song SK, Selwyn R, and Armstrong RC (2013) Oligodendrocyte lineage and subventricular zone response to traumatic axonal injury in the corpus callosum. J. Neuropathol. Exp. Neurol 72, 1106–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Flygt J, Gumucio A, Ingelsson M, Skoglund K, Holm J, Alafuzoff I, and Marklund N (2016) Human traumatic brain injury results in oligodendrocyte death and increases the number of oligodendrocyte progenitor cells. J. Neuropathol. Exp. Neurol 75, 503–15. [DOI] [PubMed] [Google Scholar]
- (37).Fuss B, Mallon B, Phan T, Ohlemeyer C, Kirchhoff F, Nishiyama A, and Macklin WB (2000) Purification and analysis of in vivo-differentiated oligodendrocytes expressing the green fluorescent protein. Dev. Biol 218, 259–274. [DOI] [PubMed] [Google Scholar]
- (38).Ruifrok AC, and Johnston DA (2001) Quantification of histochemical staining by color deconvolution. Anal. Quant. Cytol. Histol 23, 291–299. [PubMed] [Google Scholar]